
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemoproteomic identification of phosphohistidine acceptors: posttranslational activity regulation of a key glycolytic enzyme†
Solbee
Choi
 a,
Seungmin
Ahn
a,
Kyung Hyun
Cho
b,
Sung Kuk
Lee
*b and
Jung-Min
Kee
*a
a,
Seungmin
Ahn
a,
Kyung Hyun
Cho
b,
Sung Kuk
Lee
*b and
Jung-Min
Kee
*a
aDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, South Korea. E-mail: jmkee@unist.ac.kr
bSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, South Korea. E-mail: sklee@unist.ac.kr
First published on 31st March 2025
Abstract
Histidine phosphorylation, an unconventional and understudied posttranslational modification, often involves phosphohistidine (pHis) “acceptor” proteins, which bind to pHis residues and undergo phosphotransfer from pHis. While the roles of pHis acceptors are well-documented in bacterial cell signalling and metabolism, the presence and functions of additional pHis acceptors remain largely unknown. In this study, we introduce a chemoproteomic strategy leveraging a stable analogue of 3-pHis to identify 13 putative pHis acceptors in Escherichia coli. Among these, we identified phosphofructokinase-1 (PfkA), a central enzyme in glycolysis, as a pHis acceptor phosphorylated at His249 by phosphocarrier protein HPr (PtsH). This phosphorylation, modulated by carbon source availability, inhibited PfkA's kinase activity, while the pHis-specific phosphatase signal inhibitory factor X (SixA) reversed the effect, restoring the kinase function. Our findings reveal a novel regulatory mechanism in which histidine phosphorylation dynamically controls a key glycolytic enzyme, implicating a broader role for pHis in bacterial metabolism.
Introductions
Histidine phosphorylation, an underexplored form of protein phosphorylation,1 plays a role in various biological processes, including signal transduction, metabolism, and epigenetics.2–4 However, phosphohistidine (pHis) is unstable and prone to dephosphorylation and phosphoryl transfer, particularly under acidic conditions, presenting challenges for its study using conventional techniques.1,5–7Currently, the function of histidine phosphorylation is best understood within the context of cell signalling and metabolism. Bacterial histidine kinases, essential components of the two-component signal transduction system (TCS),8 are potential drug targets to combat antibiotic resistance.9–12 The phosphoenolpyruvate: sugar transferase system (PTS) features a phosphorelay where the phosphoryl group from pHis is transferred to another histidine and eventually to transported sugars.13
Recent advancements in pHis-specific antibodies14–19 and phosphoproteomics have enabled the annotation of numerous pHis sites across both prokaryotic and eukaryotic proteomes.20–22 However, the physiological roles and downstream pathways of these pHis sites remain poorly understood.
In canonical phosphorylation signalling, specialised “reader” proteins play a crucial role in mediating downstream effects. The readers specifically recognise and bind to phosphorylated amino acid residues, facilitating protein–protein interactions and signal transduction. Well-characterised examples include the Src homology 2 (SH2) domain for phosphotyrosine (pTyr),23 14-3-3 proteins for phosphoserine (pSer),24 and the forkhead-associated (FHA) domain for phosphothreonine (pThr).25 These readers form noncovalent complexes with their target phosphoproteins to mediate downstream functions (Fig. 1A).
 | ||
| Fig. 1 (A) Schematic of canonical phosphorylation readers, which bind to phosphorylated residues to mediate downstream functions. (B) Unlike readers, phosphohistidine (pHis) “acceptors” undergo phosphorylation via phosphotransfer from pHis residues. The transient interaction between pHis and its pHis acceptor presents a challenge in identifying novel pHis acceptors. (C) Our chemoproteomic strategy to capture pHis acceptors using a photoaffinity probe based on a stable pHis analogue. Figures were created in https://www.BioRender.com. | ||
In contrast, pHis-binding proteins can undergo phosphorylation through phosphotransfer from a target pHis residue. The high reactivity of pHis enables its phosphoryl transfer to a nucleophilic residue, a distinct molecular mechanism in TCS and PTS. In such cases, the pHis binders can be regarded as pHis “acceptors,” distinct from “readers” (Fig. 1B).26 To the best of our knowledge, no pHis reader lacking an acceptor function has yet been identified. Well-characterised pHis acceptors include the response regulators (pHis to phosphoaspartate (pAsp)) in the TCS and phosphorelay proteins (pHis to pHis or pHis to phosphocysteine (pCys)) in the PTS; however, the presence and functions of additional pHis acceptors remain largely unknown.
This unique phosphotransfer characteristic of pHis poses a major challenge in identifying novel pHis acceptors. For canonical phosphorylation, the interaction between a reader and a phosphorylated residue forms a relatively stable complex (Fig. 1A), facilitating the identification of binding partners by affinity enrichment or photocrosslinking.27–29
In contrast, pulldown experiments using pHis-containing baits would be far more challenging, as the interaction between the bait and the pHis acceptor during the phosphotransfer is transient, making it exceedingly difficult to capture (Fig. 1B).30
A potential solution to this challenge is to use a stable pHis analogue as bait, which can bind to pHis acceptors more reliably by preventing phosphotransfer (Fig. 1C). A similar pulldown strategy using a non-hydrolysable pTyr mimic successfully enriched SH2-containing proteins from mammalian cell extracts.31,32
In this study, we developed a chemoproteomic workflow using chemical probes derived from a stable analogue of 3-pHis to identify novel pHis acceptor proteins (Fig. 1C). This approach identified putative pHis acceptors in Escherichia coli (E. coli), including phosphofructokinase-1 (PfkA), a crucial key node enzyme in glycolysis. We also elucidated the role of pHis in regulating PfkA activity and characterised the pathways mediating PfkA phosphorylation and dephosphorylation. These findings reveal a new posttranslational regulatory mechanism controlling a crucial glycolytic enzyme, indicating a broader role for histidine phosphorylation in metabolic regulation.
