
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral ring-in-ring complexes with torsion-induced circularly polarized luminescence†
Jia
Liu
 a,
Xiujie
Han
a,
Xin
Wen
b,
Hao
Yu
a,
Bao
Li
a,
Ming
Wang
a,
Minghua
Liu
*b and
Guanglu
Wu
*a
a,
Xiujie
Han
a,
Xin
Wen
b,
Hao
Yu
a,
Bao
Li
a,
Ming
Wang
a,
Minghua
Liu
*b and
Guanglu
Wu
*a
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun, Jilin 130012, P. R. China. E-mail: guanglu@jlu.edu.cn
bKey Laboratory of Colloid, Interface, and Chemical Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: liumh@iccas.ac.cn
First published on 28th March 2025
Abstract
We introduce a class of supramolecular precursors, termed ‘folda-bonders’, which utilize macrocycles to fold and precisely align reactive groups, effectively acting as bonding facilitators. This design enables highly efficient, selective, and mild ‘click-like’ reactions, making them particularly well-suited for the modular synthesis of complex structures. In this study, we highlight the versatility of folda-bonders in the one-pot aqueous synthesis of chiral ring-in-ring complexes exhibiting torsion-induced circularly polarized luminescence (CPL). Cucurbit[8]uril macrocycles facilitate the pseudostatic pre-folding of bis(4-phenyl pyridinium) derivatives, which act as folda-bonders, enabling efficient, purification-free covalent cyclization mediated by an axially chiral fragment. The single-crystal structure, obtained directly from the product solution, confirms the formation of a chiral ring-in-ring configuration. The macrocycle-imparted rigidity, combined with the tunable flexibility of alkyl linkers, drives the emergence of distinct chiroptical properties in the ring-in-ring complexes. Remarkably, torsion within the strained shorter alkyl linker is responsible for generating CPL, whereas longer linkers retain chirality, as evidenced by CD signals, but do not exhibit CPL. These findings demonstrate the potential of integrating noncovalent and covalent strategies to design sophisticated molecular architectures with tailored functional properties.
Introduction
Ring-in-ring complexes represent a unique class of supramolecular architectures, characterized by one macrocyclic component (ring) being nested within the other.1–4 These complexes typically exhibit either perpendicular1,5–9 or parallel10–12 alignment of the mean planes of the constituent rings, requiring precise size complementarity and a delicate balance between flexibility and rigidity in both the inner and outer ring components. This distinctive structural arrangement significantly constrains the relative motion between the constituent rings, thereby conferring enhanced structural rigidity and unique physicochemical properties to the system.13For chiral macrocycles, the structural constraints imposed by the ring-in-ring architecture are expected to significantly affect circularly polarized luminescence (CPL) performance.14–16 However, constructing such rigid chiral macrocycles requires overcoming the entropy loss associated with restricting the conformational flexibility of molecular backbones.17 Conventional synthetic approaches often involves multi-step protocols, which suffer from low reaction efficiency, multiple byproducts, and notable difficulties in product purification.18,19 Therefore, developing an efficient and straightforward method for synthesizing chiral ring-in-ring complexes with tailored chiroptical properties remains a challenge.
Molecular assembly driven by noncovalent interactions can direct and facilitate covalent synthesis, enabling the construction of complex structures and functions.20–23 These noncovalent interactions often lead to the preorganization or predisposition of reactants, thereby enhancing their effective concentration and increasing the likelihood of specific reaction pathways.24–26 However, due to the inherently slow kinetics of bond-breaking and bond-forming processes in covalent synthesis, the noncovalent assemblies employed to regulate these reactions need to be relatively static (pseudostatic), maintaining a specific conformation until the reaction occurs.
Pseudostatic supramolecular complexes have been increasingly observed in cucurbituril-mediated foldamers. Urbach et al. showed that cucurbit[8]uril (CB[8]) can induce the folding of specific dipeptide segments through nanomolar affinity interactions.27,28 Later, Scherman et al. designed a synthetic foldable guest, advancing the binding affinity to the picomolar level.29 Liu et al. utilized CB[8]-mediated foldamers to precisely position heavy atoms near chromophores, achieving ultralong and efficient phosphorescence.30,31 Li further introduced reactive groups at the foldamer termini, enabling the efficient synthesis of ultra-large macrocycles.32 Inspired by these developments, we propose that pseudostatic foldamers with predisposed reactive termini offer great potential as modular and versatile precursors for chemical synthesis. By coupling them with thermodynamically controlled dynamic covalent reactions, more complex functional structures could be constructed with precision and efficiency. In this study, we introduce the term ‘folda-bonders’ to describe these macrocycle-mediated foldamers, which effectively function as bonding facilitators.
