
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward high-selectivity CO2 photoelectroreduction: mechanistic foundations, recent advances and challenges
Guosheng
Zhou
 a,
Zhenzhen
Wang
a,
Junjie
Gong
a,
Shijie
Shen
*a and
Wenwu
Zhong
*ab
a,
Zhenzhen
Wang
a,
Junjie
Gong
a,
Shijie
Shen
*a and
Wenwu
Zhong
*ab
aZhejiang Key Laboratory for Island Green Energy and New Materials, Taizhou University, Jiaojiang, 318000, Zhejiang, P. R. China. E-mail: shensj@tzc.edu.cn; zhongww@tzc.edu.cn
bSchool of Chemistry and Chemical Engineering, Shaoxing University, Shaoxing 312000, P. R. China
First published on 12th June 2025
Abstract
Photoelectrochemical carbon dioxide reduction reaction (PEC CO2RR) is a promising strategy for converting CO2 into high-value chemicals that contribute to carbon neutrality. However, CO2 reduction is often accompanied by various competitive reaction pathways in the actual reaction process, which may generate a variety of products. The selective regulation of different products not only directly affects the yield and separation cost of the target product, but also influences the energy efficiency and economic feasibility of the whole process. Improving product selectivity is essential for increasing product yield and understanding the reaction mechanism. This review systematically summarizes recent advances and challenges in achieving high selectivity in PEC CO2RR. First, the basic concept and principle of the PEC CO2RR are summarized. Next, the key factors affecting product selectivity are discussed, including catalyst design (catalyst type, modification, composition, and morphology), reaction conditions (applied voltage, light intensity and wavelength, reaction temperature, and electrolyte type) and reactor design (photoelectrode area, synergistic oxidation effect, geometric structure, and gas diffusion electrode). In addition, kinetic and thermodynamic aspects such as the CO2 adsorption model, band gap structure, and reaction free energy are also explored. Then, the research progress over the past five years on different products is described in detail, focusing on the current status and challenges in the study of C1 products and C2 products. Subsequently, the primary factors leading to the failure of PEC CO2RR are summarized, and various cooperative strategies are introduced to achieve long-term stability in product selectivity. Finally, the challenges and future directions for developing PEC CO2RR systems with enhanced selectivity are introduced. In particular, the importance of innovative catalyst design, reaction stability, reaction environment optimization, advanced equipment structure and reaction mechanism analysis for promoting PEC CO2RR in industrial applications is emphasized.
 Guosheng Zhou | Guosheng Zhou received his PhD degree from Jiangsu University, PR China in 2024. He currently serves as Lecturer in School of Materials Science and Engineering, Taizhou University. His research interests focus on photo/electro-catalytic CO2 reduction. |
 Zhenzhen Wang | Zhenzhen Wang received her PhD degree from Soochow University, PR China in 2023. She currently serves as Lecturer in School of Materials Science and Engineering, Taizhou University. Her research interests focus on photothermal water evaporation, photocatalytic and electrocatalytic water splitting. |
 Junjie Gong | Junjie Gong received her M.S. degree from Zhejiang University, PR China in 2021. She currently serves as Assistant Experimenter in School of Materials Science and Engineering, Taizhou University. Her research interests focus on the preparation, modification, and application of novel transition metal compounds for electrocatalytic water splitting. |
 Shijie Shen | Shijie Shen received his PhD degree from Institute of Physics, Chinese Academy of Sciences, PR China in 2016. He currently serves as Professor in School of Materials Science and Engineering, Taizhou University. His research interests focus on preparation and modification of novel transition metal compounds and their application in electrocatalysis. |
 Wenwu Zhong | Wenwu Zhong received his PhD degree from Beihang University, PR China in 2011. He currently serves as Professor in School of Chemistry and Chemical Engineering, Shaoxing University. His research interests include development of electrocatalytic materials and battery electrode materials. |
1 Introduction
With the rapid advancement of industrialization, the excessive consumption of fossil fuels has led to a dramatic increase in carbon dioxide (CO2) emissions, posing a serious threat to the stability of the global climate system.1–5 As one of the major greenhouse gases, the concentration of CO2 in the atmosphere has risen from 280 ppm before the Industrial Revolution to an annual average of 419 ppm in 2022.6 To address these challenges, scientists are working to develop efficient and environmentally friendly technologies to capture and convert CO2 to achieve carbon neutrality. Among these technological pathways, the photoelectrochemical CO2 reduction reaction (PEC CO2RR) is regarded as one of the ideal solutions for realizing “artificial photosynthesis” due to its ability to synergistically drive the conversion of CO2 into high-value-added carbon-based fuels by directly utilizing both solar energy and electrical energy.7–10 The technology is expected to not only reduce the concentration of CO2 in the atmosphere, but also convert renewable energy sources into chemical energy storage, providing a dual solution to address the energy crisis and the imbalance of the carbon cycle.11,12 Since 2009, the number of publications and citations on PEC CO2RR has increased year by year, indicating the progress in this field (Fig. 1a). However, the current technology is still in its infancy and faces many challenges, especially the problem of product selectivity control.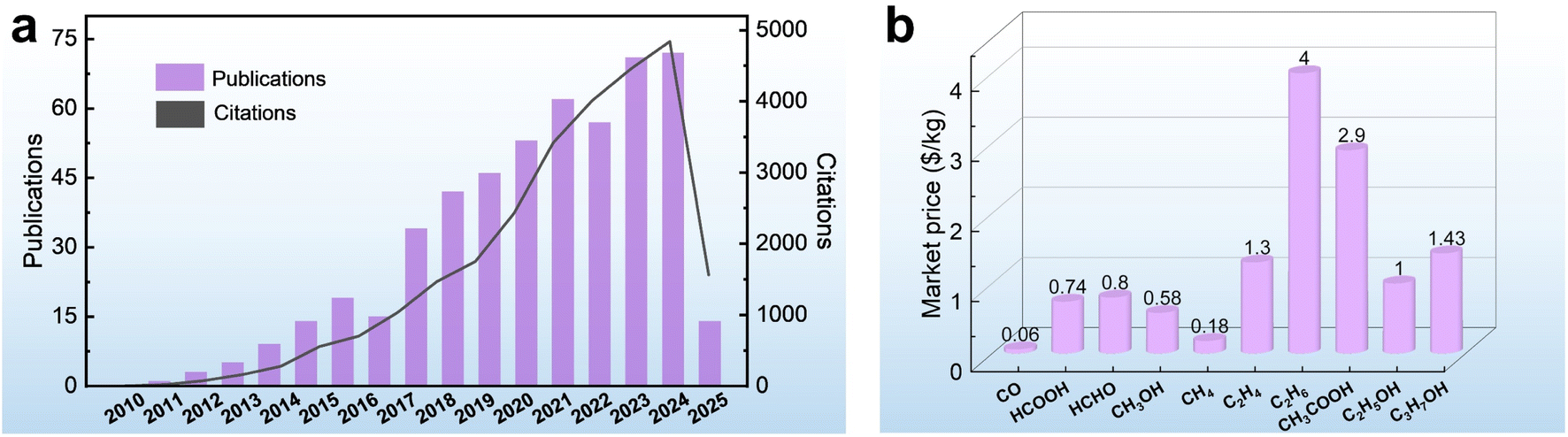 | ||
| Fig. 1 (a) The number of publications and citations related to PEC CO2RR, according to the data collected from Web of Science (the key word is “Photoelectrocatalytic CO2 Reduction”, and the data are collected up to May 2025). (b) The market prices of different products.14 | ||
The CO2RR usually involve multiple competing reduction pathways that lead to the formation of different products such as carbon monoxide (CO), formic acid (HCOOH), methanol (CH3OH), methane (CH4), acetic acid (CH3COOH), ethylene (C2H4), ethanol (C2H5OH), etc.13 There are significant differences in the market value of these products (Fig. 1b).14 For example, the price of HCOOH is 12.3 times that of CO. The differing economic values of the products are prompting researchers to focus on how to modulate the reaction pathway through catalysts to achieve highly selective product generation. From the perspective of industrial applications, a single product not only reduces the complexity of subsequent separation and purification, but also effectively reduces the cost of post-processing, making the process of product extraction and conversion more economically viable.15,16 For example, in CO2 reduction systems, the products exist as mixed gases (e.g., CO/CH4) or liquid mixtures (e.g., CH3OH/C2H5OH); additional energy-intensive processes such as distillation, adsorption, or membrane separation are required, resulting in a decrease in efficiency and economy. Therefore, achieving highly selective generation of target products is the core challenge for improving the efficiency of CO2 resource utilization, both in terms of product value and process feasibility.
The multiple barriers to selective modulation are due to the high stability of the CO2 molecule and the complexity of the multi-electron transfer process. The linear symmetric structure of CO2 and the high bond energy of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (approximately 750 kJ mol−1) necessitate overcoming significant thermodynamic barriers for its activation.7,17,18 In the photoelectrocatalytic process, CO2 first forms key intermediates (e.g., *CO2, *COOH, or *OCHO) via proton-coupled electron transfer (PCET), and then undergoes multi-step reduction and protonation reactions to generate the final product.19 In this process, the energy barriers of different reaction pathways differ by only tens to hundreds of millielectronvolts, and small energy fluctuations can lead to the diversion of reaction pathways.20 For example, the generation of C2+ products requires the adsorption of at least two CO intermediates, their migration and C–C coupling, followed by successive hydrogenation steps. The activation energy of each reaction step can be a rate-determining step. In addition, the HER is an important competing reaction that competes with the generation of the target product for protons and electrons, further increasing the difficulty of regulating product selectivity.21 For example, on the surface of copper-based catalysts, the Faraday efficiency (FE) of C2H4 can reach 61% when the applied potential is −0.74 V vs. RHE, but at the same time the hydrogen evolution reaction accounts for nearly 30%.22 The prevalence of such competing reactions makes selective modulation a delicate balance between thermodynamic driving force and kinetics.
O bond (approximately 750 kJ mol−1) necessitate overcoming significant thermodynamic barriers for its activation.7,17,18 In the photoelectrocatalytic process, CO2 first forms key intermediates (e.g., *CO2, *COOH, or *OCHO) via proton-coupled electron transfer (PCET), and then undergoes multi-step reduction and protonation reactions to generate the final product.19 In this process, the energy barriers of different reaction pathways differ by only tens to hundreds of millielectronvolts, and small energy fluctuations can lead to the diversion of reaction pathways.20 For example, the generation of C2+ products requires the adsorption of at least two CO intermediates, their migration and C–C coupling, followed by successive hydrogenation steps. The activation energy of each reaction step can be a rate-determining step. In addition, the HER is an important competing reaction that competes with the generation of the target product for protons and electrons, further increasing the difficulty of regulating product selectivity.21 For example, on the surface of copper-based catalysts, the Faraday efficiency (FE) of C2H4 can reach 61% when the applied potential is −0.74 V vs. RHE, but at the same time the hydrogen evolution reaction accounts for nearly 30%.22 The prevalence of such competing reactions makes selective modulation a delicate balance between thermodynamic driving force and kinetics.
In recent years, researchers have made remarkable progress in material design, reactor optimization and mechanistic investigation related to the regulation strategy of product selectivity. First of all, in the design of photocatalysts, it is necessary to accurately construct a suitable catalytic system based on the chemical properties of the target product.23 Through band engineering, the semiconductor bandgap structure can be adjusted to match the solar spectrum, and the electronic density distribution can be optimized by combining defect engineering, elemental doping, and surface modification.24,25 Furthermore, the design of heterojunction interfaces promotes the directional migration of photogenerated carriers, while the use of co-catalysts can enhance the stabilization of specific intermediates through localized surface plasmon resonance effects or selective adsorption properties.26–28 In addition, nanostructural morphology modification (e.g., porous frameworks, nanoflowers and core–shells) can increase active site density and improve reactant mass transfer efficiency.29–31 Secondly, in the optimization of reaction conditions, temperature, pressure, light intensity, light wavelength, and electrolyte type and parameters are systematically regulated to enhance the desired reaction pathways and inhibit the side reaction pathways by balancing the thermodynamic and kinetic parameters of the reaction, such as adjusting the rate of proton supply or decreasing the competitive activity of hydrogen precipitation to enhance the selectivity of the target products.32 Reaction device design innovation can be achieved by optimizing the photoelectrode structure, mass transfer channels and reactor configuration, anodic oxidation reaction design, etc., to achieve a synergistic enhancement of light energy absorption efficiency and reaction kinetics; it can also be combined with a visible light-responsive photocathode microbial system to achieve the effective immobilization of CO2 and highly selective generation of reduction products.32 A deep understanding of the reaction mechanism is also indispensable for guiding the experimental design. By combining in situ characterization techniques to track the dynamic evolution of intermediates in real time and leveraging quantum chemical calculations to analyse the electronic structure of active sites and the energy barrier distribution of reaction pathways, the microscopic mechanisms of selective control are revealed at the atomic level. This provides theoretical guidance for catalyst design and reaction condition optimization, driving the transition of PEC CO2RR from empirical trial-and-error to rational design.
Therefore, this review aims to systematically elucidate the factors influencing the product selectivity in PEC CO2RR and summarize the recent research progress on typical products, with a view to providing references and insights for future research. First, this review introduces the fundamental concepts of PEC CO2RR and the factors affecting product selectivity, including catalyst design, reaction condition design, and device structure design. The product selectivity of PEC CO2RR is also explored from both kinetic and thermodynamic perspectives. Subsequently, this review provides a detailed description of the research progress over the past five years on different products, with a focus on C1 products (such as CO, HCOOH, CH3OH, and CH4) and C2+ products (such as C2H4, C2H5OH, and CH3COOH), highlighting the current research status and challenges. Based on the above progress, we summarized the representative studies of PEC CO2RR products with the highest FE, as shown in Fig. 2. Finally, this review provides an outlook on the future development directions of PEC CO2RR technology, emphasizes the importance of improving product selectivity and efficiency, and highlights the areas that need to be further explored. Through systematic sorting and summarization, this review aims to provide valuable references for researchers and promote continuous innovation and advancement in PEC CO2RR technology.
 | ||
| Fig. 2 The highest FEs of different PEC CO2RR products in the past five years. Reproduced with permission from ref. 22, 107, 114, 205, 222, 230, and 254. Copyright 2022, Wiley-VCH GmbH. Copyright 2024, The Royal Society of Chemistry. Copyright 2021, Elsevier B.V. All rights reserved. Copyright 2022, Elsevier B.V. All rights reserved. Copyright 2024, Springer Nature. Copyright 2024, Springer Nature. Copyright 2023, Wiley-VCH GmbH. | ||
2 Basic principles of PEC CO2RR
2.1 Fundamentals of PEC CO2RR
PEC CO2RR is a technology that combines light and electrical energy to drive the CO2 reduction reaction. It is based on the synergistic effect of photocatalysis and electrocatalysis. Photoelectrocatalysis generates photogenerated electrons and holes through the absorption of light energy by the photocatalyst. These photogenerated carriers are subsequently separated and transported through an external circuit to participate in the reduction and oxidation reactions, respectively. In the CO2 reduction process, photogenerated electrons are transported to the reduction reaction site, where they promote the reduction of CO2. The process utilizes light energy and electrical energy, improving the reaction efficiency and product selectivity. However, photocatalytic technology has significant drawbacks: firstly, the high carrier complexation rate and the tendency of electrons and holes to recombine before migrating to the catalyst surface, resulting in low energy utilization; secondly, the reaction kinetics are slow.33 CO2 molecules exhibit strong chemical inertness and require high energy for activation, while the reduction ability of photogenerated electrons is limited, making it difficult to drive multi-electron transfer reactions (such as generating C2+ products). In contrast, electrocatalytic technology drives the reaction through an externally applied electric field, directly reducing CO2 at the catalytic sites on the electrode surface. Theoretically, applying an electrocatalytic field can regulate the electron transfer rate and the reaction path orientation to produce specific products. However, its limitations are also prominent: firstly, CO2 reduction requires energy input to overcome high thermodynamic barriers and easily triggers the competitive HER, resulting in the reduction of FE. Secondly, CO2 reduction involves multi-electron and proton transfer processes, in which the difference in adsorption strength of different intermediates (such as *COOH, *CO, etc.) makes it difficult to control product selectivity. In addition, the electrode material is prone to corrosion or poisoning under long-term high voltage or acidic/alkaline conditions, affecting the sustainability of the reaction.34,35
PEC CO2RR technology combines the advantages of photocatalysis and electrocatalysis and overcomes their limitations. Firstly, photoelectrocatalysis utilizes the synergistic effect of light and electric energy, significantly improving the reaction efficiency and energy conversion efficiency. Secondly, by designing the structure and composition of the photocatalyst, the reaction pathway can be effectively regulated to achieve high selectivity for the target products.36 In addition, the utilization of solar energy as the driving energy source is sustainable. Similar to photosynthesis in nature, PEC CO2RR technology is regarded as an artificial simulation of photosynthesis (Fig. 3).37 In photosynthesis, photosynthetic system II in chloroplasts absorbs photons and excites electron transitions to produce high-energy electrons and holes. Electrons drive the oxidation of water through the transfer chain (generating O2 and H2O), while generating ATP and NADPH (energy carrier). In the Calvin cycle, the energy provided by NADPH and ATP fixes CO2 to glucose, a process catalyzed by enzymes. Semiconductor materials (e.g. TiO2 and perovskite) absorb photons to produce electron–hole pairs, and photogenerated electrons are injected into the conduction band while holes participate in oxidation reactions (e.g., water decomposition). The electrocatalytic phase is similar to the Calvin cycle of carbon fixation. By regulating the reaction path through an external circuit, the external electric field drives the electrons to migrate to the catalytic sites, reducing CO2 to produce the target product. Compared with photosynthesis, photocatalysis has better controllability and flexibility: by designing the structure of the photocatalyst and tuning the reaction conditions, precise regulation of the reaction path and product can be achieved. In addition, photocatalytic technology can work in a wider spectral range, improving the utilization of light energy. Overall, the PEC CO2RR technology overcomes the limitations of traditional methods and shows both potential for efficiency and sustainability. By further optimizing the design and performance of the photocatalysts, photoelectrocatalysis is expected to achieve large-scale CO2 resource utilization in the future, providing an important solution to address global climate change and energy crisis.
 | ||
| Fig. 3 Schematics of (a) natural photosynthesis, and (b) PEC CO2RR. Reproduced with permission from ref. 37. Copyright 2022, Wiley-VCH GmbH. | ||
The types of devices for PEC CO2RR systems can be categorized into four main types based on the combination of optical and electrical drives (Fig. 4): photocathode-driven systems, photoanode driven systems, photoanode and photocathode-co-driven systems, and hybrid photovoltaic systems that combine electrodes with photovoltaic cells.38 Each of these four device types has its own design characteristics and functionalities suited to different experimental needs and scenarios. A photocathode-driven system is a photocatalytic system based on a photocathode, which mainly reduces CO2 to high value-added chemicals or fuels. In this system, the photocathode is usually made of a p-type semiconductor material with photocatalytic capability, and requires the assistance of an external power source to provide additional energy to overcome the high activation energy of the reaction. The advantage of this system is that it can regulate the selectivity of the product by adjusting the applied potential. It is suitable for studying the performance of photocatalysts in reduction reactions. In contrast, the photoanode-driven system uses the photoanode to decompose water to produce oxygen, while achieving CO2 reduction reaction on the photocathode. This system does not require an external power source because the photogenerated charge generated by the photoanode drives the reaction itself. Photoanodes are usually made of n-type semiconductor materials. This device is characterized by efficient use of light energy, and can achieve CO2 reduction without external energy input, which is suitable for the research of self-driven photocatalytic systems. The photo-anode and photocathode co-driven systems combines a photo-anode and a photocathode to enable simultaneous oxidation and reduction reactions. In this system, the photoanode and photocathode are responsible for the oxidation of water and the reduction of CO2, respectively, while achieving internal charge balance through the charge transport network. The advantage of this system is that it can realize the efficient utilization of light energy, and can effectively improve the overall reaction efficiency by simultaneously conducting oxidation and reduction reactions. The co-driven system is suitable for studying the performance of photocatalysts in complex reaction systems and exploring their potential for practical applications. Hybrid light systems combining electrodes with photovoltaic cells are an innovative type of device. In this system, a conventional electrode is combined with a photovoltaic cell, and the photogenerated voltage generated by the photovoltaic cell is utilized to drive the reduction reaction of CO2. Control of the reaction pathway is achieved through the regulation of the external circuit. The advantage of this system is that it can combine the adjustment of photo-generated voltage with an external power supply to achieve the dual optimization of product selectivity and reaction efficiency. The hybrid light system is suitable for exploring the synergistic effect of photoelectrocatalysis with other energy technologies and for providing new ideas for the practical application of photoelectrocatalysis. Overall, these four device types show their respective advantages and features in the CO2 reduction reaction through different driving modes and electrode designs. Based on specific experimental requirements and application scenarios, choosing an appropriate device type can effectively enhance the efficiency and product selectivity of PEC CO2RR, thus providing important technical support for realizing the resourceful utilization of CO2 and sustainable development.
 | ||
| Fig. 4 Device types of photocatalytic CO2 reduction systems. (a) The photocathode drive system, (b) the photoanode drive system, (c) the photoanode and photocathode co-drive system, and (d) the hybrid optical system combining the electrode with the photovoltaic cell. | ||
Currently, a large number of studies employ H-type reactors for testing. This reactor features a separated, static design in which the anode and cathode chambers are divided by an ion exchange membrane, offering the advantages of a simple structure and low cost. However, mass transfer in this system relies primarily on natural diffusion or stirring, which limits mass transfer efficiency.7 Furthermore, the low solubility and diffusion rate of CO2 often result in low current density and faradaic efficiency. In recent research, the PEC flow cell reactor equipped with a gas diffusion electrode (GDE) has emerged as a highly promising PEC CO2RR reactor with significant potential for future development.9 Its design aims to integrate photoelectrocatalysis with fluid dynamics to achieve efficient gas–liquid–solid three-phase interfacial reactions, overcoming the limitations of CO2 solubility and enhancing CO2 mass transfer. As shown in Fig. 5, a typical PEC flow cell can be regarded as an evolution of the photoanode-driven system, in which the photoanode decomposes water to produce oxygen while CO2 reduction occurs at the photocathode.9,39 Unlike conventional setups, the flow cell reactor operates in a continuous flow regime, integrating gas diffusion electrodes and microchannel designs. By continuously supplying CO2 and removing products via forced flow, it significantly enhances mass transfer efficiency and reaction rates, achieving current densities ranging from tens to hundreds of mA cm−2, high CO2 utilization, and high selectivity for C2+ products, thereby demonstrating excellent scalability and industrial potential. The key lies in depositing catalyst particles onto a porous gas diffusion layer composed of carbon particles or nanofibers and a carbon paper substrate to fabricate the gas diffusion electrode. This electrode effectively transports CO2 gas while isolating the electrolyte, forming an efficient gas–liquid–solid three-phase reaction interface that enhances CO2 mass transfer, enables direct power delivery to the electrode, and increases current density (Fig. 5c). Additionally, the compact design reduces the distance between the two electrodes, thereby minimizing the system's internal resistance. However, the integration of multiple components, the stability of the gas diffusion electrode, and the collection of products increase the complexity of system design, maintenance, and operation.
 | ||
| Fig. 5 Schematic of a PEC flow cell for CO2RR using a GDE. (a) Schematic of PEC flow cell for CO2RR using a photoanode or a PV-integrated oxidation catalyst and a GDE. (b) Requirements of a photoanode/oxidation catalyst. (c) The GDE consists of a CO2RR catalyst coated on a porous gas-diffusion layer. Reproduced with permission from ref. 9. Copyright 2023, Elsevier B.V. All rights reserved. | ||
Three-electrode and two-electrode systems are often used in the experimental design of PEC CO2RR. These two systems are suitable for different experimental conditions and targets. The three-electrode system consists of a working electrode (WE), a counter electrode (CE) and a reference electrode (RE). The WE is usually a conductive substrate loaded with a photocatalyst, such as FTO glass or Ti sheet. The CE uses a material with great conductivity, such as platinum, graphite or molybdenum. The RE is used to measure the potential and the commonly used materials include Ag/AgCl, Hg/Hg2SO4, and Hg/HgO. The three-electrode system is usually used in conjunction with a potentiostat to perform experiments at a constant potential. The reaction path is precisely controlled by adjusting the potential to achieve the selective control of the product. It is suitable for the situations where electrochemical measurements are required, such as current–voltage curves and electrochemical impedance spectroscopy analysis. The advantage of the three-electrode system is that it can accurately control the potential to study the electrochemical behavior of the photocatalyst. However, it is also more complex, requiring additional reference electrodes, and its long-term stability may affect the experimental results. In contrast, the two-electrode system consists of a photoanode or photocathode and a counter electrode. The photoanode is responsible for generating and transporting photogenerated electrons while the photocathode is responsible for generating and transporting photogenerated holes. Platinum or graphite is usually used as the counter electrode material. The two-electrode system is commonly used in photovoltaic devices or bias devices; the former directly uses the photogenerated voltage to drive the reaction, while the latter controls the reaction path by applying a bias voltage through an external power supply. The advantage of the two-electrode system is that the device is simple and suitable for studying the performance of the photocatalyst without external potential regulation, but its potential cannot be accurately controlled, which may affect the product selectivity.
In the PEC CO2RR device, the ion exchange membrane (IEM) plays a role in isolating the reaction solution and maintaining the charge balance.40,41 Different types of ion exchange membranes are suitable for different electrolytes. They are broadly classified into cation exchange membranes (CEMs), and anion exchange membranes (AEMs). CEMs allow cations to pass through and are suitable for acidic or neutral electrolytes, such as H2SO4, KHCO3, Na2SO4 or NaHCO3 electrolyte.42–45 AEMs allow anions to pass through and are suitable for alkaline or neutral electrolytes such as KOH, NaOH or KHCO3 electrolyte. In addition, there are solid electrolyte membranes made of metal oxides or inorganic materials, which are suitable for experimental conditions with high temperature or high stability requirements. The reasonable selection of electrolyte and ion exchange membrane types can effectively improve the efficiency and product selectivity of CO2 reduction, and provide important support for the efficient utilization of CO2.
The four basic parameters in the PEC CO2RR are as follows: photoelectrode materials, catalysts, electrolytes and external power sources. They play different roles in the reaction and have a close relationship with each other, determining the efficiency, selectivity and stability of the reaction. Photoelectrode materials are the core elements of photoelectrocatalytic reactions. They are usually made of semiconductor materials and are responsible for absorbing light energy and generating photogenerated electrons and holes. These photogenerated carriers need to be transported to the catalyst site through the surface of the material, where they then participate in the chemical reaction. The choice of photoelectrode material is very important, because it not only determines the absorption range and conversion efficiency of light energy, but also affects the separation and transmission ability of photogenerated carriers. Common photoelectrode materials include TiO2, WO3, BiVO4, Si, Fe2O3 and ZnO, which are widely used due to their excellent light absorption, stability and conductivity.46–48 Catalysts play indispensable roles in photoelectrocatalytic reactions, mainly by providing active catalytic sites and reducing the activation energy of the reaction to accelerate the reduction process of CO2. Catalysts can significantly improve the reaction rate and regulate the type of products, which is of great significance for the generation of highly selective products. The choice of catalysts depends on the reaction conditions and the expected products.49–51 In addition, the stability and durability of the catalyst are also the key factors affecting the long-term performance of the reaction. As a medium for ion transport, the electrolyte not only provides a suitable chemical environment for the reaction, but also maintains the charge balance of the entire system.52 The selection of electrolyte has an important influence on the reaction process, because it may participate in the intermediate steps of the reaction and affect the selectivity of the product. Therefore, depending on the different target products, the reasonable selection of electrolyte type and concentration is important for optimizing reaction performance. The role of external power supply in the photoelectrocatalytic system is mainly to adjust the potential of the system to ensure that the reaction is carried out under ideal conditions. By applying an appropriate voltage, the energy barrier of the reaction can be overcome, and the separation and transport of photogenerated carriers can be promoted, thereby improving the reaction efficiency. In addition, the external power supply can also be used to adjust the working mode of the device, such as switching between the self-driving mode under illumination conditions and the auxiliary driving mode under bias conditions. Proper setting of external power supply parameters, such as applied voltage and current, is essential to optimize reaction performance and achieve highly selective product generation. In summary, the performance and product selectivity of the PEC CO2RR are influenced by the photoelectrode materials, catalysts, electrolytes and external power sources. By optimizing the performance of each parameter and coordinating their interactions, the reaction efficiency and product selectivity can be significantly improved, laying a solid foundation for realizing the efficient utilization of CO2 as a resource.
The product selectivity, FE and product photocurrent density in PEC CO2 reduction are the key parameters for evaluating the reaction performance.39 Their definitions and formulae are as follows:
The product selectivity (PS) refers to the ratio of the yield of a specific reduction product (A) to that of all possible products. Usually, in some studies, only a portion of reduction products is detected to calculate the relative selectivity. Its calculation formula is shown in eqn (1):
 | (1) |
FE is used to measure how much electrical charge is used to drive the CO2 reduction reaction. Specifically, FE represents the ratio of the number of electrons used to generate a specific reduction product to the total number of electrons involved in the electrochemical reaction. FE is an important indicator for measuring the selectivity and energy efficiency of the reaction. In the PEC CO2RR, a higher FE means that more electrical energy is used for the generation of the desired reduction product rather than being consumed by other side reactions (such as water decomposition). An ideal PEC CO2RR system should have a high FE, especially in the case of highly selective generation of the target product. Its calculation formula is shown in eqn (2):
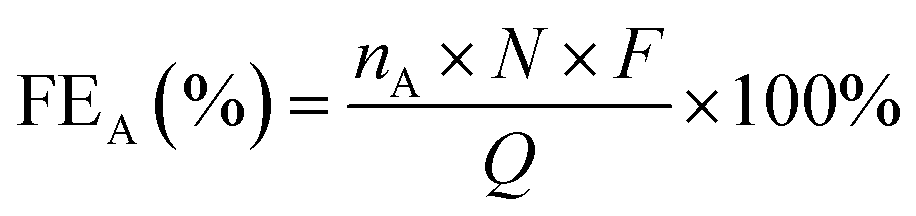 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1) and Q (C) is the total amount of charge.