Results
Design and synthesis of pPyp-BP
To identify the pHis acceptors, we designed a chemoproteomic strategy using affinity-based probes (Fig. 1C). Due to its instability and susceptibility to phosphotransfer, pHis itself is unsuitable as bait. Instead, a probe (pPyp-BP) featuring a phosphonopyrazole-based stable analogue of 3-pHis and a benzophenone photocrosslinker was designed to covalently label the putative pHis binders (Fig. 2A). This pHis analogue has been successfully utilised as a pHis-mimicking hapten to generate pHis-specific antibodies, demonstrating its structural similarity to pHis.18,19 An alkyne handle was also incorporated into the probe to enable in-gel visualisation and enrichment of labelled targets through the Cu-catalysed click reaction.33 The probe was synthesised from pyrazole (ESI Fig. S1†).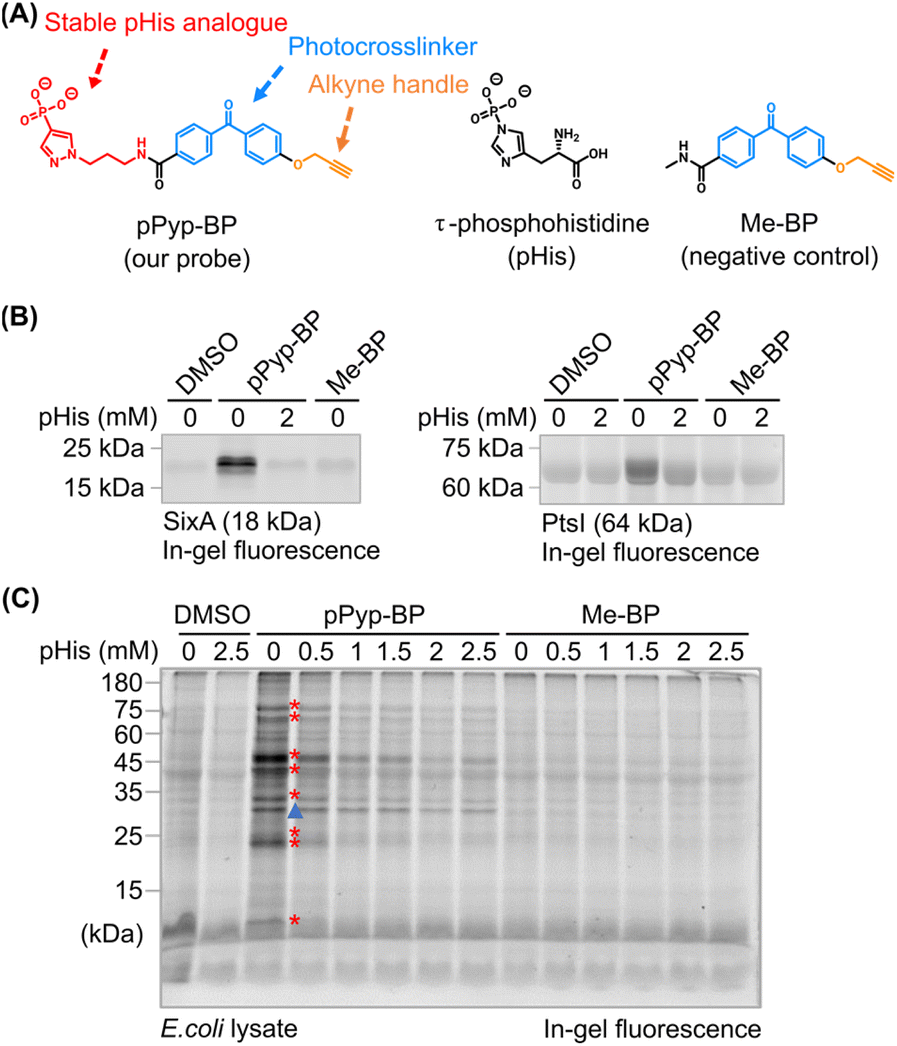 | ||
| Fig. 2 (A) Structure of the pPyp-BP probe and the negative control Me-BP. (B) Fluorescent labelling of recombinant E. coli PtsI and SixA, two well-characterized pHis acceptors. Labelling was visualised via a click reaction with TAMRA-azide, followed by in-gel fluorescence imaging. Excess pHis reduced labelling intensity, confirming the probe's specificity for pHis-binding sites. (C) Labelling of LB-cultured E. coli lysates. Protein bands with reduced labelling by pHis competition are marked with red stars, indicating probe specificity. A nonspecific band (blue triangle) was unaffected by pHis competition. Minimal labelling was observed with the Me-BP control. (Coomassie-stained gels are shown in ESI Fig. S2 and S6.†). | ||
pPyp-BP labels known pHis acceptor proteins in vitro
Next, we investigated whether pPyp-BP could effectively label known pHis acceptors. Since phosphorelay proteins in the PTS are well-established pHis acceptors, we selected E. coli phosphoenolpyruvate-protein phosphotransferase (PtsI) as a model. This PTS protein can accept the phosphoryl group from the histidine-phosphorylated phosphocarrier protein PtsH (PtsH–pHis).34,35 We also examined signal inhibitory factor X (SixA), a pHis-specific phosphatase in E. coli, as its catalytic mechanism involves phosphotransfer from the substrate pHis, forming pHis at its active site, which is subsequently hydrolysed for catalytic turnover.Both PtsI and SixA were successfully labelled via photocrosslinking with pPyp-BP, whereas no labelling was observed for Me-BP, a negative control probe lacking the pHis analogue moiety. Notably, labelling was inhibited by excess free pHis (Fig. 2B), suggesting that pPyp-BP and pHis share the binding sites. Furthermore, a pHis-accepting site mutant of PtsI (H189E) showed significantly reduced labelling, confirming that the pPyp-BP targets the pHis-binding site (ESI Fig. S3†).
The structural similarity between pHis and pTyr raised the possibility that pPyp-BP might also bind to pTyr readers.36 To address this, we performed labelling experiments using a recombinant SH2 domain (ESI Fig. S4†).37 As anticipated, only minimal background labelling was observed, which remained unaffected by the presence of pHis or pTyr.
These results indicate that pPyp-BP effectively and specifically labels pHis acceptors. However, the response regulators (E. coli OmpR and GlrR) in the TCS, other well-known pHis acceptors, were weakly labelled with pPyp-BP, but the labelling was not competed with pHis, indicating nonspecific interactions (ESI Fig. S5†). Although our probe may function for other response regulators, this finding suggests that its design may not be optimal for all pHis acceptors.
pPyp-BP labels putative pHis acceptors in E. coli
Having validated pPyp-BP with known pHis acceptors, we extended its application to identify new pHis acceptors. Numerous proteins in E. coli lysates were preferentially labelled with pPyp-BP than Me-BP (Fig. 2C). The labelling by pPyp-BP was gradually inhibited by increasing concentrations of free pHis (red stars, Fig. 2C and S7 ESI†) but not by excess pTyr, pSer, or pThr (ESI Fig. S8†), suggesting that these labelled proteins specifically interact with pHis. In contrast, some pPyp-binding proteins showed unchanged labelling (blue triangle, Fig. 2C) despite competition with free pHis, indicating they are artefacts that do not interact with pHis. The pPyp-BP labelling was also significantly reduced in urea-denatured lysates, indicating the labelling is specific for native proteins (ESI Fig. S9†).To identify the labelled proteins, we subjected the pPyp-BP-labelled lysate to a click reaction with biotin-azide, followed by streptavidin-mediated enrichment. Streptavidin-based western blots of the biotin-labelled lysates showed similar labelling patterns to in-gel fluorescence imaging (ESI Fig. S10†). Proteomic analysis, compared with a lysate labelled with Me-BP (Fig. 3A, i vs. ii), identified 170 pPyp binders (Fig. 3B and ESI Table S1†).