Herein, we report a one-pot synthesis of chiral ring-in-ring complexes by integrating macrocycle-directed folding of precursors, as a class of folda-bonders, with subsequent covalent ring closure by an axially chiral molecular fragment through dynamic covalent reactions (Fig. 1). This method not only ensures efficient production of chiral ring-in-ring complexes but also results in torsion-induced circularly polarized luminescence. The foldable precursors, alkyl chain-bridged aldehyde-functionalized bis(phenyl pyridinium) derivative (denoted as F3, where ‘3’ represents the number of methylene groups in the alkyl chain), undergo folding upon complexation with cucurbit[8]uril (CB[8]), forming a stable, pin-like folda-bonder, F3⊂CB[8],27–34 in aqueous solution (Fig. 2a and S50†). Subsequently, the resulting dialdehyde folda-bonders condense with a dihydrazide derivative in water through the formation of acylhydrazone linkers.35–38 The hydrazide molecule, BINAD, is substituted at the 2,2′-position of 1,1′-binaphthol, introducing axial chirality to the system.39–46 The two hydrazide groups adopt a specific torsional angle, allowing the molecule to react with the folda-bonder, thereby sealing their ends and forming chiral ring-in-ring complexes in a ‘click-like’ manner.47
 | ||
| Fig. 1 Schematic representation of one-pot aqueous synthesis of chiral ring-in-ring complexes. | ||
 | ||
| Fig. 2 One-pot synthesis of the chiral ring-in-ring complex. (a) Schematic representation of precursors used for the synthesis of the chiral ring-in-ring complex, (S)-BINAD-F3⊂CB[8]. (b) 1H NMR spectra (500 MHz, D2O, 298 K) of F3⊂CB[8], (S)-BINAD, and a mixture of (S)-BINAD, F3, and CB[8] before and after incubation for 5 hours (from top to bottom). Bromine counterions are omitted for clarity. | ||
Results and discussion
Mixing CB[8], F3, and (S)-BINAD in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 stoichiometry in water immediately yields a mixture of (S)-BINAD and F3⊂CB[8] at room temperature as confirmed by 1H NMR (Fig. 2b) and ESI-MS (Fig. S30†). The sharp and well-resolved proton signals for F3⊂CB[8] suggest that the complex remains relatively static at the NMR timescale. Upon the addition of trace amounts of hydrochloric acid and after incubation at 60 °C for 5 hours, the 1H NMR spectrum of the same system (the bottom spectrum in Fig. 2b) reveals a new set of signals, accompanied by the complete disappearance of the aldehyde proton signals (solid gray circle) and a shift in the binaphthyl proton signals (a, b, c, d, e, and f). These changes confirm the complete condensation of aldehyde and acylhydrazide precursors into acylhydrazone linkages. Both (S)-enantiomers and (R)-enantiomers exhibit similar behaviour. Unless otherwise specified, the following discussion will focus on the (S)-enantiomers, while results for the (R)-enantiomers are provided in the ESI.†
:1:1 stoichiometry in water immediately yields a mixture of (S)-BINAD and F3⊂CB[8] at room temperature as confirmed by 1H NMR (Fig. 2b) and ESI-MS (Fig. S30†). The sharp and well-resolved proton signals for F3⊂CB[8] suggest that the complex remains relatively static at the NMR timescale. Upon the addition of trace amounts of hydrochloric acid and after incubation at 60 °C for 5 hours, the 1H NMR spectrum of the same system (the bottom spectrum in Fig. 2b) reveals a new set of signals, accompanied by the complete disappearance of the aldehyde proton signals (solid gray circle) and a shift in the binaphthyl proton signals (a, b, c, d, e, and f). These changes confirm the complete condensation of aldehyde and acylhydrazide precursors into acylhydrazone linkages. Both (S)-enantiomers and (R)-enantiomers exhibit similar behaviour. Unless otherwise specified, the following discussion will focus on the (S)-enantiomers, while results for the (R)-enantiomers are provided in the ESI.†
The formation of a single chiral ring-in-ring complex is clearly supported by diffusion ordered spectroscopy (DOSY) analysis, where all proton signals (excluding those from the solvent) exhibit the same diffusion coefficient (D) of ca. 2.1 × 10−10 m2 s−1 (Table S1 and Fig. S19†). This D value suggests that this sole product contains only one CB[8] macrocycle, as species with two CB[8] macrocycles typically display D values smaller than 1.8 × 10−10 m2 s−1 under the same condition.48 The well-resolved signal profiles of CB[8] between 4 and 6 ppm further confirm the formation of a highly pure product that do not require additional purification. Using DMSO as an internal standard in NMR spectroscopy, the yield of the chiral ring-in-ring complex was calculated to be 96% (Fig. S10†), which highlight the high efficiency and purity of this approach.