485 C mol−1) and Q (C) is the total amount of charge.
The photocurrent density of the product (jA) refers to the current density of the product generated on the electrode per unit area. It indicates the current required per unit area on the catalyst surface to generate the target product under a specific voltage. A high photocurrent density usually means that the catalyst can more effectively convert light energy into electrical energy and drive the CO2 reduction reaction under given light conditions, thereby increasing the generation rate of the specific product. Its calculation formula is shown in eqn (3):
 | (3) |
2.2 Factors affecting product selectivity
There are many parameters that affect the selectivity of products in the PEC CO2RR. In this review, we divide them into three aspects: catalyst design, reaction conditions, and reactor design, as shown in Fig. 6. | ||
| Fig. 6 The factors affecting product selectivity: catalyst design, reaction conditions, and reactor design. | ||
The choice of catalyst type is one of the important factors determining the selectivity of CO2 reduction products. Different metal catalysts exhibit varying selectivity in PEC CO2RR, mainly due to their unique electronic structures and active sites.29,51,53–55 White et al.56 summarized the product selectivity for CO2 reduction of typical elemental electrocatalysts that are usually attached to semiconductors as cocatalysts (Fig. 7). Precious metals such as Au and Ag are ideal for producing CO due to their moderate adsorption of CO intermediates.26 By providing a suitable d-band center, they can effectively stabilize CO intermediates and reduce their generation energy barrier.21 In addition, the weak adsorption capacity of these metals for hydrogen intermediates inhibits the HER, improving the selectivity for CO. In contrast, Cu-based catalysts are more suitable for the generation of multi-carbon products, such as (C2H4 and C2H5OH) because Cu-based catalysts can promote C–C coupling reaction, which is a key step in the formation of C2+ products.57 Bi, Sn, In, etc. are promising materials for the generation of HCOOH.58 Their electronic structure exhibit a moderate binding energy with the key intermediate *OCHO for the reduction of CO2 to formic acid, which not only promotes the stability of the intermediate, but also avoids the difficulty of desorption caused by excessive adsorption, thereby reducing the reaction energy barrier. Other transition metals such as Fe, Co, Rh, etc. tend to produce a large number of hydrogen by-products during the reaction process due to their low HER overpotential.59
 | ||
| Fig. 7 The main reduction products formed on metal and carbon electrodes in a CO2-saturated aqueous electrolyte, as represented across the periodic table. Reproduced with permission from ref. 24. Copyright 2015, American Chemical Society. | ||
The modification strategy of the catalyst greatly affects the selectivity of CO2 reduction products. Modification methods such as doping, vacancy, alloying, and crystal plane control can significantly change the electronic structure and surface properties of the catalyst, thereby regulating the adsorption and reaction path of the intermediate.54,58,60–62 For example, doping metal or non-metal elements can introduce new active sites or adjust the electron density of the original sites to improve the selectivity of specific products. When CO2 is reduced using 6 wt% Re-doped CuO/TiO2-NTs, the main products are methanol and ethanol. With a lower Re doping level of 1 wt%, methanol becomes the predominant product. In the absence of Re doping, formaldehyde is the primary product.63 In alloying, Bi-based materials can enhance the electronic structure of the Bi surface by introducing elements such as In or Sn, thus promoting the reduction of CO2 to HCOOH. In addition, crystal surface control also has an important effect on product selectivity. The exposure of different crystal faces can change the surface electronic structure and the active sites of the catalyst, which can selectively enhance the adsorption and conversion of some intermediates.64 For example, the (102) crystal plane of CuSe@BiOI heterojunctions can significantly improve the reduction selectivity of CO2 to HCOOH, while the (110) crystal plane of Cu2O is more conducive to the formation of ethanol.65 In summary, the electronic structure and surface characteristics of the catalyst can be regulated by modification, so as to achieve high selectivity for specific products.
The design of composite catalysts is another important strategy to improve the selectivity of CO2 reduction. The combination of different types of catalysts can achieve synergistic effects, optimizing electron transfer and enhancing the stability of intermediates.66–68 Metal particle loading can effectively improve the electronic structure of the catalyst to improve the photoelectric separation effect, and provide active sites for the reaction. The composite of Ag and Au nanoparticles with TiO2 can enhance the selectivity of CO through interfacial charge transfer, and the LSPR effect can further improve the photocurrent density.26,69,70 In addition, heterojunction structures are often constructed to enhance the separation and transport efficiency of photogenerated carriers.71 The design of composite catalysts is not limited to intermetallic composites, but also includes organic–inorganic hybrid materials, such as Cu2O composites with polyaniline (PANI), which significantly improve the selectivity of methanol by enhancing electron transfer and CO2 adsorption capacity.72 The microbial composite catalyst combines the high selectivity of microorganisms and the high efficiency of the catalyst to achieve the efficient conversion of CO2 to specific chemicals. For example, α-Fe2O3/g-C3N4 Z-scheme heterojunction photocathodes significantly increase acetate production by facilitating electron transfer between electrodes and microorganisms.73 The design of these composite structures makes full use of the synergy of different materials and significantly improves the selectivity and efficiency of CO2 reduction, providing a new idea for the development of efficient and stable catalysts.
The morphology control of catalysts has a significant effect on the selectivity of CO2 reduction products. Because of their high specific surface area and unique surface properties, nanostructured catalysts can provide more active sites, thus enhancing the adsorption and activation of CO2.74,75 For example, Cu2O nanocubes significantly improve the selectivity of CO2 reduction to formic acid by increasing the specific surface area and optimizing the charge transfer path. In addition, hollow structure catalysts such as CdS/VS-MoS2 heterojunctions significantly improve the selectivity of C2 products by increasing the probability of internal medium collision.76 The morphology control can also affect the product selectivity by changing the exposed crystal surface of the catalyst. For example, Cu2O nanowires can selectively generate multi-carbon products by exposing specific crystal faces. In addition, two-dimensional nanosheet structures such as Bi/Bi2O2CO3 significantly improve formic acid selectivity by providing more edge sites and active centers.77 Copper nanoflower catalysts exhibit excellent CO2RR activity due to their unique layered porous structure, which not only increases local pH but also reduces competitive hydrogen generation.30 These morphology control methods not only improve the activity of the catalyst, but also enhance the selectivity of specific products by optimizing the reaction path. The morphology control improves the activity and selectivity of the catalyst, providing a new possibility for the design of efficient photoelectrocatalytic systems.
The design of catalysts has a profound influence on the selectivity of CO2 reduction products. By selecting an appropriate metal catalyst, introducing doping or modification, constructing composite catalysts and adjusting catalyst morphology, the path of CO2 reduction can be effectively regulated, so as to achieve selective generation of specific products. These design strategies not only improve the reaction efficiency, but also provide theoretical guidance and experimental basis for the development of efficient and selective CO2 reduction catalysts. Future studies should continue to explore ways to optimize these design strategies to further improve the selectivity and stability of CO2 reduction, providing strong support for achieving sustainable carbon cycle and energy conversion.
The applied voltage plays a key role in photoelectrocatalysis by regulating the separation and transport of photogenerated electrons and holes. By applying an appropriate voltage, the photo-generated carriers can be effectively separated, avoiding the recombination of electrons and holes, thereby enhancing the reaction efficiency.84 Meanwhile, regulating the applied voltage can change the reaction path and induce the reduction of CO2 to generate different target products. Ren et al.85 summarized and discussed the effect of varying the applied voltage on copper catalysts on the selectivity of CO2 reduction products. Specifically, the selectivity of the main products changed significantly across different voltage windows. When the applied potential is close to −0.7 V, the main products are CO and HCOO−, and the total FE can reach up to 75%, especially around −0.55 V. As the potential becomes negative and reaches between −0.8 V and −1.1 V, the production of C2H4 and C2H5OH increases significantly, especially for all oxide-derived copper catalysts. In addition, the process of generating syngas is very common. The ratio of CO and H2 was regulated by changing the applied voltage. The selective regulation of different CO2 reduction products can be achieved in different voltage windows by reasonably controlling the applied potential.
The light absorption and reaction efficiency of photocatalysts are directly affected by the intensity and wavelength of external light.79,81,86,87 An increase in light intensity usually increases the current density and increases the reaction rate. The role of light wavelength is also critical; different wavelengths of light are absorbed by the photocatalyst to different degrees, so selecting an appropriate light wavelength can maximize the light absorption efficiency of the photocatalyst, thereby improving the photogenerated carrier generation rate and reaction selectivity. Dong et al.88 found that the selectivity of the product did not change significantly with the increase in light intensity, that is, the FE of CO and H2 was almost the same under different light intensities, which meant that the selectivity of the reaction did not change with the change in light intensity. In addition, Sohn et al.89 studied the change in product selectivity under different lighting conditions. For ZGO/ZnO composites, the production of CO increased significantly under UVC light irradiation, while the production of CH3OH decreased. In contrast, under UVB light, the production of CH3OH increases significantly, becoming the most dominant product, followed by CO, while the production of CH4 and other C2 compounds decreases significantly.
The reaction temperature plays a crucial role in determining the reaction kinetics of PEC CO2RR.8,90,91 Light irradiation on the catalyst surface not only excites electron–hole pairs but also generates substantial heat through non-radiative relaxation, resulting in localized temperature increases that can range from several tens to over one hundred degrees Celsius.92–94 Such temperature rises can be achieved by incorporating carbon materials or utilizing the localized plasmonic effects of metal nanoparticles.95 On one hand, according to the Arrhenius equation, an increase in temperature generally accelerates the reaction rate, thereby enhancing the current density of CO2 reduction. For example, Cobb et al.96 employed a reaction setup with external heating to precisely control the system temperature. At 45 °C and 0.85 V vs. SHE, the current density increased by 60% compared to that at 20 °C. The current density for CO2 conversion to formate reached 0.47 ± 0.05 mA, and after 10 h, 42 ± 8 mmol of formate was produced. Additionally, the entropy effect at elevated temperatures leads to a reduction in Gibbs free energy, and the onset potential for water oxidation shifts slightly to lower values, thereby reducing the overall reaction energy barrier. On the other hand, temperature also affects the formation and stability of intermediates, thus altering the distribution of final products. According to the latest research results from Jing's team,97 compared to single photocatalysis, electrocatalysis, and thermal catalysis, thermally assisted photoelectrocatalysis achieves higher product yields and greater C2 product selectivity in carbon-based products, with the contribution of hot electrons to C2 product selectivity reaching 31.9%. This enhancement may be attributed to the reduced C–C coupling energy barrier between * = CO and *CHO intermediates. However, increasing the reaction temperature is not always advantageous, as factors such as apparatus tolerance, catalyst stability, and other related issues must be considered. For instance, the enzyme formate dehydrogenase (FDh) catalyst tends to lose activity above 45 °C.96 Agbo's team investigated a flow cell setup and found that rapid heat dissipation of integrated components improved the solar-to-energy conversion efficiency by up to 10%.81
The type of electrolyte has a great influence on the type and selectivity of the products by affecting the stability and reaction path of the reaction intermediates.87,98–103 Different types of electrolytes provide different chemical environments, which may affect the properties of the active site on the photocatalyst surface.104,105 Liu et al.82 investigated the impact of the proton concentration of the electrolyte on the performance of CO2 reduction by testing three representative solvents, namely aprotic solvents with (methanol–acetonitrile) and without (acetonitrile) proton donors, and a protic solvent (methanol). The addition of proton donors not only increases the partial photocurrent and selectivity of CO, but also reduces photocorrosion. However, in methanol, the HER dominates the reduction of CO2, indicating that a high concentration of protons promotes the occurrence of HER. Compared with acetonitrile, the surface state concentration in methanol–acetonitrile significantly decreases, indicating that the presence of protons activates another reaction pathway (through stable intermediates), thereby accelerating charge transfer. The additives in the electrolyte significantly change the selectivity of CO2 reduction. For example, in an electrolyte containing 1-ethyl-3-methylimidazole tetrafluoroborate ([Emim]BF4), the selectivity of the Cu2O/TiO2 nanoarray for CO2 reduction to ethanol is greatly improved, and the FE reaches 82.7%.103 This is because the cation in the ionic liquid (IL) is enriched in the cathode region under external bias, which is conducive to capturing CO2, and forming a strong interaction with the catalyst surface, improving the separation efficiency and migration rate of photogenerated charges. In addition, the differing solubilities and adsorption capacities of electrolytes for CO2 reflect their mass transfer capabilities. Gao et al.103 utilized [Emim]BF4 ionic liquid as the electrolyte, which enhanced the CO2 solubility of the system and increased the local CO2 concentration at the catalyst surface. Furthermore, the [Emim]BF4 electrolyte activates the CO2 molecules and forms an [Emim+–CO2] complex, thereby lowering the energy barrier for the reduction reaction. Throughout the reaction process, [Emim]BF4 establishes an electron transfer channel between the catalyst and CO2, significantly enhancing the selectivity and activity for the conversion to C2H5OH.
By reasonably regulating the reaction conditions and environment such as the applied voltage, the intensity and wavelength of the external light, reaction temperature and the type of electrolyte, the product selectivity in the photoelectrocatalytic CO2 reduction process can be effectively controlled, so as to improve the generation proportion of the target product and enhance the reaction efficiency. This systematic regulation strategy not only helps to improve the performance of photoelectrocatalytic technology, but also provides a new idea for the efficient selective reduction of CO2.
Reasonable design of the reactor's geometric structure can optimize the light distribution and ensure that the surface of the photocatalyst is uniformly exposed to light energy, thereby avoiding the reaction inconsistencies caused by local overheating or uneven light intensity.96 Meanwhile, the transport path of reactants in the reactor needs to be reasonably designed to ensure that CO2 or other reactants can fully contact the surface of the catalyst, thus reducing the occurrence of side reactions and increasing the proportion of target products.108 In conventional reactors, CO2 transport is primarily governed by diffusion, leading to a near-zero concentration of gaseous CO2 in the static electrolyte.109 As a result, achieving high conversion efficiency and product selectivity is extremely challenging. In Fig. 8a, the electrolyte in the PEC device is static, so diffusion serves as the primary transport mechanism, followed by the generation of ionic currents carried by H+, HCO3−, and other major ionic species. Acid–base reactions occur during the diffusion and transport of CO2 (aq), H+, and HCO3− (Fig. 8c). The pH gradient between the anode and cathode, ion transport, and acid–base buffering reactions constitute the main sources of transport losses. To address this, Hu's team109 designed a series of BiVO4 photoanodes arranged in parallel and inclined toward Si photocathodes, creating a boundary layer flow effect (Fig. 8b). This configuration enables continuous flow of gaseous CO2 reactants to the catalyst, thereby enhancing CO2 mass transfer. Specifically, as shown in Fig. 8d, the establishment of boundary laminar flow alters the electrolyte flow direction from the photoanode to the photocathode, and the resulting increase in H+ flux further reduces the pH gradient. Importantly, the continuous flux of CO2 is also significantly improved. The photoanode generates H+, which acidifies HCO3−, resulting in the in situ production of gaseous CO2 that rapidly reaches the photocathode catalyst. The customized PEC device significantly increased CO selectivity from 3% to 21%, approaching the theoretical limit of 30% predicted by the multiphysics model. Meanwhile, a solar-to-fuel (STF) efficiency of 0.71% was achieved.
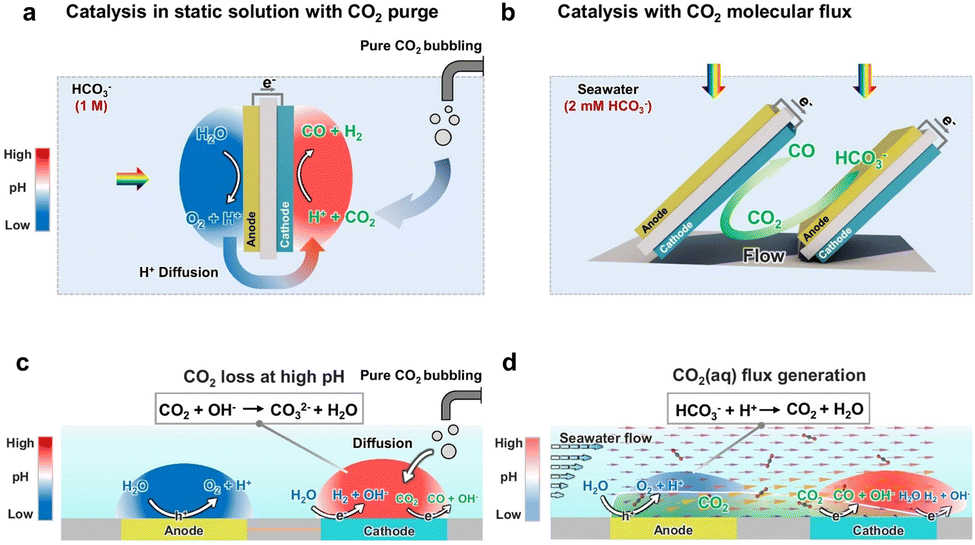 | ||
| Fig. 8 (a) Schematic diagram and (c) mechanism diagram of the traditional PEC device using static solution. (b) A schematic diagram and (d) mechanism diagram of CO2 flux catalysis with clear boundary layer shear flow. Reproduced with permission from ref. 109. Copyright 2025, Springer Nature. | ||
As a cutting-edge PEC device, the PEC flow cell incorporates a GDE to overcome the low solubility of CO2 in aqueous electrolytes, thereby significantly improving both the reaction rate and the selectivity for specific products.39 However, due to the unique structure of the reactor, the mass transfer of CO2 within the GDE is influenced by the electrolyte flow rate, the CO2 gas flow rate, and the CO2 gas pressure.110 As shown in Fig. 9a, a low electrolyte flow rate is insufficient to effectively remove accumulated molecules near the catalyst layer, resulting in low mass transfer efficiency at the catalyst surface. Conversely, an excessively high electrolyte flow rate increases hydraulic pressure, leading to the gradual infiltration of the electrolyte into the catalyst layer and significantly hindering mass transfer within the gas layer. Only at an appropriate electrolyte flow rate can the balance between hydraulic and gas pressures at the catalyst surface be maintained, optimally establishing the three-phase interface within the catalyst layer. In addition to the electrolyte flow rate, the CO2 gas flow rate is also a critical parameter. Fig. 9b illustrates that a continuous increase in the CO2 gas flow rate enhances the mass transfer of gaseous reactants at the catalyst surface. However, an excessively high CO2 flow rate reduces the residence time of CO2 molecules, limiting their effective adsorption, catalytic reaction, and product desorption on the catalyst surface. Furthermore, CO2 pressure significantly affects mass transfer, although it is often not specifically reported during testing. As shown in Fig. 9c, excessively low gas pressure hinders the passage of CO2 molecules through the GDE, reducing their contact with the catalyst. In contrast, excessive gas pressure disrupts the three-phase interface, disturbing the equilibrium between the adsorption and desorption of reactants and products. Maintaining optimal gas pressure is essential for sustaining an effective three-phase interface and optimizing photocathode performance. Therefore, the design of the reactor also affects the selectivity of the product by regulating the contact time of the gas–liquid interface and the flow state of the reaction medium. Appropriately prolonging the contact time or optimizing the flow state can increase the residence time of the reactant on the catalyst surface, thereby improving the selectivity of the product. However, excessive extension of the contact time may lead to an increase in by-products, so it is necessary to find a balance point in the design to achieve efficient and selective generation of the target product.
 | ||
| Fig. 9 Diagrams of the surface of the gas diffusion photocathode at different (a) electrolyte flow rates, (b) gas flow rates and (c) gas pressures. Reproduced with permission from ref. 110. Copyright 2024, Wiley-VCH GmbH. | ||
In summary, the design of PEC CO2RR technology requires comprehensive consideration of many factors such as the area and structure of optical devices and the design of reactors. By optimizing these design parameters, the utilization of light energy and enhancement of CO2 mass transfer can be maximized, the charge transfer efficiency can be improved, and the reactant contact and reaction path can be optimized, so as to achieve effective regulation of product selectivity and further improve the overall performance and efficiency of the reaction.
2.3 Dynamics and thermodynamics of product selectivity
The product selectivity of PEC CO2RR is affected by both kinetic and thermodynamic factors. The CO2 adsorption model, band gap structure and reaction free energy are the key influencing factors.The first step in CO2 reduction is the adsorption and activation of CO2 on the surface sites of the catalyst, so the adsorption model also plays an important role in determining the reaction pathway and product selectivity (Fig. 10a).111,112 It directly affects the activation of CO2 molecules and the subsequent reaction path by adjusting the electronic structure and active site properties of the catalyst surface.113 The CO2 molecule reacts with the surface atoms of the catalyst through the chemical adsorption process, resulting in the mutual repulsion between C and O atoms, forming a curved CO2 molecule (CO2−). The formation of *CO2− reduces the energy barrier of electron absorption, thus accelerating the electron transfer process from the electrode to *CO2−. The adsorption of CO2 can be divided into three main modes: C coordination, O coordination and mixed coordination. Specifically, O coordination forms a stable η2(O, O) coordination structure with the O atom on the catalyst surface. This adsorption mode is usually conducive to the formation of HCOOH.90,114 In contrast, C coordination or mixed coordination forms an incompletely activated M–CO2 structure by binding to metal or semiconductor sites on the catalyst surface. This adsorption mode tends to promote the further reduction of CO2 molecules to produce C1 products such as CO. Mixed coordination refers to CO2 binding to multiple active sites on the catalyst surface through carbon and oxygen atoms to form a more complex adsorption structure. This adsorption mode may promote a variety of reaction pathways, including the formation of C1 products and C2+ products. In addition, the adsorption mode is not only affected by the catalyst material, but also closely related to the reaction conditions. These factors work together to determine the activation degree of CO2 molecules and the selectivity of their subsequent reaction pathways. Therefore, the adsorption characteristics of CO2 on the catalyst surface not only determine the path selectivity of the subsequent reaction, but also further affect the selectivity of the final product by regulating the stability of the reaction intermediate and the reaction barrier.
 | ||
| Fig. 10 (a) Common methods of CO2 activation. Reproduced with permission from ref. 112. Copyright 2023, American Chemical Society. (b) The bias potentials, conduction band positions and valence band positions of various CO2 reduction reactions, and the calculated semiconductor oxidation and reduction potentials. Reproduced with permission from ref. 115. Copyright 2024, published by Elsevier B.V. | ||
The band gap structure is an important feature of the energy band characteristics of a photocatalyst. The band alignment directly determines the energy distribution of photogenerated electrons and holes (Fig. 10b).115 When the semiconductor material absorbs photon energy, the electrons in the valence band transition to the conduction band to form a carrier pair. At this time, the band gap width between the conduction band bottom and the valence band top not only determines the light absorption range, but also affects the driving force of the subsequent redox reaction through the initial kinetic energy of the carrier. The band gap size of the photocatalyst affects the excitation process of electrons and holes after illumination, as well as their energy matching, which determines the required energy input and the energy demand of the generated product to a certain extent. This energy transfer characteristic causes catalysts with different band gap structures to exhibit significant selectivity differences during photocatalytic reactions. Compared with the reduction potential system of the standard hydrogen electrode, it can be found that the formation of various carbon-based products corresponds to a specific energy threshold. The existence of this energy gradient requires that the conduction band position of the catalytic material must exceed the theoretical reduction potential of the target product to achieve effective conversion. That is to say, the main purpose of tuning the band structure of the photocatalyst is to control the reaction from a thermodynamic perspective. The conduction band minimum (CBM) must be sufficiently negative to provide the potential required for CO2 reduction.15 Different reduction products require different reduction potentials, so the position of the CBM must match the potential of the desired product. For example, the standard potential for the reduction of CO2 to CO is approximately −0.11 V vs. NHE (pH = 0), and a CBM < −0.11 V is more favorable for this process. Furthermore, by adjusting the band position, certain reaction pathways can be “screened” while others can be suppressed. For instance, if the CBM is positioned between the reduction potentials for the formation of CO and HCOOH, only the generation of CO is allowed, while the formation of HCOOH is inhibited. The band position of a semiconductor can be precisely regulated through band engineering strategies (such as doping,116 introduction of vacancies,117 and construction of heterostructures118,119) to achieve the required potential for generating the target product.
However, it is quite challenging to regulate product selectivity solely through the band structure, because many reduced products have standard reduction potentials lower than that of CO, and the standard reduction potentials of different products are often very close to each other. The surface of the catalyst supported by semiconductors possesses its own inherent “adsorption/desorption energies for reaction intermediates” (for example, gold tends to desorb CO, while copper can catalyze C–C coupling), which ultimately determines the selectivity of the catalyst toward different reaction pathways.120 An ideal semiconductor–catalyst combination can provide an appropriate driving force via the semiconductor CBM, while the catalyst surface offers suitable adsorption and activation capabilities to achieve a synergistic effect. When regulating the bandgap between the semiconductor and catalyst to achieve product selectivity, the following two principles should be considered. (1) Appropriate thermodynamic driving force: the reduction level of the electrode should match the reduction potential of the target product. In other words, the conduction band position needs to be sufficiently negative to provide the required electron energy for the reduction reaction of the product, thereby effectively driving the formation of the specific product. (2) Synergistic interfacial kinetics: the energy levels between the photocathode and the catalyst also need to be properly aligned. Only when the energy level alignment is appropriate can the photogenerated charge carriers efficiently transfer from the semiconductor to the catalyst surface, thereby promoting the catalytic reaction. If the energy levels are mismatched, the transfer efficiency of charge carriers decreases, leading to reduced catalytic activity and product selectivity. Therefore, by rationally designing the energy level structures of the semiconductor and catalyst, it is possible to effectively regulate the reaction pathway and achieve precise control over product selectivity.
The change in Gibbs free energy has a profound influence on product selectivity. The formation paths of different reduction products correspond to significantly different thermodynamic driving forces. Standard Gibbs free energy changes (ΔG0) for overall CO2 reduction with different products are presented in Table 1.24 Taking the two-electron transfer product as an example, the formation path of CO involves the formation and desorption of *COOH intermediates. The standard free energy change is 257.4 kJ mol−1, which shows a slight energy advantage over the 270.4 kJ mol−1 generated by HCOOH. This subtle energy difference can be significantly amplified by variations in the electronic structure of the catalyst surface. For example, metal catalysts such as gold and silver can effectively promote the selective formation of CO by optimizing the adsorption strength of *COOH intermediates, while metal catalysts such as Bi and Sn can effectively promote the selective formation of HCOOH. For deeply reduced products requiring multiple electron transfer, such as methanol (ΔG = 702.8 kJ mol−1) and methane (ΔG = 919.6 kJ mol−1), the reaction path involves a continuous proton-electron coupling transfer process, and the free energy changes at each step together form a complex energy barrier network. The formation mechanism of multi-carbon products shows a higher dimension of free energy correlation. Taking the synthesis of C2H4 as an example, it involves 12 electron transfer steps, and its standard free energy change exceeds 1332 kJ mol−1, which involves the dimerization of CO intermediates, the formation of carbon–carbon bonds and the subsequent hydrogenation process. There may be energy bottlenecks at each stage of such reactions. For example, the coverage and bonding strength of CO intermediates on the surface of copper-based catalysts directly affect the feasibility of the carbon–carbon coupling step. Meanwhile, the high free energy state of some intermediates in the reaction path may trigger competitive side reactions, resulting in a shift in product distribution to a more thermodynamically favorable direction. This requires effective stabilization of key intermediates of different products. The Gibbs free energy difference of the key intermediates (such as *CO, *CHO, etc.) in different reaction paths directly affects the reaction kinetics. The formation of CO requires a stable *CO intermediate, while the formation of CH4 involves the formation of a more stable *CHO intermediate. The stability of these intermediates directly influences the reaction pathway. If the Gibbs free energy of the *CO intermediate is low, the reaction tends to generate CO; if the Gibbs free energy of *CHO intermediate is lower, it is more conducive to the formation of CH4. For C2+ products, the key is the change in Gibbs free energy of the C–C coupling process. This process usually requires the interaction of two C1 intermediates (such as *CHO or *CO), and its energy demand determines the difficulty of the coupling reaction and the selectivity of the product. If the energy barrier for coupling some C1 intermediates is low, a specific C2+ product may be preferentially generated.
| Reaction | ΔG0 (kJ mol−1) |
|---|---|
| CO2(g) → CO(g) + 1/2O2(g) | 257.4 |
| CO2(g) + H2O(l) → HCOOH(l) + 1/2O2(g) | 270.4 |
| CO2(g) + H2O(l) → HCHO(l) + O2(g) | 521.9 |
| CO2(g) + 2H2O(l) → CH3OH(l) + 3/2O2(g) | 702.8 |
| CO2(g) + 2H2O(l) → CH4(g) + 2O2(g) | 919.6 |
| 2CO2(g) + 2H2O(l) → CH3COOH(l) + 2O2(g) | 963.8 |
| 2CO2(g) + 2H2O(l) → C2H4(g) + 3O2(g) | 1332 |
| 2CO2(g) + 3H2O(l) → C2H5OH(l) + 3O2(g) | 1327 |
In summary, the process of CO2 reduction is very complex. Both kinetic and thermodynamic factors determine the product selectivity of PEC CO2RR. In this section, the band gap structure, adsorption characteristics and thermodynamic parameters of the reaction path of the optimized photocatalyst are briefly described, so as to provide experience for the selective formation of the target product and the improvement of the reaction efficiency.