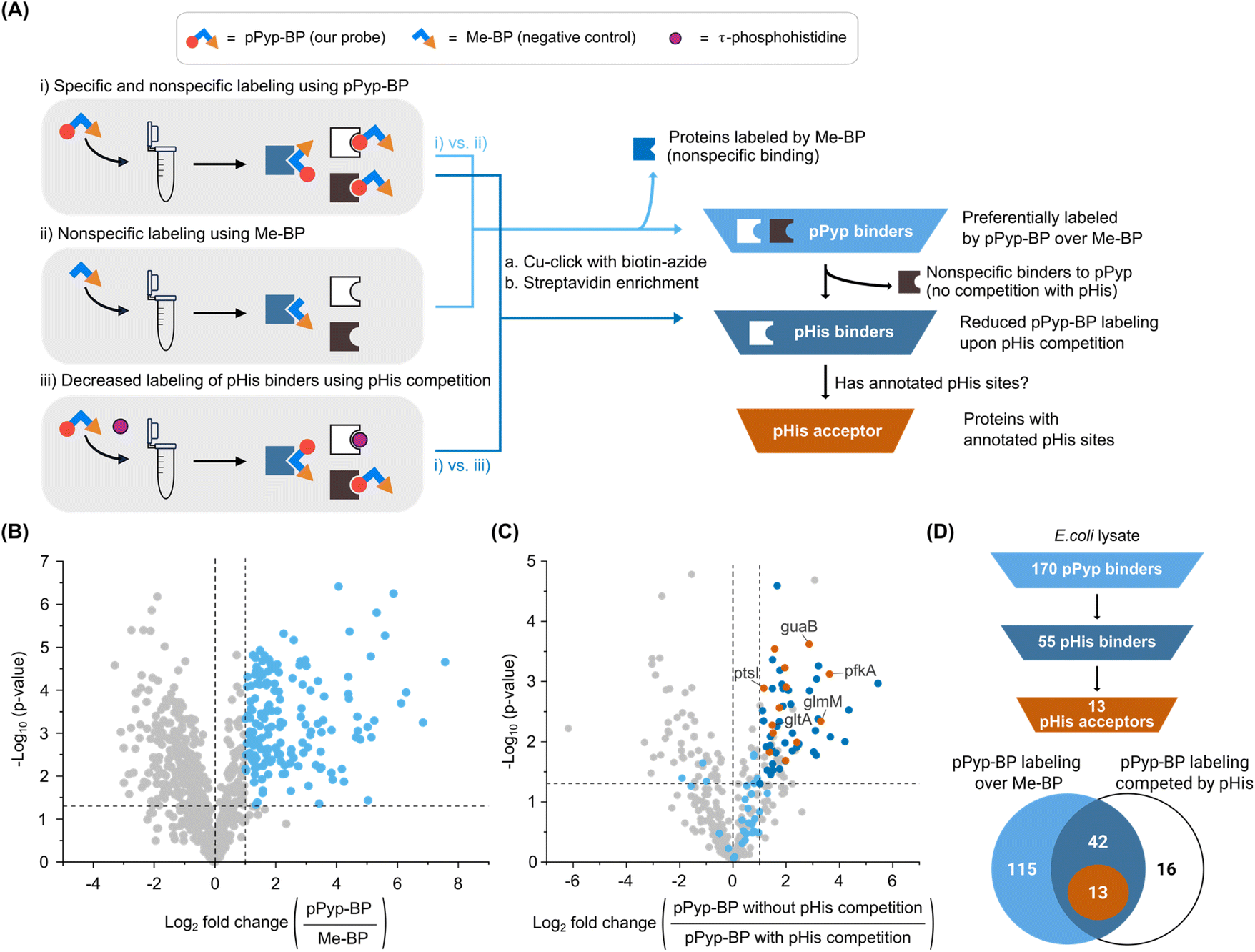 | ||
| Fig. 3 (A) Overview of the chemoproteomic workflow to identify pHis acceptors. Probe-labelled E. coli lysates were subjected to a Cu-click reaction with biotin-azide and streptavidin agarose bead enrichment. The pPyp-BP probe photocrosslinks with pPyp binders, which are distinguished from nonspecific binders to the negative control Me-BP (experiments i vs. ii). Competitive proteome labelling with pPyp-BP in the presence of excess pHis identifies pHis binders (experiments i vs. iii) by eliminating proteins that interact with pPyp-BP but do not recognize native pHis. Proteins with annotated pHis sites are subsequently designated as pHis acceptors. (B) Identification of putative pPyp binders in E. coli grown in LB medium. Light blue dots represent 170 proteins preferentially labelled by pPyp-BP over Me-BP. (C) Identification of putative pHis acceptors in LB-grown E. coli. A total of 77 proteins that exhibited significantly reduced pPyp-BP labelling in the presence of pHis made the cutoff, indicating pHis specificity. Among them, 55 proteins (dark blue and orange dots) also showed preferential labelling by pPyp-BP over Me-BP (as in panel B) and were designated as pHis binders. Of these, 13 proteins (orange dots) with previously annotated pHis sites were further classified as putative pHis acceptors. (D) Selection process for pHis acceptors using data from panels B and C. The Venn diagram is color-coded to match the dot classifications in the volcano plots. | ||
Some of these binders bound to pPyp but not to native pHis (Fig. 2C, blue triangle). To eliminate such artefacts, we performed a competitive pPyp-BP labelling in the presence of excess pHis (Fig. 3A, i vs. iii). Among the 170 pPyp binders, 55 proteins showed reduced labelling under these conditions, indicating competition with pHis (Fig. 3C and Supplementary Table S2†). These 55 proteins were identified as putative pHis binders (Fig. 3D).