This purification-free system allows for the preparation of single crystals through solvent evaporation direct from the product solution. The single crystal structure, as shown in Fig. 3a, confirms the formation of a ring-in-ring complex, where the folda-bonder F3⊂CB[8] is sealed by the 1,1′-binaphthyl derivative through two acylhydrazone linkages. In the crystal of the resulting ring-in-ring complex, the dihedral angle between the 2 and 2′ positions of the 1,1′-binaphthyl moiety is 77°, significantly compressed compared to the typical angle of 110° observed in freely rotating 1,1′-binaphthyl derivatives such as 1,1′-binaphthyl-2,2′-dicarboxylate (Fig. 3b).49 This structural strain propagates to the neighboring bis(4-phenyl pyridinium) moieties, causing the two 4-phenyl pyridinium units to adopt a twisted alignment with a small crossing angle of 18°, as illustrated in the top view of crystal structure in Fig. 3a and depicted in Fig. 3b.
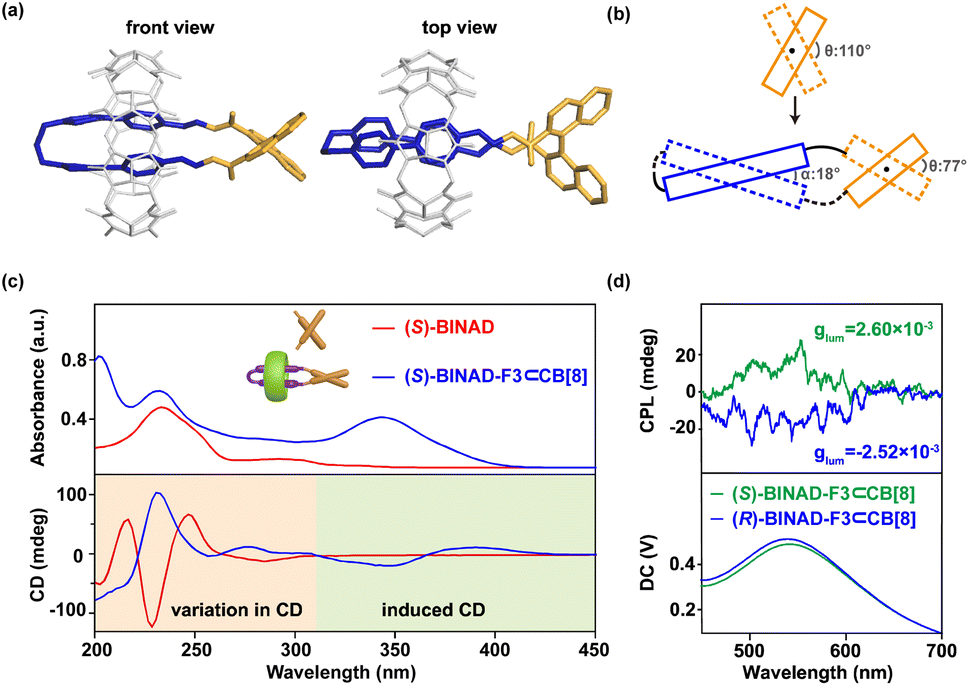 | ||
| Fig. 3 Structural and chiroptical analysis of the ring-in-ring complex. (a) Crystal structure of (S)-BINAD-F3⊂CB[8] (CB[8] in gray; F3 moiety in blue; and BINAD moiety in orange. Hydrogen atoms, counterions, and solvents are omitted for clarity). (b) Illustration of the structural chirality induced by the attachment to an axial chiral moiety. (c) UV-vis and CD spectra for (S)-BINAD and (S)-BINAD-F3⊂CB[8]. Similar spectra for (R)-enantiomers exhibit the same profiles, except for inverted signal signs in CD spectra (Fig. S35†). (d) CPL spectra and glum measured for both (S)- and (R)-BINAD-F3⊂CB[8] (conditions: H2O, 100 μM, 25 °C, λex = 350 nm). | ||
The twisted alignment is also supported by the 1H NMR spectra (Fig. 2b), where the number of signals for the 4-phenyl pyridinium units doubles upon completion of the reaction. Each signal is identified and assigned using 2D correlation spectroscopy (COSY) and 2D nuclear Overhauser spectroscopy (NOESY), confirming the asymmetric chemical environment arising from the crossing pattern (Fig. S51 and S52†). Signal splitting is also observed at around 5 ppm for the methylene groups connected to 4-phenyl pyridinium units. This complex is further confirmed by a cluster of m/z peaks observed in ESI-MS (Fig. S30†), corresponding to the ionized product with a charge state of +2 or +3. These findings demonstrate the formation of a well-defined chiral ring-in-ring complex, where the outer ring stabilizes the highly strained and twisted inner ring.
CB[8] is essential not only for promoting cyclization but also for stabilizing the resulted cyclic structure. In the absence of CB[8], the reaction between BINAD and F3 yields a complex mixture, as evidenced by multiple imine proton signals in the 1H NMR spectrum (Fig. S49†). Quantitative analysis shows that only ∼54% of the imine protons correspond to cyclic products, while ∼46% originate from linear oligomers or other byproducts. Furthermore, adding excess DMADA (Fig. S47†),50 a high-affinity guest for CB[8] (Ka > 1011 M−1), to (S)-BINAD-F3⊂CB[8] complex fully displaces CB[8] (Fig. S48†). Once CB[8] is removed, the system re-equilibrates, and the cyclic product loses its thermodynamic bias, reverting to a mixture of species probably due to the dynamic and reversible nature of the acylhydrazone linkages (Fig. S49†).