3 Research progress on selectivity of CO2 reduction products
3.1 C1 products
| Catalyst | Electrolyte | Light source | Product and performance | Ref. |
|---|---|---|---|---|
| CuBi2O4–Bi2O3 | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 86.03% at 0.1 V vs. RHE | 122 |
| Bi/ZnO@ZnSe | 0.5 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 88.9% at −0.9 V vs. RHE | 123 |
| Au25 nanocluster inside a porphyrin nanoring | 0.5 M KHCO3 | 300 W Xe lamp | FE = 92.1% at −0.67 V vs. RHE | 124 |
| CuGaS2/PPy | 0.1 M KHCO3 | 300 W Xe lamp with a 420 nm cutoff filter | FE = 6% at −0.6 V vs. Ag/AgCl | 125 |
| HJ-Si-Ag | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 97.4% at −0.2 V vs. RHE | 126 |
| Cu2O/C/PTFE | 0.1 M KHCO3 | 300 W Xe lamp of 100 mW cm−2 | FE = ∼70% at −0.7 V vs. RHE | 127 |
| In-doped GaN | 0.5 M KHCO3 | 300 W Xe lamp | FE = ∼45% at −0.75 V vs. RHE | 128 |
| Molecular Re/porous Si | phenol/MeCN | AM 1.5G of 100 mW cm−2 400–1100 nm | FE = 90% at −1.7 V vs. Fc+/Fc | 129 |
| CoPc/K7HNb6O19 | 0.5 M KHCO3 | 300 W Xe lamp | FE = 98.6% at −0.85 V vs. RHE | 130 |
| Ni–NC/Si | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 100% at −0.6 V vs. RHE | 131 |
| ZnO/ZnTe | 0.1 M TBAPF6-MeOH | AM 1.5G of 100 mW cm−2 | FE = 93.88% at −2.58 V vs. Fc+/Fc | 132 |
| Fe2O3/Cu2O/Cu | 0.1 M TBAPF6-MeOH | AM 1.5G of 100 mW cm−2 | FE > 89% at −0.8 to −1.2 V vs. Fc+/Fc | 133 |
| ZnTe/SnS2 | 0.1 M TBAPF6-MeOH | AM 1.5G of 100 mW cm−2 | FE = 87% at −1.78 V vs. Fc+/Fc | 134 |
| Ag-modified CuBi2O4 | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 92% at −0.2 V vs. RHE | 69 |
| CuBDC-xNH2/CF | 0.1 M KHCO3 | 300 W Xe lamp with a 420 nm cutoff filter | FE = 51.5% at −0.3 V vs. RHE | 135 |
| STA-GO/CoPc | 0.1 M KHCO3 | 300 W Xe lamp with a 400 nm cutoff filter | FECO = 86% at −0.28 V vs. RHE; FEHCOOH = 8% at −0.62 V vs. RHE | 136 |
| Cu2O–SnO2 | 0.1 M KHCO3, 0.1 M KOH | AM 1.5G, 450 W Xe lamp | FECO = 37%; FEHCOOH = 26% | 137 |
| PPy/Cu2O/FeOOH | 0.1 M Bu4NPF6 | AM 1.5G | FE = 90% at −1.8 V vs. Ag/Ag+ | 138 |
| Cu2O–SnO2 | 0.1 M KHCO3, 0.1 M KOH | AM 1.5G of 100 mW cm−2 | FECO = 35.47% and FEHCOOH = 19.58% at −0.2 V vs. RHE | 139 |
| Ni–Mn–Cu–FeAl HELO | 0.5 M KHCO3, 1 M KOH | 300 W Xe lamp | 909.55 μmol g−1 h−1 at −0.8 V vs. RHE | 140 |
| Re supramolecular | 0.05 M NaHCO3 | AM 1.5G of 100 mW cm−2 | FE = 94%, −0.2 V vs. Ag/AgCl | 141 |
| Re@NH2-frac-MOF | 0.1 M TBAPF6-MeOH | 300 W Xe lamp with a 400 nm cutoff filter | FE = 97.9% at −0.7 V vs. Ag/AgCl | 142 |
| Cu@Au/SiNW | 0.1 M KHCO3 | Simulated solar light | FE = 72% at −1 V vs. Ag/AgCl | 99 |
| p-Si/TiO2/TaOx/Cu | 0.1 M KHCO3 | AM 1.5G | FE = ∼65% at −1.26 V vs. RHE | 143 |
| CoII(BrqPy) | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 100% at −0.11 V vs. RHE | 144 |
| ZnGa2O4/ZnO | 0.1 M NaHCO3 | 365 nm LED | FE = 28.7% at −1.4 vs. Ag/AgCl | 89 |
| Ag/Si or Cu/Si | 1 M KOH | AM 1.5G of 100 mW cm−2 | Ag, FE = 90%; Cu, FEC2+ = 53% | 145 |
| In/Cu2O | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 82% at −0.7 V vs. RHE | 146 |
| Cupric porphyrin | 0.1 M TBAPF6-MeOH | AM 1.5G of 100 mW cm−2 | FE = 87% at −2.5 V vs. Fc+/Fc | 147 |
| Au/GaN/Si | 0.5 M KHCO3 | 100 mW cm−2 | FE = ∼90% at 0.17 V vs. RHE | 148 |
| Au-NR | 0.5 M KHCO3, 1 M KOH | AM 1.5G of 100 mW cm−2 | FE = 85% at −1 V vs. RHE | 70 |
| Mn(TPP)Cl | 0.1 M [NBu4][BF4]-MeCN | Halogen lamp of 90 mW cm−2 | FE = 48.2% at −1 vs. Ag/AgCl | 149 |
| CuGa3Se5 | 0.1 M KHCO3 | Fiber-coupled LED | FE = ∼80% at −0.4 V vs. RHE | 150 |
| CsAgBr2-PNC | Na2CO3 | 150 W Xe lamp | FE = 50% at −0.6 V vs. Ag/AgCl | 151 |
| AgCl/GaNNW/Si | 0.1 M KHCO3 | 300 mW cm−2 | FE = 80% at −0.4 V vs. RHE | 88 |
| Au/TiO2/n + p-Si | 0.1 M KHCO3 | AM 1.5G | FE = 86% at −0.8 V vs. RHE | 152 |
| [Cd(DPNDI)(TDC)]n | 0.1 M [n-Bu4N]PF6–MeCN | 100 W Hg lamp with a UV cut off filter | FE = 78% at −1.3 V vs. Fc+/Fc | 153 |
| CoPor-N3 | Saturated KHCO3 | 300 W Xe lamp | FE = 96% at −0.5 V vs. RHE | 154 |
| Au–TiO2/InP | 0.1 M KHCO3 | AM 1.5G | FE = 84.2% at −0.11 V vs. RHE | 155 |
| Au | 0.1 M TBAPF6-10 M H2O | LED white of 300 mW cm−2 | FE = 90% at −1.68 V vs. Ag/AgCl | 156 |
| ZnTe | 0.1 M KHCO3 | 455 nm LED | FE = 50.4% at −1.0 V vs. RHE | 157 |
| Si/viologen polymer/Au | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 25% at 1.2 V vs. RHE | 158 |
| Single-atom Co–N5 | 0.5 M K3BO3 | AM 1.5G of 100 mW cm−2 | FE = 90.6% at 1.23 V vs. RHE | 159 |
| Si/Al-PMOF(Co) | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 80.7% at −0.4 V vs. RHE | 160 |
| Cu(In,Ga)S2 | 0.1 M KHCO3 | 1 sun illumination | FECO = 30% and FEHCOOH = 14% at −0.4 V vs. RHE | 161 |
| ZnTe | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FECO = 45% and FEHCOOH = 15% at −0.6 V vs. RHE | 162 |
| Ox-Au | 0.5 M KHCO3 | 1 Sun illumination | FE = ∼98% at −0.5 V vs. RHE | 163 |
A large number of studies have confirmed that Ag-based and Au-based catalysts show significant advantages in the CO generation path, which is mainly due to the unique characteristics of their electronic structures. Specifically, the suitable d-band center position enables the catalyst to produce a moderate adsorption effect on the key intermediates (*COOH and *CO), which not only guarantees the kinetic stability of the intermediates, but also avoids the problem of the increased desorption energy barrier caused by excessive adsorption. Combined with its weak surface adsorption characteristics (especially low binding energy to CO), the energy required for desorption is significantly reduced, thereby promoting the efficient release of CO. In addition, the weak adsorption capacity of Ag and Au for the hydrogen intermediate (*H) effectively inhibits the kinetic process of the competitive HER. The localized charge transfer between Au and TiO2 in the Au–TiO2 heterogeneous interface system enhances the binding strength of the positively charged Au interface site to the CO* intermediate, thereby optimizing the thermodynamic path of CO2 photoreduction to CO.155 The experimental results show that the FE of CO reaches 84.2% when the Au–TiO2/InP nanorod array photocathode is applied at −0.11 V vs. RHE bias under simulated AM 1.5G illumination. However, limited by the efficiency of the mass transfer process and the complex electric field environment around the active site, the activity and selectivity of the existing catalytic system still face obvious bottlenecks. Bi et al.70 innovatively used a continuous flow cell reactor coupled with plasma Au nanorods (NRs) to successfully achieve efficient PEC CO2 to CO conversion. Benefiting from the fast mass transfer process of flow field optimization and the local CO2 enrichment effect on the surface of Au NRs, the photocurrent density of the system reached 1.0 mA cm−2, which was nearly 5 times higher than that of the traditional H-type reactor (0.17 mA cm−2). At the same time, the localized surface plasmon resonance (LSPR) effect of Au NRs increased the CO FE by about 20% under illumination. For atomically precise gold nanocluster systems, their size-dependent quantum effects and ligand assembly strategies have shown unique potential in the field of CO2 directional conversion, but the agglomeration phenomenon that tends to occur during the catalytic process seriously restricts its stability. The research group designed a host–guest composite structure in which pyridinethiol-functionalized Au25 nanoclusters were embedded in six zinc porphyrin unit nanorings.124 The assembly not only significantly improved the stability of gold clusters, but also showed excellent activity and selectivity in the photochemical CO2 reduction to CO and singlet oxygen generation reaction. The FE of CO reached 92.1% at −0.67 V vs. RHE. Compared with Au, the cost advantage of Ag made it more practical. By combining plasmonic Ag nanoparticles with semiconductor photocatalysts, the carrier separation efficiency can be effectively enhanced. Ag nanoparticles modified CuBi2O4 inverse opal photocathode (CuBi2O4–IOs–Ag) was prepared by the sacrificial template method.69 The three-dimensional ordered structure optimizes both mass transfer kinetics and light capture efficiency. The ohmic contact between Ag nanoparticles and CuBi2O4 significantly improves the surface charge distribution, inhibits carrier recombination and optimizes kinetic behavior. The experimental results showed that the CO FE reached 92% at 0.2 V vs. RHE bias, which was 1.6 times higher than that of the original CuBi2O4 film. The crystal phase regulation of Ag nanoparticles also had a key effect on the catalytic performance. A hexagonal close-packed (hcp) crystalline Ag nanocrystal co-catalytic layer was constructed on the heterojunction silicon (Si) substrate (Fig. 11a–c).126 The unconventional hcp phase accelerated the charge transfer process of the CO2RR, and the light absorption/catalytic layer decoupling design of the Si substrate avoided parasitic light absorption. The CO FE of the photocathode exceeded 90% in a wide potential window (−0.2 V vs. RHE), up to 97.4%. At the CO2/CO equilibrium potential, the photocurrent density for CO production was −2.7 mA cm−2 (0.1 M KHCO3 electrolyte), and the FE was 96%, which was superior to that of the precious metal Au-based reference system. In addition, the ion valence regulation strategy of Ag has attracted much attention in the field of photoelectrocatalysis. The team systematically studied the CO2RR performance of GaN nanowire (NW)/n+–p–Si heterojunction photocathodes loaded with different silver halide (AgX, X = Cl, Br, I) cocatalysts.88 The nitrogen-terminated GaN-NW as a chemically stable substrate could effectively load silver halide, and the halogen element significantly improved the catalytic activity by reducing the energy barrier for *COOH intermediate formation. The experimental results showed that the CO FE was more than 80% at −0.4 V vs. RHE, and the constant potential is stable at ∼0.2 V vs. RHE.
 | ||
| Fig. 11 (a) Diagrammatic representation of various synthetic methodologies for co-catalyst deposition on front-side-illuminated photocathodes. (b) Structural configuration of HJ-Si photocathodes designed for CO2 conversion. (c) FE of CO production on different catalysts under controlled electrochemical conditions. Reproduced with permission from ref. 126. Copyright 2024, published by PNAS. (d) Energy band diagram of a photoelectrode for CO2 reduction to CO. (e) Adjusting the surface states by reducing disorder. Reproduced with permission from ref. 132. Copyright 2024, Elsevier B.V. All rights reserved. (f) Schematic presentation for Re@NH2-frac-MOF. (g) Plot of the product obtained after 6 h of PEC CO2RR with different catalysts and its proportion to the total mass of the catalyst. Reproduced with permission from ref. 142. Copyright 2023, Wiley-VCH GmbH. | ||
In addition to precious metal systems such as Au and Ag, researchers are working on the development of low-cost and high-abundance metal-based photoelectrocatalytic materials such as Cu, Bi, Ni, Fe and Zn to build a more economical and sustainable CO2 conversion system. The In/Cu2O core–shell nanowire Schottky junction constructed by physical vapor deposition exhibited a space charge region reconstruction effect, which significantly optimized the photogenerated carrier dynamics.146 Experiments showed that the heterostructure increased electron mobility by 2.3 times, achieved a CO yield of 75.94 μmol cm−2 h−1 at −0.7 V vs. RHE bias, and maintained 12 hours of continuous operation (FE = 82%). Combined with in situ Fourier transform infrared spectroscopy (in situ FTIR) analysis, it was confirmed that the introduction of In promoted the CO desorption kinetics by enhancing the adsorption energy of the COOH* intermediate (ΔEads = −1.2 eV). Gao's team138 innovatively constructed a three-level functional Cu2O-based photocathode system: (1) the Cu2O structure was used for efficient light collection, and the light capture efficiency was increased to 92%; (2) the FeOOH hole transport layer enabled directional carrier migration; (3) the PPy modification layer provided continuous multi-electron transfer channels. The integrated system achieved a record CO yield of 46.17 mmol h−1 at −2.0 V vs. Ag/AgCl voltage, a half-cell solar-to-fuel conversion efficiency (hSTC) of 1.58%, a quantum efficiency breakthrough of 3.08%, and extended continuous operation stability to 7 h. In view of the limitations imposed by semiconductor/electrolyte interface states on energy conversion efficiency, the surface modification of ZnTe with an ultra-thin ZnO layer in the ZnO/ZnTe gradient band heterojunction reduces energy disorder and improves the electron utilization rate in the CO2 reduction reaction (Fig. 11d and e).132 In a CO2-saturated acetonitrile system containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6), the photocathode achieved a current density of −7.70 mA cm−2 and a CO selectivity of 93.88% at a potential of −2.58 V vs. Fc+/Fc. The precise construction of single-atom catalysts provided a new paradigm for the regulation of active sites. The Ni single atom anchored on a silicon nanowire substrate enabled CO2 reduction to achieve nearly 100% CO selectivity at −0.6 V vs. RHE potential, and the TOF value (3.2 × 10−2 s−1) was two orders of magnitude higher than that of the nanoparticle system.131 The introduction of single-atom nickel sites provided sufficient active sites for CO2 reduction, thereby improving the selectivity of CO formation.
Molecular catalysts are a class of catalysts with well-defined active sites and precisely tailored structures.164 The active center is usually composed of mononuclear or multinuclear metal ions (such as Fe, Co, Ni, etc.) or specific organic functional groups, and is precisely regulated by the coordination group and the surrounding chemical environment. In the PEC CO2RR system, such catalysts can act as photosensitizers or catalytically active sites, directly participate in the transport of photogenerated carriers and the chemical conversion of CO2 molecules, and generally exhibit excellent CO product selectivity. In the past five years, the molecular catalysts commonly used in the PEC CO2RR are molecular Re catalyst,142 molecular Co catalyst,130 molecular Mn catalyst,149 molecular Pd catalyst.141 The low-temperature calcination process realized the coupling of cobalt phthalocyanine (CoPc) with oxygen vacancy-enriched K7HNb6O19, and successfully constructed a Z-scheme heterojunction structure and applied it to the photoelectrocatalytic reduction of CO2 to CO.130 Experiments showed that the heterojunction catalyst exhibits >90% FE and significantly improved turnover frequency (TOF) in a wide potential window of −0.65 to −1 V (vs. RHE). Its excellent PEC performance stemmed from two key designs: first, the intrinsic electric field formed by the Z-scheme heterojunction interface significantly enhanced the separation efficiency of photogenerated electron–hole pairs; second, the oxygen vacancies in K7HNb6O19 not only optimized the activation path of CO2 adsorption, but also effectively inhibited the agglomeration of CoPc molecules through the steric hindrance effect, thereby improving the structural stability of the catalyst. Density functional theory calculations further confirmed that the synergistic effect of oxygen vacancies and CoPc significantly reduced the CO2 activation energy barrier. Li's research group144 innovatively immobilized the cobalt complex CoII(BrqPy) (BrqPy = 4′,4′-bis(4-bromophenyl) −2,2′:6′,2′′:6′′,2′′′-quaternary pyridine) on the surface of a multi-walled carbon nanotube (CNT) modified TiO2 protective layer on an n-type Si electrode (Si|TiO2) via π–π stacking. Under standard solar light intensity, the composite catalytic system was continuously operated in a neutral electrolyte for 2 h to obtain a photocurrent density of 1.5 mA cm−2, and achieve 100% CO selectivity and FE. The performance improvement was attributed to the high dispersion of molecular active sites and CNT-mediated fast charge transfer channels. In order to further improve the loading efficiency of molecular catalysts, Lee's research group142 developed a lattice-guided anisotropic etching strategy based on 2D MOF-5 nanocrystals, and successfully constructed a carrier material with hierarchically ordered nano-mesoporous structure for the stable loading of Re(bpy)(CO)3Cl complexes (Fig. 11f and g). The highly exposed active sites at the boundary of the fractal pores enabled the catalyst to achieve a CO yield of 74.8 μmol mg−1 h−1 and a FECO of 97.9% at −0.7 V (vs. Ag/AgCl), while maintaining a continuous and stable photocurrent output for 24 h. The important role of microscopic pore engineering in improving the loading density and interfacial mass transfer efficiency of molecular catalysts was revealed.
In the process of CO formation, although there is a problem that the competitive HER leads to a decrease in product selectivity, the controllable preparation of syngas can be realized by reasonably regulating the yield ratio of CO to H2. Syngas is a key chemical raw material composed of H2 and CO, and its molar ratio regulation plays a decisive role in downstream industrial applications.173 The performance of PEC CO2RR for syngas is summarized in Table 3. For example, the molar ratio of H2/CO in the Fischer–Tropsch synthesis process needs to be 2:1, while in methanol synthesis the ratio should be regulated in the range of 2:1–3:1. Based on this, the dynamic adjustment of syngas components through reaction condition optimization and catalyst structure design has become the focus of current research. The band-matched SnO/g-C3N4 p–n heterojunction catalyst exhibited significantly improved light absorption efficiency (λ > 420 nm) and photogenerated carrier separation efficiency.42 Under an applied bias of −1.5 V vs. RHE, the system achieved a total FE of 70%, and the H2/CO ratio could be linearly changed in the range of 1.5–10 by voltage regulation. As shown in Fig. 12a–c, the Cu2O–SnOx hybrid nanowire (NW) photocathode achieved a total FE of 90.32% at −0.35 V (vs. RHE), and the CO/H2 ratio could be precisely controlled in the range of 2.2:1 to 4.6:1.168 The mechanistic study showed that the electrochemical deposition of SnO x on the surface of Cu2O NWs significantly improved the charge transport kinetics and CO2 adsorption capacity (about 3.8 times), and photoexcitation accelerated the formation kinetics of *COOH intermediates, thus promoting the efficient desorption of CO. As shown in Fig. 12d–f, Ag and AgX (X = Cl, Br, I) cocatalysts were loaded on the surface of the GaN NWs/Si heterojunction photocathode.88 Compared with the pure Ag system, AgCl and AgBr increased the CO selectivity from 62% to 82%, and the AgX/GaN/Si system exhibited an onset potential shift of 0.2 V (vs. RHE) and a current density of > 20 mA cm−2 at −0.6 V (CO/H2 > 2). It is worth noting that the AgBr/GaN/Si cathode in the flow cell achieved a photocurrent density of 92 mA cm−2 under 3 solar intensity (300 mW cm−2), and the CO/H2 ratio remained stable for 12 h. The Ag3Cu/TiO2/ZnTe metal–insulator–semiconductor (MIS) photocathode system exhibited a significant and stable photocurrent density of 5.10 mA cm−2 at 0.20 V (vs. RHE) under AM 1.5G illumination, and its CO:H2 generation ratio could reach up to 6.8, showing excellent syngas composition control ability.169
| Photocathode | Electrolyte | Light source | Product and performance | Ref. |
|---|---|---|---|---|
| PCE10:EH-IDTBR | 0.5 M KHCO3 | AM 1.5G of 100 mW cm−2 | (Syngas) 15 mA cm−2 at 1.23 V vs. RHE | 83 |
| Sn/SnOx–Cu2O/Ga2O3/TiO2 | 100 mM SnCl2 | AM 1.5G | FEsyngas = 62%, CO:H2 ratio: 0.5; FEHCOOH = 32% at −1 V vs. RHE |
165 |
| SnO/g-C3N4 | Na2SO4 | 100 mW cm−2 | FEsyngas = 70% at −1.3 V vs. RHE | 42 |
| Au/TiO2/GaN/Si | 0.5 M KHCO3 | AM 1.5G of 100 mW cm−2 | FEsyngas = 100% at 0.17 V vs. RHE, CO:H2 ratio: ∼1 |
166 |
| Au | 2 M KOH | AM1.5G | CO:H2 ratio: 10–20 |
81 |
| Cu92In8|GE|BiOI | 0.5 M KHCO3 | AM 1.5G of 100 mW cm−2 | CO:H2 ratio: 3.7 at −0.15 V vs. RHE |
167 |
| Cu2O–SnOx | 0.5 M NaHCO3 | 300 W xenon lamp with a 420 nm cutoff filter providing a light intensity of 200 mW cm−2 | FEsyngas = 90.32% at −0.35 V vs. RHE, CO/H2: 2.2–4.6 | 168 |
| AgCl/GaNNW/Si | 0.1 M KHCO3 | 300 mW cm−2 | CO/H2 ratio > 4 with a high current density >13 mA cm−2 | 88 |
| Ag/CuxO@rGO/Mo | 0.1 M KHCO3 | AM 1.5G of 200 mW cm−2 | CO:H2 ratio: 0.65–1.84 |
80 |
| Ag3Cu/TiO2/ZnTe | 0.1 M KHCO3 | AM 1.5G | CO:H2 ratio: 6.8 at −0.2 V vs. RHE |
169 |
| CuIn0.3Ga0.7S2 | MeOH–MeCN | AM 1.5G | CO:H2 ratio: 6.24 at −1.86 V vs. Fc+/Fc |
82 |
| Ag | 0.5 M KHCO3 | Visible light (100 mW cm2) | (Syngas) 124.47 μmol (cm−2 h) at 1.57 V vs. RHE | 170 |
| 1 M KOH | ||||
| FeNiOx/WO3/W | 0.5 M Na2SO4 | 300 W Xe lamp with a 420 nm cutoff filter | CO:H2 ratio: 0.5 at −1.05 V vs. RHE |
171 |
| ZnS–Cu0.8Ag0.2GaS2 | 0.1 M KHCO3 | 300 W Xe-arc lamp (>420 nm) | FEsyngas = 95% at 0 V vs. RHE | 172 |
 | ||
| Fig. 12 (a) Schematic illustration of the Cu2O/SnOx heterojunction for PEC CO2 reduction. (b) Total current densities and partial current densities. (c) FE of different products and the proportions of syngas. Reproduced with permission from ref. 168. Copyright 2021 Wiley-VCH GmbH. (d) Preparation mechanism diagram of the AgX catalyst (X = Cl, Br, I). (e) Energy band diagram. (f) CO/H2 ratio diagram of different photocathodes at different potentials from 0.2 to −0.6 V vs. RHE. Reproduced with permission from ref. 88. Copyright 2022, American Chemical Society. | ||
In photoelectrocatalytic carbon dioxide reduction systems, the CO formation pathway has become one of the main research directions in this field due to its low reaction energy barrier and excellent selectivity. Studies have shown that noble metal catalysts (such as Ag and Au) can significantly reduce the reaction activation energy and optimize product selectivity due to their moderate adsorption characteristics of key intermediates (*COOH and *CO), thus exhibiting excellent performance in CO selective generation. In recent years, the research focus has gradually shifted to non-precious metal-based catalyst systems such as Cu, Bi and Ni, aiming to reduce production costs and improve process sustainability through material innovation. It is noteworthy that the construction of single-atom catalysts and the precise loading strategy of molecular catalysts have been proven to be effective technical approaches to achieve near 100% FE. By rationally designing the photoelectrocatalytic system, such as optimizing the surface characteristics of the catalyst, improving the light absorption capacity and enhancing the charge separation efficiency, the efficiency and stability of CO generation can be significantly improved. Although the competitive formation of H2 and CO may affect the product selectivity, the ratio of CO to H2 can be precisely controlled by adjusting the reaction conditions and catalyst design, so as to meet the needs of downstream processes such as syngas. In general, the generation of CO in PEC CO2RR has significant advantages. With the further optimization of catalyst performance and the improvement of reaction conditions, this technology is expected to achieve greater breakthroughs in efficiency and selectivity in the future.
| Catalyst | Electrolyte | Light source | Product and performance | Ref. |
|---|---|---|---|---|
| VS-ZnIn2S4/TiN | 0.1 M KHCO3 | AM 1.5G of 200 mW cm−2 | FE = 81% at −0.45 V vs. RHE, 1173.2 μM h−1 cm−2 | 174 |
| In–Bi2O3/Cu2O | 0.5 M KHCO3, 0.5 M KOH | 300 W Xe lamp with a 420 nm cutoff filter | FE = 97.8% at −0.87 V vs. RHE | 175 |
| STO/a-R TNTAs | 0.5 M Na2SO4 | 300 W Xe lamp of 1240 mW cm−2 | FE = 90.70% | 33 |
| FeOOH/p–nCu2O/Co:CdS | 0.5 M KHCO3 | AM 1.5G, 350–800 nm | FE = 82.9% at −0.75 V vs. RHE | 176 |
| Bi-doped InOCl | 0.5 M NaHCO3 | 300 W Xe lamp | FE = 89.9% at −0.9 V vs. RHE | 177 |
| Bi | 0.5 M KHCO3 | LED lamp, 450 nm of 100 mW cm−2 | FE = 79.1% at −1.8 V vs. Ag/AgCl | 178 |
| CuBi-MOF/Si | 0.5 M KHCO3 | 300 W Xe lamp with a 420 nm filter providing an intensity of 200 mW cm−2 | FE = 95% at −0.3 V vs. RHE | 179 |
| 2D Bi/Bi2O2CO3 | 0.5 M KHCO3 | Xe lamp | FE = 92.68% at −0.95 V vs. RHE | 77 |
| LD-Bi | 0.5 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 93.8% at −2.2 V vs. RHE | 180 |
| 4:1-BiVO4/ZIF-8 | 0.1 M KHCO3, 0.05 M KOH | 300 W Xe lamp | FE = 91.24% at −0.9 V vs. RHE | 181 |
| Formate dehydrogenase | 0.067 M NaHCO3 | AM 1.5G, 3 sun | FE = 97% | 96 |
| CuSe@BiOI | 0.5 M KHCO3 | AM 1.5G of 150 mW cm−2 | FE = 80.5% at −1.2 V vs. RHE | 65 |
| Sn–Bi2O3 | 0.5 M KHCO3, 0.5 M KOH | AM 1.5G of 100 mW cm−2 | FE = 94% | 182 |
| Bi–Sn/SiNW | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 88.67% at −1.02 V vs. RHE | 183 |
| PCN-n-SiOx | 0.1 M KHCO3 | AM 1.5G | 283.0 μmol L−1 at −0.8 V vs. OCP | 184 |
| Bi2Se3/Bi2O3 | 0.5 M KHCO3, 0.5 M KCl | 300 W Xe lamp | FE = 81.33% at −0.9 V vs. RHE | 185 |
| Cu8%-BiVO4 | 1 M Na2SO4 | 300 W Xe lamp | FE = 87.15% at −1 V vs. RHE | 186 |
| TiO2/Ti3CN Mxene | 0.1 M KHCO3 | 300 W Xe lamp | 45.6 μM cm−2 h−1 at −0.8 V vs. SCE | 187 |
| Bi/GaN/Si | 0.5 M KHCO3, 1 M KOH | AM 1.5G of 100 mW cm−2 | FE = 85.2% at −0.2 V vs. RHE | 188 |
| Hybrid CuGaO2 | 0.3 M BMIM·TfO-MeCN | 450 W Xe lamp with an AM 1.5G filter | FE = 81% at −1.5 mA cm−2 | 189 |
| CuFe2O4/CFs-DSAC | 1.5 M KHCO3 | Visible light | FE = 52% at −0.8 V vs. RHE | 190 |
| Bi@ZFOPEC | 0.1 M KHCO3 | 300 W Xe lamp | FE = 61.2% at −0.65 V vs. RHE | 60 |
| Bi/GaN/Si | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 98% at −0.3 V vs. RHE | 191 |
| Bi–Bi2O3/ZnO/p-Si | 0.1 M KHCO3 | AM 1.5G of 200 mW cm−2 | FE = 84.3% at −0.95 V vs. RHE | 192 |
| TOAgx | 0.5 M K2SO4 | 300 W Xe lamp | FE = 73% at −1.2 V vs. Ag/AgCl | 193 |
| 10-Bi2S3/ZIF-8 | 0.5 M NaHCO3 | 300 W Xe lamp with a 420 nm cutoff filter | FE = 74.2% at −0.7 V vs. RHE | 194 |
| g-C3N4/SnO2 | 0.5 M NaHCO3 | 300 W Xe lamp | FE = 58% at −0.5 V vs. RHE | 195 |
| Bi2CuO4 | 0.5 M KHCO3, 0.5 M Na2SO4 | AM1.5G filter | 273.56 μmol cm−2 h−1 at −0.9 V vs. RHE | 196 |
| CoPcS/GO-COO | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 83.9% at −1.0 V vs. Ag/AgCl | 197 |
| Cu–Bi2Se3 | 1 M Na2SO4 | 300 W Xe lamp | FE = 65.31% at −0.5 V vs. RHE | 198 |
| Cu2O-AF-PSi | 0.2 M Na2SO4 | 1 Sunlight | FE = 61% at −0.3 V vs. Ag/AgCl | 199 |
| Cu–SnO2/ZIF-8 | 0.5 M NaHCO3 | 300 Xe lamp | FE = 68.96% at −0.364 V vs. RHE | 200 |
| Ni@In/SiNW | 0.10 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 87% at −1.2 V vs. RHE | 31 |
| IO-TiO2|FDH | 86 mM MOPS, 50 mM NaHCO3, 50 mM CsCl | AM 1.5G of 100 mW cm−2 | FE = 83% at 0.4 V vs. RHE | 100 |
| Cu6Sn5 | 0.05 M H2SO4 | AM 1.5G of 100 mW cm−2 | FE = 61% at −0.91 V vs. SHE | 201 |
| Si/dT/Bi | 0.10 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 82.7% at −0.2 V vs. RHE | 202 |
| BiVO-/ZIF-8 | 0.5 M NaHCO3 | 300 W Xe lamp | FE = 91.24% at −0.9 V vs. RHE | 119 |
| Mn complexes | 2 M triethylamine and 2 M isopropanol-MeCN | 1 Sun illumination | FE = 96% at −1.75 V vs. Fc+/Fc | 203 |
| Cu2O/Ga2O3/TiO2/In | 0.10 M KHCO3 | AM 1.5G | FE = 62.1% at −0.9 V vs. Ag/AgCl | 204 |
| OPV|IO-TiO2|FDH + CA | 0.05 M NaHCO3–KCl | AM 1.5G | FE = ∼100% at 0.6 V vs. RHE | 205 |
| ITO-CF|FDHNvH | 0.1 M MOPS-DMSO-NAD + -acetophenone | AM 1.5G of 100 mW cm−2 | FE = 99% at −0.25 V vs. RHE | 206 |
| CNX-ITO|FDH | 100 mM NaHCO3, 50 mM KCl, and 7.5 mM MBA | AM 1.5G of 100 mW cm−2 | FE = 94% | 207 |
| Ultra-thin TiO2 | 0.10 M KHCO3 | 1 Sun illumination | FE = ∼98% at −0.5 V vs. RHE | 208 |
As a star candidate material in the field of photoelectrocatalytic reduction of CO2 to formate, Bi-based materials have excellent properties due to their unique electronic structure characteristics. The binding energy between the 5p orbital of Bi and the key intermediate OCHO is in the optimal range, which can not only promote the stabilization of the intermediate by moderate adsorption, but also avoid the difficulty of product desorption caused by excessive adsorption, thus effectively reducing the reaction energy barrier. In addition, the weak adsorption of H on the Bi surface significantly inhibits the HER, leading to the preferential flow of electrons to the CO2 reduction path. At present, the main research systems include pure Bi-based, Bi–Cu-based, Bi–Sn-based, etc., and performance optimization is achieved through strategies such as alloying, doping control, and heterostructure construction. In terms of interface engineering research, Dong et al.191 revealed the enhancement mechanism of interfacial electron interaction on catalytic activity by constructing a bismuth nanoparticle (Bi-NP)/gallium nitride nanowire (GaNNW) interface system (Fig. 13a–c). Theoretical calculations confirmed that the Bi-GaN binary system can optimize the synthesis pathway of HCOOH and enhance the electron transfer efficiency from GaNNWs to Bi–NPs. The experimental results showed that the optimized Bi/GaN/Si photocathode has a FEHCOOH of 98% under standard AM 1.5G illumination conditions and −0.3 V vs. RHE potential. The nanosheet structure of Bi/Bi2O2CO3 composite films prepared in situ significantly improved the reaction kinetic parameters.77 At −0.95 V vs. RHE potential, the FE of HCOOH reached 92.68%, and the performance remained stable over 10 h. It is worth noting that its highest bias photon current efficiency (1.19%) and cathode energy efficiency (61%) indicate that the catalyst has excellent energy conversion ability. In addition, the interaction between Bi and Cu elements also contributes to the formation of HCOOH. Cu2O@Bi-300/Si photocathodes were prepared using Bi-MOF as a template.179 The characterization showed that the bridging effect of Cu–O–Bi bonds and the synergistic effect of coordinated unsaturated Bi sites lead to excellent carrier separation efficiency. Under the condition of −0.3 V vs. RHE, the formation rate of formate reached 101.1 μmol cm−2 h−1 (FE = 95%), which was 10.2 times higher than that of the Bi-300/Si system, and the stability was maintained for 50 h. Combined with in situ FT-IR data and DFT calculations, it was confirmed that Cu2O-induced electron-rich Bi sites can enhance the chemical adsorption of CO2 and stabilize key intermediates. The CuSe@BiOI heterojunction system was regulated using an epitaxial growth strategy, and its BiOI (102) anisotropic charge transport properties significantly improved the charge separation efficiency.65 In 0.5 M KHCO3 electrolyte, the system exhibited a formate selectivity of 80.5%. In-based and SN-based materials have also demonstrated potential for HCOOH production, and are often combined with Bi-based materials to enhance HCOOH selectivity. As shown in Fig. 13d and e, the integrated system comprising an InGaP/GaAs/Ge photoanode and a Sn-modified BiOx cathode achieved 100 h of continuous operation in a non-auxiliary two-electrode system, with an average FE of 88%, a yield of 17.3 mmol L−1 h−1, a solar-fuel conversion efficiency (STF) of 12%, and an electrical energy efficiency (EE) of 60%.182 The mechanism analysis showed that the metal-semiconductor heterostructure formed by the Sn–BiOx interface can optimize the electron transport path. At the potential of −1.02 V vs. RHE, the FE of formate reached 88.67%, and the yield was 80.07 μmol h−1 cm−2.69 The band structure analysis confirmed that the cascade band structure formed by Bi–Sn alloy and Si nanowires significantly promotes photogenerated electron migration. The In-doped Bi2O3/Cu2O foam photocathode had a three-dimensional hierarchical nanoflower structure, which maintained FEHCOOH ≥ 90% in a wide potential window of −0.87 ∼ −1.17 V vs. RHE, with a peak efficiency of 97.8% (J = 14.41 mA cm−2).175 The oxygen vacancies induced by In doping and the Bi2O3/Cu2O heterojunction synergistically enhance the efficiency of intermediate adsorption and charge separation.
 | ||
| Fig. 13 Schematic diagram of (a) PEC CO2 RR and (b) energy band diagram. (c) FE of HCOOH with different samples. (d) Mechanistic demonstration of the PEC device during the CO2RR. Reproduced with permission from ref. 191. Copyright 2022, The Royal Society of Chemistry. (e) FE of HCOOH and H2 production rates with the Sn–Bi2O3 NCs cathode. Reproduced with permission from ref. 182. Copyright 2024, Springer Nature. (f) The device for CO2RR to HCOO−. (g) Cross-over plots under 3-sun irradiation. (h) Product detection from PEC experiments. Reproduced with permission from ref. 96. Copyright 2024, published by Elsevier Inc. | ||
In addition to Bi-based materials, the material systems based on Cu, In, Sn, and Cd can also achieve highly selective catalytic conversion of HCOOH products through component optimization and structural regulation. The bifunctional Cu–SnO2/ZIF-8 catalyst constructed by the combination of hydrophobic ZIF-8 and hydrophilic Cu–SnO2 enables the direct use of gas phase CO2 to improve the molecular activation efficiency and effectively inhibit the HER.200 Under an overpotential of −364 mV vs. Ag/AgCl, the FE of formic acid increased to 68.96%, and a peak current density of 12.8 mA cm−2 was obtained at −1.4 V vs. Ag/AgCl. The FeOOH/Cu2O/Co:CdS multistage heterojunction photocathode constructed by sequential electrodeposition combined with chemical bath deposition showed significantly enhanced photovoltage, which was 1.9 times higher than that of the existing photocathode, and the FE of CO2 reduction to formic acid reached 82.9%.176 The Co:CdS quantum dots on the outer layer of the photocathode significantly promoted formic acid formation by introducing additional hybrid orbitals that enhance the binding energy to the key intermediate *OCHO. In addition, doping Co into the composite material containing CdS introduces impurity levels, which not only reduce the band gap of the photocathode, but also improve its visible light absorption capacity, thus significantly improving the photochemical properties. Wei et al.174 proposed a new strategy for constructing VS-ZnIn2S4/TiN-x foliated heterojunctions. Due to its plant cell-like morphological characteristics and enhanced electron mobility from the heterojunction interface to surface active sites, the system demonstrated excellent catalytic performance with a product formation rate of 1173.2 μM h−1 cm−2.