The identified pHis binders included both potential pHis readers and acceptors. Since pHis acceptors are targets for phosphoryl transfer, they are expected to be phosphoproteins (Fig. 1B).26 Approximately 24% of the identified hits (13 out of 55) possessed previously annotated pHis sites22 (Tables 1 and ESI S2†) and were classified as pHis acceptors (Fig. 3D). Among these, PtsI – a known pHis acceptor – was identified, validating the reliability of our chemoproteomic approach. However, response regulators of the TCS, another class of known pHis acceptors, were not detected, which is consistent with our in vitro results (ESI Fig. S5†). SixA, one of our model pHis acceptors, was also not detected, presumably due to its low abundance (20–60 copies per cell).38
| Protein symbol | Protein name (UniProt) | Reported pHis site22,44 | Log 2 fold change |
|---|---|---|---|
| PfkA | ATP-dependent 6-phosphofructokinase isozyme 1 | His249 | 3.63 |
| GlmM | Phosphoglucosamine mutase | His102, His387 | 3.30 |
| GuaB | Inosine-5′-monophosphate dehydrogenase | His88 | 2.87 |
| GltX | Glutamate-tRNA ligase | His128, His130, His131 | 2.40 |
| AceE | Pyruvate dehydrogenase E1 component | His459 | 2.01 |
| Rho | Transcription termination factor Rho | His294 | 1.98 |
| GroEL | 60 kDa chaperonin | His400 | 1.95 |
| NrdA | Ribonucleoside-diphosphate reductase 1 subunit alpha | His331 | 1.75 |
| PykA | Pyruvate kinase II | His46 | 1.57 |
| ClpB | Chaperone protein ClpB | His566, His567 | 1.52 |
| SucC | Succinyl-CoA ligase [ADP-forming] subunit beta | His3 | 1.48 |
| GltA | Citrate synthase | His110, His114, His122, His229, His283 | 1.37 |
| PtsI | Phosphoenolpyruvate-protein phosphotransferase | His188 (ref. 45) | 1.16 |
Recombinant PfkA is selectively targeted by pPyp-BP
Next, phosphofructokinase-1 (PfkA), the top hit in our chemoproteomic analysis (Table 1), was selected for further validation. PfkA catalyses the phosphorylation of fructose 6-phosphate (F6P) to fructose 1,6-bisphosphate (F1,6BP), the first committed step in glycolysis. This reaction is a key regulatory point that determines overall glycolytic flux.39–41 Therefore, investigating this enzyme could reveal a potential mechanistic link between histidine phosphorylation and central metabolic pathways. Although the allosteric regulation of PfkA by small molecule metabolites is well established,42 its modulation through posttranslational modification has yet to be elucidated.We first repeated the photocrosslinking using recombinant PfkAin vitro. Notably, PfkA was efficiently photolabeled with pPyp-BP but not with Me-BP. Moreover, the labelling gradually decreased with increasing concentrations of free pHis (Fig. 4A). These results indicate that pPyp-BP targets PfkA at its pHis-recognition site. Subsequent LC-MS/MS analysis of the probe-labelled PfkA identified Met60 and Phe73 as the labelling sites (Fig. 4B and ESI Table S3†), both located near the substrate-binding pocket and His249, a known pHis site (ESI Fig. S11†).22,43 This finding suggests that if PfkA is a pHis acceptor, a pHis donor might bind PfkA at this site, leading to His249 phosphorylation.
 | ||
| Fig. 4 (A) pPyp-BP labelling of recombinant PfkA decreased with increasing concentrations of pHis, indicating labelling specificity. (B) LC-MS/MS spectrum of PfkA labelled with pPyp-BP, identifying Met60 as the modification site. (C) LC-MS/MS spectrum confirming histidine phosphorylation at His249 in PfkA. (D) Schematic representation of the PTS phosphorelay: The phosphoryl group of phosphoenol pyruvate (PEP) is sequentially transferred to histidine residues of PtsI, PtsH, and EIIA, ultimately phosphorylating the incoming sugar (glucose) for glycolysis. (E) PfkA was histidine-phosphorylated by the PtsH/PtsI phosphorelay system. Minimal PfkA phosphorylation occurred without PtsH, confirming PtsH–pHis as the pHis donor. Reduced phosphorylation of the PfkA H249A mutant confirms His249 as the primary pHis site. Both PfkA–pHis and PtsI–pHis were dephosphorylated by SixA, suggesting the possibility of indirect PfkA dephosphorylation via unphosphorylated PtsI/PtsH. (F) SixA directly dephosphorylated PfkA–pHis without requiring PtsH or PtsI. (Coomassie-stained gels shown in ESI Fig. S12.†). | ||
PfkA His249 is phosphorylated by PtsH and dephosphorylated by SixA in vitro
To confirm PfkA as a pHis acceptor, we incubated it with free pHis (ESI Fig. S13†). Western blot analysis demonstrated PfkA was indeed histidine-phosphorylated, and LC-MS/MS analysis confirmed phosphorylation at His249, a previously annotated pHis site (Fig. 4C and ESI Table S4†).22,44 No additional phosphosites were detected, highlighting the unique reactivity of His249.While promising, free pHis as an amino acid monomer is unlikely an endogenous phosphoryl donor for PfkA. We next examined proteins within the PTS, which are well-established pHis donors and acceptors (Fig. 4D).13 Notably, PTS-mediated phosphoryl transfer can extend to non-PTS pHis acceptors, such as transcriptional regulators, to regulate carbohydrate metabolism.46
Gratifyingly, we observed histidine phosphorylation of PfkA following incubation with histidine-phosphorylated PtsH (PtsH–pHis), which was phosphorylated by PtsI–pHis (Fig. 4E). However, PtsI–pHis alone could not phosphorylate PfkA. Mutation of PfkA His249 to Ala reduced pHis signal by 78%, confirming His249 as the primary phosphorylation site in vitro (Fig. 4E).
We then investigated whether SixA, the only known pHis-specific phosphatase in E. coli,47,48 could dephosphorylate PfkA–pHis. Notably, PfkA–pHis phosphorylated by PtsI/PtsH was efficiently dephosphorylated by SixA in vitro (Fig. 4E). Western blot analysis revealed that PtsI–pHis was also dephosphorylated by SixA, suggesting that dephosphorylation of PfkA–pHis might be mediated indirectly via nonphosphorylated PtsI/PtsH. However, SixA directly dephosphorylated PfkA–pHis even in the absence of PtsI/PtsH (Fig. 4F).