Circular dichroism (CD) response, particularly in binaphthyl-type systems, is well known to be highly sensitive to the dihedral angle between the two twist naphthalene units.51–55 Even moderate alteration in this angle can cause dramatic changes in CD intensity. As illustrated by the (S)-enantiomer, the CD spectrum of (S)-BINAD in Fig. 3c exhibits a positive maximum at 250 nm, a large negative maximum at 230 nm, and a small negative minimum at 280 nm, corresponding to the axial chirality of (S)-1,1′-binaphthyl. In contrast, the aqueous solution of ring-in-ring complexes produced from (S)-BINAD displays an almost opposite CD profile below 310 nm. This significant change arises from the compression of the torsional angle (θ) of BINAD by nearly 35° (from 110° to 77°) upon ring formation, which significantly alters the chiroptical properties (Fig. 3b and c). Additionally, a new exciton type Cotton effect with a negative valley and positive peak at 347 and 391 nm emerges, indicating that the chirality of the closure moiety is effectively transmitted to the folded segment. The exciton coupling further demonstrates that the two aromatic ring inside the CB[8] has a strong interaction. The (R)-BINAD-F3⊂CB[8] reveals a mirror image Cotton effect, verified the enantiomer nature (Fig. S39†).
It is further found that this highly constrained chiral ring-in-ring complex can exhibit circularly polarized luminescence. Upon photoexcitation at 350 nm (Fig. 3d), opposite handedness CPL is observed at the emission band (around 533 nm) for both (S)– and (R)-chiral complexes with a dissymmetry factor, glum values, of 2.6 × 10−3. This value is relatively large compared to typical pure organic emitters.56 Importantly, the rigidity of the chiral ring-in-ring complex is crucial to ensuring a large glum for CPL. By extending the alkyl bridge of bis(4-phenyl pyridinium) precursor from three methylene units (as in F3) to four (F4), five (F5), and six (F6) units (Fig. 4a), all new folda-bonders are able to generate chiral ring-in-ring complexes in a high conversion manner similar to F3 (Fig. S11–S13†). These complexes all exhibit induced CD signatures with gabs values also similar to that of F3 (Fig. S40†). However, CPL is no longer detectable in these systems. The increased length and flexibility of the alkyl bridges (Fig. 4c–e and S41†) leads to the diminish of CPL. These results suggests that the decreasing rigidity with longer spacers impacts the excited-state chiroptical properties (CPL) more significantly than the ground-state CD response (Fig. S42†). Crystal structure (Fig. 3a) reveals that the short spacer in F3 imposes structural strain, leading to a compressed BINAD dihedral angle and inducing a helical twist between the two stacked 4-phenyl pyridinium units (twist angle: 18°, Fig. 3b). This reduced co-linearity57 of the 4-phenyl pyridinium stacking, combined with the constrained BINAD geometry, enhances chiral conformational stability in the excited state and enables CPL activity in this chiral ring-in-ring complex.58–63
 | ||
| Fig. 4 Complex rigidity modulated by alkyl linker length. (a) Molecular structures of (S)-BINAD-Fn⊂CB[8], where n denotes the number of methylene units in the linker chain. (b) 1H NMR spectra (500 MHz, D2O, 298 K) of (S)-BINAD-Fn⊂CB[8] (n = 3–6), showing signal shifting and broadening corresponding to the variation in linker length. (c–e) CPL spectra of (S)- and (R)-BINAD-Fn⊂CB[8] (n = 4–6), demonstrating the absence of detectable CPL signals in complexes with flexible linkers. Bromine counterions are omitted for clarity. | ||
The influence of the bridged alkyl chain is evident in two key aspects (Fig. 4a). First, the extended alkyl chain reduces structural tension, leading to a less twisted alignment of bis(4-phenyl pyridinium) units. This is reflected in the gradual convergence of the chemical shifts for the two types of hydrogen atoms in different environments near the pyridine nitrogen, as indicated by the changes in the 1H NMR signals (lake blue and orange hollow circle). Second, the increased flexibility of the linkage enhances the axial sliding freedom of the folded bis(4-phenyl pyridinium) units, resulting in faster exchange dynamics. This is supported by the broader and more diffuse NMR peaks, particularly evident in the spectrum of F6. The disappearance of the CPL signal is primarily attributed to the increased flexibility of the system, which induces dynamic changes in chiral conformation and destabilizes the coupling between the emissive units. Interestingly, binding studies reveal that longer, more flexible chains exhibit higher CB[8] affinities (Fig. S50†), suggesting that CPL loss arises from internal conformational dynamics rather than guest–host association kinetics. This underscores the unique sensitivity of CPL to disentangle these two closely related factors.