Organic catalyst systems, including organic polymers, molecular catalysts, biological enzyme catalysts, etc., show high reduction selectivity toward HCOOH products. The composite photocatalyst of polymer carbon nitride (PCN) and defective nano-silica (n-SiOx) has a higher specific surface area, improved photoresponse and reduced charge recombination.184 The synergistic effect of the PCN-n-SiOx interface promoted photoinduced charge transfer from PCN to n-SiOx, which inhibited carrier recombination. The resulting PCN-n-SiOx composite was three times more effective in reduction of CO2 to methanol and formic acid. In addition, during the CO2 reduction process, the composite selectively produced 283.0 μmol L−1 formic acid, which was five times higher than that produced using bare PCN (57.0 μmol L−1). Nandal et al. combined cobalt tetrasulfonamide phthalocyanine (CoPcS) with carboxylated graphene oxide.197 The chemical connection of the CoPcS complex unit to the carboxylated GO carrier effectively provided enough interfacial area to promote the adsorption of CO2 and increase CO2 concentration, thus achieving a higher conversion rate. Under simulated sunlight irradiation, a high current density (−1.5 mA cm−2) was obtained at a voltage of −1.0 V vs. Ag/AgCl, and the selective formation rate of formate was 2.35 mmol h−1 cm−2. As shown in Fig. 13f–h, Cobb et al.96 innovatively used formate dehydrogenase (FDh) as a model catalyst to achieve the selective conversion of CO2 to formate under minimum overpotential. The catalytic activity of the FDh cathode and BiVO4 photoanode was facilitated by solar heating, which allowed the non-biased semi-artificial FDH-TE-BIVO4 device to produce HCOOH with a FE of 97% under 3 Suns of solar irradiation.
The formation pathway of HCOOH has the advantages of simple reaction steps and a low thermodynamic barrier. Its formation mechanism mainly depends on the stabilization of key intermediates *OCHO or *COOH in the CO2 reduction process, and then the formation of HCOOH. And the selective formation of formic acid faces less competition from by-products. Therefore, the available results show high selectivity. The Bi-based catalysts (including their alloying structure, heterojunction complex and doping modification) have become promising materials to achieve highly selective formic acid generation (FE > 90%). Other materials such as Cu base, In-base and Sn-base also show good formic acid selectivity, and some materials can achieve FEs close to 90% after reasonable design. It is worth noting that systematic studies of non-metallic catalysts (such as organic polymers, molecular catalysts and bioenzyme catalysts) have also confirmed their significant selective advantages, providing an important research direction for the development of efficient and low-cost catalytic systems. In general, with the continuous development of catalyst design, photoelectric catalytic CO2 reduction to formic acid has shown considerable application potential.
| Catalyst | Electrolyte | Light source | Product and performance | Ref. |
|---|---|---|---|---|
| CuO-graphene-ZnFe2O4–TiO2 | 0.04 M NaHCO3 | UV light (λ = 254 nm) | FE = 44.8% | 210 |
| BiOBr | KHCO3 | — | FE = 60.68% at 1.2 V vs. RHE | 211 |
| Macroporous conjugated polymer | 0.10 M KHCO3 | AM 1.5G | FE = 85% at 0 V vs. RHE | 212 |
| TNT/Cu2O/Au | 0.1 M Na2SO4 | 300 W Xe lamp | FEMeOH = 43% and FEMethane = 27% at 0.2 V vs. Ag/AgCl | 213 |
| g-C3N4/ZnO | 0.1 M KHCO3 | UV LED light (365 nm, 4 W) of 80 mW cm−2 | FE = 23% at −1 V vs. RHE | 214 |
| CNT/CoPc-NH2 | 0.1 M KHCO3 | 300 W Xe lamp | FE = 20% at −10.7 V vs. RHE | 215 |
| BiVO4 | 0.5 M NaHCO3 | 100 W LED | (MeOH) 22 and (HAc) 5.5 mmol cm−2 at −1.0 V vs. Ag/AgCl | 216 |
| CoPc/CN | [BMMIm]Br | 300 W Xe lamp in the 350–780 nm range | (MeOH) 6465.9 and (EtOH) 218.6 mmol cm−2 at −1.2 V vs. RHE | 101 |
| Cu2O/PANI/SiPY | 0.5 M CuSO4 | Visible light (70 W) | FE = 57.66% at −1.3 V vs. SCE | 72 |
| Mo/CuGaS2/CdS/TiO2 | 0.1 M Na2SO4 | AM 1.5G of 100 mW cm−2 | FE = 65% at −0.7 V vs. RHE | 217 |
| Ti/TiO2/PDA/phosphorene | 0.1 M Na2SO4 | 300 W Xe lamp | 458 μmol L−1 at −0.8 V vs. Ag/AgCl | 104 |
| rGO-CuO/Cu | 0.5 M Na2SO4 | Xe lamp of 100 mW cm−2 | 3.3 g L−1 h−1 at −0.8 V vs. Ag/AgCl | 218 |
| Ag/α-Fe2O3 | 0.1 M KHCO3 | AM 1.5G | FE = 51% at −0.5 V vs. RHE | 209 |
| Re doped CuO/TiO2 NTs | 0.1 M NaHCO3 | 300 W Xe lamp | (MeOH) 19.9 and (EtOH) 7.5 mmol 5 h−1 at −0.4 V vs. RHE | 63 |
| TiO2-NT/GNR | 0.1 M Na2SO4 | 150 W Xe lamp | FEMeOH = 84.17% and FEEtOH = 13.78% at 1.0 V vs. Ag/AgCl | 219 |
| CuFeO2/CuInS2 | 0.1 M CH3COONa-10 mM pyridine | Xe lamp of 100 mW cm−2 | FE = 87% at −0.65 V vs. SCE | 220 |
| Ti/TiO2NT-Ru3(BTC)2 | 0.1 M Na2SO4 | 300 W Xe lamp | 314 μmol L−1 at −0.5 V vs. Ag/AgCl | 221 |
| Au/α-Fe2O3/RGO | 0.1 M KOH | A Xe lamp (cutoff range: 400–450 nm) | FE = 91% at −0.6 V vs. RHE | 222 |
| Cu/Cu2O–Cu(BDC) MOF | 0.1 M Na2SO4 | 300 W Xe lamp | 225 μmol L−1 h−1 at 0.1 V vs. RHE | 223 |
| Au@TNT | 0.5 M KHCO3, 0.5 M Na2SO4 | AM 1.5G | (MeOH) 2.5 and (EtOH) 2 mM h−1 at 1 V vs. Ag/AgCl | 224 |
| Cu | 0.1 M NaHCO3 | 470 nm light source | FE = 73% at −0.4 V vs. NHE | 225 |
| rGO/Sn3O4/SnO2 | 0.5 M Na2SO4 | 300 W Xe lamp | FE = 45% at −0.3 V vs. Ag/AgCl | 226 |
| Ag–TiO2/RGO | 1.0 M KOH | UV-vis light | FE = 60.5% at −0.7 V vs. RHE | 227 |
| Cu@Cu2O/TiO2/Ti3C2 | 0.1 M KHCO3 | 300 W Xe lamp | (MeOH) 1276.2 and (EtOH) 960.5 μMg−1 h−1 at −0.8 V vs. SCE | 118 |
Early studies showed that TiO2, as a typical semiconductor material, can effectively promote the CO2RR due to its excellent photocatalytic properties, especially in catalyzing the conversion of CO2 to CH2OH.24 With advances in photocatalysis technology, TiO2 nanotubes (TNTs) have been developed as electrode substrate materials due to their unique structural advantages. The ordered nanoarray structure of TNTs has both a high specific surface area and unique nanoscale porosity characteristics, which not only greatly increases the density of active sites, but also promotes the separation efficiency of photogenerated electron–hole pairs by constructing directional charge transport channels, thereby systematically improving the catalytic performance. Further studies have shown that surface modification with transition metals or precious metal doping can effectively regulate the energy band structure of TNTs, and optimize the selectivity and kinetics of the CO2 reduction reaction path. A Ru3(BTC)2 metal–organic framework (MOF) film was constructed on the Ti/TiO2 nanotube substrate to develop a new photocathode system. The design cleverly combines the porous adsorption properties of the Ru based MOF with the photoresponse advantage of anatase type TiO2, showing enhanced photocurrent response characteristics under UV-visible light irradiation.221 The experimental data showed that under optimal reaction conditions, 314 μmol L−1 methanol could be generated by the photocatalytic reaction of the composite electrode for 3 h, with a yield 2.3 times higher than that of the traditional photocatalytic system, which confirmed the effectiveness of the interface engineering strategy. Zanoni's research group213 innovatively introduced a polydopamine (PDA) interface layer as an electron transport medium by constructing a heterogeneous structure of Cu2O–Au/TiO2 nanotubes. The results showed that the π-conjugated electron system of PDA can significantly promote the interfacial charge transfer, which enabled the cube-like catalyst to exhibit better performance than the octahedral catalyst, achieving the selective co-production of methanol (43%) and methane (27%). This finding provides an important experimental basis for the study of morphology–property correlation. The plasma silver–titanium dioxide/reduced graphene oxide (Ag–TiO2/RGO) nanohybrid system designed by Banat's team227 showed excellent performance. The material prepared by hydrothermal synthesis has clear structural characteristics: 4 nm silver nanoparticles and 7 nm TiO2 nanoparticles are uniformly loaded on the surface of micron-scale two-dimensional RGO nanosheets. The photochemical test showed that the photocathode achieved a current density of 23.5 mA cm−2 and a methanol yield of 85 μmol L−1 cm−2 at an initial potential of −0.7 V under UV-visible irradiation in a 1.0 M KOH electrolyte system. The quantum efficiency was 20% and FE was 60.5%. Otgonbayar et al.210 constructed a CuO–graphene–ZnFe2O4–TiO2 quaternary heterojunction that exhibited unique advantages. The results showed that the material achieved 44.08% FE in the buffer electrolyte system under ultraviolet irradiation, and maintained 42.2% FE in an electrolyte system with NaHCO3. The mechanism study showed that the gradient band arrangement built in the heterojunction effectively promotes carrier separation. Notably, the significant effect of proton concentration on the FE value reveals the critical role of electrolyte engineering in optimizing the reaction system.
The graphene oxide (GO) based catalytic system has shown unique advantages in the field of methanol synthesis. As shown in Fig. 14a and b, Liu's research team228 innovatively integrated adenine functionalized graphene oxide (A-GO) as an interface modification layer on the surface of the Cu2O photocathode, and constructed an A-GO/Cu2O composite catalytic system. The A-GO interface layer significantly optimized charge transport kinetics at the electrode interface through its abundant π–π conjugated network, thereby simultaneously improving the photoelectrochemical activity and cyclic stability of the system. The photocurrent density of the A-GO/Cu2O photocathode at −1.1 V vs. Ag/AgCl potential was 2.74 mA cm−2, which was 2.23 times higher than that of the bare Cu2O and GO/Cu2O systems, and 1.56 times higher than that of the bare Cu2O and GO/Cu2O systems, respectively. The mechanism study showed that the Lewis base site in the adenine molecule and the N atom in the pyrimidine ring could cooperatively regulate the adsorption energy barrier of CO2 intermediates, thus optimizing the PEC CO2RR path. The system achieved a 69.25% FE of methanol products in the continuous reaction, which confirmed the effectiveness of the interface engineering strategy of biomolecules and graphene oxide. In addition, Banat's team222 developed a PEC electrolyzer based on the Au/α-Fe2O3/RGO photocathode and Ru/RGO anode. This innovative design synchronized the bi-functional reaction of CO2 reduction at the cathode to methanol and the oxidation of furfural (FF) at the anode to produce 2-furanoic acid (2-FA) and 5-hydroxyfuranoic acid (5-HFA). The photocathode performance test showed that under visible light irradiation and −0.6 V bias, the system achieved a quantum efficiency of 21.5%, a stable methanol yield of 63 μmol L−1 h−1 cm−2, and a FE record of 91%. The simultaneous anodic reaction showed that the conversion rate of furfural was as high as 82%, and the yield of 2-FA and 5-HFA was 37% higher than that of conventional electrochemical system. This mechanism improves the overall energy efficiency of the system and provides a new paradigm for PEC system design.
 | ||
| Fig. 14 (a) A schematic diagram of the charge transfer mechanism of A-GO/Cu2O in the PEC CO2RR. (b) FE of CH3OH production. Reproduced with permission from ref. 228. Copyright 2021, Elsevier B.V. All rights reserved. (c) Schematic depiction of solar CH3OH production by the MCN photoelectrode with enhanced photogenerated electron flux. (d) Photocurrent density generated by solar irradiation of MCN-CA and MCN-C. (e) In situ FT-IR spectra of cobalt–phthalocyanine catalyst assembly. (f) Schematic illustration of the cooperative CO2-to-CH3OH conversion mechanism for cobalt–phthalocyanine catalyst assembly. Reproduced with permission from ref. 212. Copyright 2024, American Chemical Society. | ||
A variety of organic catalytic systems also show significant potential in selectivity. Xiao's team215 successfully realized the efficient synthesis of methanol by constructing a micro-space environment using the CoPc-NH2 molecular catalyst. The silicon micropillar array structure significantly improved the catalyst loading and interface bonding strength while maintaining the light absorption efficiency by expanding the electrode surface area, and enriched the gaseous intermediates near the active site of the catalyst through the confinement effect. The introduction of superhydrophobic coating effectively inhibited the occurrence of side reactions, and realized the local enrichment of reaction intermediates through capillary action. After the single-electron reduction process of the molecular catalyst, the semiconductor–catalyst interface structure changed from an adaptive junction to an embedded junction, and the interface reconstruction provided sufficient thermodynamic driving force for the CO2 reduction reaction. The above synergistic effect enabled the system to achieve a 20% methanol FE during the CO2 electroreduction process, with a partial current density of 3.4 mA cm−2 which was 17 times higher than that of the planar silicon photoelectrode reference system. Inspired by the porous structure of photosynthetic organelles, Shan's research team212 developed a novel photoelectrode based on a single-pore macroporous conjugated polymer network (MCN) (Fig. 14c–f). The material achieved supramolecular assembly of photocatalytic components through rich functional groups, overcoming the geometric limitations of traditional inorganic materials. A stable interface was formed between MCN and the photocatalyst through a strong chemical bond network. The results showed that the Co-based catalyst and MCN composite system can produce highly reducing electrons under light conditions, and realize the CO2 to CH3OH conversion (conversion efficiency 70%), and the process can run for more than 100 h, with only a slight decline in activity. It is worth noting that when the electrode area was enlarged from 1 cm2 to 100 cm2, the system still maintained a stable photocurrent output of 0.25 A, and continued to generate CH3OH at 85% conversion efficiency, which confirmed the feasibility of large-scale application. In the field of organic–inorganic hybrid materials, Hocine's team72 found that the combination of polyaniline (PANI) and cuprous oxide (Cu2O) significantly improved the CO2 reduction performance of the PEC system. This enhancement effect was due to the enhancement of CO2 chemisorption capacity on the photocathode surface and the enhancement of photogenerated e−/h+ on the separation efficiency. The electrochemical test showed that the application of potential could significantly regulate the stability of photocurrent. When the Cu2O/PANI/SiPY heterojunction was used as the photocathode, a methanol FE of 57.66% was obtained at −1.2 V (vs. SCE) bias. Chidambaram's research group214 prepared a composite catalyst of g-C3N4 organic material supported by ZnO inorganic nanorods. At −1 V bias, the photocurrent density of the composite was 2.8 times higher than that of bare g-C3N4. The reduction efficiency of PEC CO2 to methanol was 2.86 times greater than that of the bare ZnO catalyst. The performance enhancement was mainly due to three factors: (1) the higher specific surface area provides abundant active sites; (2) increased carrier generation rate; (3) reduced e−/h+ pair recombination probability. Compared with a single component, g-C3N4/ZnO composites showed significant advantages in methanol yield.
Therefore, as a liquid C1 reduction product, methanol has been increasingly studied in recent years. Although the methanol generation process is relatively complex, it still has certain advantages compared with other high value-added products. In recent years, researchers have significantly improved the efficiency of methanol generation and FE by optimizing catalyst designs, such as the use of TiO2 nanotubes, Cu-based catalysts, and graphene oxide carriers. In addition, some organic catalysts and organo–inorganic hybrid materials also showed excellent catalytic activity in methanol generation. In particular, the precise regulation of the microspace environment may be a key factor in achieving selective methanol generation.
| Catalyst | Electrolyte | Light source | Product and performance | Ref. |
|---|---|---|---|---|
| AgxCu100x-Si MP | 0.1 M KHCO3 | AM 1.5G | FEsyngas = 16.7% and FEmethane = 9% | 229 |
| Si/TiO2/trzpOs | 0.5 M Na2SO4 | AM 1.5G of 100 mW cm−2 | FE = 91.8% at 0 V vs. RHE | 230 |
| Cl–Cu2O/ZnO | 0.1 M KHCO3 | AM 1.5G | FE = 88.6% at −0.3 V vs. RHE | 102 |
| PbS/CsPbBr3-PNC | 0.1 M TBAPF6 | 300 W Xe lamp | (CO) 2.94 and (methane) 0.36 μmol cm−2 h−1 at −0.6 V vs. Ag/AgCl | 231 |
| CuBDC/CF | 0.1 M KHCO3 | 300 W Xe lamp | FE = 5% at −0.1 V vs. RHE | 232 |
| Cu–Ag | 0.1 M KHCO3 | 100 mW cm−2 | FECO = 79.8% at 1.0 V vs. RHE; FE = 59.3% at −1.4 V vs. RHE | 233 |
| Au/p-GaN | 0.05 M KHCO3 | Visible light | FE = 32.9% at −1.3 V vs. Ag/AgCl | 234 |
In recent years, studies have shown that Cu-based catalysts are commonly used catalysts for the selective generation of CH4. As shown in Fig. 15a and b, Liu's team102 achieved a methane faradaic efficiency of 88.6% and catalytic stability for more than 5 hours by constructing a chlorine-element-modified Cu2O/ZnO heterostructure system. The study revealed that the Cl− ions at the heterointerfaces effectively inhibited the photocorrosion phenomenon of Cu2O through passivation, and the stabilized Cu+ active sites significantly enhanced the hydrogenation process of the CO intermediate. Density functional theory calculations showed that compared with the energy barrier of 0.344 eV required for simple CO desorption, the CO intermediate in this heterostructure system tended to form the *CHO intermediate state through a low-energy barrier path of 0.220 eV. This characteristic explains the underlying reason why its methane selectivity is significantly better than the 60% benchmark value reported in earlier literature. In the study of bimetallic systems, the Cu–Ag composite catalyst also showed the advantage of regulating the selectivity of CH4.229 When the thickness of the Cu film decreased, its grain boundary density increased, and the Ag layer (3 nm) deposited on the surface showed a discrete island-like distribution feature.233 The uncoordinated Cu atoms at the grain boundaries are prone to spontaneous oxidation in an ambient atmosphere, and this interfacial oxidation phenomenon directly led to a decrease in the faradaic efficiency of CO and CH4. It is worth noting that when the thickness of the Cu layer reached more than 80 nm, the grain boundary oxidation process was effectively suppressed. At this time, the thin film catalyst showed catalytic performance comparable to that of the bulk Cu–Ag material. The optimized Cu (100 nm)–Ag (3 nm) composite film achieved a CO selectivity of 79.8% and a CH4 selectivity of 59.3% at −1.0 V vs. RHE and −1.4 V vs. RHE potentials, respectively, showing significant bifunctional catalytic characteristics. Further research has been extended to the photoelectrochemical system. By constructing a patterned array of Cu–Ag thin films on the surface of the p-type silicon photocathode and introducing a SiO2 passivation layer, the research team successfully developed a new type of composite photocatalyst. Under standard light intensity (100 mW cm−2), this structure simultaneously improved the selective production efficiency of CO and CH4, providing an innovative device design scheme for solar-driven CO2 conversion.
 | ||
| Fig. 15 (a) Schematic illustration of the CCZO, (b) FE and total passed charge at different applied potentials for CCZO. Reproduced with permission from ref. 102. Copyright 2022, Elsevier B.V. All rights reserved. (c) Solar-driven CO2-to-CH4 conversion on the Si/TiO2 electrode in the presence of the [Os] complex. (d) FE of Si/TiO2, Si/TiO2/przpOs and Si/TiO2/trzpOs at 0 V vs. RHE. Reproduced with permission from ref. 230. Copyright 2024, Springer Nature. | ||
To significantly improve the selectivity of methane products in the photoelectrochemical carbon dioxide reduction reaction (PEC CRR), researchers have introduced molecular organic catalyst systems into this field. As shown in Fig. 15c and d, Jian's team230 innovatively used two osmium-based complexes—pyrazole-functionalized osmium complex (przpOs) and triazole-functionalized osmium complex (trzpOs)—as molecular catalysts for the selective reduction of CO2 to prepare CH4. Kinetic studies showed that przpOs and trzpOs exhibit high catalytic rate constants of 0.544 s−1 and 6.41 s−1, respectively, in the catalytic reduction of CO2, and this difference revealed the significant regulatory effect of the ligand structure on the reaction kinetics. Under AM1.5G standard solar irradiation, the optimally designed Si/TiO2/trzpOs composite photocathode shows excellent catalytic performance. The faradaic efficiency of its main product, CH4, reached 91.8%, and a photocurrent density of −14.11 mA cm−2 was achieved at 0.0 V vs. RHE potential. The nitrogen sites on the bipyrazole and triazole ligands effectively stabilized the adsorption configuration of the key intermediate *COOH through multiple coordination, promoting the reaction path toward deep hydrogenation to generate CH4, thereby endowing the system with ultra-high CO2-to-CH4 selectivity. It is worth noting that the comprehensive performance indicators of this system (including faradaic efficiency and current density) have reached the cutting-edge level among silicon-based photocathode materials for CO2 reduction, fully verifying the application potential of molecular catalysts in photoelectrochemical methane synthesis. In addition, Khezri's research group235 successfully achieved a methane faradaic efficiency of 74% by regulating the dopamine polymerization time to 15 hours to form a polymer coating layer and combining with the surface modification of copper nanoparticles, which further expanded the design idea of organic–inorganic hybrid catalysts in the field of CO2 reduction.
In PEC CO2RR, the selective production of CH4 is more challenging than the conversion of CO2 to CO because the generation of methane involves more complex multi-step reactions, including the breaking of the C–O bond and the formation of the C–H bond. Although Cu-based catalysts show certain advantages in the selective generation of methane, the problem of excessive generation of CO intermediates still needs to be overcome. Further modification of Cu-based catalysts (such as constructing bimetallic systems, heterostructures, and other modification methods) is still needed to significantly improve the faradaic efficiency of methane and improve the stability of the catalyst. In addition, organic catalysts also show great potential in improving the selectivity of methane. Therefore, in-depth studies on catalyst design and reaction mechanisms are needed, especially by regulating the surface structure of the catalyst and the generation path of intermediates, and it is expected that more efficient and selective methane generation can be achieved in the future.
3.2 C2+ products
In the process of PEC CO2RR, the controllable synthesis of C2+ products faces more significant technical bottlenecks compared to C1 products. The challenge stems from the complexity of the multistep reaction pathways and the dual effects of thermodynamic and kinetic co-constraints.19 On the one hand, in the CO2 reduction reaction, the generation of C2+ products often competes with side reactions such as the formation of CO, hydrogenation reactions, etc. Therefore, these side reactions need to be suppressed and the appropriate reaction pathways need to be promoted to improve the selectivity of C2+ products. Especially, the efficiency and selectivity of C–C coupling directly affect the yield of C2+ products. The intermediates generated during the reaction (such as *CH2 and C2H4O) have low stability and are prone to desorption or further decomposition into C1 products, making the selectivity difficult to control. On the other hand, the formation of C2+ products requires multiple consecutive PCET steps (usually requiring more than 8 electrons to participate) and involves high-energy barrier C–C coupling reactions (such as the dimerization of *CO to form the *OCCO intermediate), imposing higher demands on the design of the active sites of the catalyst and the efficiency of electron transfer.| Catalyst | Electrolyte | Light source | Product and performance | Ref. |
|---|---|---|---|---|
| S. ovata/p-Si | Bacterial medium, 0.1 M glycerol in 1 M KOH | Red light (740 nm) of 20 mW cm−2 | FE = 80% at 0 V vs. RHE | 107 |
| CO3O4/TiO2 | 0.5 M Na2SO4 | 300 W Xe lamp | (HAc) 7, (MeOH) 6.5 and (HCOOH) 5.2 mg L−1 cm−1 h−1 | 240 |
| BiOI–PdCu | 0.1 M KHCO3 | 420 nm LED lamp | (HCOOH) 0.14 μmol g−1 h−1 at −0.12 V vs. NHE; (HAc) 0.18 μmol g−1 h−1 at 0.11 V vs. NHE | 239 |
| Ag/Cu2O | 0.1 M Na2SO4 | 455 nm LED lamp (100 W cm−2) | FE = 54% at −0.4 V vs. Ag/AgCl | 241 |
| CuO/g-C3N4-microorganisms | — | 100 W iodine–tungsten lamp | 5.1 g L−1 at −1.05 V vs. RHE | 242 |
| Cu2O/Ag | 0.1 M KHCO3 | AM 1.5G | FE = 47.7% at −0.7 V vs. RHE | 238 |
| α-Fe2O3/g-C3N4-microorganisms | — | 100 W iodine–tungsten lamp | 0.33 g per L per day at −0.9 V vs. Ag/AgCl | 73 |
| Zn–Cu2O | 0.1 M KHCO3, 0.5 M Na2SO4 | 300 W Xe lamp of 200 mW cm−2 | FE = 58.1% at −0.5 V vs. Ag/AgCl | 237 |
| TiO2/Cu2O | 0.1 M KHCO3 | 300 W Xe lamp | 20 μmol cm−2 at −0.4 V vs. RHE | 243 |
| Cu2O–TiO2 | 0.1 M KHCO3, 1 M NaOH | 300 W Xe lamp | FE = 61.9% | 236 |
Cu-based catalysts are a common choice in the catalysis of C2+ products. In the reduction reaction of Hac, Cu-based catalysts often exist in the form of metal oxides or alloys. Studies have shown that when applied to the reduction reaction of HAc, copper-based materials often present their catalytic activity in the form of metal oxides or alloys. Giusi et al.236 reported the successful construction of a Cu2O–TiO2 heterogeneous structure catalytic system through ultrasound-assisted coprecipitation. Experimental data showed that in the bare Cu2O/GDL electrode system, the production rates of formic acid and acetic acid were 31.8 and 80.6 μmol h−1 g−1, while the catalytic performance of the Cu2O–TiO2/GDL composite electrode was significantly improved, with the corresponding yields reaching 0.69 and 2.59 mmol h−1 g−1. Among them, the FE of acetic acid production was further increased to 61.9%. This performance enhancement can be attributed to the synergistic electrocatalytic effect at the Cu2O–TiO2 heterogeneous interface. This structure effectively promotes the formation of C–C bonds by optimizing the electron transfer path, providing a new approach for the sustainable synthesis of C2+ chemicals and fuels. Further studies showed that the coupling system of a Au-loaded nitrogen-doped TiO2 nanoarray photoanode and a Zn-doped Cu2O dark cathode realized the efficient conversion of carbon dioxide to acetic acid at a potential of 0.5 V (vs. Ag/AgCl), with a FE of 58.1% (carbon selectivity of 91.5%).237 The analysis showed that the Zn dopant improved the catalytic selectivity in the Cu2O lattice through a dual mechanism: on the one hand, it regulated the local electronic structure, and on the other hand, it modified the configuration of the active sites. The two synergistically promote the formation of the key intermediate *CH2/*CH3 and its C–C coupling process. At the same time, the efficient carrier separation ability of the nitrogen-doped TiO2-based photoanode provides sufficient electron supply for the multi-electron transfer CO2 reduction reaction. As shown in Fig. 16a–c, another innovative study used plasma metal (Ag) to modify the Cu2O nanowire array to construct a photoelectrochemical catalytic system with enhanced charge separation efficiency.238 Experimental results showed that the acetic acid production rate of the Cu2O/Ag composite photoelectrode reached 212.7 μmol cm−2 h−1 under a bias of 0.7 V (vs. RHE) under light conditions, and its FE was 47.7%, which was 4.8 times higher than that under dark conditions (44.4 μmol cm−2 h−1), confirming the key role of the plasma effect in strengthening the surface catalytic reaction. In the area of multi-metal composite catalyst research, the catalytic performances of three composite materials, BiOI–Pd, BiOI–Cu, and BiOI–PdCu, were systematically compared.239 Electrochemical tests showed that at a potential of −0.85 V (vs. RHE), the Faraday current density of the BiOI–PdCu composite electrode reached 3.15 mA cm−2, which was significantly better than that of the BiOI–Pd (2.06 mA cm−2) and BiOI–Cu (2.15 mA cm−2) systems. It is worth noting that this bimetallic composite catalyst showed a unique light response characteristic under visible light irradiation, and its main reduction products were formic acid and acetic acid, providing an important reference for the development of a photo-electrochemical catalytic system.
 | ||
| Fig. 16 (a) Schematic diagram of reaction pathways. (b) Charge transport diagram of Cu2O/Ag for PEC CO2RR. (c) FE of products on the cathode under different applied potentials. Reproduced with permission from ref. 238. Copyright 2022, The Royal Society of Chemistry (d) Photocatalytic mechanism of MES. Comparison of (e) current density and (f) acetate yield. Reproduced with permission from ref. 242. Copyright 2021, published by Elsevier B.V. | ||
Combining the reaction device with a microbial system is an effective research strategy to achieve the synthesis of HAc. The research team successfully constructed a self-biased microbial photoelectrochemical cell (LMPC) based on the synergy of a gas diffusion photoelectrode cathode and a microbial anode.240 In this system, while achieving the oxidative degradation of anode pollutants, the process of reducing CO2 to liquid products at the photoelectrode cathode was simultaneously completed. Experimental data showed that after 10 hours of operation under light conditions, the production rate of acetic acid reached 70.4 ± 4.8 mg L−1 cm, which was 1.7 times higher than that of the unmodified photoelectrode cathode system. It is worth noting that this process was accompanied by the generation of a significant amount of methanol and formic acid by-products, and the selectivity of the acetic acid product was approximately 37%. It should be particularly pointed out that this technical approach does not directly utilize the acetic acid synthesis capability of microorganisms, but rather indirectly improves the system performance by enhancing the carrier separation efficiency through the microbial degradation effect. In the field of visible light-responsive photoelectrode microbial electrosynthesis (MES), recent studies have made significant progress. The Z-scheme heterojunction system constructed based on α-Fe2O3/g-C3N4 exhibits excellent photogenerated electron–hole separation characteristics.73 The unique energy band structure of α-Fe2O3, characterized by a low Fermi level facilitated the combination of anode transferred electrons and photogenerated holes, thereby providing an additional electrochemical driving force for the improvement of MES performance. More notably, the introduction of α-Fe2O3 significantly improved the electron transfer efficiency at the electrode–microorganism interface. Experimental results showed that the acetic acid yield of this composite material reached 0.33 g per L per d, which is 3 times higher than that achieved with the traditional carbon felt cathode system. This finding provides a new idea for the design of MES photoelectrode cathodes. Further innovative research showed that the CuO/g-C3N4 photoelectric material can be directly integrated into the MES system employing a mixed culture biocatalyst (Fig. 16d–f).242 This composite material not only exhibited excellent light absorption characteristics, but also achieved an ideal level of electron–hole separation efficiency. Mechanism analysis revealed that CuO/g-C3N4 mainly enhances the electron supply of electroautotrophic microorganisms through the direct electron transfer mechanism (rather than the traditional hydrogen-mediated indirect pathway), while the photogenerated holes produce a synergistic enhancement effect by capturing anode electrons. This photoelectrode cathode significantly reduced the charge transfer resistance of the system, improved the electron transfer rate, and ultimately achieved a significant 2.6-fold increase in the acetic acid yield by optimizing the biocatalyst loading and regulating the microbial community structure of the cathode.