Histidine phosphorylation of PfkA His249, mediated by PtsH and SixA, regulates the kinase activity in E. coli
With promising in vitro results, we next investigated the histidine phosphorylation of PfkA in E. coli. His6-tagged PfkA was recombinantly expressed and purified from wild-type (WT) or sixA deletion (ΔsixA) strains of E. coli MG1655. Gratifyingly, PfkA from the ΔsixA strain showed robust histidine phosphorylation, whereas PfkA from the WT strain was unphosphorylated (Fig. 5A), indicating that SixA is the primary endogenous phosphatase for PfkA–pHis. Additionally, PfkA H249A mutant from the ΔsixA strain lacked pHis (Fig. 5B), verifying His249 as the in vivo phosphorylation site.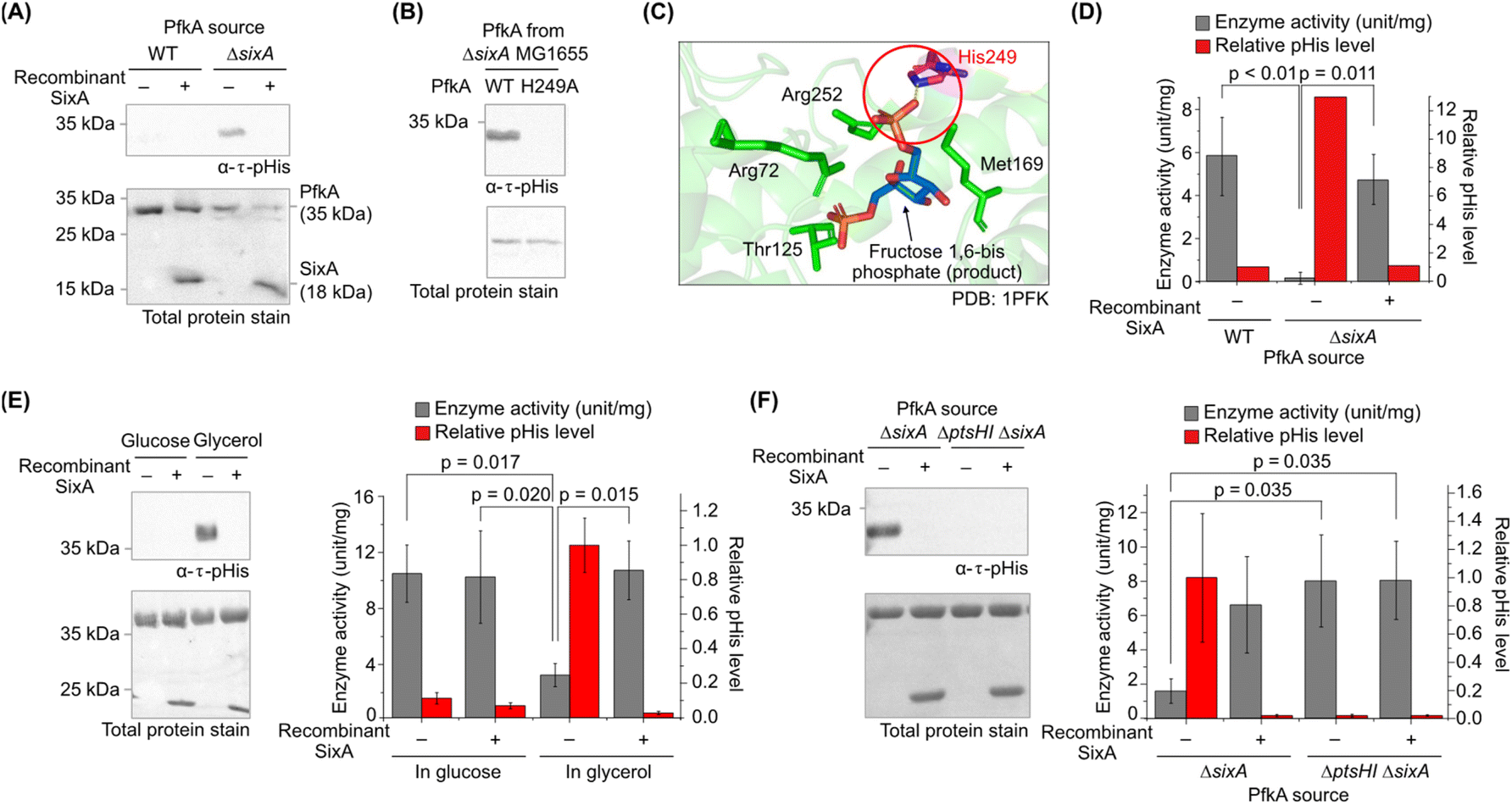 | ||
| Fig. 5 (A) Western blot analysis comparing PfkA isolated from wild-type (WT) or sixA deletion (ΔsixA) E. coli MG1655 strains which were cultured in minimal media supplemented with glycerol. PfkA from the ΔsixA strain exhibited a higher pHis level than the WT strain, and treatment with recombinant SixA dephosphorylated PfkA–pHis. (B) The PfkA H249A mutant isolated from the ΔsixA strain lacked phosphorylation, confirming His249 as the in vivo phosphorylation site. Cells were cultured in minimal media supplemented with glycerol. (C) Crystal structure of PfkA's active site (PDB: 1PFK), showing His249 interacting with the phosphoryl group of fructose 1,6-bisphosphate. (D) Quantification of pHis levels (from Figure 5A) and PfkA kinase activity from WT and ΔsixA strains. PfkA–pHis from the ΔsixA strain exhibited reduced enzyme activity and elevated histidine phosphorylation, while the addition of recombinant SixA restored the kinase activity and reduced pHis levels, demonstrating inverse correlation between these parameters. (E) Histidine phosphorylation and kinase activity of PfkA isolated from the ΔsixA strain cultured in glucose-versus glycerol-containing minimal medium. PfkA from glucose-grown bacteria showed negligible histidine phosphorylation and higher kinase activity, whereas PfkA from glycerol-cultured E. coli showed the opposite, implicating the PTS involvement in PfkA phosphorylation. (F) Significant reduction in PfkA phosphorylation and higher enzyme activity in the ΔptsHI ΔsixA strain indicated that PtsH is responsible for PfkA phosphorylation in vivo. In all cases, the addition of recombinant SixA fully dephosphorylated PfkA and restored kinase activity. Cells were cultured in minimal media supplemented with glycerol. (Coomassie-stained gels and uncropped full membrane images are shown in ESI Fig. S14.† Raw kinetic data for the PfkA activity assays are provided in ESI Table S5.†). | ||
Next, we investigated the functional impact of His249 phosphorylation. His249, located within the substrate-binding pocket, forms a crucial hydrogen bond with its substrate F6P or product F1,6BP (Fig. 5C).43 Mutation of His249 to Glu substantially reduced enzyme activity by decreasing its affinity for F6P, likely due to electrostatic repulsion between the phosphoryl group and the glutamate residue.49 Therefore, we hypothesised that phosphorylation of His249 would similarly impair enzyme activity.
To test this, we performed in vitro kinase activity assays50 using PfkA purified from different cellular conditions, reflecting the in vivo phosphorylation states (ESI Table S5†). PfkA isolated from the ΔsixA strain showed a 13-fold increase in pHis level and a 97% reduction in kinase activity compared with PfkA from the WT strain (Fig. 5D). Notably, treatment with recombinant SixA restored PfkA activity by dephosphorylating PfkA–pHis (Fig. 5D), thereby demonstrating the reversible inhibitory effect of histidine phosphorylation.
Finally, we evaluated PtsH as the endogenous phosphorylation donor of PfkA, as our in vitro results suggested. In E. coli, PtsH is well-known to exist as PtsH–pHis under non-PTS carbon sources, such as glycerol, but remains unphosphorylated in glucose media.51 Notably, PfkA from glycerol-grown E. coli showed higher histidine phosphorylation and significantly lower kinase activity than PfkA from glucose-fed E. coli (Fig. 5E, left). Again, recombinant SixA restored PfkA activity by dephosphorylating PfkA–pHis (Fig. 5E, right). The catalytically inactive SixA H8A mutant failed to dephosphorylate PfkA–pHis or restore its enzyme activity, confirming that SixA's effect is strictly dependent on its phosphatase activity (ESI Fig. S15†). Furthermore, the deletion of the ptsHI genes abrogated PfkA phosphorylation, indicating that PtsH is responsible for PfkA phosphorylation in vivo (Fig. 5F).
Discussion
This study identified novel pHis acceptors in E. coli by developing a novel chemoproteomics workflow based on a stable pHis analogue. Despite the established significance of known pHis acceptors, the presence and function of additional pHis acceptors have remained unknown. Capturing the transient interaction during phosphotransfer has been a key challenge in discovering new pHis acceptors (Fig. 1B), which our approach successfully addresses. Given that pHis acceptors participate in phosphotransfer via nucleophilic attack, future probes may be improved by incorporating electrophilic warheads or transition-state analogues to target the transfer event.Our findings demonstrate that the enzymatic activity of E. coliPfkA is posttranslationally regulated through histidine phosphorylation mediated by PtsH–pHis in the PTS (Fig. 4D). Beyond the canonical role in sugar uptake, PTS proteins also regulate other non-PTS proteins depending on their phosphorylation state. For example, unphosphorylated PtsH, also known as HPr, binds to and modulates the activity of several metabolic enzymes in E. coli, including pyruvate kinase, glucosamine-6-phosphate deaminase, glycogen phosphorylase, and phosphofructokinase-2 (PfkB), an isoenzyme of PfkA.52–54
In contrast, the regulatory functions of histidine-phosphorylated PTS remain less understood. Most identified pHis acceptors of PTS proteins are transcription regulators.46 This study broadens the functional scope of PTS by demonstrating that PfkA, a key glycolytic enzyme, serves as a pHis acceptor for PtsH–pHis. Glycerol kinase (GlpK) in Firmicutes has also been reported to undergo histidine phosphorylation in vitro, mediated by PtsI and PtsH, but whether this phosphorylation occurs in vivo is unclear.55
The robust phosphorylation of overexpressed PfkA-His6 in this study suggests that PtsI/PtsH possesses a phosphorylation capacity well beyond what is required for regulating endogenous PfkA. This raises the intriguing possibility that PtsI/PtsH may phosphorylate additional cellular targets beyond PfkA, potentially expanding its regulatory role. Further investigations will be necessary to explore this possibility.
PfkA is a unique pHis acceptor as it lacks previously known pHis-accepting domains such as HPr, EII, PTS regulation domains (PRD),56 and the receiver domain (REC) of response regulators.57 AlphaFold3 modelling58 of the complex between PtsH–pHis and PfkA revealed that the pHis15 of PtsH–pHis and His249 of PfkA are well-positioned for phosphotransfer. This spatial arrangement was absent in the predicted interaction between PfkA and unphosphorylated PtsH (ESI Fig. S16†), suggesting a pHis-dependent interaction. Further structural studies of this interaction will be necessary to fully understand this mechanism.
We also demonstrated that SixA dephosphorylates and reactivates of PfkA–pHis. Previously, SixA was only known to target the phosphotransfer protein NPr48 and the histidine kinase ArcB,47 which are involved in nitrogen assimilation and anaerobic responses, respectively. Our findings suggest that SixA has a broader role in regulating central metabolism in E. coli.
Based on our findings, we propose a mechanistic model for how histidine phosphorylation of PfkA regulates glycolysis (Fig. 6). In the absence of glucose, elevated PtsH–pHis levels lead to PfkA phosphorylation at His249, leading to reduced enzymatic activity. As PfkA catalyses the first committed step in glycolysis, its inhibition would decelerate the glycolytic pathway.41 In glucose-rich conditions, both PtsH and PfkA remain unphosphorylated, thereby facilitating glycolysis.
 | ||
| Fig. 6 A mechanistic model depicting the regulation of glycolysis by the interaction between PtsH and PfkA in E. coli. In glucose-rich conditions, PtsH remains dephosphorylated, allowing PfkA to retain its enzymatic activity. In this active state, PfkA phosphorylates F6P, facilitating its entry into the downstream Embden–Meyerhof–Parnas pathway (EMPP). Conversely, in the absence of glucose, PtsH is phosphorylated to PtsH–pHis, which phosphorylates PfkA at His249 to form PfkA–pHis. This phosphorylation inactivates PfkA, preventing F6P processing and redirecting the metabolic flux to alternative pathways. | ||
This regulatory mechanism may extend beyond E. coli. The PTS is widespread in bacteria, and sequence alignment reveals that His249 of PfkA is highly conserved across both Gram-positive and Gram-negative bacteria (ESI Fig. S17†). Notably, the homologous histidine (His250) in PfkA of Staphylococcus aureus is also a pHis site,59 suggesting that PfkA may undergo similar posttranslational regulation in Gram-positive bacteria.
In E. coli, several enzymes are regulated by phosphorylation at serine, threonine, or tyrosine residues.60 Our findings indicate that histidine phosphorylation similarly modulates enzyme activity. Notably, 11 of the 13 putative pHis acceptors identified are metabolic enzymes (Table 1), highlighting potential links between histidine phosphorylation and various metabolic pathways. Further studies are needed to determine whether the activities of these enzymes are regulated by histidine phosphorylation.
Our work also has practical applications in metabolic engineering. Phosphofructokinases are targeted to regulate metabolic flux through glycolysis, particularly through the Embden–Meyerhof–Parnas pathway (EMPP). Deletion of pfkA and/or pfkB in E. coli has been utilised to redirect metabolic flux toward the pentose phosphate pathway (PPP) and the Entner–Doudoroff pathway; however, these deletion mutants suffer from reduced growth rates.61,62 While we have not directly measured metabolic changes in this study, insights from this study may offer an alternative approach for dynamically regulating PfkA activity and glycolytic flux in E. coli.
While this proof-of-concept study focused on E. coli, our method can be extended to eukaryotes, where numerous pHis sites have been identified.21 In mammals, histidine kinases, including nucleoside diphosphate kinases (NDPKs),63 and pHis phosphatases, such as phospholysine phosphohistidine inorganic pyrophosphate phosphatase (LHPP) and phosphohistidine phosphatase 1 (PHPT1), are implicated in cancer development and metastasis.64,65 The identification of novel pHis acceptors in eukaryotes will help further elucidate the physiological functions of pHis.
Conclusions
We developed a chemoproteomic workflow utilising a stable pHis analogue as bait to identify pHis acceptors in E. coli. This study validated PfkA, a key glycolytic enzyme, as a bona fidepHis acceptor, of which catalytic activity is dynamically modulated by histidine phosphorylation. The identification of additional putative pHis acceptors, many of which are metabolic enzymes, hints at the potential of histidine phosphorylation as a regulatory mechanism in bacterial metabolism, warranting further investigations.As the number of annotated pHis sites in the proteome grows, future studies should aim to elucidate the regulatory mechanisms and physiological roles of these phosphosites. This proof-of-concept study provides a foundation for addressing these challenges.
Data availability
Additional experimental details and data are provided in the ESI.† The proteomics raw data have been deposited to the ProteomeXchange Consortium (PRIDE, PXD053006 and PXD056526).Author contributions
S. C., S. A., and J.-M. K. designed research and analysed data. S. C., S. A. performed the experiments. K. H. C. prepared the mutant E. coli strains. S. K. L. and J.-M. K. supervised the research. S. C., S. A., and J.-M. K. wrote the manuscript with inputs from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Research Foundation of Korea (RS-2022-NR069957, RS-2023-00208026 and RS-2023-00274785) for financial support. This manuscript is dedicated in memory of Prof. Hee-Yoon Lee.Notes and references
- J.-M. Kee and T. W. Muir, ACS Chem. Biol., 2012, 7, 44–51 CrossRef CAS PubMed.
- S. R. Fuhs and T. Hunter, Curr. Opin. Cell Biol., 2017, 45, 8–16 CrossRef CAS PubMed.
- K. Adam, J. Ning, J. Reina and T. Hunter, Int. J. Mol. Sci., 2020, 21, 5848 CrossRef CAS PubMed.
- P. G. Besant and P. V. Attwood, Biochem. Soc. Trans., 2012, 40, 290–293 CrossRef CAS PubMed.
- M. V. Makwana, R. Muimo and R. F. Jackson, Lab. Invest., 2018, 98, 291–303 CrossRef CAS PubMed.
- A. M. Marmelstein, J. Moreno and D. Fiedler, Top. Curr. Chem., 2017, 375, 22 CrossRef PubMed.
- A. Hauser, M. Penkert and C. P. Hackenberger, Acc. Chem. Res., 2017, 50, 1883–1893 CrossRef CAS PubMed.
- N. S. Papon and M. Ann, Curr. Biol., 2019, 29, R724–R725 CrossRef CAS PubMed.
- A. E. Bem, N. Velikova, M. T. Pellicer, P. v. Baarlen, A. Marina and J. M. Wells, ACS Chem. Biol., 2015, 10, 213–224 CrossRef CAS PubMed.
- D. Lee, Y. Lee, S. H. Shin, S. M. Choi, S. H. Lee, S. Jeong, S. Jang and J.-M. Kee, Bioorg. Chem., 2023, 130, 106232 CrossRef CAS PubMed.
- C. A. Fihn and E. E. Carlson, Curr. Opin. Microbiol., 2021, 61, 107–114 CrossRef CAS PubMed.
- K. E. Wilke, S. Francis and E. E. Carlson, J. Am. Chem. Soc., 2012, 134, 9150–9153 CrossRef CAS PubMed.
- J. Deutscher, F. M. D. Aké, M. Derkaoui, A. C. Zébré, T. N. Cao, H. Bouraoui, T. Kentache, A. Mokhtari, E. Milohanic and P. Joyet, Microbiol. Mol. Biol. Rev., 2014, 78, 231–256 CrossRef PubMed.
- J.-M. Kee, B. Villani, L. R. Carpenter and T. W. Muir, J. Am. Chem. Soc., 2010, 132, 14327–14329 CrossRef CAS PubMed.
- J.-M. Kee, R. C. Oslund, D. H. Perlman and T. W. Muir, Nat. Chem. Biol., 2013, 9, 416–421 CrossRef CAS PubMed.
- S. R. Fuhs, J. Meisenhelder, A. Aslanian, L. Ma, A. Zagorska, M. Stankova, A. Binnie, F. Al-Obeidi, J. Mauger, G. Lemke, J. R. Yates III and T. Hunter, Cell, 2015, 162, 198–210 CrossRef CAS PubMed.
- M. V. Makwana, C. dos Santos Souza, B. T. Pickup, M. J. Thompson, S. K. Lomada, Y. Feng, T. Wieland, R. F. Jackson and R. Muimo, ChemBioChem, 2023, 24, e202300182 CrossRef CAS PubMed.
- J.-M. Kee, R. C. Oslund, A. D. Couvillon and T. W. Muir, Org. Lett., 2015, 17, 187–189 CrossRef CAS PubMed.
- M. Lilley, B. Mambwe, M. J. Thompson, R. F. Jackson and R. Muimo, Chem. Commun., 2015, 51, 7305–7308 RSC.
- R. C. Oslund, J.-M. Kee, A. D. Couvillon, V. N. Bhatia, D. H. Perlman and T. W. Muir, J. Am. Chem. Soc., 2014, 136, 12899–12911 CrossRef CAS PubMed.
- G. Hardman, S. Perkins, P. J. Brownridge, C. J. Clarke, D. P. Byrne, A. E. Campbell, A. Kalyuzhnyy, A. Myall, P. A. Eyers and A. R. Jones, EMBO J., 2019, 38, e100847 CrossRef CAS PubMed.
- C. M. Potel, M.-H. Lin, A. J. Heck and S. Lemeer, Nat. Methods, 2018, 15, 187–190 CrossRef CAS PubMed.
- T. Pawson, G. D. Gish and P. Nash, Trends Cell Biol., 2001, 11, 504–511 CrossRef CAS PubMed.
- A. J. Muslin, J. W. Tanner, P. M. Allen and A. S. Shaw, Cell, 1996, 84, 889–897 CrossRef CAS PubMed.
- D. Durocher, J. Henckel, A. R. Fersht and S. P. Jackson, Mol. Cell, 1999, 4, 387–394 CAS.
- S. Ahn, H. Jung and J. M. Kee, ChemBioChem, 2021, 22, 319–325 CrossRef CAS PubMed.
- A. Uezu, H. Okada, H. Murakoshi, C. D. d. Vescovo, D. D. R. Yasuda and S. H. Soderling, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, E2929–E2938 CAS.
- A. E. H. Elia, L. C. Cantley and M. B. Yaffe, Science, 2003, 299, 1228–1231 CAS.
- H. R. Christofk, N. Wu, L. C. Cantley and J. M. Asara, J. Proteome Res., 2011, 10, 4158–4164 CrossRef CAS PubMed.
- G. M. Clore and V. Venditti, Trends Biochem. Sci., 2013, 38, 515–530 CAS.
- K. Tsumagari, T. Niinae, A. Otaka and Y. Ishihama, Proteomics, 2022, 22, e2100144 Search PubMed.
- M. Höfener, S. Heinzlmeir, B. Kuster and N. Sewald, Proteome Sci., 2014, 12, 41 Search PubMed.
- C. G. Parker and M. R. Pratt, Cell, 2020, 180, 605–632 CrossRef CAS.
- N. D. Meadow, R. L. Mattoo, R. S. Savtchenko and S. Roseman, Biochemistry, 2005, 44, 12790–12796 CrossRef CAS.
- Y.-J. Seok, B. R. Lee, P.-P. Zhu and A. Peterkofsky, Proc. Natl. Acad. Sci. U.S.A., 1996, 93, 347–351 CrossRef CAS PubMed.
- T. E. Mcallister, K. A. Horner and M. E. Webb, ChemBioChem, 2014, 15(8), 1088–1091 CrossRef CAS PubMed.
- K. Machida, C. M. Thompson, K. Dierck, K. Jablonowski, S. Kärkkäinen, B. Liu, H. Zhang, P. D. Nash, D. K. Newman and P. Nollau, Mol. Cell, 2007, 26, 899–915 CrossRef CAS PubMed.
- A. Schmidt, K. Kochanowski, S. Vedelaar, E. Ahrné, B. Volkmer, L. Callipo, K. Knoops, M. Bauer, R. Aebersold and M. Heinemann, Nat. Biotechnol., 2016, 34, 104–110 CrossRef CAS PubMed.
- B. Buschmeier, W. Hengstenberg and J. Deutscher, FEMS Microbiol. Lett., 1985, 29, 231–235 CrossRef CAS.
- H. W. Hellinga and P. R. Evans, Nature, 1987, 327, 437–439 CrossRef CAS PubMed.
- W. D. Hollinshead, S. Rodriguez, H. G. Martin, G. Wang, E. E. Baidoo, K. L. Sale, J. D. Keasling, A. Mukhopadhyay and Y. J. Tang, Biotechnol. Biofuels, 2016, 9, 212 CrossRef PubMed.
- I. Auzat, G. Le Bras and J.-R. Garel, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5242–5246 CrossRef CAS PubMed.
- S. A. Berger and P. R. Evans, Biochemistry, 1992, 31, 9237–9242 CrossRef CAS PubMed.
- M.-H. Lin, N. Sugiyama and Y. Ishihama, Sci. Signaling, 2015, 8, rs10 Search PubMed.
- L. F. García-Alles, K. Flükiger, J. Hewel, R. Gutknecht, C. Siebold, S. Schürch and B. Erni, J. Biol. Chem., 2002, 277, 6934–6942 CrossRef PubMed.
- J. Deutscher, C. Francke and P. W. Postma, Microbiol. Mol. Biol. Rev., 2006, 70, 939–1031 CrossRef CAS PubMed.
- M. Matsubara and T. Mizuno, FEBS Lett., 2000, 470, 118–124 CrossRef CAS PubMed.
- J. E. Schulte, M. Roggiani, H. Shi, J. Zhu and M. Goulian, J. Biol. Chem., 2021, 296, 100090 CrossRef CAS PubMed.
- A. W. Fenton, N. M. Paricharttanakul and G. D. Reinhart, Biochemistry, 2004, 43, 14104–14110 CrossRef CAS PubMed.
- D. Kotlarz and H. Buc, Methods Enzymol., 1982, 90, 60–70 CAS.
- M. Choe, Y.-H. Park, C.-R. Lee, Y.-R. Kim and Y.-J. Seok, Sci. Rep., 2017, 7, 43431 CrossRef CAS PubMed.
- I. A. Rodionova, Z. Zhang, J. Mehla, N. Goodacre, M. Babu, A. Emili, P. Uetz and M. H. Saier, J. Biol. Chem., 2017, 292, 14250–14257 Search PubMed.
- Y.-J. Seok, M. Sondej, P. Badawi, M. S. Lewis, M. C. Briggs, H. Jaffe and A. Peterkofsky, J. Biol. Chem., 1997, 272, 26511–26521 CAS.
- S. Chowdhury, S. Hepper, M. K. Lodi, M. H. Saier Jr and P. Uetz, Proteomes, 2021, 9, 16 Search PubMed.
- V. Charrier, E. Buckley, D. Parsonage, A. Galinier, E. Darbon, M. Jaquinod, E. Forest, J. Deutscher and A. Claiborne, J. Biol. Chem., 1997, 272, 14166–14174 CrossRef CAS.
- D. B. Greenberg, J. Stülke and M. H. Saier, Res. Microbiol., 2002, 153, 519–526 CrossRef CAS PubMed.
- R. B. Bourret, Curr. Opin. Microbiol., 2010, 13, 142–149 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard and J. Bambrick, et al. , Nature, 2024, 630, 493–500 CrossRef CAS PubMed.
- N. Prust, P. C. v. Breugel and S. Lemeer, Mol. Cell. Proteomics, 2022, 21, 100232 CrossRef CAS.
- E. Schastnaya, Z. Raguz Nakic, C. H. Gruber, P. F. Doubleday, A. Krishnan, N. I. Johns, J. Park, H. H. Wang and U. Sauer, Nat. Commun., 2021, 12, 5650 CrossRef CAS.
- Y. E. Kim, K. H. Cho, I. Bang, C. H. Kim, Y. S. Ryu, Y. Kim, E. M. Choi, L. K. Nong, D. Kim and S. K. Lee, Biotechnol. Biofuels Bioprod., 2022, 15, 120 CrossRef CAS PubMed.
- B. S. Sekar, E. Seol, S. M. Raj and S. Park, Biotechnol. Biofuels, 2016, 9, 95 CrossRef PubMed.
- P. V. Attwood and R. Muimo, Lab. Invest., 2018, 98, 283–290 CrossRef CAS PubMed.
- S. K. Hindupur, M. Colombi, S. R. Fuhs, M. S. Matter, Y. Guri, K. Adam, M. Cornu, S. Piscuoglio, C. K. Y. Ng, C. Betz, D. Liko, L. Quagliata, S. Moes, P. Jenoe, L. M. Terracciano, M. H. Heim, T. Hunter and M. N. Hall, Nature, 2018, 555, 678–682 CrossRef CAS PubMed.
- A. Xu, J. Hao, Z. Zhang, T. Tian, S. Jiang, J. Hao, C. Liu, L. Huang, X. Xiao and D. He, Lung Cancer, 2010, 67, 48–56 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures, experimental details, and synthesis of all compounds used in the study. See DOI: https://doi.org/10.1039/d5sc01024a |
| This journal is © The Royal Society of Chemistry 2025 |