In addition to the differences in NMR and CPL behavior, the three-methylene linkage induces a significant bathochromic shift in emission (maximum at 533 nm) spectra of the resulting ring-in-ring complexes (Fig. 5c). This shift gradually diminishes (emission maximum below 500 nm) as the linker length increases (Fig. S43 and S44†). These variations in photophysical properties are attributed to the torsional flexibility of the 1,1′-binaphthyl groups. The axial torsional flexibility allows the two 4-phenyl pyridinium units to adapt to the most relaxed geometry based on the length of the bridged linkage. For the complex with the shortest three-methylene linker, the highly rigid inner-ring scaffold enhances the coupling between the π-stacked chromophores, resulting in a pronounced bathochromic shift in emission.64–66
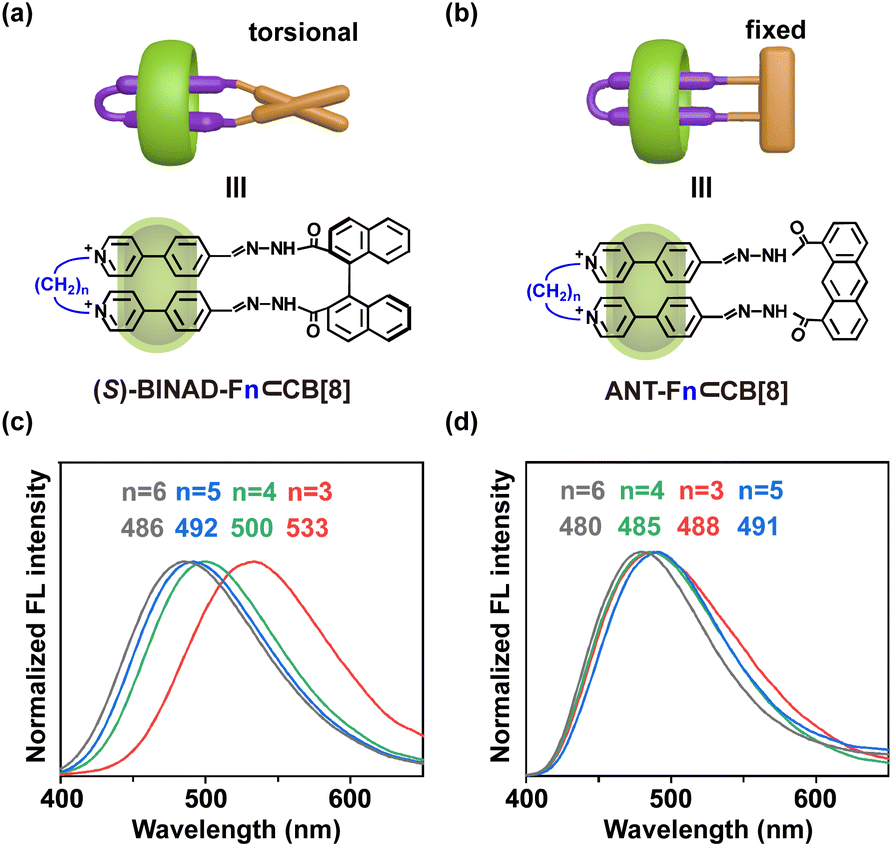 | ||
| Fig. 5 Influence of torsional or fixed closure moieties on emissions. Schematic representations of ring-in-ring complexes for (a) those bearing a torsional and chiral closure moieties, (S)-BINAD-Fn⊂CB[8], and (b) those bearing rigid and achiral closure moieties, ANT-Fn⊂CB[8]. (c) (S)-BINAD-Fn⊂CB[8] complexes exhibit significant shifts in emission wavelength corresponding to variations in linkage length. (d) ANT-Fn⊂CB[8] complexes show minimal variation in emission wavelength with changes in linkage length. Bromine counterions are omitted for clarity. | ||
When the 1,1′-binaphthyl groups are replaced with non-rotatable 1,8-anthracene units, ANT-Fn⊂CB[8], the ring-in-ring complexes are still formed with high conversion rates (Fig. S14–S17†). However, the emission spectra of these complexes show minimal variation with changes in linkage length (Fig. 5d, S45 and S46†). Furthermore, since 1,8-anthracene lacks chirality, the resulting ring-in-ring complexes exhibit neither CD nor CPL characteristics. These results highlight the critical role of high rigidity and axial torsion-induced chirality in the effective generation of CPL.