Therefore, for this product, acetic acid, the catalytic process using Cu-based catalysts is similar to the generation process of other C2+ products to achieve efficient C–C coupling. Although its production can be achieved, the reaction environment of Cu-based catalysts requires strict regulation, often accompanied by the generation of a large number of additional products, resulting in a low selectivity of Hac. However, this product is special as some microorganisms can directly achieve its production, and it can be considered to be combined with the photoelectric system to stimulate microbial production with electrons. Therefore, constructing a photoelectrode microbial electrosynthesis (MES) system is a potential method for the highly selective reduction of carbon dioxide to acetic acid.
| Catalyst | Electrolyte | Light source | Product and performance | Ref. |
|---|---|---|---|---|
| Cu-cluster/GaN | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 61% at −0.74 V vs. RHE | 22 |
| Cu2O | 0.1 M KHCO3 | 100 W Xe lamp | FE = 57% at −1.4 V vs. RHE | 106 |
| Cu–TiO2 | 1 M KOH | UV LED (365 nm) | FE = 46.6% at −1.8 V vs. Ag/AgCl | 46 |
| CuNP/SiNW | 0.1 M KHCO3 | AM 1.5G | FE = 25% at −0.5 V vs. RHE | 244 |
| Cu2O | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 60% at −1.2 V vs. Fc+/Fc | 2 |
| Cu2Ta4O11 | 0.1 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 13.9% at 0 V vs. RHE | 245 |
In the research on photoelectrocatalysts for ethylene generation, copper-based materials have always been dominant. Roh et al.244 constructed a coupled system consisting of a silicon nanowire (SiNW) photocathode and a copper nanoparticle (CuNP) ensemble, which exhibited selectivity for CO2 conversion to C2H4 at −0.50 V vs. RHE, with a faradaic efficiency close to 25% and achieved a partial current density exceeding 2.5 mA cm−2. Notably, further stability tests showed that the CuNP/SiNW composite system could maintain stable CO2 reduction performance under constant potential illumination conditions for 50 hours. The above results experimentally confirmed the feasibility of the nanowire/Cu catalyst system as a modular platform, which could not only achieve efficient separation of photogenerated carriers but also promote the complex reaction pathways of C2H4 products through interface regulation. Particularly, copper oxides, where active sites formed by low-valence copper can efficiently promote C–C coupling, may be one of the important factors promoting ethylene formation. Liu et al.2 proposed a charge separation strategy based on the Ag co-catalyst, by constructing a Z-scheme heterojunction to accelerate photogenerated electron transfer and selectively extract holes, thereby effectively suppressing the self-reduction of Cu2O under light conditions and hole-induced oxidative degradation. The optimized photocathode showed a stable CO2 reduction photocurrent response, with its ethylene faradaic efficiency increased to about 60%, and no significant attenuation after continuous operation for several hours, while the bare Cu2O electrode experienced significant performance attenuation within a few minutes. Recent breakthroughs in interface engineering strategies have further promoted the development of copper-based catalysts for ethylene generation. As shown in Fig. 17, Zhang et al.22 developed an in situ interface coupling architecture of low – coordinated copper cluster catalysts and GaN nanowire photocathodes, achieving a synergistic improvement with 61% faradaic efficiency for ethylene and 14.2 mA cm−2 partial current density, with system stability extended to 116 hours. The researchers revealed the dynamic self-optimization mechanism of the Ga–N–O interface: this interface not only stabilized the active cuprous oxide species on the copper cluster surface (as an efficient site for C–C coupling) but also regulated the CO hydrogenation process through the hydrogen spillover effect of GaN, thereby guiding the specific coupling pathway of *CHO intermediates.
 | ||
| Fig. 17 (a) Schematic diagram of the in situ construction of copper clusters and photocathodes. (b) FE of products at different photocathodes. (c) Energy barriers at different steps in the free energy diagram. (d) Possible reaction mechanism of CO2RR to C2H4. Reproduced with permission from ref. 22. Copyright 2024, Springer Nature. | ||
Cu-based catalysts play an important role in ethylene generation, especially in improving selectivity by regulating the catalyst surface structure and promoting C–C coupling reactions. In recent years, researchers have made some progress through innovative catalyst design, such as the combination of Cu nanoparticles and silicon nanowires, low-coordinated copper cluster catalysts, etc., and some studies have achieved a faradaic efficiency as high as 61% and made significant breakthroughs in stability. However, there are still very few related studies, and future research should continue to focus on the stability and selectivity of catalysts.
| Catalyst | Electrolyte | Light source | Product and performance | Ref. |
|---|---|---|---|---|
| CuxS-ReS2/TiO2NTs | 0.1 M KHCO3 | 250 W Xe lamp of 200 mW cm−2 | 6 μmol cm−2 h−1 at −0.8 V vs. RHE | 247 |
| FeS2/TiO2 | 0.1 M NaHCO3 | 300 W Xe lamp | 1170 μmol L−1 cm−2 at −0.7 V vs. RHE | 248 |
| C@SiC | 0.5 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 87.8% | 249 |
| BiFeO3 | 0.5 M KHCO3 | 300 W Xe lamp | FE = 23.2% at −0.7 V vs. RHE | 250 |
| ZnO/Cu2O/Cu | 0.1 M NaSO4, 0.1 M KHCO3 | 365 nm UVA-LED and 430 nm vis-LED | FEEtOH = 16% and FEMeOH = 14% at 0.5 V vs. RHE | 79 |
| B-gC3N4/ZIF-67 | 0.1 M NaHCO3 | Visible light | 32.1096 μmol cm−2 h−1 at −1 V vs. NHE | 251 |
| CuFeO2/CuO | H2O-TEOA (9:1) |
500 W Xe lamp of 100 mW cm−2 | FE = 66.73% at −0.6 V vs. Ag/AgCl | 252 |
| CuS/MgFe2O4/Graphite | 0.1 M KHPO4 | 20 W LED lamp, visible light | FE = 35% | 253 |
| GO/SiC | 0.5 M KHCO3 | 300 W Xe lamp | FE = 99% at −0.05 V vs. Ag/AgCl | 254 |
| Cu2O/TiO2 | [Emim]BF4 | AM 1.5G | FE = 82.7% at −0.9 V vs. RHE | 103 |
| Cu2ZnSnS4/TiO2NTs | 0.1 M NaHCO3 | 250 W Xe lamp | 7.0 mmol cm−2 −0.6 V vs. RHE | 255 |
| CuxO/GO-Cu-MOF | 0.1 M KHCO3–Na2SO4 (4:1) |
AM 1.5G | FE = 43% at −0.5 V vs. Ag/AgCl | 256 |
| Si/ZnO/Cu2O | 0.1 M KHCO3 | Simulated sunlight | FE = 60% at −0 V vs. RHE | 257 |
| Zn-TPY-TTF CPG | 0.2 M Na2SO4, 0.5 M KHCO3 | AM 1.5G of 100 mW cm−2 | FE = 13.9% at 0 V vs. RHE | 258 |
| Carbazole-BODIPY | 0.1 M TBAPF6-MeCN | 50 W halogen lamp | FE = 34.79% at −1.15 V vs. NHE | 259 |
| Ag/Cu2O/g-C3N4 | 0.5 M KHCO3 | 300 W Xe lamp | FE = 44.0% at −1.0 V vs. RHE | 260 |
| CuO–MoO3/TiO2NTs | 1 M NaHCO3 | 300 W Xe lamp of 200 mW cm−2 | FE = 89% at −0.5 V vs. SCE | 261 |
It is worth mentioning that the TiO2 photocatalyst not only has the catalytic ability to reduce carbon dioxide for methanol preparation, but also shows significant application potential in the field of ethanol synthesis. The research team successfully constructed the FeS2/TiO2 p–n heterojunction photoelectrode system by using the electrochemical anode oxidation–electrodeposition coupled sulfidation process.248 By systematically exploring the effects of the FeS2 loading amount and the applied bias voltage on the photoelectrochemical performance, the experiment found that the photocurrent polarity inversion phenomenon is significantly correlated with the FeS2 loading amount, and the essence of this phenomenon lies in the dynamic evolution of the energy band structure at the semiconductor/electrolyte interface. Through surface site analysis, it is confirmed that the active center for ethanol generation is localized on the surface of TiO2 nanotubes rather than at the FeS2 interface. The introduction of FeS2 not only realizes the expansion of the visible light response range, but also, more importantly, forms a p–n heterojunction structure with TiO2 to optimize the space charge layer. Under the synergy of −0.7 V bias voltage and ultraviolet-visible light (UV-vis), the FeS2/TiO2 sample with an electrodeposition time optimized to 15 minutes showed the optimal ethanol yield, reaching 1170 μmol L−1 cm−2 after a reaction time of 3.5 hours. Compared with pure TiO2, the ethanol yield of the FeS2/TiO2 system was significantly improved due to the synergistic effect of the optimized loading amount, enhanced UV-vis light capture ability, and efficient separation–migration of carriers at the p–n junction interface. Similarly, the TNTs also showed application potential in the field of ethanol synthesis. The Cu2ZnSnS4/TiO2NTs composite electrode was prepared by the pulse electrodeposition combined with the high temperature sulfidation process.255 After parameter optimization, the light response current density of this material reached 7 mA cm−2, and the ethanol yield remained stable at 7.0 mmol cm−2 during a 5 h photoelectrocatalytic reaction. The characterization analysis showed that no new phase is generated during the catalytic process, and the electrode still maintained a stable output performance at 33.2% of the initial photocurrent density after the reaction.
Cu-based catalysts are also a major class of materials for producing ethanol. As shown in Fig. 18a and b, Kan et al.257 designed and constructed a Si/ZnO/Cu2O p–n–p heterojunction potential well structure with an electron tunnelling effect to achieve the selective photoelectrochemical CO2 reduction to ethanol. This heterojunction was formed by the controllable growth of n-type ZnO nanosheets between the defect-rich p-type Cu2O nanoparticles and the nanoporous p-type Si substrate. Driven by the built – in potential of approximately 0.6 V, the photogenerated electrons could be captured and enriched in the n-ZnO layer under low applied bias conditions, and then directionally migrated to the defect energy level of Cu2O through the tunneling effect. When the Si/ZnO/Cu2O photoelectrode was used for the photoelectrochemical CO2 reduction in the aqueous phase system under simulated sunlight irradiation, its onset potential relative to the reversible hydrogen electrode was positively shifted to 0.2 V. Due to the confined electron energy level distribution characteristics at the heterojunction interface, the product selectivity significantly shifted from the traditional CO or formate to ethanol, and the FE of ethanol exceeded 60% at 0 V vs. RHE. Regarding the photocorrosion problem of Cu-based catalysts, Lu et al.252 developed a 0D/1D CuFeO2/CuO nanowire heterojunction array with a high specific surface area. This heterojunction exhibited excellent photoelectrochemical CO2 reduction activity at −0.6 V vs. Ag/AgCl, and the FE of ethanol could reach up to 66.73%. The analysis showed that the built-in electric field at the heterojunction interface promoted the directional migration of photogenerated holes from the valence band of CuO to the valence band of CuFeO2, while the photogenerated electrons transferred from the conduction band of CuFeO2 to the conduction band of CuO. This synergistic mechanism significantly improves the efficiency of the highly selective reduction of CO2 to ethanol. Leng et al.79 constructed a ZnO/Cu2O/Cu composite photocathode by wet chemical oxidation combined with the hydrothermal method. The experimental results showed that at −0.5 V vs. RHE bias, the yields of CH3OH and C2H5OH were 10.0 μmol L−1 and 5.6 μmol L−1, respectively.
 | ||
| Fig. 18 (a) Schematic illustration of the designed defect nanomaterial structure for selective PEC CO2RR to ethanol. (b) FE of ethanol. Reproduced with permission from ref. 257. Copyright 2023, Wiley-VCH GmbH (c) Mechanistic demonstration of the PEC CO2RR. (d) Comparison of PEC CO2RR properties of different samples. Reproduced with permission from ref. 254. Copyright 2023, Wiley-VCH GmbH. | ||
In addition to copper-based materials, recent studies have shown that some non-copper-based materials also have the potential to catalyse the reduction of CO2 to prepare ethanol. The spin polarization effect of the BiFeO3 catalyst improved the electron migration and utilization efficiency, successfully achieving the highly selective catalytic preparation of CO2 to ethanol.250 In the photoelectrocatalytic system, this material showed an excellent ethanol yield (7.05 μmol cm−2 h−1), and under a – 0.7 V vs. RHE bias voltage, the ethanol FE reached 23.2%. The characterization results showed that the external electric field induces the formation of a multi-electric field coupling effect in BiFeO3, effectively enhancing the carrier separation efficiency and optimizing the surface reaction path, thereby significantly improving the CO2 reduction performance. Feng et al.249 developed a carbon@silicon carbide (C@SiC) composite catalyst, which showed excellent CO2 photoelectroreduction performance under simulated solar radiation conditions, with a CO2 conversion rate of 487 μmol gcat−1 h−1 and an ethanol selectivity of up to 87.8%. The study confirmed that the optimal ratio of sp2/sp3 hybridized carbon in the carbon layer not only significantly promoted the efficient transfer of photogenerated electrons from SiC to the carbon layer, but also optimized the C–C coupling kinetics of the key intermediates, ultimately achieving the efficient conversion of CO2 to ethanol. As shown in Fig. 18c and d, Feng et al.254 carried out the photoelectrochemical reduction of CO2 on the graphene/silicon carbide (GO/SiC) catalyst. The FE of ethanol was >99%, and the CO2 conversion rate reached as high as 17.1 mmol g−1 h−1. Experimental and theoretical studies showed that the optimal interface layer helped the photogenerated electrons transfer from the SiC substrate to the monolayer graphene coating, which was beneficial for the efficient conversion of CO2 to C2H5OH.
It can be seen that TiO2 materials play a special role in the generation of alcohol materials; however achieving high selectivity for ethanol still needs further exploration. Cu-based catalysts also occupy an important position in the generation of ethanol, especially through the design of heterojunctions and improvement of the electron transfer efficiency to improve product selectivity. Non-copper materials such as BiFeO3 and C@SiC catalysts also show good ethanol generation ability and have high selectivity. In the future, it is necessary to further optimize and regulate the structure of TiO2 and Cu-based materials to reduce the generation of by – products, and at the same time, it is necessary to continuously explore new materials with high selectivity for ethanol production.
In order to generate oxalic acid, Bergamini et al.262 employed a CsPbBr3/graphite composite system to achieve the directional synthesis of oxalic acid, obtaining an oxalic acid FE of 43.9% at a potential of −0.8 V vs. Ag/AgCl. Pawar et al.263 proposed the construction of an N-heterocyclic polymer coordination activation system to regulate the thermodynamic stability and reaction path of CO2 molecules. Studies have shown that by precisely selecting electrode materials and applying bias voltage parameters, the kinetics and thermodynamics of the photoelectrochemical CO2 reduction reaction could be doubly regulated. A solar-driven photoelectrocatalytic system based on a multilayer Cu/rGO/PVP/Nafion hybrid cathode achieved the selective generation of formaldehyde and acetaldehyde; however, the product distribution analysis showed the existence of significant side reactions, and the overall selectivity toward C2 products remained low. In addition, other C2 products were also generated, and the FE was usually expressed as the overall efficiency of C2. The CuNx/CuO heterojunction photoelectrode achieved a photocurrent density of −1.0 mA cm−2 at 0.2 V vs. RHE, which is 2.5 times higher than that of the pure CuO system.264 The total FE of this composite system for C2 products reached 15.2% at the same potential. Density functional theory calculations confirmed that the asymmetric d–p orbital hybrid Cu–N sites anchored on the CuO substrate can significantly reduce the C–C coupling free energy (ΔG = 0.86 eV), and this effect arises from the optimized regulation of the adsorption strength of *OCCO and *COCH2 intermediates. In addition, the local charge redistribution characteristics of the Cu–N sites enhance the electrical conductivity of the system, thereby improving the photogenerated electron transfer efficiency and photocurrent density. Cao et al.265 developed a NiMoO4/ZnO-3 heterojunction composite photoelectrode to achieve a synthesis rate of oxygen-containing compounds of 29.2 mmol cm−2 h−1, with a C2 product selectivity reaching 72.6%. The photoelectrochemical conduction rate was 3 times higher than that of a single photo/electrocatalytic system, indicating that the photo-electric synergy significantly enhances the reaction kinetics. Mechanism studies showed that the Ni–Mo dual active centers achieved the efficient and selective synthesis of C2 products by reducing the C–C coupling energy barrier. Wan et al.68 constructed a silicon nanowire array-supported copper-coordinated covalent triazine framework (Si@CuCTF6) heterostructure, which showed an ultra-high selectivity of 95.6% for multi-carbon products and an apparent quantum efficiency of 0.89% for carbon-based products. Synchronous radiation characterization confirmed that the active site stemmed from the synergistic effect of the coordination structure of the triazine ring nitrogen atom and the bipyridine unit Cu–N, and its ordered porous framework structure was conducive to the mass transfer of reactants and the stability of intermediates. Zhuang et al.266 reported a NiCO2O4@h-BN-6 organic–inorganic heterojunction system, which achieved 100% selective synthesis of C2 products for the first time. The detection of N–H species confirms that the nitrogen active site can efficiently adsorb/activate the proton–electron pair to generate highly active hydrogen atoms. Verification experiments showed that this material will oxidize the intermediate to acetic acid, with the boron atom existing as a hole in the semiconductor.
In particular, metal sulfide composite materials show significant potential in the catalytic synthesis of C2 products. Liu et al.76 successfully constructed a CdS/VS-MoS2 hollow heterojunction structure with sulfur vacancy (VS) enrichment characteristics. Benefiting from the improved mass transfer efficiency of the reaction medium in the confined cavity structure, the FE of the C2 product on its inner surface reached 67.0%. It was confirmed that the efficient capture of the H intermediate by surface S atoms could promote the kinetics of CO2 reduction. Cao et al.93 synthesized a porous Cu2S/MoS2-VS octahedral semiconductor heterostructure by using the MOF template method and revealed its topological confinement effect and photothermal synergy mechanism. Experiments showed that the introduction of sulfur vacancies significantly enhances the chemical adsorption capacity of CO2, and the formation of the heterointerfaces increases the charge transfer rate by 3.2 times. Based on DFT calculations, VS could reduce the activation energy for the formation of key intermediates. Liu et al.92 designed and prepared a series of CdIn2S4–N/C biomimetic catalytic systems by loading CdIn2S4 on the surface of nitrogen-doped carbon spheres to construct a plant cell-like structure for application in the photoelectrothermal synergistic catalysis of CO2. The hierarchical porous structure of the nitrogen-doped carbon matrix and the LSPR effect jointly endow the catalyst with excellent CO2 reduction performance. Among them, the CdIn2S4–N/C2-800 optimized sample showed excellent catalytic performance, with a carbon-based product generation rate of 168.5 μM h−1 cm−2, and the electron selectivity for C2 products reached 66.9%. Theoretical calculations showed that pyridinic and pyrrolic nitrogen doping sites could reduce the energy barrier of the rate-determining step of CO2 reduction. Two key intermediates, CO and OC–COH, were proposed based on this to construct the C2 product generation path based on the carbene (CO) coupling mechanism.
In addition to C2+ products, some three-carbon (C3) and higher carbon-containing compounds have been reported successively. Guo et al.267 developed a gallium-doped cuprous oxide catalyst (Ga/Cu2O), which showed excellent C2+ product selectivity in the CO2 electroreduction reaction. At a potential of −1.8 V (vs. RHE), the total FE for C2+ products reached 20%, with ethanol (CH3CH2OH) and propanol (CH3CH2CH2OH) contributing 6.5% and 6.64%, respectively. Experimental characterization confirmed that Ga doping induces the generation of an oxygen vacancy concentration gradient in Cu2O through the lattice substitution mechanism, promoting the rapid separation of photogenerated carriers, and at the same time strengthening the adsorption and activation process of CO2 molecules through electronic structure modulation. Compared with undoped Cu2O, the coverage of the *CO intermediate on the surface of Ga/Cu2O is significantly increased, and the C–C coupling energy barrier is reduced, which clarified the mechanism of the enhanced selectivity of C2+ products from a kinetic perspective. In addition, the p–n heterojunction Si@WO3-NS photocatalyst showed an apparent quantum efficiency of 0.49%, which is nearly 25 times higher than that of pure silicon nanowires (0.02% AQE), with a C2+ product selectivity of 62.7%.268 This performance improvement is attributed to the hierarchical structural design of the biomimetic plant thylakoid, which effectively optimizes the spatial transmission path of photogenerated carriers. Ponnamma's team269 further used a high-sensitivity detection technology to identify long-chain hydrocarbons such as octane (C8H18) and heptane (C7H16) in the products. In a transition metal (Fe, Co and Ni) doped TiO2 system prepared by the sol–gel method, the bandgap regulation effect of Fe–TiO2 is the most significant, and its bandgap value increases in the order of Fe–TiO2 < Co–TiO2 < Ni–TiO2. The gas chromatography characterization results of the reduction products confirmed the generation of hydrocarbons, and Fe–TiO2 showed the highest product abundance due to its optimal bandgap characteristics. It should be particularly noted that the quantitative analysis of multi-carbon gaseous products by gas chromatography requires high-sensitivity detection conditions or an extended retention time, and technical limitations often hinder actual detection.
Therefore, compared with C2 products such as ethylene, ethanol, and acetic acid, the generation of other products (such as ethane, propionic acid, propylene, etc.) faces significant challenges, and the difficulty mainly stems from the steep increase in the thermodynamic energy barrier and the complexity of the kinetic path: thermodynamically, more electron and proton migrations are required (for example, C3 products often require > 12 electrons and 12 protons), and kinetically, it involves the coordinated regulation of multiple steps of C–C coupling, proton migration, and intermediate stabilization. This imposes more stringent requirements on the design of the active sites of the catalyst (such as multi-metal synergy or confined microenvironments). It is worth mentioning that the ability of metal sulfide composite materials to reduce CO2 to C2 products is not limited to photoelectrocatalysis, and there are also a large number of reports in the field of photocatalysis. Thus, metal sulfide composite materials deserve attention. In addition, the precise detection of multi-carbon products also limits the accurate assessment of selectivity.
4 Reaction durability
Although considerable attention has been paid to activity and selectivity, the stability of product selectivity, which is often overlooked, is equally crucial for industrial applications.123,161,270 Catalyst failure can result in increased overpotential, reduced current, or diminished selectivity for the target product. A decline in catalyst activity or selectivity during operation not only lowers energy utilization efficiency, but also leads to the formation of by-products, increases the cost of separation and purification, and may even compromise the economic viability of the process. Therefore, enhancing both the selectivity and stability of products is essential for realizing the practical application of PEC CO2RR.Compared with the stable operation of electrocatalytic CO2 reduction (EC CO2RR), which can last from tens to hundreds of hours, the actual stable operation time of PEC CO2RR is often much shorter, typically ranging from several hours to just over ten hours.201,271 Significant differences in stability exist between EC CO2RR and PEC CO2RR, primarily due to differences in energy input methods and material systems. Electrocatalytic CO2reduction relies solely on an applied voltage to drive the reaction and does not involve strong light irradiation. As a result, its stability issues are mainly related to catalyst degradation in the electrochemical environment, such as metal ion dissolution, surface reconstruction, electrode passivation, and corrosion of materials by electrolytes. In contrast, PEC CO2RR not only faces the same electrochemical stability challenges as EC CO2RR, but also contends with additional degradation mechanisms induced by light exposure. Semiconductor photoabsorbers are typically incorporated into PEC CO2RR systems. These materials are susceptible to photocorrosion or photo-induced reconstruction under intense illumination, leading to structural damage and performance degradation. Additionally, the accumulation of photogenerated carriers can trigger side reactions or accelerate material degradation, while chemical instability at heterojunctions or interfaces can further compromise the overall stability of the system. Due to the more complex architecture of PEC CO2RR devices, which require the coordinated operation of both the catalyst and the light absorber, failure in any single component can significantly impact overall performance and increase the difficulty of maintenance and repair. As a result, the stability of PEC CO2RR is generally lower than that of EC CO2RR, and its actual operational lifetime is often shorter. In general, the failure modes of PEC CO2RR are mainly attributed to photocorrosion, catalyst leaching, and structural collapse. Improving the stability of PEC CO2RR requires not only attention to the catalyst itself, but also enhanced protection and optimization of the light absorber and interface structures. This can be achieved through a range of collaborative strategies, such as introducing protective layers, accelerating carrier separation, developing semiconductor materials with improved resistance to photocorrosion, and refining reactor design, with the goal of achieving long-term and efficient CO2 reduction.
Introducing a protective layer into PEC CO2RR systems is an effective strategy to enhance the stability of the catalyst. Protective layers are typically applied to the surfaces of semiconductors or catalysts to prevent photocorrosion and chemical degradation of materials under intense illumination and electrochemical conditions. Moreover, a well-designed protective layer can facilitate efficient charge transfer and suppress interfacial carrier recombination, thereby maintaining high catalytic activity and product selectivity. Common protective layer materials include metal oxides (such as TiO2 and Al2O3), carbon materials, and conductive polymers.243 These materials not only exhibit excellent chemical stability, but also enable precise control over the catalytic interface by tuning their thickness and structure. For example, Bergamini et al.262 sputtered Pt or C onto CsPbBr3 to fabricate a hybrid photoelectrode with a uniform protective layer of consistent thickness. Owing to the presence of Pt and C layers, the instability of perovskite in aqueous environments was mitigated, and the resulting hybrid electrode retained its ability to absorb visible light. Prabhakar et al.161 also synthesized CIGS films by vulcanizing Cu–In–Ga metal laminates through sputtering. Their analysis suggests that the stability of CO2 reduction may be attributed to a self-protection effect, formed in situ by the oxides and hydroxides of Ga and In during operation. In addition to sputtering, oxide coatings represent another promising approach. For example, Andrei et al.167 extended the operational stability of their device to an impressive 72 h by integrating a BiOI photocatalyst into a robust oxide-based structure sealed with graphite paste (Fig. 19a). Xu et al.272 coated the organic compound pyridine as a co-catalyst onto Cu2O, which enhanced the selectivity of the Cu2O surface for CO2 reduction while maintaining the stability of the Cu2O (110) surface. Protective layers are thus of great significance for stabilizing precious metals and improving catalytic performance. Ziarati et al.124 reported a host–guest system composed of atomically precise gold nanoclusters incorporated within zinc porphyrin nanorings. Structural characterization revealed strong binding between the pyridine thiol-based Au25 nanocluster and the nanoring, which consists of six zinc porphyrin units, thereby enhancing the stability of the gold nanocluster. This system also demonstrated significant activity and selectivity in the photochemical conversion of CO2 to CO and in the generation of singlet oxygen.
 | ||
| Fig. 19 (a) Schematic depiction of the BiOI photocathode with its components. (b) The stability test. Reproduced with permission from ref. 167. Copyright 2022, Springer Nature. (c) Schematic diagram of solar-driven bias-free PEC-CO2RR system for formic acid production. (d) The stability test. Reproduced with permission from ref. 201. Copyright 2025, Wiley-VCH GmbH. | ||
Accelerating carrier separation is also a widely adopted strategy to mitigate photocorrosion and enhance the stability of PEC CO2RR systems. Efficient carrier transfer can substantially mitigate adverse effects, such as structural damage resulting from carrier-induced redox reactions. For example, to address the photocorrosion of Cu2O materials, Zhang et al.127 coated Cu2O with a carbon layer and employed a C electron transport layer to accelerate electron transfer, thereby alleviating Cu2O corrosion. Jia et al.138 designed an efficient photocathode system featuring a photonic crystal structure based on Cu2O for enhanced light harvesting. The hole transport layer (HTL: FeOOH) mediates and directs interfacial charge transfer from Cu2O to FTO, while a polypyrrole (PPy) layer serves as a continuous multi-electron transfer agent for CO2 reduction. This integrated photocathode system demonstrated operational stability for 7 h. Liu et al.2 proposed a protection scheme utilizing a silver catalyst to accelerate the transfer of photogenerated electrons and employing a Z-scheme to extract holes from heterojunctions. The resulting photocathode exhibited a stable photocurrent for CO2 reduction under hydrogen equilibrium conditions, achieving a faradaic efficiency of approximately 60% for ethylene over several hours, whereas unprotected Cu2O degraded within minutes. It is noteworthy that the reaction system designed by Yao et al.201 achieved unbiased photoelectrochemical CO2 reduction to HCOOH, with a yield of up to 172.9 μmol h−1 cm−2 over 85 h of long-term testing, outperforming traditional solar-driven systems (Fig. 19b). The oxygen evolution reaction (OER) at the anode was replaced by the sulfur oxidation reaction (SOR, E = 0.48 V), which proceeds at a lower reaction rate and effectively suppresses photocorrosion, thereby protecting the anode material.
Currently, research on the development of semiconductor materials resistant to photocorrosion and the optimization of reaction devices remains ongoing, and both areas require further advancement in the future. In the development of new semiconductor materials, Liu et al.82 employed a bare CuIn0.3Ga0.7S2 photocathode to drive PEC CO2RR. Under standard 1 sun illumination, the current density exceeded 2 mA cm−2 (at −2 V vs. Fc+/Fc), and the selectivity for CO production (CO/total products) reached as high as 87%. The system also exhibited long-term stability for syngas production, operating for more than 44 h. Özdemir et al.259 synthesized a cross-linked boron-dipyrrole methylene (BODIPY) photocatalyst containing a carbazole group. During the course of the reaction, both the turnover number and turnover frequency increased significantly. At a potential of −1.15 V, the faradaic efficiency reached 34.79%. This BODIPY system demonstrated both high activity and long-term stability. Optimizing the reaction device is another effective strategy to enhance system stability. Jung et al.110 developed a continuous-flow PEC device with a gas-permeable photocathode. By precisely controlling parameters such as the flow rate and pressure of the reaction gas and the flow rate of the electrolyte, both the magnitude of the photogenerated current and the stability of the system could be optimized. Compared to a conventional H-cell using the same photocatalyst material under optimal conditions, the continuous PEC flow reactor achieved a 10-fold increase in the faradaic efficiency for CO, a 30-fold increase in the production rate, and a 16-fold improvement in stability.
5 Conclusion and outlook
In recent years, significant progress has been made in the field of PEC CO2RR in achieving high selectivity for various products, including CO, HCOOH, CH3OH, CH4, CH3COOH, C2H4, C2H5OH and so on. A large number of parameters in the reaction process affect the selectivity of the products. We mainly focus on the effects of the catalyst design, reaction conditions, and the reactor design on product selectivity. The catalyst design is the most widely studied and the most deeply studied aspect at present. It involves aspects such as the type of catalyst, the modification strategy, the composite structure, and the morphology control to achieve the regulation of the active sites on the catalyst surface for the efficient and selective generation of the products. If the design of the catalyst is regarded as the regulation at the micro level, then the design of reaction conditions and the environment represents the regulation at the macro level. The regulation of the catalytic reaction on the catalyst surface can be achieved by changing the external conditions, such as applying voltage, applying external light (including light intensity and light wavelength), reaction temperature, and filling the electrolyte. In addition, the device structure design also affects the absorption of light energy by the photoelectrode, the charge transfer efficiency, and the kinetic mass transfer problem of CO2 molecules on the catalyst surface. Although many aspects influencing selectivity need to be considered, there are still patterns to follow. In addition, the thermodynamics and kinetics of the reduction process should also be comprehensively considered during the design to optimize the design of the catalyst, the reaction environment, and the device structure, thereby achieving the regulation of selectivity.Through a systematic review of the research on PEC CO2RR in the past five years, we summarize the optimal FE of the main products (Fig. 20). Among the C1 products, the faradaic efficiency of CO and HCOOH is particularly outstanding, with the highest performance reaching nearly 100%. In addition to catalysts with precious metals such as Au and Ag, single-atom catalysts and molecular catalysts can show 100% FE for CO production. The synthesis of HCOOH is dominated by enzyme-based formate dehydrogenase materials and requires precise regulation. In addition, syngas, as a special product combination, has attracted much attention due to its wide application in the industry, and its generation efficiency and adjustable proportion range have also been significantly improved. For the liquid product methanol, important progress has also been made, and the FE of methanol has exceeded 90%. It is worth noting that the high selectivity for methanol in some studies is attributed to catalysts with graphene oxide as the carrier, and it is speculated that there may be a suitable environment for methanol generation on graphene oxide, which still needs further exploration. However, methane is rarely studied, and individual research results show that the FE of methane can reach 90%, and related research still needs to be further advanced. Among the C2+ products, research has mainly focused on the reduction of acetic acid, ethylene, and ethanol, and Cu-based catalysts are the most commonly used, but due to the formation of a large number of by-products, it is difficult to improve the selectivity. The generation of acetic acid involves a diversified strategy, in which a combination of Cu2O and TiO2 as well as cathodic microbial electrosynthesis seem to be good options. In the current research, Cu-based catalysts are the main choice for ethylene production, and new catalyst types still need to be further developed. Ethanol has shown the most significant research progress among the C2+ products, involving various types of catalysts, such as TiO2, Cu-based materials, and non-Cu-based materials (BiFeO3 and SiC). Among them, the application of GO/SiC materials enables the selectivity for ethanol reach almost 100%. For other C2+ products, although they can be produced using various types of catalysts, their generation is difficult and the selectivity efficiency is low. In general, the high selectivity and generation efficiency of C1 products are still important goals in the current research on PEC CO2RR, while C2+ products represent the potential for future technological development. Finally, it is important to conduct in-depth investigations into the limited reaction stability of PEC systems, which severely hinders the achievement of high product selectivity. One possible cause is the material instability induced by light irradiation; however, this mechanism requires further detailed exploration. The construction of catalyst coating layers and the enhancement of rapid charge carrier separation have already demonstrated significant improvements in reaction stability, with reaction times extended to approximately 80 h.
 | ||
| Fig. 20 The highest FE of different PEC CO2RR products reported in the last five years. | ||
The PEC CO2RR technology provides an innovative approach for achieving carbon neutrality and the synthesis of high value-added chemicals. However, research in this field is still in its infancy. The precise control of product selectivity still faces multiple challenges at the material, mechanism, and system levels, and a significant amount of work is still required to improve the conversion efficiency and product selectivity.
5.1 Precise regulation of catalysts
In the PEC CO2RR, the selectivity and efficiency of the catalyst are directly affected by the properties and structure of the catalyst itself. Although metal catalysts such as copper-based catalysts have made significant progress in generating C2 products (such as ethylene and ethanol), their selectivity and stability still need to be further improved. Therefore, precisely regulating the microstructure, surface active sites, and electronic properties of the existing catalyst is crucial for optimizing the catalytic performance. By adjusting the size, morphology, and surface functional groups of the metal particles, as well as their coordination with other elements, the product selectivity and FE can be significantly improved. In addition, rationally designing the electronic coupling effect between metals and optimizing the adsorption characteristics of the catalyst surface are also the keys to achieving high selectivity.5.2 Improvement of reaction stability
Reaction stability is one of the core challenges in achieving efficient and sustainable conversion. Currently, photoelectrocatalytic systems commonly suffer from instability issues caused by photocorrosion, interfacial mismatches, and fluctuations in reaction conditions, which severely affect long-term operation and product selectivity. Future research directions for improving reaction stability include the development of novel catalyst materials with enhanced resistance to photocorrosion, the construction of efficient interface engineering strategies, and the optimization of light absorption, charge carrier separation, and transport pathways through advanced device design. For example, employing multilayer heterojunction structures, introducing protective layers or self-healing mechanisms, and integrating advanced device architectures such as microfluidic systems are expected to significantly enhance the overall stability and efficiency of PEC CO2RR, thereby advancing their progress toward practical application.5.3 Device design and reaction environment design
The regulation of device design and the reaction environment has an equally important impact on the efficiency and selectivity of the PEC CO2RR reaction. By optimizing the structure of the photoelectrocatalytic device, such as the shape, thickness and porosity of the electrodes, as well as the geometric structure of the device, the contact area between reactants can be expanded and the photocurrent density can be increased, thereby promoting the reaction. At the same time, the regulation of the reaction environment, such as the pH of the reaction solution, temperature, and the selection of the electrolyte, can also significantly affect the reaction path and product distribution. By precisely adjusting these parameters, it is possible to achieve the highly selective generation of different products. In the future, the development of reactors with high stability and operability will have a positive impact on the large-scale application of the PEC CO2RR technology. In theoretical calculations, the corresponding microenvironment can be included in simulation, but its influence is usually ignored.5.4 Deep analysis of the multi-scale reaction mechanism
A thorough understanding of the mechanism of the PEC CO2RR reaction is the basis for improving the performance of the catalyst, optimizing the reaction path, and enhancing the selectivity of the products. Currently, although some studies have revealed details of some reaction mechanisms, the formation of intermediates in the reaction process, the kinetics of electron–proton transfer, and the active sites on the catalyst surface are still not completely clear. In the future, in-depth studies on each stage of the reaction process should be strengthened, especially by using high-resolution characterization techniques (such as in situ spectroscopy, electron microscopy technology, etc.) to track the evolution process of intermediates, thereby providing a scientific basis for the design and optimization of the catalyst. In addition, with the development of computational chemistry, the use of computational simulations to further reveal the reaction path and catalytic mechanism can accelerate catalyst design and reaction optimization.This review mainly expounds the factors influencing the selectivity of the PEC CO2RR products and summarizes the research progress on commonly studied products, in order to provide insights and ideas for future efforts to improve product selectivity. It is expected that in the near future, the practical application of photoelectrocatalytic CO2 to achieve C1 products at the industrial scale and significant improvements in the selectivity and efficiency of C2+ product generation will be realized.
Data availability
Data availability is not applicable to this article as no new data were created or analyzed in this study.Author contributions
All of the authors contributed to the manuscript preparation. G. Zhou conceptualized the work, performed the investigations and wrote the manuscript. Z. Wang and J. Gong provided resources and edited the manuscript. S. Shen and W. Zhong were involved in funding acquisition and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52072255 and 52271184), Science Fund for Distinguished Young Scholars of Zhejiang Province (LR22E020003), Natural Science Foundation of Zhejiang Province, China (LY23E020001), and High-level Talents Special Support Program of Taizhou.Notes and references
- S. Bhattacharjee, M. Rahaman, V. Andrei, M. Miller, S. Rodríguez-Jiménez, E. Lam, C. Pornrungroj and E. Reisner, Photoelectrochemical CO2-to-fuel conversion with simultaneous plastic reforming, Nat. Synth., 2023, 2, 182–192 CrossRef CAS.
- G. Liu, F. Zheng, J. Li, G. Zeng, Y. Ye, D. M. Larson, J. Yano, E. J. Crumlin, J. W. Ager, L. Wang and F. M. Toma, Investigation and mitigation of degradation mechanisms in Cu2O photoelectrodes for CO2 reduction to ethylene, Nat. Energy, 2021, 6, 1124–1132 CrossRef CAS.
- Y. Boyjoo, Y. Jin, H. Li, G. Zhao, H. Guo and J. Liu, Nanoengineering of photocatalytic electrode materials toward net zero emissions, Cell Rep. Phys. Sci., 2023, 4, 101391 CrossRef CAS.
- J. Han, X. Bai, X. Xu, X. Bai, A. Husile, S. Zhang, L. Qi and J. Guan, Advances and challenges in the electrochemical reduction of carbon dioxide, Chem. Sci., 2024, 15, 7870–7907 RSC.
- Y. Feng, X. Wang, J. Ma, N. Wang, Q. Liu, K. Suenaga, W. Chen, J. Zhang, Y. Zhou and J. Wang, A solid-solution with asymmetric Ni-O-Cr sites for boosting protonation toward anodic oxidation, Adv. Energy Mater., 2024, 14, 2401501 CrossRef CAS.
- C. P. Consortium, Toward a Cenozoic history of atmospheric CO2, Science, 2023, 382, eadi5177 CrossRef PubMed.
- X. Jiang, R. Chen, Y. Chen and C. Lu, Research progress of photoelectrochemical conversion of CO2 to C2+ products, Chem. Synth., 2024, 4, 46 CAS.
- J. Lv, J. Xie, A. G. A. Mohamed, X. Zhang, Y. Feng, L. Jiao, E. Zhou, D. Yuan and Y. Wang, Solar utilization beyond photosynthesis, Nat. Rev. Chem., 2023, 7, 91–105 CrossRef PubMed.
- J. H. Lee, D. Hansora and J. S. Lee, Turning CO2 into valuables with sunlight only, Chem, 2023, 9, 1632–1635 CAS.
- B. Shan, S. Vanka, T. Li, L. Troian-Gautier, M. K. Brennaman, Z. Mi and T. J. Meyer, Binary molecular-semiconductor p–n junctions for photoelectrocatalytic CO2 reduction, Nat. Energy, 2019, 4, 290–299 CrossRef CAS.
- T. Chen, R. Ramachandran, S. Chen, G. Anushya, A. G. Al-Sehemi, V. Mariyappan, S. Alargarsamy, M. M. Alam, T. C. Mahesh and P. Kalimuthu, An Overview of Semiconductor Electrode Materials for Photoelectrochemical Water Splitting and CO2 Conversion, Int. J. Electrochem. Sci., 2024, 19, 100542 CrossRef CAS.
- J. Tian, W. Xu, J. Gao, T. Zhu, G. Xu, Z. Zhong and F. Su, Electrocatalytic and Photocatalytic CO2 Methanation: From Reaction Fundamentals to Catalyst Developments, ChemCatChem, 2024, 16, e202301406 CrossRef CAS.
- J. Wang, L. Yuan, P. Zhang, J. Mao, J. Fan and X. L. Zhang, Advances in zeolitic-imidazolate-framework-based catalysts for photo-/electrocatalytic water splitting, CO2 reduction and N2 reduction applications, Nanoscale, 2024, 16, 7323–7340 RSC.
- B. Rhimi, M. Zhou, Z. Yan, X. Cai and Z. Jiang, Cu-based materials for enhanced C2+ product selectivity in photo-/electro-catalytic CO2 reduction: challenges and prospects, Nano-Micro Lett., 2024, 16, 64 CrossRef CAS PubMed.
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Product selectivity of photocatalytic CO2 reduction reactions, Mater. Today, 2020, 32, 222–243 CrossRef CAS.
- J. Li, J. Ren, S. Li, G. Li, M. M.-J. Li, R. Li, Y. S. Kang, X. Zou, Y. Luo and B. Liu, Potential industrial applications of photo/electrocatalysis: Recent progress and future challenges, Green Energy Environ., 2024, 9, 859–876 CrossRef CAS.
- Y. Li, S. Li and H. Huang, Metal-enhanced strategies for photocatalytic and photoelectrochemical CO2 reduction, Chem. Eng. J., 2023, 457, 141179 CrossRef CAS.
- H.-R. Zhu, H.-M. Xu, C.-J. Huang, Z.-J. Zhang, Q.-N. Zhan, T.-Y. Shuai and G.-R. Li, Recent advances of the catalysts for photoelectrocatalytic oxygen evolution and CO2 reduction reactions, Chin. J. Catal., 2024, 62, 53–107 CrossRef CAS.
- G. H. Han, J. Bang, G. Park, S. Choe, Y. J. Jang, H. W. Jang, S. Y. Kim and S. H. Ahn, Recent advances in electrochemical, photochemical, and photoelectrochemical reduction of CO2 to C2+ products, Small, 2023, 19, 2205765 CrossRef CAS PubMed.
- L. Fei, L. Lei, T. J. Meyer and D. Wang, Dye-Sensitized Photocathodes Assembly and Tandem Photoelectrochemical Cells for CO2 Reduction, Acc. Mater. Res., 2024, 5, 124–135 CrossRef CAS.
- Z. Wang, S. Shen, J. Wang and W. Zhong, Modulating the D-Band Center of Electrocatalysts for Enhanced Water Splitting, Chem.–Eur. J., 2024, 30, e202402725 CrossRef CAS PubMed.
- B. Zhang, P. Zhou, Z. Ye, I. A. Navid, Y. Pan, Y. Xiao, K. Sun and Z. Mi, Interfacially coupled Cu-cluster/GaN photocathode for efficient CO2 to ethylene conversion, Nat. Synth., 2024, 3, 1567–1576 CrossRef CAS.
- J. F. de Brito, G. G. Bessegato, J. A. L. Perini, L. D. de Moura Torquato and M. V. B. Zanoni, Advances in photoelectroreduction of CO2 to hydrocarbons fuels: Contributions of functional materials, J. CO2 Util., 2022, 55, 101810 CrossRef.
- D. Li, K. Yang, J. Lian, J. Yan and S. Liu, Powering the world with solar fuels from photoelectrochemical CO2 reduction: basic principles and recent advances, Adv. Energy Mater., 2022, 12, 2201070 CrossRef CAS.
- J. Wang, W. He, Y. Zong, Y. Tang, J. Wang and R. Ma, Electronic redistribution induced by interaction between ruthenium nanoparticles and Ni–N (O)–C sites boosts alkaline water electrolysis, Chem. Commun., 2024, 60, 9444–9447 RSC.
- J. Liu, C. Xia, S. Zaman, Y. Su, L. Tan and S. Chen, Surface plasmon assisted photoelectrochemical carbon dioxide reduction: progress and perspectives, J. Mater. Chem. A, 2023, 11, 16918–16932 RSC.
- X. Zhang, J. Wang, J. Song, T. Sun, M. Wang, J. Zhu and Y. Yu, Constructing Nanoneedle Arrays of Heterostructured RuO2-Co3O4 with Tip-effect-induced Enrichment of Reactants for Enhanced Water Oxidation, Chem. Commun., 2025, 61, 8723–8726 RSC.
- P. Uthirakumar, D. Kim, V. Dao, C. Kai, C. Yun, Y. J. Jang and I.-H. Lee, Introducing Cu2O (111) phase on Cu (OH) 2 nanorods integrated Pd-cocatalyst for boosting acetone selectivity via photoelectrochemical CO2RR, Environ. Res., 2025, 265, 120423 CrossRef CAS PubMed.
- G. Ding, C. Li, L. Chen and G. Liao, Emerging porphyrin-based metal-organic frameworks for photo (electro) catalytic CO2 reduction, Energy Environ. Sci., 2024, 17, 5311–5335 RSC.
- V. Andrei, I. Roh, J.-A. Lin, J. Lee, Y. Shan, C.-K. Lin, S. Shelton, E. Reisner and P. Yang, Perovskite-driven solar C2 hydrocarbon synthesis from CO2, Nat. Catal., 2025, 8, 137–146 CrossRef CAS.
- W. C. Ma, M. C. Xie, S. J. Xie, L. F. Wei, Y. C. Cai, Q. H. Zhang and Y. Wang, Nickel and indium core-shell co-catalysts loaded silicon nanowire arrays for efficient photoelectrocatalytic reduction of CO2 to formate, J. Energy Chem., 2021, 54, 422–428 CrossRef CAS.
- M. Kan, Q. Wang, S. Hao, A. Guan, Y. Chen, Q. Zhang, Q. Han and G. Zheng, System engineering enhances photoelectrochemical CO2 reduction, J. Phys. Chem. C, 2022, 126, 1689–1700 CrossRef CAS.
- Y. Cao, C. Zhang, W. Wang, Y. Liu, Y. Tao, J. Fan, M. Chen, D. Zhang and G. Li, Binary junctions enhance electron storage and potential difference for photo-assisted electrocatalytic CO2 reduction to HCOOH, Appl. Catal., B, 2024, 349, 123867 CrossRef CAS.
- Z. Zhao, H. Wang, Q. Yu, S. Roy and X. Yu, Photo-/electrocatalytic approaches to CO2 conversion on Cu2O-based catalysts, Appl. Catal., A, 2023, 119445 CrossRef CAS.
- Q. Wang, Y. Zhang, Y. Liu, K. Wang, W. Qiu, L. Chen, W. Li and J. Li, Photocorrosion behavior of Cu2O nanowires during photoelectrochemical CO2 reduction, J. Electroanal. Chem., 2022, 912, 116252 CrossRef CAS.
- Y. Wang, G. Zhou, Y. Xu, Y. Cheng, M. Song, J. Jin, X. Liu and Z. Lu, Hydrothermal Synthesis of Inorganic Imprinted Bi4Ti3O12 Nanosheets for Efficient Selective Photocatalytic Degradation of Ciprofloxacin, Chem. Res. Chin. Univ., 2025, 1–10 Search PubMed.
- S. H. Xu, Q. Shen, J. G. Zheng, Z. M. Wang, X. Pan, N. J. Yang and G. H. Zhao, Advances in Biomimetic Photoelectrocatalytic Reduction of Carbon Dioxide, Adv. Sci., 2022, 9, 2203941 CrossRef CAS.
- K. Xu, Q. Zhang, X. Zhou, M. Zhu and H. Chen, Recent Progress and Perspectives on Photocathode Materials for CO2 Catalytic Reduction, Nanomaterials, 2023, 13, 1683 CrossRef CAS PubMed.
- L. Zuo, Y. Deng, L. Chen, T. He, J. Yang and J. Zhang, Fundamental Insights into Photoelectrochemical Carbon Dioxide Reduction: Elucidating the Reaction Pathways, ACS Catal., 2024, 14, 16795–16833 CrossRef CAS.
- P. Zuo, Z. Xu, Q. Zhu, J. Ran, L. Ge, X. Ge, L. Wu, Z. Yang and T. Xu, Ion exchange membranes: constructing and tuning ion transport channels, Adv. Funct. Mater., 2022, 32, 2207366 CrossRef CAS.
- J. Ran, L. Wu, Y. He, Z. Yang, Y. Wang, C. Jiang, L. Ge, E. Bakangura and T. Xu, Ion exchange membranes: New developments and applications, J. Membr. Sci., 2017, 522, 267–291 CrossRef CAS.
- J. Li, J. Zheng, X. Liu, Y. Yang, X. Han and Z. Huang, Regulating the photoelectrocatalytic reduction efficiency of CO2 to syngas via SnO enhanced g-C3N4 based pn heterojunction, Opt. Mater., 2023, 138, 113703 CrossRef CAS.
- H. Du, T. Sun, M. Wang, Y. Tang, Y. Yu and J. Wang, Impact of harmful ions in seawater on electrolysis catalysts: challenges and mitigation strategies, Chem. Commun., 2025, 61, 5719–5730 RSC.
- H. Zhu, J. J. Wang, Z. Xu, Y. Tan and J. Wang, Pd Nanoparticle Size-Dependent H* Coverage for Cu-Catalyzed Nitrate Electro-Reduction to Ammonia in Neutral Electrolyte, Small, 2024, 20, 2404919 CrossRef CAS.
- A. Parzuch, K. Kuder, K. Nikiforow, P. Wróbel, G. Kaproń, K. Bieńkowski and R. Solarska, Efficient Photoelectrochemical Reduction of CO2 in Seawater with Cheap and Abundant Cu2O/Al2O3/TiO2 Electrode, Materials, 2025, 18, 620 CrossRef CAS.
- I. Merino-Garcia, S. Castro, A. Irabien, I. Hernandez, V. Rodriguez, R. Camarillo, J. Rincon and J. Albo, Efficient photoelectrochemical conversion of CO2 to ethylene and methanol using a Cu cathode and TiO2 nanoparticles synthesized in supercritical medium as photoanode, J. Environ. Chem. Eng., 2022, 10, 107441 CrossRef CAS.
- B. Zhou, J. Li, X. Dong and L. Yao, GaN nanowires/Si photocathodes for CO2 reduction towards solar fuels and chemicals: advances, challenges, and prospects, Sci. China:Chem., 2023, 66, 739–754 CAS.
- X. Zhang, R. Tang, H. Sun, Z. Lin, W. Liang, F. Li, R. Zheng and J. Huang, Copper-tin composite bimetallic nanoparticles on bismuth vanadate nanoplate photocathodes for photoelectrochemical carbon dioxide reduction, Mater. Today Energy, 2025, 50, 101861 CrossRef CAS.
- Z. Wang, Y. Wang, S. Ning and Q. Kang, Zinc-based materials for photoelectrochemical reduction of carbon dioxide, Energy Fuels, 2022, 36, 11380–11393 CrossRef CAS.
- H. Wu, D. Zhang, B. X. Lei and Z. Q. Liu, Metal oxide based photoelectrodes in photoelectrocatalysis: advances and challenges, ChemPlusChem, 2022, 87, e202200097 CrossRef CAS PubMed.
- Y. Pan, R. Abazari, B. Tahir, S. Sanati, Y. Zheng, M. Tahir and J. Gao, Iron-based metal–organic frameworks and their derived materials for photocatalytic and photoelectrocatalytic reactions, Coord. Chem. Rev., 2024, 499, 215538 CrossRef CAS.
- L. K. Putri, B. J. Ng, W. J. Ong, S. P. Chai and A. R. Mohamed, Toward excellence in photocathode engineering for photoelectrochemical CO2 reduction: Design rationales and current progress, Adv. Energy Mater., 2022, 12, 2201093 CrossRef CAS.
- H. Kumar, T. Nike, A. Kumar, D. Kaushal, V. Chauhan and M. Kumar, Recent advances in photoelectrochemical potential improvement of CuBi2O4: Energy applications, Inorg. Chem. Commun., 2025, 114584 CrossRef CAS.
- J. Wang, J. Xiong, H. Guo and Y. Wei, An overview of carbon dioxide photo/electrocatalyzed by titanium dioxide catalyst, ChemCatChem, 2024, 16, e202400707 CrossRef CAS.
- J. C. Yu, X. Hao, L. X. Mu, W. S. Shi and G. W. She, Photoelectrocatalytic Utilization of CO: A Big Show of Si-based Photoelectrodes, Chem.–Eur. J., 2024, 30, e202303552 CrossRef CAS PubMed.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Light-Driven Heterogeneous Reduction of Carbon Dioxide: Photocatalysts and Photoelectrodes, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- L. Xie, Y. Jiang, W. Zhu, S. Ding, Y. Zhou and J.-J. Zhu, Cu-based catalyst designs in CO2 electroreduction: precise modulation of reaction intermediates for high-value chemical generation, Chem. Sci., 2023, 14, 13629–13660 RSC.
- C. B. Hiragond, J. Kim, H. Kim, D. Bae and S. I. In, Elemental-Doped Catalysts for Photoelectrochemical CO2 Conversion to Solar Fuels, Sol. RRL, 2024, 2400022 CrossRef CAS.
- T. Jin, S. Shen, A. Xu, J. Pan, G. Zhou and W. Zhong, Triggering Intrinsic Electrochemical Hydrogen Evolution Activity within Heusler Alloys via Electronegativity-Induced Charge Rearrangement, Small, 2025, 21, 2500667 CrossRef CAS PubMed.
- T. Ouyang, Y. Ye, C. Tan, S. Guo, S. Huang, R. Zhao, S. Zhao and Z. Liu, 1D α-Fe2O3/ZnO junction arrays modified by Bi as photocathode: high efficiency in photoelectrochemical reduction of CO2 to HCOOH, J. Phys. Chem. Lett., 2022, 13, 6867–6874 CrossRef CAS PubMed.
- P. Devi, R. Verma and J. P. Singh, Advancement in electrochemical, photocatalytic, and photoelectrochemical CO2 reduction: Recent progress in the role of oxygen vacancies in catalyst design, J. CO2 Util., 2022, 65, 102211 CrossRef CAS.
- X.-D. Wang, J. Wang, Y.-J. Dong, H.-Y. Chen and D.-B. Kuang, Halide perovskite-based photoelectrodes for solar-to-chemical synthesis, Trends Chem., 2025, 7, 186–201 CrossRef CAS.
- Y. H. Lu, H. Z. Cao, S. H. Xu, W. Y. Feng, G. Y. Hou, Y. P. Tang, H. B. Zhang and G. Q. Zheng, CO2 photoelectroreduction with enhanced ethanol selectivity by high valence rhenium-doped copper oxide composite catalysts, J. Colloid Interface Sci., 2021, 599, 497–506 CrossRef CAS.
- M. Aresta, T. Baran, D. Caringella, S. G. Wasse, Y. I. Abubakar, A. Petrucciani, A. Norici, E. Mesto, E. De Giglio and R. Comparelli, Orientational-growth of Cu2O {111} or {110} facets induced by frustules for CO2-H2O specific photo (electro) chemical conversion into methanol/ethanol under visible-light, J. CO2 Util., 2025, 95, 103071 CrossRef CAS.
- X. Gong, S. Fan, Q. Yang, J.-L. Yang, X. Qi, H. Shen, D. Ren and M. Wang, Porous two-dimensional CuSe@ BiOI isotype heterojunction with highly exposed (1 0 2) facets for efficient photoelectrocatalytic CO2 reduction and photodetection, Chem. Eng. J., 2024, 152773 CrossRef CAS.
- X. Tan, H. Zhu, C. He, Z. Zhuang, K. Sun, C. Zhang and C. Chen, Customizing catalyst surface/interface structures for electrochemical CO2 reduction, Chem. Sci., 2024, 15, 4292–4312 RSC.
- N. Nawaz, S. Perveen, K. Ramalingam, K. Bieńkowski, P. Wrobel, M. Pisarek, S. Prabhakaran, N. G. Szwacki, S. Pitchaimuthu and R. Solarska, Insights of Cu2O/Zn5 (OH) 8Cl2 photocathode architecture for an efficient photoelectrochemical CO2 reduction to multi-carbon products, Chem. Eng. J., 2025, 162272 CrossRef CAS.
- W. Wan, F. Meng, S. Chen, J. Wang, C. Liu, Y. Wei, C. He, L. Fan, Q. Zhang and W. Ye, Triazine-COF@ Silicon nanowire mimicking plant leaf to enhance photoelectrocatalytic CO2 reduction to C2+ chemicals, Green Energy Environ., 2025, 10, 422–432 CrossRef CAS.
- G. Liu, R. Cai, Z. Lv, G. Ma, J. Li, J. Jin, X. Zhong and F. Li, Ameliorating the carrier dynamics behavior via plasmonic Ag-modified CuBi2O4 inverse opal for the efficient photoelectrocatalytic reduction of CO2 to CO, J. Catal., 2023, 424, 130–139 CrossRef CAS.
- X. Bi, H. Wang, Z. Yang, Y. Zhao, Z. Ma, T. Liu and M. Wu, Localized surface plasmon resonance enhanced continuous flow photoelectrocatalytic CO2 conversion to CO, Energy Fuels, 2022, 36, 7206–7212 CrossRef CAS.
- Y. Wei, J. Wang, C. He, Z. Hu, C. Zhuang, N. Ullah, F. Meng, Q. Zhang and H. Jing, Flower-like TiO2/TiB2 heterojunction for selective photoelectrocatalytic CO2 conversion to carboxylic acids–Boron-assisted oxidation mechanism, Chem. Eng. J., 2025, 505, 159347 CrossRef CAS.
- L. Talbi, I. Bozetine, S. A. Boussaa, K. Benfadel, D. Allam, N. Rahim, Y. O. Mohamed, M. Leitgeb, C. Torki and S. Hocine, Photoelectrochemical properties of Cu2O/PANI/Si-based photocathodes for CO2 conversion, Emerging Mater. Res., 2023, 12, 78–91 CrossRef.
- T. Li, K. Zhang, T.-s. Song and J. Xie, α-Fe2O3/g-C3N4 Z-scheme heterojunction photocathode to enhance microbial electrosynthesis of acetate from CO2, ACS Sustainable Chem. Eng., 2022, 10, 17308–17317 CrossRef CAS.
- M. Wang, M. Langer, R. Altieri, M. Crisci, S. Osella and T. Gatti, Two-dimensional layered heterojunctions for photoelectrocatalysis, ACS Nano, 2024, 18, 9245–9284 CrossRef CAS PubMed.
- X. Wang, Z. Zhao, K. Zahra, J. Li and Z. Zhang, Sub-nanomaterials for Photo/Electro-catalytic CO2 Reduction: Achievements, Challenges, and Opportunities, Chem. Res. Chin. Univ., 2023, 39, 580–598 CrossRef CAS.
- C. Liu, Y. Xiao, W. Wan, Y. Wei, Y. Cao, L. Hong, Y. Wang, J. Chen, Q. Zhang and H. Jing, Different behaviors on the external and inner surface of hollow CdS/VS-MoS2 heterojunctions in photoelectrocatalytic CO2 reduction via SH-assisted mechanism, Appl. Catal., B, 2023, 325, 122394 CrossRef CAS.
- X. Guo, X. Wang, X. Guan, J. Li, C. Zhang, Y. Bai and X. Zhang, In situ fabrication of 2D Bi/Bi2O2CO3 nanosheets anchored on Bi substrate for highly-efficient photoelectrocatalytic CO2 reduction to formate, Appl. Surf. Sci., 2024, 664, 160216 CrossRef CAS.