Conclusions
In conclusion, this work combines pseudostatic noncovalent assemblies with efficient dynamic covalent reactions in a one-pot synthesis to construct complexes with unique chiral ring-in-ring structures and torsion-associated CPL features. The reversible nature of dynamic covalent bonds, coupled with noncovalent predisposition, ensures the formation of thermodynamically stable, single products without the need for purification. In this study, acylhydrazone serves as the dynamic covalent linkage, though other dynamic covalent bonds could be explored in the future to examine their synergy with noncovalent preorganization. The use of torsional flexible closure units enables precise control over emission and CPL properties of chiral ring-in-ring complexes, underscoring the critical role of rigidity in effective CPL generation.This work also highlights the versatility of folda-bonders in ‘click-like’ modular synthesis. As simple supramolecular precursors, folda-bonders offer a fresh approach to chemical synthesis, leveraging pseudostatic preassembly to precisely arrange reactive groups. This strategy enables the formation of well-defined reactive entities and provides a flexible toolkit for constructing complex structures with high precision.
Data availability
Additional experimental details as well as the data supporting this article, including materials and methods, 1H NMR spectra, 13C NMR spectra, COSY spectra, DOSY spectra, NOESY spectra, ESI-MS, UV-vis, FL, CD, CPL, ITC, and single-crystal data, have been included as part of the ESI.† Crystallographic data for (S)-BINAD-F3⊂CB[8] has been deposited at the CCDC under CCDC 2404494.Author contributions
Jia Liu: conceptualization, investigation, major experiments, writing original draft, writing – review & editing. Xiujie Han: single-crystal cultivation, writing original draft. Xin Wen: CPL measurements and analysis. Hao Yu: single-crystal data collection. Bao Li: single-crystal structure analysis. Ming Wang: single-crystal data collection, writing – review & editing. Minghua Liu: conceptualization, CPL measurements and analysis, writing original draft, writing – review & editing, supervision. Guanglu Wu: conceptualization, writing original draft, writing – review & editing, supervision. All authors contributed to data analysis and manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22171103, G. W. and 52321006, M. L.), the National Key R&D Program of China (2021YFA1501600, G. W.), and the Natural Science Foundation of Jilin Province (***202301002, G. W.). This work was carried out with the support of the Shanghai Synchrotron Radiation Facility, beamline BL17B (proposal 2023-NFPS-PT-500640). We extend our gratitude to Prof. Kun Liu for granting us access to their CD facilities and to Dr Xuman Chen for the insightful discussions.References
- S.-H. Chiu, A. R. Pease, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2002, 41, 270–274 CAS.
- W.-X. Gao, H.-J. Feng, B.-B. Guo, Y. Lu and G.-X. Jin, Chem. Rev., 2020, 120, 6288–6325 CAS.
- T. Wang, X. He, J. Q. Xu, D. K. Shen, L. Y. Mei, G. Y. Xu, P. F. Wei and B. Z. Tang, CCS Chem., 2025, 7, 105–115 CAS.
- S. Hoshino, K. Ono and H. Kawai, Front. Chem., 2022, 10, 885939 CAS.
- A. I. Day, R. J. Blanch, A. P. Arnold, S. Lorenzo, G. R. Lewis and I. Dance, Angew. Chem., Int. Ed., 2002, 41, 275–277 CAS.
- C. Alvariño, C. Platas-Iglesias, V. Blanco, M. D. García, C. Peinador and J. M. Quintela, Dalton Trans., 2016, 45, 11611–11615 Search PubMed.
- Z. W. Wang, L. Mei, C. X. Guo, S. Huang, W.-Q. Shi, X. W. Li, W. Feng, X. P. Li, C. Yang and L. H. Yuan, Angew. Chem., Int. Ed., 2023, 135, e202216690 Search PubMed.
- Y. M. Zhang, Y. Liu, Y. Y. Wu, C. Q. Ge, B. D. Li, Y. X. Wei, Y. W. Qian, Q. Li, F. H. Huang and H. Li, CCS Chem., 2025, 7, 50–58 CAS.
- W. Shi, Y. N. Hu, L. Leanza, Y. Shchukin, P. A. Hoffmann, M.-H. Li, C. B. Ning, Z.-Y. Cao, Y.-Q. Xu, P. Du, M. von Delius, G. M. Pavan and Y. Z. Xu, Angew. Chem., Int. Ed., 2025, e202421459 CAS.
- F. Vögtle and W. M. Müller, Angew. Chem., Int. Ed., 1979, 91, 676–677 CrossRef.
- S.-Y. Kim, I.-S. Jung, E. Lee, J. Kim, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 2119–2121 CrossRef CAS.
- T. Kawase, Y. Nishiyama, T. Nakamura, T. Ebi, K. Matsumoto, H. Kurata and M. Oda, Angew. Chem., Int. Ed., 2007, 46, 1086–1088 CAS.
- H. Wu, Y. Wang, L. O. Jones, W. Q. Liu, B. Song, Y. P. Cui, K. Cai, L. Zhang, D. K. Shen, X.-Y. Chen, Y. Jiao, C. L. Stern, X. P. Li, G. C. Schatz and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 16849–16860 CAS.
- W. B. Huang, Y. X. Zhu, K. Zhou, L. T. Chen, Z. J. Zhao, E. G. Zhao and Z. K. He, Chem.–Eur. J., 2024, 30, e202303667 CAS.
- T. Ikai, N. Mishima, T. Matsumoto, S. Miyoshi, K. Oki and E. Yashima, Angew. Chem., Int. Ed., 2024, 63, e202318712 CAS.
- T. J. Li, X. F. Zhu, G. H. Ouyang and M. H. Liu, Mater. Chem. Front., 2023, 7, 3879–3903 CAS.
- V. Marti-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CAS.
- H. Wang, Y. Q. Tang, H. Krishna Bisoyi and Q. Li, Angew. Chem., Int. Ed., 2023, 62, e202216600 CAS.
- K. Sato, M. Hasegawa, Y. Nojima, N. Hara, T. Nishiuchi, Y. Imai and Y. Mazaki, Chem.–Eur. J., 2021, 27, 1323–1329 CAS.
- C. O. Dietrich-Buchecker, J. P. Sauvage and J. P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CAS.
- M. C. O'Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. W. Claridge, A. Saywell, M. O. Blunt, J. N. O'Shea, P. H. Beton, M. Malfois and H. L. Anderson, Nature, 2011, 469, 72–75 Search PubMed.
- Z. Ashbridge, E. Kreidt, L. Pirvu, F. Schaufelberger, J. H. Stenlid, F. Abild-Pedersen and D. A. Leigh, Science, 2022, 375, 1035–1041 CAS.
- L. F. Tan, M. Sun, H. X. Wang, J. S. Wang, J. Kim and M. Lee, Nat. Synth., 2023, 2, 1222–1231 CAS.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CAS.
- Q. X. Shi, D. Masseroni and J. Rebek Jr, J. Am. Chem. Soc., 2016, 138, 10846–10848 CAS.
- Q.-Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS PubMed.
- Z. Hirani, H. F. Taylor, E. F. Babcock, A. T. Bockus, C. D. Varnado Jr, C. W. Bielawski and A. R. Urbach, J. Am. Chem. Soc., 2018, 140, 12263–12269 CrossRef CAS PubMed.
- P. Suating, L. B. Kimberly, M. B. Ewe, S. L. Chang, J. M. Fontenot, P. R. Sultane, C. W. Bielawski, D. A. Decato, O. B. Berryman, A. B. Taylor and A. R. Urbach, J. Am. Chem. Soc., 2024, 146, 7649–7657 CrossRef CAS PubMed.
- Z. H. Huang, A. S. Groombridge, G. L. Wu, M. Olesinska, X. Y. Chen, J. A. McCune and O. A. Scherman, J. Am. Chem. Soc., 2024, 146, 24244–24249 CrossRef CAS.
- X.-K. Ma, W. Zhang, Z. X. Liu, H. Y. Zhang, B. Zhang and Y. Liu, Adv. Mater., 2021, 33, e2007476 CrossRef PubMed.
- S.-H. Li, X. F. Xu, Y. Zhou, Q. Zhao and Y. Liu, Org. Lett., 2017, 19, 6650–6653 CrossRef CAS PubMed.
- Q. Chen, Y. Lei, G. C. Wu, Q. Li, Y. J. Pan and H. Li, Chem. Sci., 2022, 13, 798–803 RSC.
- W. S. Jeon, A. Y. Ziganshina, J. W. Lee, Y. H. Ko, J.-K. Kang, C. Lee and K. Kim, Angew. Chem., Int. Ed., 2003, 42, 4097–4100 CrossRef CAS PubMed.
- G. L. Wu, F. Li, B. H. Tang and X. Zhang, J. Am. Chem. Soc., 2022, 144, 14962–14975 CAS.
- T. Y. Jiao, G. C. Wu, Y. Zhang, L. B. Shen, Y. Lei, C. Y. Wang, A. C. Fahrenbach and H. Li, Angew. Chem., Int. Ed., 2020, 59, 18350–18367 CrossRef CAS PubMed.
- I. Neira, A. Blanco-Gomez, J. M. Quintela, M. D. Garcia and C. Peinador, Acc. Chem. Res., 2020, 53, 2336–2346 CrossRef CAS PubMed.
- Y. X. Chen, Y. Lei, L. Tong and H. Li, Chem.–Eur. J., 2022, 28, e202102910 CrossRef CAS PubMed.
- Y. H. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 CAS.
- Y. T. Sang, J. L. Han, T. H. Zhao, P. F. Duan and M. H. Liu, Adv. Mater., 2020, 32, 1900110 CAS.
- X. M. Chen, S. Zhang, X. Chen and Q. Li, ChemPhotoChem, 2022, 6, e202100256 CAS.
- X. F. Yang, X. Q. Gao, Y.-X. Zheng, H. Kuang, C.-F. Chen, M. H. Liu, P. F. Duan and Z. Y. Tang, CCS Chem., 2023, 5, 2760–2789 CAS.
- N. X. Han, J. J. Ma, H. Yu, J. J. Shi, Q. X. Bai, X. Jiang, Z. Zhang, P. S. Wang, J. Yu and M. Wang, CCS Chem., 2023, 6, 1264–1277 Search PubMed.
- R. Fu, Q.-Y. Zhao, H. Han, W.-L. Li, F.-Y. Chen, C. Tang, W. Zhang, S.-D. Guo, D.-Y. Li, W.-C. Geng, D.-S. Guo and K. Cai, Angew. Chem., Int. Ed., 2023, 62, e202315990 CAS.
- Y. B. Yu, Y. N. Hu, C. B. Ning, W. D. Shi, A. Yang, Y. Zhao, Z.-Y. Cao, Y. Z. Xu and P. W. Du, Angew. Chem., Int. Ed., 2024, 136, e202407034 Search PubMed.
- S. Di Noja, F. Amato, F. Zinna, L. Di Bari, G. Ragazzon and M. Prato, Angew. Chem., Int. Ed., 2022, 61, e202202397 CAS.
- H. T. Z. Zhu, L. Pesce, R. Chowdhury, W. C. Xue, K. Wu, T. K. Ronson, R. H. Friend, G. M. Pavan and J. R. Nitschke, J. Am. Chem. Soc., 2024, 146, 2379–2386 CAS.
- A. Blanco-Gómez, M. Díaz-Abellás, I. Montes de Oca, C. Peinador, E. Pazos and M. D. García, Chem.–Eur. J., 2024, 30, e202400743 Search PubMed.
- G. L. Wu, D. E. Clarke, C. Wu and O. A. Scherman, Org. Biomol. Chem., 2019, 17, 3514–3520 RSC.
- B. Ibragimov, K. Beketov, K. Makhkamov and E. Weber, Perkin. Trans., 1997, 2, 1349–1352 RSC.
- S. M. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 15959–15967 CrossRef CAS PubMed.
- N. Berova, L. D. Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- T. Kinuta, N. Tajima, M. Fujiki, M. Miyazawa and Y. Imai, Tetrahedron, 2012, 68, 4791–4796 CrossRef CAS.
- H. K. Zhang, X. Y. Zheng, R. T. K. Kwok, J. Wang, N. L. C. Leung, L. Shi, J. Z. Sun, Z. Y. Tang, J. W. Y. Lam, A. J. Qin and B. Z. Tang, Nat. Commun., 2018, 9, 4961–4970 CrossRef PubMed.
- N. Zhao, W. W. Gao, M. Zhang, J. F. Yang, X. Y. Zheng, Y. Li, R. R. Cui, W. Yin and N. Li, Mater. Chem. Front., 2019, 3, 1613–1618 RSC.
- Y. Nojima, M. Hasegawa, N. Hara, Y. Imai and Y. Mazaki, Chem. Commun., 2019, 55, 2749–2752 RSC.
- X. N. Li, Y. J. Xie and Z. Li, Adv. Photon. Res., 2021, 2, 2000136 CrossRef CAS.
- D. Hartmann, S. E. Penty, M. A. Zwijnenburg, R. Pal and T. A. Barendt, Angew. Chem., Int. Ed., 2025, e202501122 Search PubMed.
- T. Kimoto, N. Tajima, M. Fujiki and Y. Imai, Chem.–Asian J., 2012, 7, 2836–2841 CrossRef CAS PubMed.
- Y. X. Wang, Y. Z. Li, S. Liu, F. Li, C. J. Zhu, S. H. Li and Y. X. Cheng, Macromolecules, 2016, 49, 5444–5451 CrossRef CAS.
- Y. Nojima, M. Hasegawa, N. Hara, Y. Imai and Y. Mazaki, Chem.–Eur. J., 2021, 27, 5923–5929 Search PubMed.
- K. Zhang, J. Y. Zhao, N. Zhang, J.-F. Chen, N. Wang, X. D. Yin, X. Y. Zheng and P. K. Chen, J. Mater., 2022, 10, 1816–1824 Search PubMed.
- Y. Zhang, C. Y. Zhao, J.-R. Chen, G. H. Ouyang and M.-J. Lin, Chem. Mater., 2024, 36, 4806–4812 CAS.
- Y. Sheng, D. Shen, W. J. Zhang, H. X. Zhang, C. J. Zhu and Y. X. Cheng, Chem.–Eur. J., 2015, 21, 13196–13200 CrossRef CAS PubMed.
- D. Bialas, E. Kirchner, M. I. S. Röhr and F. Würthner, J. Am. Chem. Soc., 2021, 143, 4500–4518 CAS.
- C. Kaufmann, D. Bialas, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2018, 140, 9986–9995 CAS.
- Y. L. Wu, J. W. Zhou, B. T. Phelan, C. M. Mauck, J. F. Stoddart, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 14265–14276 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2404494. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00624d |
| This journal is © The Royal Society of Chemistry 2025 |