- Y. Zhang, C. Ye, J. Duan, H. Feng, D. Liu and Q. Li, Solar-Driven Carbon Dioxide Reduction: A Fair Evaluation of Photovoltaic-Biased Photoelectrocatalysis and Photovoltaic-Powered Electrocatalysis, Front. Energy Res., 2022, 10, 956444 CrossRef.
- H. Leng, Z. Li, W. Li, Z. Lv, J. Guo, H. You, Y. Jia, G. Zhang and L. Wang, Synergy of dual photoelectrodes for simultaneous antibiotic degradation and CO2 reduction by Z-scheme PEC system, Sep. Purif. Technol., 2024, 338, 126504 CrossRef CAS.
- C. Xu, X. Zhang, M.-N. Zhu, L. Zhang, P. Sui, R. Feng, Y. Zhang and J. Luo, Accelerating photoelectric CO2 conversion with a photothermal wavelength-dependent plasmonic local field, Appl. Catal., B, 2021, 298, 120533 CrossRef CAS.
- T. A. Kistler, M. Y. Um, J. K. Cooper, I. D. Sharp and P. Agbo, Exploiting heat transfer to achieve efficient photoelectrochemical CO2 reduction under light concentration, Energy Environ. Sci., 2022, 15, 2061–2070 RSC.
- Y. Liu, M. Xia, D. Ren, S. Nussbaum, J.-H. Yum, M. Grätzel, N. Guijarro and K. Sivula, Photoelectrochemical CO2 Reduction at a Direct CuInGaS2/Electrolyte Junction, ACS Energy Lett., 2023, 8, 1645–1651 CrossRef CAS PubMed.
- C. W. S. Yeung, V. Andrei, T. H. Lee, J. R. Durrant and E. Reisner, Organic Semiconductor-BiVO4 Tandem Devices for Solar-Driven H2O and CO2 Splitting, Adv. Mater., 2024, 36, 2404110 CrossRef CAS PubMed.
- H. J. Jang, J. H. Yang, J. Y. Maeng, M. H. Joo, Y. J. Kim, C. K. Rhee and Y. Sohn, Photoelectrochemical CO2 reduction products over sandwiched hybrid Ga2O3: ZnO/Indium/ZnO nanorods, Front. Chem., 2022, 10, 814766 CrossRef CAS PubMed.
- D. Ren, J. H. Fong and B. S. Yeo, The effects of currents and potentials on the selectivities of copper toward carbon dioxide electroreduction, Nat. Commun., 2018, 9, 925 CrossRef PubMed.
- J. Jin, G. Cao, Y. Liu, Y. Shu, Z. Deng, W. Sun and X. Yang, Metal-organic-frameworks passivated CuBi2O4 photocathodes boost CO2 reduction kinetics, Mater. Rep.: Energy, 2023, 3, 100229 CAS.
- Z. Otgonbayar, C.-M. Yoon and W.-C. Oh, Photoelectrocatalytic CO2 reduction with ternary nanocomposite of MXene (Ti3C2)-Cu2O-Fe3O4: comprehensive utilization of electrolyte and light-wavelength, Chem. Eng. J., 2023, 464, 142716 CrossRef CAS.
- W. J. Dong, P. Zhou, Y. Xiao, I. A. Navid, J. L. Lee and Z. Mi, Silver halide catalysts on GaN nanowires/Si heterojunction photocathodes for CO2 reduction to syngas at high current density, ACS Catal., 2022, 12, 2671–2680 CrossRef CAS.
- H. J. Jang, J. H. Yang, J. Y. Maeng, M. H. Joo, Y. J. Kim, S.-M. Hong, C. K. Rhee and Y. Sohn, CO2 reduction by photocatalytic and photoelectrocatalytic approaches over Eu (III)-ZnGa2O4 nanoparticles and Eu (III)-ZnGa2O4/ZnO nanorods, J. CO2 Util., 2022, 60, 101994 CrossRef CAS.
- Z. Masoumi, M. Tayebi, M. Tayebi, S. A. Masoumi Lari, N. Sewwandi, B. Seo, C.-S. Lim, H.-G. Kim and D. Kyung, Electrocatalytic reactions for converting CO2 to value-added products: recent progress and emerging trends, Int. J. Mol. Sci., 2023, 24, 9952 CrossRef CAS PubMed.
- Z. Zhang, X. Ding, X. Yang, W. Tu, L. Wang and Z. Zou, Shedding light on CO2: Catalytic synthesis of solar methanol, EcoMat, 2021, 3, e12078 CrossRef CAS.
- C. Liu, L. Fan, F. Meng, Y. Wei, C. Zhuang, J. Wang, S. Chen, W. Ye, Q. Zhang and H. Jing, Photoelectrothermocatalytic reduction of CO2 to glycol via CdIn2S4-N/C hollow heterostructure mimicking plant cell, Chem. Eng. J., 2024, 485, 149707 CrossRef CAS.
- Y. Cao, C. Zhuang, J. Wang, S. Chen, Q. Zhang, W. Ye and H. Jing, Photoelectrothermocatalytic reduction of CO2 to glycol via Cu2S/MoS2-Vs octahedral heterostructure with synergistic mechanism, J. Colloid Interface Sci., 2024, 666, 141–150 CrossRef CAS PubMed.
- W. Yang, J. Bao, L. Sun, Z. Mo, M. Du, Y. Shi and Y. Xu, Multiphysics Modeling of Heat and Mass Transfer for Photoelectrochemical CO2 Reduction to CO, Ind. Eng. Chem. Res., 2025, 64, 3022–3033 CrossRef CAS.
- S. Chakraborty and S. C. Peter, Solar-Fuel Production by Photodriven CO2 Reduction: Facts, Challenges, and Recommendations, ACS Energy Lett., 2025, 10, 2359–2371 CrossRef CAS.
- S. J. Cobb, C. Pornrungroj, V. Andrei, V. M. Badiani, L. Su, R. R. Manuel, I. A. Pereira and E. Reisner, A photoelectrochemical-thermoelectric device for semi-artificial CO2 fixation employing full solar spectrum utilization, Device, 2024, 2, 100505 CrossRef.
- J. Wang, Z. Hu, N. Ullah, Y. Wei, C. Zhuang, C. He, C. Liu, Y. Cao, F. Meng and W. Ye, The bi-pn heterojunction of TiO2@ NC-nanospheres with microchannels mimicking plant cell for photoelectro-thermocatalytic CO2 reduction—thermoelectron quantum tunneling effect, Chem. Eng. J., 2025, 160633 CrossRef CAS.
- M. B. Akbar, Y. Wang, X. Zhang and T. He, Study on photoelectrochemical CO2 reduction over Cu2O, J. Photochem. Photobiol., A, 2023, 437, 114483 CrossRef CAS.
- Y. Horiuchi, K. Miyazaki, M. Tachibana, K. Nishigaki and M. Matsuoka, Solar light-driven selective photoelectrochemical CO2 reduction to CO in aqueous media using Si nanowire arrays decorated with Au and Au-based metal nanoparticles, Res. Chem. Intermed., 2023, 49, 1131–1146 CrossRef CAS.
- E. Edwardes Moore, V. Andrei, A. R. Oliveira, A. M. Coito, I. A. Pereira and E. Reisner, A Semi-artificial Photoelectrochemical Tandem Leaf with a CO2-to-Formate Efficiency Approaching 1%, Angew. Chem., Int. Ed., 2021, 133, 26507–26511 CrossRef.
- P. Li, Y. Lin, Z. Qi and D. Yan, Efficient photoelectrocatalytic CO2 reduction to CH3OH via porous g-C3N4 nanosheets modified with cobalt phthalocyanine in ionic liquids, J. Mater. Chem. A, 2023, 11, 21078–21088 RSC.
- S.-T. Guo, Z.-Y. Tang, Y.-W. Du, T. Liu, T. Ouyang and Z.-Q. Liu, Chlorine anion stabilized Cu2O/ZnO photocathode for selective CO2 reduction to CH4, Appl. Catal., B, 2023, 321, 122035 CrossRef CAS.
- Y. Gao, X. Wang, H. Guo, L. Liu, H. Wang and W. Cui, Ionic liquids enhanced highly efficient photoelectrochemical reduction of CO2 to ethanol over Cu2O/TiO2 nanoarrays, Mol. Catal., 2023, 543, 113161 CrossRef CAS.
- F. A. Sayao, X. Ma, M. V. B. Zanoni and A. Lachgar, Modulating the photoelectrocatalytic conversion of CO2 to methanol and/or H2 O to hydrogen at a phosphorene modified Ti/TiO2 electrode, J. Mater. Chem. C, 2022, 10, 11276–11285 RSC.
- J. Y. Zheng, A. U. Pawar and Y. S. Kang, Preparation of C3N4 Thin Films for Photo-/Electrocatalytic CO2 Reduction to Produce Liquid Hydrocarbons, Catalysts, 2022, 12, 1399 CrossRef CAS.
- M. Chen, Y.-h. Sun, D. Zhou, Y. Yan, L. Sun, H.-B. Cheng, Z. Chen, C.-M. Tang, L. Chang and J.-Q. Xu, Efficient CO2 reduction to C2 products in a Ce-TiO2 photoanode-driven photoelectrocatalysis system using a Bnanometer Cu2O cathode, Appl. Catal., A, 2024, 687, 119966 CrossRef CAS.
- J. Kim, J. Lin, J. Kim, I. Roh, S. Lee and P. Yang, A red-light-powered silicon nanowire biophotochemical diode for simultaneous CO2 reduction and glycerol valorization, Nat. Catal., 2024, 7, 977–986 CrossRef CAS.
- M. M. May and K. Rehfeld, Negative emissions as the new frontier of photoelectrochemical CO2 reduction, Adv. Energy Mater., 2022, 12, 2103801 CrossRef CAS.
- B. Liu, Z. Qian, X. Shi, H. Su, W. Zhang, A. Kludze, Y. Zheng, C. He, R. Yanagi and S. Hu, Solar-driven selective conversion of millimolar dissolved carbon to fuels with molecular flux generation, Nat. Commun., 2025, 16, 1558 CrossRef CAS PubMed.
- H. Jung, A. Jamal, I. Gereige, T. T. Nguyen, J. W. Ager and H. T. Jung, Continuous Flow Photoelectrochemical Reactor with Gas Permeable Photocathode: Enhanced Photocurrent and Partial Current Density for CO2 Reduction, Adv. Sci., 2025, 12, 2411348 CrossRef CAS PubMed.
- D.-d. Hu, R.-t. Guo, J.-s. Yan, S.-h. Guo and W.-g. Pan, Metal–organic frameworks (MOFs) for photoelectrocatalytic (PEC) reducing carbon dioxide (CO2) to hydrocarbon fuels, Nanoscale, 2024, 16, 2185–2219 RSC.
- L. Qiu, H. Li and L. He, Incorporating catalytic units into nanomaterials: Rational design of multipurpose catalysts for CO2 valorization, Acc. Chem. Res., 2023, 56, 2225–2240 CrossRef CAS PubMed.
- L. Yang, A. U. Pawar, R. P. Sivasankaran, D. Lee, J. Ye, Y. Xiong, Z. Zou, Y. Zhou and Y. S. Kang, Intermediates and Their Conversion into Highly Selective Multi-Carbons in Photo-/Electrocatalytic CO2 Reduction Reaction, J. Mater. Chem. A, 2023, 11, 19172–19194 RSC.
- O. Christensen, A. Bagger and J. Rossmeisl, The Missing Link for Electrochemical CO2 Reduction: Classification of CO vs. HCOOH Selectivity via PCA, Reaction Pathways, and Coverage Analysis, ACS Catal., 2024, 14, 2151–2161 CrossRef CAS.
- H. Lu and L. Wang, Unbiased photoelectrochemical carbon dioxide reduction shaping the future of solar fuels, Appl. Catal., B, 2024, 123707 CrossRef CAS.
- Z. Xiao, H. Zhang, X. Tan, F. Ye, Y. Zhang, J. Gu, J. Li, K. Sun, S. Zhang and J. J. Zou, Comprehensive Insight into External Field-Driven CO2 Reduction to CO: Recent Progress and Future Prospects, Adv. Energy Mater., 2025, 2500988 CrossRef CAS.
- H. Du and Z. Shao, A Review on Modulating Oxygen Vacancy Defect of Catalysts to Promote CO2 Reduction Reaction to CO, Energy Fuels, 2025, 39, 5672–5690 CrossRef CAS.
- Y. Liu, Y. Xu, X. Pang, B. Zhang, Y. Gao, Z. Hu, Y. Miao, W. Tian and H. Jing, The cooperative pn heterojunction and Schottky junction in Cu-coated Cu2O/TiO2/Ti3C2 for efficiently photoelectrocatalytic CO2 reduction to ethanol, J. Alloys Compd., 2025, 1017, 179012 CrossRef CAS.
- Z. Yang, J. Yang, H. Yang, F. Gao, C. Nan, R. Chen, Y. Zhang, X. Gao, Y. Yuan and Y. Jia, Photoelectrocatalytic CO2 Reduction to Formate Using a BiVO4/ZIF-8 Heterojunction, ChemPlusChem, 2025, 90, e202400452 CrossRef PubMed.
- N. Zhang and Y. Xiong, Photoelectrochemical CO2 fixation to C2 hydrocarbons, Sci. China:Chem., 2025, 1–2 Search PubMed.
- G. Zhou, X. Liu, Y. Xu, S. Feng, Z. Lu and Z. Q. Liu, Enhancing d/p-2π* Orbitals Hybridization via Strain Engineering for Efficient CO2 Photoreduction, Angew. Chem., Int. Ed., 2024, 63, e202411794 CrossRef CAS PubMed.
- G. Liu, G. Ma, H. Mu, J. Li, F. Li, M. Zhu and J. Zhang, Enhanced photoelectrocatalytic CO2 reduction to CO via structure-induced carrier separation in coral-like CuBi2O4-Bi2O3, Sep. Purif. Technol., 2025, 358, 130319 CrossRef CAS.
- G. Ma, H. Mu, Z. Lv, J. Guo, M. Zhu, Y. Li, X. Wang, J. Li and F. Li, Coaxially Bi/ZnO@ ZnSe array photocathode enables highly efficient CO2 to C1 conversion via long-lived high-energy photoelectrons, ChemSusChem, 2025, 18, e202401436 CrossRef CAS PubMed.
- A. Ziarati, H. Gotfredsen, A. Rosspeintner, J. Zhao, H. L. Anderson and T. Bürgi, Encapsulation of an Au25 Nanocluster inside a Porphyrin Nanoring Enhances Singlet Oxygen Generation and Photo-Electrocatalytic CO2 Reduction, Angew. Chem., Int. Ed., 2024, e202414908, DOI:10.1002/anie.202414908.
- T. Takayama, A. Iwase and A. Kudo, Enhancing Photocathodic Performances of Particulate-CuGaS2-Based Photoelectrodes via Conjugation with Conductive Organic Polymers for Efficient Solar-Driven Hydrogen Production and CO2 Reduction, ACS Appl. Mater. Interfaces, 2024, 16, 36423–36432 CrossRef CAS PubMed.
- Q. Wang, B. Liu, S. Wang, P. Zhang, T. Wang and J. Gong, Highly selective photoelectrochemical CO2 reduction by crystal phase-modulated nanocrystals without parasitic absorption, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2316724121 CrossRef CAS PubMed.
- X. Zhang, J. Wang, Y. Liu, J. Sun, B. Xu and T. Li, Photoelectrocatalytic Reduction of CO2 to CO via Cu2O/C/PTFE Nanowires Photocathodes, ChemPhotoChem, 2024, 8, e202400005 CrossRef CAS.
- M. Zhang, W. Luo, S. Gu, W. Xu, Z. Lu and F. Wang, Photoelectrochemical catalytic CO2 reduction enhanced by In-doped GaN and combined with vibration energy harvester driving CO2 reduction, Appl. Catal., A, 2024, 683, 119859 CrossRef CAS.
- X. Jia, E. Stewart-Jones, J. L. Alvarez-Hernandez, G. P. Bein, J. L. Dempsey, C. L. Donley, N. Hazari, M. N. Houck, M. Li and J. M. Mayer, Photoelectrochemical CO2 Reduction to CO Enabled by a Molecular Catalyst Attached to High-Surface-Area Porous Silicon, J. Am. Chem. Soc., 2024, 146, 7998–8004 CrossRef CAS PubMed.
- C. Liu, Z. Bian, W. Wang, H. Li, D. Dang and Y. Bai, Photoelectrochemically converting CO2 over Z-scheme heterojunction CoPc/K7HNb6O19 with high Faraday efficiency, Appl. Surf. Sci., 2024, 663, 160192 CrossRef CAS.
- J. Kang, W. He, K. Wang, Y. Chen, Y. Liu, Y. Li and W. Li, Single-atom nickel sites boosting Si nanowires for photoelectrocatalytic CO2 conversion with nearly 100% selectivity, Chem. Commun., 2024, 60, 6039–6042 RSC.
- Y. Wei, Y. Zhu, P. Li, X. Gao, Z. Yu, S. Liu, N. Li, Y. Shen and M. Wang, Surface states modulation of ZnTe via ultrathin ZnO layer as efficient photocathodes for CO2 reduction reaction, Appl. Catal., B, 2024, 347, 123760 CrossRef CAS.
- Y. Zhu, Y. Wei, P. Li, S. Liu, J. Zhang, L. Tian, P. Gao, Y. Zhang, J. Li and D. Wang, Type-II heterojunction photocathode for CO2 reduction and light-assisted metal–CO2 batteries, J. Mater. Chem. A, 2024, 12, 5133–5144 RSC.
- X. Gao, N. Li, P. Li, Y. Wei, Q. Huang, K. Akhtar, E. M. Bakhsh, S. B. Khan, Y. Shen and M. Wang, ZnTe/SnS2 heterojunction for photo-electrocatalysis of CO2 to CO, Electrochim. Acta, 2024, 144603 CrossRef CAS.
- C. Lei, M. Luo, H. Li, Q. Shen, X. Liu, H. Jia and J. Xue, Amino group promoted photoelectrocatalytic CO2 reduction activity observed in mixed-linker Cu-based metal–organic frameworks, J. Mater. Sci.:Mater. Electron., 2023, 34, 1232 CrossRef CAS.
- B. Shang, C. L. Rooney, D. J. Gallagher, B. T. Wang, A. Krayev, H. Shema, O. Leitner, N. J. Harmon, L. Xiao and C. Sheehan, Aqueous photoelectrochemical CO2 reduction to CO and methanol over a silicon photocathode functionalized with a cobalt phthalocyanine molecular catalyst, Angew. Chem., Int. Ed., 2023, 62, e202215213 CrossRef CAS PubMed.
- M. Zoli, H. Guzmán, A. Sacco, N. Russo and S. Hernández, Cu2O/SnO2 Heterostructures: Role of the Synthesis Procedure on PEC CO2 Conversion, Materials, 2023, 16, 4497 CrossRef CAS PubMed.
- Y. Jia, Z. Tian and J. Gao, An effective integrated Cu2O photocathode to boost photoelectrocatalytic CO2 conversion, J. Mater. Chem. A, 2023, 11, 11411–11425 RSC.
- M. Zoli, D. Roldán, H. Guzmán, M. Castellino, A. Chiodoni, K. Bejtka, N. Russo and S. Hernández, Facile and scalable synthesis of Cu2O-SnO2 catalyst for the photoelectrochemical CO2 conversion, Catal. Today, 2023, 413, 113985 CrossRef.
- X. Liu, X. Wang, B. Yang, J. Zhang and J. Lu, High-entropy layered oxides nanosheets for highly efficient photoelectrocatalytic reduction of CO2 and application research, Nano Res., 2023, 16, 4775–4785 CrossRef CAS.
- J. K. Karlsson, F. J. Cerpentier, R. Lalrempuia, M. V. Appleby, J. D. Shipp, D. Chekulaev, O. Woodford, J. A. Weinstein, M. T. Pryce and E. A. Gibson, Ruthenium–rhenium and ruthenium–palladium supramolecular photocatalysts for photoelectrocatalytic CO2 and H+ reduction, Sustainable Energy Fuels, 2023, 7, 3284–3293 RSC.
- S. Dutta, A. Gurumoorthi, S. Lee, S. Jang, N. Kumari, Y. Hong, W. Choi, C. Son and I. Lee, Sculpting In-plane Fractal Porous Patterns in Two-Dimensional Metal-Organic Framework (MOF) Nanocrystals for Photoelectrocatalytic CO2 Reduction, Angew. Chem., Int. Ed., 2023, e202303890 CAS.
- R. R. Prabhakar, R. Lemerle, M. Barecka, M. Kim, S. Seo, E. N. Dayi, I. Dei Tos and J. W. Ager, TaOx electron transport layers for CO2 reduction Si photocathodes, J. Mater. Chem. A, 2023, 11, 13588–13599 RSC.
- Z. Wen, S. Xu, Y. Zhu, G. Liu, H. Gao, L. Sun and F. Li, Aqueous CO2 Reduction on Si Photocathodes Functionalized by Cobalt Molecular Catalysts/Carbon Nanotubes, Angew. Chem., Int. Ed., 2022, 61, e202201086 CrossRef CAS PubMed.
- B. Liu, T. Wang, S. Wang, G. Zhang, D. Zhong, T. Yuan, H. Dong, B. Wu and J. Gong, Back-illuminated photoelectrochemical flow cell for efficient CO2 reduction, Nat. Commun., 2022, 13, 7111 CrossRef CAS PubMed.
- Q. Wang, Y. Zhang, Y. Liu, K. Wang, W. Qiu, L. Chen, J. Li and W. Li, Core–Shell In/Cu2O Nanowires Schottky Junction for Enhanced Photoelectrochemical CO2 Reduction under Visible Light, Ind. Eng. Chem. Res., 2022, 61, 16470–16478 CrossRef CAS.
- Z. Wei, Q. Mu, R. Fan, Y. Su, Y. Lu, Z. Deng, M. Shen and Y. Peng, Cupric porphyrin frameworks on multi-junction silicon photocathodes to expedite the kinetics of CO2 turnover, Nanoscale, 2022, 14, 8906–8913 RSC.
- S. Chu, R. T. Rashid, Y. Pan, X. Wang, H. Zhang and R. Xiao, The impact of flue gas impurities and concentrations on the photoelectrochemical CO2 reduction, J. CO2 Util., 2022, 60, 101993 CrossRef CAS.
- K. M. Alenezi, Metalloporphyrin catalysed photoelectrochemical reduction of CO2 at p-type silicon semiconducting electrode, Int. J. Electrochem. Sci., 2022, 17, 220147 CrossRef CAS.
- Y. Lai, N. B. Watkins, C. Muzzillo, M. Richter, K. Kan, L. Zhou, J. A. Haber, A. Zakutayev, J. C. Peters and T. Agapie, Molecular coatings improve the selectivity and durability of CO2 reduction chalcogenide photocathodes, ACS Energy Lett., 2022, 7, 1195–1201 CrossRef CAS.
- N. H. Makani, M. Singh, T. Paul, A. Sahoo, J. Nama, S. Sharma and R. Banerjee, Photoelectrocatalytic CO2 reduction using stable lead-free bimetallic CsAgBr2 halide perovskite nanocrystals, J. Electroanal. Chem., 2022, 920, 116583 CrossRef CAS.
- K. Wang, N. Fan, B. Xu, Z. Wei, C. Chen, H. Xie, W. Ye, Y. Peng, M. Shen and R. Fan, Steering the Pathway of Plasmon-Enhanced Photoelectrochemical CO2 Reduction by Bridging Si and Au Nanoparticles through a TiO2 Interlayer, Small, 2022, 18, 2201882 CrossRef CAS PubMed.
- B. Ding, B. Chan, N. Proschogo, M. B. Solomon, C. J. Kepert and D. M. D'Alessandro, A cofacial metal–organic framework based photocathode for carbon dioxide reduction, Chem. Sci., 2021, 12, 3608–3614 RSC.
- T. Wang, L. Guo, H. Pei, S. Chen, R. Li, J. Zhang and T. Peng, Electron-Rich Pincer Ligand-Coupled Cobalt Porphyrin Polymer with Single-Atom Sites for Efficient (Photo)Electrocatalytic CO2 Reduction at Ultralow Overpotential, Small, 2021, 17, 2102957 CrossRef CAS PubMed.
- G. Liu, P. R. Narangari, Q. T. Trinh, W. Tu, M. Kraft, H. H. Tan, C. Jagadish, T. S. Choksi, J. W. Ager, S. Karuturi and R. Xu, Manipulating Intermediates at the Au–TiO2 Interface over InP Nanopillar Array for Photoelectrochemical CO2 Reduction, ACS Catal., 2021, 11, 11416–11428 CrossRef CAS.
- J. Wang, L. Wu, S. Liu, S. Liu, H. Ji, C. Chen, Y. Zhang and J. Zhao, Bias distribution and regulation for efficient oxygen atom transfer in photoelectrochemical cells coupled with styrene epoxidation and CO2 reduction, J. Catal., 2025, 443, 115941 CrossRef CAS.
- C. P. Muzzillo, Y. Lai, J. A. Haber and A. Zakutayev, Chloride Treatments Improve Zinc Telluride Absorbers for Photoelectrochemical Carbon Dioxide Reduction, ACS Appl. Energy Mater., 2025, 8, 983–990 CrossRef CAS PubMed.
- C. J. Sheehan, S. Suo, S. Jeon, Y. Zheng, J. Meng, F. Zhao, Z. Yang, L. Xiao, S. Venkatesan and A. S. Metlay, Electron Transfer Energetics in Photoelectrochemical CO2 Reduction at Viologen Redox Polymer-Modified p-Si Electrodes, J. Am. Chem. Soc., 2025, 147, 9629–9639 CrossRef CAS PubMed.
- C. Wang, L. Geng and Y. Bi, Highly Active Oxygen Evolution Integrating with Highly Selective CO2-to-CO Reduction, Nano-Micro Lett., 2025, 17, 184 CrossRef CAS PubMed.
- K. Wang, Y. Liu, M. Liu and W. Li, Isolated Co atoms anchored Al-oxo chain based-porphyrin framework for highly selective CO2 reduction over Si nanowire photocathode, Appl. Catal., B, 2025, 371, 125260 CrossRef CAS.
- R. R. Prabhakar, S. Shukla, H. Li, R. S. Kim, W. Chen, J. Beaudelot, J. D’Haen, D. R. Santos, P. M. Vereecken and G.-M. Rignanese, Origin of photoelectrochemical CO2 reduction on bare Cu (In, Ga) S2 (CIGS) thin films in aqueous media without co-catalysts, EES Catal., 2025, 3, 327–336 RSC.
- G. Zeng, G. Liu and G. Panzeri, Surface Composition Impacts Selectivity of ZnTe Photocathodes in Photoelectrochemical, ACS Energy Lett., 2025, 10(1), 34–39 CrossRef CAS PubMed.
- K. Peramaiah, P. Varadhan, V. Ramalingam, B. Khan, P. K. Das, H. Huang, H. C. Fu, X. Yang, V. Tung and K. W. Huang, Unassisted photoelectrochemical CO2 reduction by employing III–V photoelectrode with 15% solar-to-fuel efficiency, Carbon Energy, 2025, 7, e669 CrossRef CAS.
- S. S. Saund, M. K. Gish, J. Choate, T. H. Le, S. C. Marinescu and N. R. Neale, Design Strategies for Coupling CO2 Reduction Molecular Electrocatalysts to Silicon Photocathodes, ACS Mater. Au, 2025, 5, 569–579 CrossRef CAS PubMed.
- M. Xia, L. Pan, Y. Liu, J. Gao, J. Li, M. Mensi, K. Sivula, S. M. Zakeeruddin, D. Ren and M. Grätzel, Efficient Cu2O Photocathodes for Aqueous Photoelectrochemical CO2 Reduction to Formate and Syngas, J. Am. Chem. Soc., 2023, 145, 27939–27949 CrossRef CAS PubMed.
- S. Chu, P. Ou, R. T. Rashid, Y. Pan, D. Liang, H. Zhang and J. Song, Efficient photoelectrochemical conversion of CO2 to syngas by photocathode engineering, Green Energy Environ., 2022, 7, 545–553 CrossRef CAS.
- V. Andrei, R. A. Jagt, M. Rahaman, L. Lari, V. K. Lazarov, J. L. MacManus-Driscoll, R. L. Hoye and E. Reisner, Long-term solar water and CO2 splitting with photoelectrochemical BiOI–BiVO4 tandems, Nat. Mater., 2022, 21, 864–868 CrossRef CAS PubMed.
- Y. Zhang, D. Pan, Y. Tao, H. Shang, D. Zhang, G. Li and H. Li, Photoelectrocatalytic reduction of CO2 to syngas via SnOx-enhanced Cu2O nanowires photocathodes, Adv. Funct. Mater., 2022, 32, 2109600 CrossRef CAS.
- P. Wen, H. Li, X. Ma, R. Lei, X. Wang, S. M. Geyer and Y. Qiu, A colloidal ZnTe quantum dot-based photocathode with a metal–insulator–semiconductor structure towards solar-driven CO2 reduction to tunable syngas, J. Mater. Chem. A, 2021, 9, 3589–3596 RSC.
- C. Zhang, C. Chen, J. Mao, D. Wang, P. Luan, Q. Song, Y. Xie, Y. Wang, Y. Zhao and Y. Zhang, Boosting syngas production in photoelectrochemical CO2 reduction through organic molecule interaction with copper photoanodes, Nano Res., 2025, 18, 94907306 CrossRef.
- D. Pan, Y. Wang, Y. Tao, C. Zhang, K. Xu, H. Li, G. Li and H. Li, Efficient carbon recycling for syngas generation through a Dual-Photoelectrode artificial photosynthesis system, Chem. Eng. J., 2025, 160559 CrossRef CAS.
- T. Takayama, A. Iwase and A. Kudo, Improvement of Performance to form Syngas utilizing Water and CO2 over Particulate-Cu0.8Ag0.2 GaS2-Based Photocathode by Surface Comodification with ZnS and Ag, Sustainable Energy Fuels, 2025, 9, 1709–1716 RSC.
- B. Fabre and G. Loget, Silicon photoelectrodes prepared by low-cost wet methods for solar photoelectrocatalysis, Acc. Mater. Res., 2023, 4, 133–142 CrossRef CAS.
- Y. Wei, C. He, N. Ullah, Y. Cao, C. Zhuang, B. Wang, J. Wang, Z. Hu, D. Ma and W. Ye, Leaf-like MSX/TiN heterojunction photocathodes mimicking plant cell–An effective strategy to enhance photoelectrocatalytic carbon dioxide reduction and systematic mechanism investigation, J. Colloid Interface Sci., 2025, 678, 1–12 CrossRef CAS PubMed.
- L. Li, B. Nan, N. N. Xu, G. Bai, R. He, Y. Liu and J. Qiao, Sunlight-promoted CO2 electroreduction with staggered pn heterojunction by indium-doped bismuth 3D nanoflower structure on oxidized copper foam as self-standing photoelectric cathode over a wide potential window, Appl. Catal., B, 2025, 360, 124489 CrossRef CAS.
- S. Liu, Z. Guo, Y. Yang, P.-d. Wu, Z. Li, K. Wang, H. Zhang, H. Li and S. Yang, Cobalt-doped CdS quantum dots enhanced photoelectroreduction of CO2 to formic acid with high selectivity, Environ. Chem. Lett., 2024, 22, 463–470 CrossRef CAS.
- Y. Jia, H. Yang, R. Chen, Y. Zhang, F. Gao, C. Nan, J. Yang and X. Gao, Efficient photoelectrocatalytic reduction of CO2 to formate via Bi-doped InOCl nanosheets, J. Alloys Compd., 2024, 987, 174220 CrossRef CAS.
- J. A. Abarca, I. Merino-Garcia, G. Díaz-Sainz, M. Perfecto-Irigaray, G. Beobide, A. Irabien and J. Albo, Fabrication and optimization of perovskite-based photoanodes for solar-driven CO2 photoelectroreduction to formate, Catal. Today, 2024, 429, 114505 CrossRef CAS.
- W. Li, J. Hong, J. Shang, H. Yamashita, C. Wei and Y. Hu, In situ construction of CuBi-MOF derived heterojunctions with electron-rich effects enhances localized CO2 enrichment integrated with Si photocathodes for CO2 reduction, Appl. Catal., B, 2025, 365, 124890 CrossRef CAS.
- W. Cheng, Y. Wang, S. Guo, Q. Cheng, H. Zhao and L. Gao, Photoanode driven photoelectrocatalytic system for CO2 reduction to formic acid based on lattice-dislocated Bi nanosheets cathode, New J. Chem., 2024, 48, 3110–3119 RSC.
- Z. Yang, J. Yang, H. Yang, F. Gao, C. Nan, R. Chen, Y. Zhang, X. Gao, Y. Yuan and Y. Jia, Photoelectrocatalytic CO2 Reduction to Formate Using a BiVO4/ZIF-8 Heterojunction, ChemPlusChem, 2024, e202400452, DOI:10.1002/cplu.202400452.
- B. Khan, M. B. Faheem, K. Peramaiah, J. Nie, H. Huang, Z. Li, C. Liu, K.-W. Huang and J.-H. He, Unassisted photoelectrochemical CO2-to-liquid fuel splitting over 12% solar conversion efficiency, Nat. Commun., 2024, 15, 6990 CrossRef CAS PubMed.
- W. Shen, Z. Yang, J. Wang, J. Cui, Z. Bao, D. Yu, M. Guo, G. Xu and J. Lv, Bi–Sn Co-Catalyst-Modified p-Si Nanowire Array Photocathodes for Photoelectrocatalytic CO2 Reduction to Formate, ACS Sustainable Chem. Eng., 2023, 11, 13451–13457 CrossRef CAS.
- T. Bhoyar, B. M. Abraham, N. R. Manwar, A. Gupta, N. Mameda, S. Tonda, D. Vidyasagar and S. S. Umare, Defective nano-silica loaded polymeric carbon nitride for visible light driven CO2 reduction and dye degradation, Catal. Commun., 2023, 179, 106692 CrossRef CAS.
- H. Yang, F. Gao, W. Zhou, N. Gao, D. Zhang, Z. Li and C. Nan, Efficient CO2 reduction to formate in a photoanode-driven photoelectrocatalysis system using a Bi2Se3/Bi2O3 nanocomposite cathode, Appl. Surf. Sci., 2023, 623, 157097 CrossRef CAS.
- F. Gao, H. Yang, C. Nan, W. Zhou, N. Gao, Y. Jia, Y. Zhang and R. Chen, Efficient CO2 reduction to formate using a Cu-doped BiVO4 electrocathode in a WO3 photoanode-assisted photoelectrocatalytic system, J. Electroanal. Chem., 2023, 930, 117146 CrossRef CAS.
- Y. Xu, F. Wang, S. Lei, Y. Wei, D. Zhao, Y. Gao, X. Ma, S. Li, S. Chang and M. Wang, In situ grown two-dimensional TiO2/Ti3CN MXene heterojunction rich in Ti3+ species for highly efficient photoelectrocatalytic CO2 reduction, Chem. Eng. J., 2023, 452, 139392 CrossRef CAS.
- Y. Pan, H. Zhang, B. Zhang, F. Gong, J. Feng, H. Huang, S. Vanka, R. Fan, Q. Cao and M. Shen, Renewable formate from sunlight, biomass and carbon dioxide in a photoelectrochemical cell, Nat. Commun., 2023, 14, 1013 CrossRef CAS PubMed.
- R. Miró, H. Guzmán, C. Godard, A. Gual, F. Zammillo, T. J. Schubert, B. Iliev, A. Chiodoni, S. Hernández and M. D. de los Bernardos, Solar-driven CO2 reduction catalysed by hybrid supramolecular photocathodes and enhanced by ionic liquids, Catal. Sci. Technol., 2023, 13, 1708–1717 Search PubMed.
- G. Bharath, A. Hai, K. Rambabu, M. A. Haija and F. Banat, Sustainable electrochemical process for recovery of metal ions in synthetic mining wastewater and their utilization in photocathodic CO2 reduction into formic acid, Resour., Conserv. Recycl., 2023, 190, 106778 CrossRef CAS.
- W. J. Dong, I. A. Navid, Y. Xiao, T. H. Lee, J. W. Lim, D. Lee, H. W. Jang, J.-L. Lee and Z. Mi, Bi catalysts supported on GaN nanowires toward efficient photoelectrochemical CO2 reduction, J. Mater. Chem. A, 2022, 10, 7869–7877 RSC.
- Q. Zhang, X. Zhou, Z. Kuang, Y. Xue, C. Li, M. Zhu, C.-Y. Mou and H. Chen, A bismuth species-decorated ZnO/p-Si photocathode for high selectivity of formate in CO2 photoelectrochemical reduction, ACS Sustainable Chem. Eng., 2022, 10, 2380–2387 CrossRef CAS.
- S. Mubarak, D. Dhamodharan, H.-S. Byun, D. K. Pattanayak and S. Arya, Efficient photoelectrocatalytic conversion of CO2 to formic acid using Ag-TiO2 nanoparticles formed on the surface of nanoporous structured Ti foil, J. Ind. Eng. Chem., 2022, 113, 124–131 CrossRef CAS.
- S. Ren, H. Yang, D. Zhang, F. Gao, C. Nan, Z. Li, W. Zhou, N. Gao and Z. Liang, Excellent performance of the photoelectrocatalytic CO2 reduction to formate by Bi2S3/ZIF-8 composite, Appl. Surf. Sci., 2022, 579, 152206 CrossRef CAS.
- E. Zhang, J. Tang, Z. Li and Y. Zhou, Insight into the Synergistic Collaboration of g-C3N4/SnO2 Composites for Photoelectrocatalytic CO2 Reduction, ChemElectroChem, 2022, 9, e202200134 CrossRef CAS.
- H.-z. Wang, Y.-z. Zhao, Z.-x. Yang, X.-z. Bi, Z.-l. Wang and M.-b. Wu, Oxygen-incorporated carbon nitride porous nanosheets for highly efficient photoelectrocatalytic CO2 reduction to formate, N. Carbon Mater., 2022, 37, 1135–1142 CrossRef CAS.
- N. Nandal, N. R. Manwar, B. M. Abraham, P. K. Khatri and S. L. Jain, Photoelectrochemical reduction of CO2 promoted by a molecular hybrid made up of Co (II) Pc on graphene oxide under visible light illumination, Energy Fuels, 2022, 36, 3760–3770 CrossRef CAS.
- W. Zhou, H. Yang, N. Gao, D. Zhang, Z. Li, F. Gao and C. Nan, Vacancies and electronic effects enhanced photoelectrochemical activity of Cu-doped Bi2Se3 for efficient CO2 reduction to formate, J. Alloys Compd., 2022, 903, 163707 CrossRef CAS.
- M. T. Galante, P. V. Santiago, V. Y. Yukuhiro, L. A. Silva, N. A. Dos Reis, C. T. Pires, N. G. Macedo, L. S. Costa, P. S. Fernandez and C. Longo, Aminopolysiloxane as Cu2O photocathode overlayer: photocorrosion inhibitor and low overpotential CO2-to-formate selectivity promoter, ChemCatChem, 2021, 13, 859–863 CrossRef CAS.
- D. Zhang, H. Yang, Y. Li, Z. Li, N. Gao, W. Zhou and Z. Liang, High-performance Photoelectrocatalytic Reduction of CO2 by the hydrophilic–hydrophobic composite Cu-SnO2/ZIF-8, Int. J. Electrochem. Sci., 2021, 16, 150951 CrossRef CAS.
- Y. Yao, Z. Wu, Z. Zhao, Z. Sun, T. Li, Z. Li, X. Lu and Z. Chen, Architecting a Bias-Free Photoelectrochemical CO2 Reduction System for Sustainable Formic Acid, Adv. Sci., 2025, 2415774 CrossRef CAS PubMed.
- J. Xing, J. Shen, Z. Wei, Z. Zheng, Y. Cao, C. Chen, P. Y. Olu, W. Dong, Y. Peng and M. Shen, Dual Effect of Oxygen Vacancy-Enriched TiO2 Interlayer in Si Photocathode for Enhanced Photoelectrochemical CO2 Reduction to HCOOH, Small, 2025, 21, 2502226 CrossRef CAS PubMed.
- Y. H. Hong, X. Jia, E. Stewart-Jones, A. Kumar, J. C. Wedal, J. L. Alvarez-Hernandez, C. L. Donley, A. Gang, N. J. Gibson and N. Hazari, Photoelectrocatalytic reduction of CO2 to formate using immobilized molecular manganese catalysts on oxidized porous silicon, Chem, 2025, 102462 Search PubMed.
- Y. Liu, B. Chen, Y. Liu, Y. Ren, J. Wang, H. Qiu, Y. Chen, J. Shi and F. Wang, Photoelectrochemical CO2 reduction to formic acid using as cuprous oxide-based photocathodes, Fuel, 2025, 387, 134168 CrossRef CAS.
- Y. Liu, C. W. S. Yeung and E. Reisner, Photoelectrochemical comproportionation of pre-treated PET plastics and CO2 to formate, Energy Environ. Sci., 2025 10.1039/D5EE00689A.
- T. Bouwens, S. J. Cobb, C. W. Yeung, Y. Liu, G. Martins, I. A. Pereira and E. Reisner, Semiartificial Photoelectrochemistry for CO2-Mediated Enantioselective Organic Synthesis, J. Am. Chem. Soc., 2025, 147, 13114–13119 CrossRef CAS PubMed.
- M. Rahaman, C. Pulignani, M. Miller, S. Bhattacharjee, A. Bin Mohamad Annuar, R. R. Manuel, I. A. Pereira and E. Reisner, Solar-Driven Paired CO2 Reduction–Alcohol Oxidation Using Semiartificial Suspension, Photocatalyst Sheet, and Photoelectrochemical Devices, J. Am. Chem. Soc., 2025, 147, 8168–8177 CrossRef CAS PubMed.
- M. Amiri, M. Ahmadi, N. Khossossi, P. Gonugunta, K. Roohi, B. Kooi, M. Ramdin, P. R. Anusuyadevi, T. Tätte and N. Kongi, Ultra-thin defective TiO2 films as photocathodes for selective CO2 reduction to formate, J. Catal., 2025, 116022 CrossRef CAS.
- L. Wang, G. Qi and X. Liu, Ag/α-Fe2O3 nanowire arrays enable effectively photoelectrocatalytic reduction of carbon dioxide to methanol, J. Power Sources, 2021, 507, 230272 CrossRef CAS.
- Z. Otgonbayar and W.-C. Oh, Comprehensive-designed graphene-based quaternary nanocomposite and its synergistic effect towards photoelectrocatalytic CO2 reduction under different electrolytes, Fuel, 2024, 364, 131161 CrossRef CAS.
- X. Zhao, Z. Li, X. Zhang, X. Guan, C. Zhang, P. Tian, H. Yue, L. Wang, Y. Bai and Y. Wang, In situ preparation of Mo: BiVO4 photoanodes coupled with BiOBr cathode for efficient photoelectrocatalytic CO2 reduction performance, Sol. RRL, 2024, 8, 2301070 CrossRef CAS.
- Y. Fang, Y. Gao, Y. Wen, X. He, T. J. Meyer and B. Shan, Photoelectrocatalytic CO2 Reduction to Methanol by Molecular Self-Assemblies Confined in Covalent Polymer Networks, J. Am. Chem. Soc., 2024, 146, 27475–27485 CrossRef CAS PubMed.
- J. A. L. Perini, L. D. M. Torquato, J. F. de Brito, G. A. Andolpho, M. A. Gonçalves, L. D. De Angelis, L. D. Germano, S. I. C. de Torresi, T. C. Ramalho and M. V. B. Zanoni, Solar-driven CO2 conversion to methane and methanol using different nanostructured Cu2O-based catalysts modified with Au nanoparticles, J. Energy Chem., 2024, 91, 287–298 CrossRef CAS.
- P. Saravanan, P. P. Gotipamul, K. DamodarReddy, C. H. Campos, A. Selvaraj, R. V. Mangalaraja and S. Chidambaram, Synthesis of isolated ZnO nanorods on introducing g-C3N4 for improved photoelectrocatalytic methanol production by CO2 reduction, Inorg. Chem. Commun., 2024, 170, 113313 CrossRef CAS.
- B. Shang, F. Zhao, S. Suo, Y. Gao, C. Sheehan, S. Jeon, J. Li, C. L. Rooney, O. Leitner and L. Xiao, Tailoring Interfaces for Enhanced Methanol Production from Photoelectrochemical CO2 Reduction, J. Am. Chem. Soc., 2024, 146, 2267–2274 CrossRef CAS PubMed.
- R. M. e Silva, E. H. Dias, F. Escalona-Durán, W. Alnoush, J. A. de Oliveira, D. Higgins and C. Ribeiro, Unveiling BiVO4 photoelectrocatalytic potential for CO 2 reduction at ambient temperature, Mater. Adv., 2024, 5, 4857–4864 RSC.
- J. F. de Brito, M. A. S. Andrade Jr, M. V. B. Zanoni and L. H. Mascaro, All-solution processed CuGaS2-based photoelectrodes for CO2 reduction, J. CO2 Util., 2022, 57, 101902 CrossRef.
- R. Shah, M. Mastar, L. Minggu, W. Wong and R. Yunus, Potential of methanol production from the photoelectrochemical reduction of CO2 on rGO-CuO/Cu composite, Mater. Today: Proc., 2022, 57, 1123–1126 CAS.
- M. K. R. De Souza, E. d. S. F. Cardoso, G. V. Fortunato, M. R. Lanza, C. E. Nazário, M. V. B. Zanoni, G. Maia and J. C. Cardoso, Combination of Cu-Pt-Pd nanoparticles supported on graphene nanoribbons decorating the surface of TiO2 nanotube applied for CO2 photoelectrochemical reduction, J. Environ. Chem. Eng., 2021, 9, 105803 CrossRef CAS.
- F. Liu, J. Z. Zhu and J. L. Yuan, CuFeO/CuInS Composite Thin Film Photocathode Prepared by Template Method for CO Conversion Into Methanol, J. Electrochem. Soc., 2021, 168, 066505 CrossRef CAS.
- K. Irikura, J. A. L. Perini, J. B. S. Flor, R. C. G. Frem and M. V. B. Zanoni, Direct synthesis of Ru3(BTC)2 metal-organic framework on a Ti/TiO2NT platform for improved performance in the photoelectroreduction of CO2, J. CO2 Util., 2021, 43, 101364 CrossRef CAS.
- B. G, K. Rambabu, A. Hai, N. Ponpandian, J. E. Schmidt, D. D. Dionysiou, M. Abu Haija and F. Banat, Dual-functional paired photoelectrocatalytic system for the photocathodic reduction of CO2 to fuels and the anodic oxidation of furfural to value-added chemicals, Appl. Catal., B, 2021, 298, 120520 CrossRef CAS.
- B. C. e Silva, K. Irikura, R. C. G. Frem and M. V. B. Zanoni, Effect of Cu (BDC-NH2) MOF deposited on Cu/Cu2O electrode and its better performance in photoelectrocatalytic reduction of CO2, J. Electroanal. Chem., 2021, 880, 114856 CrossRef.
- A. G. K. Raj, C. Murugan and A. Pandikumar, Efficient photoelectrochemical reduction of carbon dioxide into alcohols assisted by photoanode driven water oxidation with gold nanoparticles decorated titania nanotubes, J. CO2 Util., 2021, 52, 101684 CrossRef.
- K. M. R. Karim, M. Tarek, S. M. Sarkar, R. Mouras, H. R. Ong, H. Abdullah, C. K. Cheng and M. M. R. Khan, Photoelectrocatalytic reduction of CO2 to methanol over CuFe2O4@PANI photocathode, Int. J. Hydrogen Energy, 2021, 46, 24709–24720 CrossRef CAS.
- F. C. Romeiro, B. C. Silva, A. S. Martins, M. V. B. Zanoni and M. O. Orlandi, Superior performance of rGO-tin oxide nanocomposite for selective reduction of CO2 to methanol, J. CO2 Util., 2021, 46, 101460 CrossRef CAS.
- G. Bharath, J. Prakash, K. Rambabu, G. D. Venkatasubbu, A. Kumar, S. Lee, J. Theerthagiri, M. Y. Choi and F. Banat, Synthesis of TiO2/RGO with plasmonic Ag nanoparticles for highly efficient photoelectrocatalytic reduction of CO2 to methanol toward the removal of an organic pollutant from the atmosphere, Environ. Pollut., 2021, 281, 116990 CrossRef CAS PubMed.
- X. Zhong, Y. Song, A. Cui, X. Mu, L. Li, L. Han, G. Shan and H. Liu, Adenine-functionalized graphene oxide as a charge transfer layer to enhance activity and stability of Cu2O photocathode for CO2 reduction reaction, Appl. Surf. Sci., 2022, 591, 153197 CrossRef CAS.
- H. Chaliyawala, S. Bastide, C. Cachet-Vivier, N. Ilic, T. Bourouina, F. Marty, K. Bah and E. Torralba, AgxCu100−x Decorated Si Micropillars as Photocathodes for the Reduction of CO2, ChemElectroChem, 2025, 12, e202400405 CrossRef CAS.
- X.-Y. Li, Z.-L. Zhu, F. W. Dagnaw, J.-R. Yu, Z.-X. Wu, Y.-J. Chen, M.-H. Zhou, T. Wang, Q.-X. Tong and J.-X. Jian, Silicon photocathode functionalized with osmium complex catalyst for selective catalytic conversion of CO2 to methane, Nat. Commun., 2024, 15, 5882 CrossRef CAS PubMed.
- X. Wu, R. Xu, X. Li, R. Zeng and B. Luo, Amino acid-assisted preparation of homogeneous PbS/CsPbBr3 nanocomposites for enhanced photoelectrocatalytic CO2 reduction, J. Phys. Chem. C, 2022, 126, 15744–15751 CrossRef CAS.
- M. Luo, H. Li, Z. Wang, Q. Shen, J. Xue, X. Liu and H. Jia, In-situ growth of MOF nanosheets with controllable thickness on copper foam for photoelectrocatalytic CO2 reduction, J. Mater. Sci.:Mater. Electron., 2022, 33, 14568–14580 CrossRef CAS.
- W. J. Dong, J. W. Lim, D. M. Hong, J. Kim, J. Y. Park, W. S. Cho, S. Baek and J. Lee, Grain boundary engineering of Cu–Ag thin-film catalysts for selective (photo) electrochemical CO2 reduction to CO and CH4, ACS Appl. Mater. Interfaces, 2021, 13, 18905–18913 CrossRef CAS PubMed.
- T. Manabe, K. Akai, Y. Einaga, R. Toyoshima and H. Kondoh, In Situ Vibrational Spectroscopic Study for Photoelectrochemical CO2 Reduction over the Au/p-GaN Catalyst: The Role of HCO3–for Selective Reaction, J. Phys. Chem. C, 2025, 129, 5140–5147 CrossRef CAS.
- L. Dekanovsky, J. Plutnar, J. Sturala, J. Brus, J. Kosina, J. Azadmanjiri, D. Sedmidubsky, Z. k. Sofer and B. Khezri, Multifunctional Photoelectroactive Platform for CO2 Reduction toward C2+ Products–Programmable Selectivity with a Bioinspired Polymer Coating, ACS Catal., 2022, 12, 1558–1571 CrossRef CAS.
- D. Giusi, F. Tavella, M. Miceli, C. Ampelli, G. Centi, D. Cosio, C. Genovese and S. Perathoner, Synergetic Electrocatalytic Effects of Cu2O-TiO2 Heterostructures in a Solar Driven PEC Device for CO2 Reduction to> C1 Chemicals, Chem. Eng. Trans., 2021, 86, 1405–1410 Search PubMed.
- X. Wang, C. Gao, J. Low, K. Mao, D. Duan, S. Chen, R. Ye, Y. Qiu, J. Ma, X. Zheng, R. Long, X. Wu, L. Song, J. Zhu and Y. Xiong, Efficient photoelectrochemical CO2 conversion for selective acetic acid production, Sci. Bull., 2021, 66, 1296–1304 CrossRef CAS PubMed.
- Y. Zhang, Q. Wang, K. Wang, Y. Liu, L. Zou, Y. Zhou, M. Liu, X. Qiu, W. Li and J. Li, Plasmonic Ag-decorated Cu2O nanowires for boosting photoelectrochemical CO2 reduction to multi-carbon products, Chem. Commun., 2022, 58, 9421–9424 RSC.
- J. M. Mora-Hernandez, L. A. A. Herrera, L. F. Garay-Rodriguez, L. M. Torres-Martínez and I. Hernandez-Perez, An enhanced photo (electro) catalytic CO2 reduction onto advanced BiOX (X= Cl, Br, I) semiconductors and the BiOI–PdCu composite, Heliyon, 2023, 9, e20605 CrossRef CAS PubMed.
- J. Wu, G. Zhao, S. Yin, S. Tian, J. Qi, S. Wang, D. Li and Y. Feng, Light-assisted microbial photoelectrochemical cell with a gas diffusion photocathode for effective CO2 reduction and simultaneous sewage treatment, Appl. Catal., B, 2025, 361, 124630 CrossRef CAS.
- E. Landaeta, N. I. Kadosh and Z. D. Schultz, Mechanistic study of plasmon-assisted in situ photoelectrochemical CO2 reduction to acetate with a Ag/Cu2O nanodendrite electrode, ACS Catal., 2023, 13, 1638–1648 CrossRef CAS.
- T.-s. Song, T. Li, R. Tao, H. F. Huang and J. Xie, CuO/g-C3N4 heterojunction photocathode enhances the microbial electrosynthesis of acetate through CO2 reduction, Sci. Total Environ., 2022, 818, 151820 CrossRef CAS PubMed.
- M. B. Akbar, Y. Gong, Y. Wang, A. R. Woldu, X. Zhang and T. He, Role of TiO2 coating layer on the performance of Cu2O photocathode in photoelectrochemical CO2 reduction, Nanotechnology, 2021, 32, 395707 CrossRef CAS PubMed.
- I. Roh, S. Yu, C.-K. Lin, S. Louisia, S. Cestellos-Blanco and P. Yang, Photoelectrochemical CO2 Reduction toward Multicarbon Products with Silicon Nanowire Photocathodes Interfaced with Copper Nanoparticles, J. Am. Chem. Soc., 2022, 144, 8002–8006 CrossRef CAS PubMed.
- A. Köche, K. Hong, S. Seo, F. Babbe, H. Gim, K. H. Kim, H. Choi, Y. Jung, I. Oh and G. V. Krishnamurthy, Copper Tantalate by a Sodium-Driven Flux-Mediated Synthesis for Photoelectrochemical CO2 Reduction, Small Methods, 2025, 2401432 CrossRef PubMed.
- A. Kaliyaperumal, P. Gupta, Y. S. S. Prasad, A. K. Chandiran and R. Chetty, Recent progress and perspective of the electrochemical conversion of carbon dioxide to alcohols, ACS Eng. Au, 2023, 3, 403–425 CrossRef CAS.
- C. Huazhen, Y. Yihang, F. Wenyu and Z. Huibin, Crafting CuxS-ReS2 semiconductor with enhanced adsorption capacity to facilitate photoelectrocatalytic ethanol production from CO2, Int. J. Hydrogen Energy, 2024, 69, 68–78 CrossRef.
- Y. Tian, L. Long, H. Wang, J. Zhang, D. Lu, M. Zhang and J. Liu, Efficient Photoelectrocatalytic Reduction of CO2 to Selectively Produce Ethanol Using FeS2/TiO2 p–n Heterojunction Photoelectrodes, ACS Appl. Mater. Interfaces, 2024, 16, 52299–52308 CrossRef CAS PubMed.
- G. Feng, G. Li, J. Mao, X. Dong, S. Li, C. Zhu, G. Wu, A. Chen, Y. Wei and X. Liu, Photoelectrocatalytic CO2 conversion over carbon@ silicon carbide composites, Catal. Today, 2024, 430, 114519 CrossRef CAS.
- B. Liu, J. Y. Li, L. P. Tan, X. C. Zhang, X. Guo, X. K. Wang and C. M. Zhang, Photoelectroreduction CO2 to Ethanol Over BiFeO3 with Synergistic Effect of Self-Polarization and External Electric Field, Sol. RRL, 2024, 8, 2400401 CrossRef CAS.
- M. R. Atta, M. S. Shaharun, M. M. R. Khan, A. F. Al-Mahmodi, A. A. Almashwali and I. Bangash, ZIF-67 hybridization and boron doping to enhance the photo-electrocatalytic properties of g-C3N4, Int. J. Hydrogen Energy, 2024, 53, 925–934 CrossRef CAS.
- M. Lu, D. Jia, H. Xue, J. Tian and T. Jiang, 0D/1D CuFeO2/CuO nanowire heterojunction arrays for improved photoelectrocatalytic reduction of CO2 to ethanol, J. Alloys Compd., 2023, 960, 170626 CrossRef CAS.
- M. Mehravaran, S. Aber and K. Asadpour-Zeynali, Combining the bioelectricity generation with Photo-Electrocatalytic reduction of CO2 for pollutants degradation and ethanol generation, J. Electroanal. Chem., 2023, 941, 117541 CrossRef CAS.
- G. Feng, S. Wang, S. Li, R. Ge, X. Feng, J. Zhang, Y. Song, X. Dong, J. Zhang, G. Zeng, Q. Zhang, G. Ma, Y. D. Chuang, X. Zhang, J. Guo, Y. Sun, W. Wei and W. Chen, Highly Selective Photoelectroreduction of Carbon Dioxide to Ethanol over Graphene/Silicon Carbide Composites, Angew. Chem., Int. Ed., 2023, 62, e202218664 CrossRef CAS PubMed.
- H. Cao, W. Zheng, L. Zhang, W. Feng and H. Zhang, Preparation of Cu2ZnSnS4@ TiO2 nanotubes by pulsed electrodeposition for efficiently photoelectrocatalytic reduction of CO2 to ethanol, Int. J. Hydrogen Energy, 2023, 48, 32342–32355 CrossRef CAS.
- N. Nandal, P. K. Prajapati, B. M. Abraham and S. L. Jain, CO2 to ethanol: A selective photoelectrochemical conversion using a ternary composite consisting of graphene oxide/copper oxide and a copper-based metal-organic framework, Electrochim. Acta, 2022, 404, 139612 CrossRef CAS.
- M. Kan, C. Yang, Q. Wang, Q. Zhang, Y. Yan, K. Liu, A. Guan and G. Zheng, Defect-Assisted Electron Tunneling for Photoelectrochemical CO2 Reduction to Ethanol at Low Overpotentials, Adv. Energy Mater., 2022, 12, 2201134 CrossRef CAS.
- D. Maity, S. Barman, F. A. Rahimi, R. Jena, T. N. Das, P. Verma, S. Biswas, A. Dey, A. Ghosh and T. K. Maji, A Coordination Polymer Gel as Dual Electrode Material: Photoelectrochemical Water Oxidation Coupled Dark CO2 Reduction to Ethanol, Adv. Energy Mater., 2025, 2404976 CrossRef CAS.
- M. Özdemir, S. Uluçay, S. Altınışık, B. Köksoy, B. Yalçın and S. Koyuncu, A New Strategy for Photo-Electrochemical Reduction of Carbon Dioxide Using a Carbazole-BODIPY Based Metal-Free Catalyst, Adv. Sustainable Syst., 2025, 9, 2400812 CrossRef.
- H. Qi, X. Han, J. Fu, J. Ma, X. Jia, M. Zhao, X. Wang, Z. Fu, J. Zhang and X. Zhao, Reduction of CO2 to C2 products by tandem actives sites of a novel 2D/3D structured photoelectrocatalyst Ag/Cu2O/g-C3N4, J. Environ. Chem. Eng., 2025, 13, 115058 CrossRef CAS.
- L. Fei, Z. Wanjun, J. Liequn and C. Huazhen, Selective photoelectrocatalytic reduction of CO2 to ethanol with a CuO–MoO3/TiO2 NTs composite photoelectrode, New J. Chem., 2025, 49, 6702–6712 RSC.
- L. Bergamini, N. Sangiorgi, A. Gondolini, M. Rancan, G. Bottaro, L. Armelao and A. Sanson, CsPbBr3/platinum and CsPbBr3/graphite hybrid photoelectrodes for carbon dioxide conversion to oxalic acid, Sol. Energy, 2023, 254, 213–222 CrossRef CAS.
- A. U. Pawar, U. Pal, J. Y. Zheng, C. W. Kim and Y. S. Kang, Thermodynamically controlled photo-electrochemical CO2 reduction at Cu/rGO/PVP/Nafion multi-layered dark cathode for selective production of formaldehyde and acetaldehyde, Appl. Catal., B, 2022, 303, 120921 CrossRef CAS.
- K. Wang, Y. Liu, Q. Wang, Y. Zhang, X. Yang, L. Chen, M. Liu, X. Qiu, J. Li and W. Li, Asymmetric Cu-N sites on copper oxide photocathode for photoelectrochemical CO2 reduction towards C2 products, Appl. Catal., B, 2022, 316, 121616 CrossRef CAS.
- Y. Cao, Y. Wei, W. Wan, C. Liu, C. Zhuang, C. Gong, L. Nan, Q. Zhang, H. Gao and J. Chen, Photoelectrochemical reduction of CO2 catalyzed by a 3D core–shell NiMoO 4@ ZnO heterojunction with bicentre at the (111) plane and thermal electron assistance, J. Mater. Chem. A, 2023, 11, 4230–4237 RSC.
- C. Zhuang, C. He, J. Wang, N. Ullah, Z. Hu, F. Meng, Q. Zhang, Z. Yi and H. Jing, Building organic-inorganic heterojunction NiCo2O4/h-BN toward artificial photosynthesis—Boron as hole, Appl. Catal., B, 2025, 361, 124678 CrossRef CAS.
- X. Guo, C. Wang, Z. Yang and Y. Yang, Boosting C2+ production from photoelectrochemical CO2 reduction on gallium doped Cu2O, Chem. Eng. J., 2023, 471, 144539 CrossRef CAS.
- W. Wan, Q. Zhang, Y. Wei, Y. Cao, J. Hou, C. Liu, L. Hong, H. Gao, J. Chen and H. Jing, pn Heterojunctions of Si@ WO3 mimicking thylakoid for photoelectrocatalytic CO2 reduction to C2+ products—morphology control, Chem. Eng. J., 2023, 454, 140122 CrossRef CAS.
- S. Yempally, A. M. Nassar, C. N. Lakshmi, N. Singh, S. R. Sanapala, M. Al-Ejji, S. A. Zaidi and D. Ponnamma, Enhanced photo-electro catalytic CO2 conversion using transition metal doped TiO2 nanoparticles, J. Photochem. Photobiol., A, 2024, 456, 115792 CrossRef CAS.
- Y. Yu, J. Ma, L. Zhang, T. Sun, M. Wang, J. Zhu and J. Wang, Selective electrooxidation of 5-hydroxymethylfurfural to value-added 2, 5-furanodiformic acid: mechanism, electrolyzer system, and electrocatalyst regulation, Chem. Commun., 2025, 61, 7751–7769 RSC.
- D. Suri, S. Das, S. Choudhary, G. Venkanna, B. Sharma, M. A. Afroz, N. K. Tailor, R. Joshi, S. Satapathi and K. Tripathi, Enigma of Sustainable CO2 Conversion to Renewable Fuels and Chemicals Through Photocatalysis, Electrocatalysis, and Photoelectrocatalysis: Design Strategies and Atomic Level Insights, Small, 2025, 2408981 CrossRef CAS PubMed.
- L. Xu and S. Xu, Investigation of Pyridine as a Cocatalyst for the CO2 Reduction Reaction on the Cu2O Cathode Surface, ACS Catal., 2024, 14, 9554–9564 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |