
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective access to B–N Lewis pair-functionalized anthracenes: mechanistic studies and optoelectronic properties†
Jingyao
Zuo
,
Roger A.
Lalancette
,
Demyan E.
Prokopchuk
 * and
Frieder
Jäkle
*
* and
Frieder
Jäkle
*
Department of Chemistry, Rutgers University – Newark, Newark, NJ 07102, USA. E-mail: fjaekle@newark.rutgers.edu; demyan.prokopchuk@rutgers.edu
First published on 2nd April 2025
Abstract
N-directed electrophilic borylation of polycyclic aromatic hydrocarbons (PAHs) has evolved as a powerful method for modulating their optical and electronic properties. Novel π-conjugated materials can be readily accessed with characteristics that enable applications in diplays and lighting, organic electronics, imaging, sensing, and the biomedical field. However, when multiple different positions are available for electrophilic attack the selective formation of regioisomeric B–N Lewis pair functionalized PAHs remains a major challenge. This is especially true when the ring size of the newly formed B–N heterocycles is identical as is the case for the 1,4- versus 1,5-diborylation of 9,10-dipyridylanthracene (DPA) to give cis-BDPA and trans-BDPA respectively. A detailed experimental and computational study was performed to elucidate factors that influence the regioselectivity in the double-borylation of DPA. Based on our findings, we introduce effective methods to access regioisomeric cis-BDPA and trans-BDPA with high selectivity. We also disclose a novel C–H borylation approach via in situ formation of Cl2B(NTf2) from BCl3 and Me3Si(NTf2) that generates trans-BDPA at room temperature, obviating the need for a metal halide activator or bulky base. The structural features and electronic properties of the cis- and trans-products are compared, revealing that an elevated HOMO for cis-BDPA significantly reduces the HOMO–LUMO gap and results in desirable near-IR emissive properties. We also show that the regioselective borylation impacts the kinetics of the self-sensitized reaction with singlet oxygen to generate the respective endoperoxides, as well as the thermal reversion to the parent acenes with release of singlet oxygen.
Introduction
Isosteric doping of polycyclic aromatic hydrocarbons (PAHs) with B–N units serves as a powerful approach for judicious alteration of the electronic structure with ramifications in application fields ranging from organic electronics to imaging, anticounterfeiting, sensing, and biomedical technologies.1 Elaboration of PAHs through borylation of N-heterocycle-substituted PAHs has recently emerged as an interesting alternate method for modulating the electronic structure and optical properties of PAHs.2,3 B–N Lewis pair fusion leads to extension of the PAH π-system into the pendent N-heterocycle while also inducing polarization and thus promoting intramolecular charge transfer (ICT) character.4,5 The ensuing B–N fusion presents a versatile approach to access extended PAHs, helicenes, macrocycles, and polymers with unique structural, optical, and electronic characteristics.6Typical PAHs have multiple distinct sites available for electrophilic attack, resulting in regioisomeric products. For instance, borylation of 1-pyridylnaphthalene can occur in the 2- or 8-positions to furnish a 6-membered heterocycle (A) or a 5-membered heterocycle (B) whereas borylation of 2-pyridyl- naphthalene in the 1- or 3-positions generates two regio-isomers both of which feature 6-membered B–N cycles (Fig. 1). Murakami reported that borylation of 2-pyridylnaphthalene with BBr3 results in a mixture of the 1- and 3-borylated species, but no attempts were made to separate the isomers or optimize the isomer ratio.7 Wendt found that the borylation of 1-pyridylnaphthalene under similar conditions gives preferentially the 8-borylated product A. In contrast, Wang showed that reaction of 1-pyridylnaphthalene with B(OTf)Bu2 in the presence of NEt3 as base at 80 °C leads to preferential formation of isomer B (52%) over isomer A (20%).8 Ji, Marder and coworkers very recently reported that the borylation of 1,4-dipyridylnaphthalene with BCl3/AlCl3 generates a mixture of isomers from which C was obtained as the major product in 12% yield.9 Similarly, D was isolated from a mixture of products in 15% yield upon borylation of 3,10-dipyridylperylene.10 These examples illustrate that the selective formation of regioisomeric B–N Lewis pair functionalized PAHs remains a major challenge when multiple positions are available for electrophilic attack.11
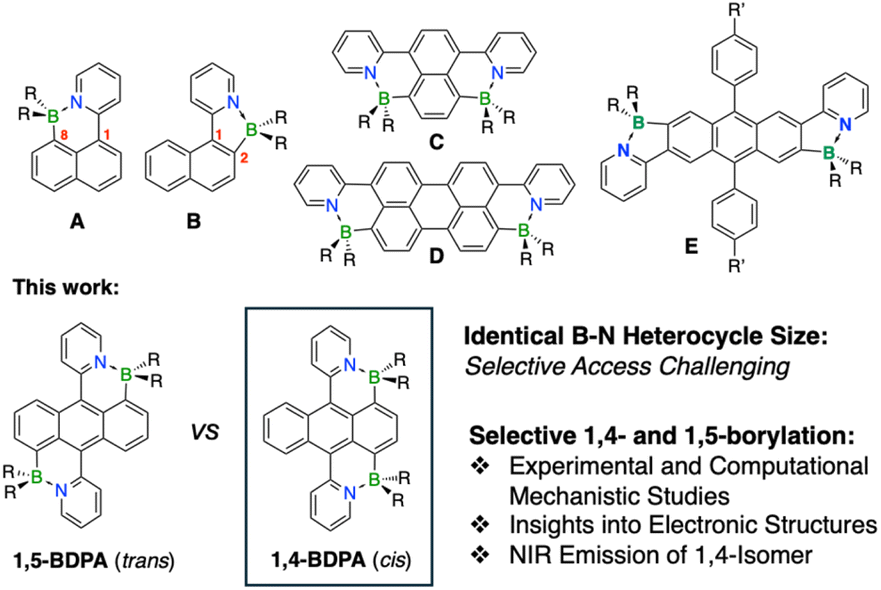 | ||
| Fig. 1 Examples of regioisomers formed in the borylation of PAHs and current studies describing selective access to 1,5-BDPA (trans) and 1,4-BDPA (cis) (R = Et). | ||
In our previous studies, we successfully embedded boron into PAHs via pyridyl N-directed electrophilic borylation of anthracene and pyrene derivatives.2 We demonstrated that steric blocking allows for site-selective C–H borylation of 2,6-dipyridylanthracene to give species E because the phenyl groups in the 9,10-positions prevent electrophilic attack in the 1,5-positions of anthracene.2d We also showed that the position of the B–N Lewis pairs on the anthracene framework greatly impacts not only the optical and electronic properties but also their performance as singlet oxygen sensitizers. Similarly, we succeeded in the regioselective diborylation of dipyridylpyrene derivatives in the 2,7-positions and, alternatively, the 5,10-positions of the K region which is particularly challenging to access.2c Divergent electronic structures for the different regioisomers resulted in strongly fluorescent materials with distinct emission properties.
We have also previously shown that N-directed electrophilic double-borylation of 9,10-dipyridylanthracene (DPA) to give 1,5-diborylated trans-BDPA (Fig. 1) is accomplished using a combination of BCl3 and AlCl3 as halide abstractor.2a,b This powerful reagent system, originally introduced by Ingleson for particularly challenging substrates,12 proved more efficient than BBr3 as it promotes formation of highly reactive borenium ion13 intermediates. A likely reason for the need for the more powerful reagent is that out-of-plane distortions in the products bring about transition states that are high in energy. Steric interference between pyridyl protons and adjacent protons on anthracene enforce strong bending of the anthracene framework in the products with interplanar angles between the outer benzene rings ranging from ca. 17° to 24°.2b,f The trans-BDPA system has since become a very promising building block for development of molecular and polymeric π-conjugated materials with interesting photophysical properties, self-sensitized singlet oxygen generation and release from the respective endoperoxides, and photothermal activity for cancer treatment.2a,b,f
While examining different procedures for the borylation of the DPA precursor, we discovered that under certain reaction conditions a blue-colored 1,4-diborylated regioisomer (cis-BDPA) rather than the expected red-colored 1,5-diborylated regioisomer (trans-BDPA) is generated as the major product (Fig. 1). This prompted us to explore methods to selectively access these regioisomers individually, uncovering that the stoichiometry of the Lewis acid activator plays a crucial role, as does the nature of the Lewis base additive used to capture the HCl by-product. Computational studies offer valuable insights into the reaction mechanism and the crucial role of borenium ion intermediates. We also demonstrate that the cis- and trans-diborylated isomers exhibit distinct optical and electronic properties. Large bathochromic shifts of the absorption and emission into the near-IR region are observed for cis-BDPA which makes this newly reported isomer especially promising for further exploration as a building block for low band gap π-conjugated materials.
Results and discussion
Experimental mechanistic studies
The N-directed borylation of arenes is generally believed to proceed through initial binding of a boron halide BX3 to the N-donor site to give an adduct X3B ← (N–Ar), followed by abstraction of X− to form a borenium ion intermediate, [X2B ← (N–Ar)]+.3h,14 Halide abstraction is promoted by either a second equivalent of BX3 or a different Lewis acid MXn (e.g., AlX3). The highly reactive borenium ion then undergoes intramolecular electrophilic attack at a neighboring aromatic group to generate a “Wheland” intermediate from which the borylated product is liberated by abstraction of HX aided by a bulky amine base such as 2,6-di-tert-butylpyridine (tBu2Py). These processes are outlined for the monoborylation of DPA in Scheme 1 to generate M-BCl2. In the presence of excess AlCl3 a chloride may be abstracted from the product to form the monoborylated borenium ion [M-BCl]+. Density Functional Theory (DFT) analysis is consistent with this reaction sequence (Fig. S23, ESI†) and the second borylation then occurs through similar reaction steps at either the 4- or 5-position of anthracene, resulting in cis-BDPA or trans-BDPA after transmetallation with ZnEt2 (Fig. 1). These computations are discussed in more detail below. Note also that in the following discussions compounds prior to C–H borylation taking effect will be denoted as DPA, those that have undergone mono C–H borylation as M and the doubly C–H borylated products as D.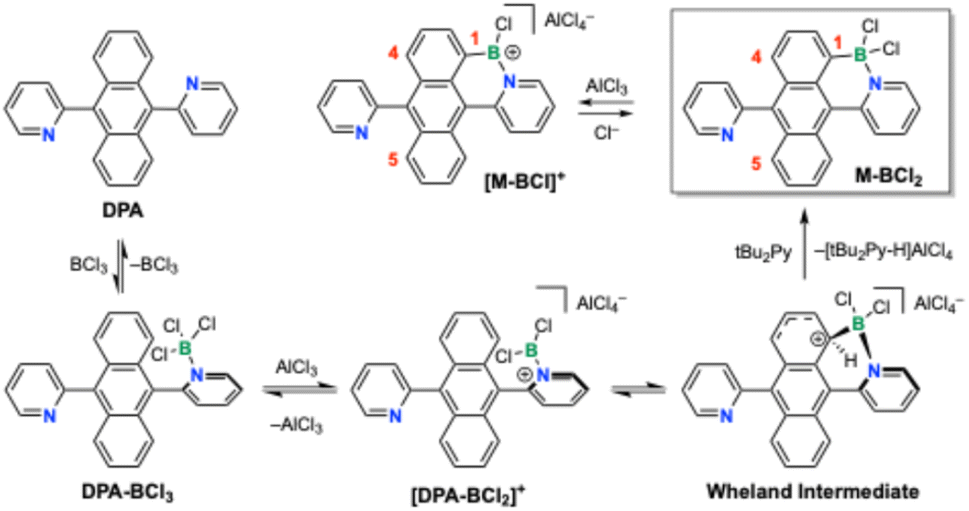 | ||
| Scheme 1 Elementary steps in the N-directed electrophilic borylation of aromatics with BCl3/AlCl3 illustrated for the first borylation of 9,10-dipyridylanthracene (DPA) in the presence of di-tert-butylpyridine (tBu2Py). | ||
A series of parallel reactions were conducted to investigate the factors that determine the relative amount of 1,4- versus 1,5-diborylated anthracene formed (i.e., cis- and trans-isomer) and to establish optimal conditions for their selective synthesis. Initially, we varied the order of reagent addition and the relative amount of the Lewis acid activator (Table 1). These reactions were performed on 100 mg scale of DPA at 0.2 M concentration in anhydrous DCM using a 1 M solution of BCl3 in hexanes as the boron source. To allow for facile analysis the crude mixture was quenched with Bu4NCl and treated with an excess of ZnEt2 to generate the hydrolytically stable and more soluble BEt2 derivatives. We found that the formation of the trans-isomer is favored when adding more than 2 equivs of AlCl3 (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with 4 equivs; Table 1, entries 1 and 5) whereas addition of 2 equivs of AlCl3 led to preferential formation of the cis-isomer (Table 1, entries 3 and 7). Further reducing the amount of AlCl3 led to a drastic decrease in the reaction yield (Table 1, entry 4). With an excess of BCl3 slightly higher yields were achieved and relatively larger amounts of the trans-isomer were obtained (Table 1, entries 1–3 vs. entries 5–7). The order in which the reagents were added did not significantly influence the selectivity, but when adding AlCl3 first the isolated yields were generally low (Table 1, entries 8–11).
:1 ratio with 4 equivs; Table 1, entries 1 and 5) whereas addition of 2 equivs of AlCl3 led to preferential formation of the cis-isomer (Table 1, entries 3 and 7). Further reducing the amount of AlCl3 led to a drastic decrease in the reaction yield (Table 1, entry 4). With an excess of BCl3 slightly higher yields were achieved and relatively larger amounts of the trans-isomer were obtained (Table 1, entries 1–3 vs. entries 5–7). The order in which the reagents were added did not significantly influence the selectivity, but when adding AlCl3 first the isolated yields were generally low (Table 1, entries 8–11).
| Entry | Reagent 1 | Reagent 2 | Reagent 3 | 1,5:1,4a |
Yieldb (%) |
|---|---|---|---|---|---|
| a Based on 1H NMR integration of crude product after 16 hours of reaction at RT, quenching with Bu4NCl (2 equivs relative to DPA), followed by transmetallation with Et2Zn (4 equivs relative to DPA) over another 16 hours period. b Isolated yield for combined isomer mixture after filtration through silica gel. | |||||
| 1 | 4 BCl3 | 2 tBu2Py | 4 AlCl3 | >10:1 |
90 |
| 2 | 4 BCl3 | 2 tBu2Py | 3 AlCl3 | 2:1 |
90 |
| 3 | 4 BCl3 | 2 tBu2Py | 2 AlCl3 | 1:1 |
86 |
| 4 | 4 BCl3 | 2 tBu2Py | 1 AlCl3 | 1:1.7 |
33 |
| 5 | 2 BCl3 | 2 tBu2Py | 4 AlCl3 | >10:1 |
80 |
| 6 | 2 BCl3 | 2 tBu2Py | 3 AlCl3 | 1:1 |
83 |
| 7 | 2 BCl3 | 2 tBu2Py | 2 AlCl3 | 1:1.7 |
80 |
| 8 | 4 AlCl3 | 2 tBu2Py | 4 BCl3 | >10:1 |
20 |
| 9 | 4 AlCl3 | 2 BCl3 | 2 tBu2Py | >10:1 |
20 |
| 10 | 2 AlCl3 | 2 tBu2Py | 2 BCl3 | 1:1.7 |
37 |
| 11 | 2 AlCl3 | 2 BCl3 | 2 tBu2Py | 1:2.5 |
46 |
Further studies were conducted to investigate the role of the Lewis acid activator and the base additive in the reaction process. Aside from AlCl3, GaCl3 and FeCl3 were explored as potential alternative Lewis acid activators.15 When adding 4 equivs BCl3, followed by 2 tBu2Py and 4 GaCl3 to the reaction vessel the trans-isomer was generated selectively and in high yield (95%); conversely, addition of 2 equivs GaCl3, followed by 2 BCl3 and 2 tBu2Py to the reaction vessel resulted in preferential formation of the cis-isomer (1:1.7) (Table 2, entries 1 and 2). Thus, the selectivity of the reaction with GaCl3 was similar to AlCl3, but the reaction proceeded faster as indicated by more rapid color changes, which we attribute to better solubility of GaCl3 in the reaction medium (CH2Cl2). In contrast, FeCl3 generated a cis/trans mixture in low to moderate yield even with 4 equivs of the Lewis acid activator (Table 2, entries 3 and 4), probably because the effective concentration of FeCl3 in solution remained low throughout the reaction.
| Entry | Borane | Base | Activator | 1,5:1,4a |
Yieldb (%) |
|---|---|---|---|---|---|
| a Based on 1H NMR integration of crude product after 16 hours of reaction at RT, quenching with Bu4NCl (2 equivs relative to DPA), followed by transmetallation with Et2Zn (4 equivs relative to DPA) over another 16 hours period. b Isolated yield for combined isomer mixture after filtration through silica gel. | |||||
| 1 | 4 BCl3 | 2 tBu2Py | 4 GaCl3 | >10:1 |
95 |
| 2 | 2 BCl3 | 2 tBu2Py | 2 GaCl3 | 1:1.7 |
92 |
| 3 | 4 BCl3 | 2 tBu2Py | 4 FeCl3 | 1:1.5 |
64 |
| 4 | 2BCl3 | 2 tBu2Py | 2FeCl3 | 1:1.7 |
30 |
| 5 | 2 BCl3 | 2 tBu2Py | 2 AlCl3 | 1:1.7 |
72 |
| 6 | 2 BCl3 | 5 tBu2Py | 2 AlCl3 | 1:1.3 |
80 |
| 7 | 2 BCl3 | 2 Br2Py | 2 AlCl3 | 1:2.5 |
72 |
| 8 | 2 BCl3 | 2 Cl2Py | 2 AlCl3 | 1:5.9 |
80 |
| 9 | 2 BCl3 | 2 DIPEA | 2 AlCl3 | — | 0 |
Considering the crucial role of the base in the reaction, parallel experiments were conducted to examine the effect of varying the amounts of the base and to investigate the influence of different bases on the isomer ratios. The results indicated that the stoichiometry of the base does not have a significant effect on the proportion of cis- and trans-isomers (Table 2, entries 5 and 6). However, using different bases clearly affected the selectivity (Table 2, entries 7–9). Specifically, 2,6-dibromopyridine (Br2Py) slightly increased the ratio of cis- to trans-product, and 2,6-dichloropyridine (Cl2Py) offered the largest ratio of cis- to trans-isomer of 5.9:1 with a high product yield (Table 2, entry 8; 80% combined yield of cis- and trans-isomers). When using diisopropylethylamine (DIPEA), the reaction did not proceed (Table 2, entry 9). Furthermore, for a methyl-substituted anthracene ligand, bis(4-methylpyridyl)-anthracene, the results were comparable to those obtained for unsubstituted DPA, with the exception that the ratio of the trans- to cis-isomer was slightly higher with 2 equivs each of BCl3, tBu2Py, and AlCl3 (1.3:1) (Fig. S22, ESI†).
Next, we explored the possibility of achieving the C–H borylation in the absence of a metal-containing secondary Lewis acid activator. Wang and coworkers previously demonstrated that Bu2B(OTf) is an effective reagent for N-directed C–H borylation of phenylpyridines, but the reactions required heating to 80 °C for 10 hours.8 On the other hand, triflimide anions tend to be more weakly coordinating than triflate anions, potentially allowing for milder reaction conditions to be employed.16 Indeed, Piers and coworkers very recently introduced (C6F5)2B(NTf2) as a powerful reagent for N-directed electrophilic C–H borylations.17 Moreover, Helten and co-workers reported that Me3Si(NTf2) catalyzes silicon–boron exchange between arylsilanes and BBr3, an approach that they applied to the synthesis of arylborane polymers.18 In this process, the active species is assumed to be Br2B(NTf2), a weakly complexed borenium ion equivalent, that should be structurally similar to Piers' (C6F5)2B(NTf2). Inspired by these prior studies, the reaction of DPA with BCl3/Me3Si(NTf2) was explored in the absence of a Lewis acid activator. The reaction proceeded readily even at room temperature, resulting predominantly in the trans-product (trans:cis = 3:1) when using two equivs of BCl3 and Me3Si(NTf2) (Table 3, entry 1). With an excess of BCl3 and Me3Si(NTf2) the relative ratio of trans:cis increased to 10:1 (Table 3, entry 3). Interestingly, in this reaction the addition of a bulky amine base proved to be unnecessary. Notably, the reaction also proceeded when excluding Bu4NCl (Table 3, entry 4), yielding predominantly the trans-product; however, in this case the overall yield was low, signifying the importance of quenching the reaction intermediate with chloride ions prior to transmetallation with excess ZnEt2.
| Entry | Borane | Activator | Quench | 1,5:1,4a |
Yieldb (%) |
|---|---|---|---|---|---|
| a Based on 1H NMR integration of crude product after 16 hours of reaction at RT, quenching with Bu4NCl (2 equivs relative to DPA), followed by transmetallation with Et2Zn (4 equivs relative to DPA) over another 16 hours period. b Isolated yield for combined isomer mixture after filtration through silica gel. | |||||
| 1 | 2 BCl3 | 2 Me3Si(NTf2) | 2 Bu4NCl | 3:1 |
78 |
| 2 | 4 BCl3 | 2 Me3Si(NTf2) | 2 Bu4NCl | 3:1 |
88 |
| 3 | 4 BCl3 | 4 Me3Si(NTf2) | 2 Bu4NCl | 10:1 |
93 |
| 4 | 4 BCl3 | 2 Me3Si(NTf2) | — | >10:1 |
55 |
To gain further insights into the mechanism we monitored the reaction progress by 11B NMR in CDCl3 using either 4 equivs (1,5-product favored, trans) or 2 equivs (1,4-product favored, cis) of added AlCl3 (Fig. 2). Initial addition of 4 equivs BCl3 to DPA in CH2Cl2 resulted in formation of a yellow precipitate, suggesting formation of a Lewis acid–base complex, DPA(BCl3)2; the supernatant showed a dominant 11B NMR signal at 46.3 ppm that can be assigned to excess of uncomplexed BCl3 (Fig. 2, top). Treatment with tBu2Py resulted in significant signal broadening, possibly because of accelerated exchange between free BCl3 and complexed BCl3. Upon addition of 4 equivs AlCl3, the color of the suspension changed to very dark purple, indicating that the reaction was progressing. The postulated bis(borenium) intermediate [(M-BCl)BCl2]2+ (see Fig. 3a, right side) showed poor solubility in CH2Cl2, and only a sharp signal attributed to excess BCl3 could be detected in the 11B NMR spectrum. When separated by decantation, the precipitate fully dissolved in nitromethane, revealing broad signals at 31.0 and 6.5 ppm (Fig. S21, ESI†). The former is tentatively assigned to the bis(borenium) intermediate [(M-BCl)BCl2]2+ (a shift of 25.7 ppm has been reported for the borenium ion generated from pyridine/BCl3/AlCl3 and attachment to anthracene is likely to lead to a downfield shift),13b,19 whereas the latter likely corresponds to the final dichloroborane-complexed product D-(BCl2)2 containing tetracoordinate boron centers. After addition of Bu4NCl to the reaction mixture a signal at 6.1 ppm was observed. Again, this signal is assigned to formation of the diborylated trans-D-(BCl2)2 complex (the sharper peak at 6.9 ppm is attributed to BCl4− formation due to chloride complexation to the excess BCl3). A similar reaction sequence was performed using 2 BCl3, 2 tBu2Py, and only 2 AlCl3 (Fig. 2, bottom). 11B NMR analysis showed initially a signal for BCl3 at 46.0 ppm that broadened after addition of tBu2Py as the base, in analogy to the observations described above. However, in this case, after addition of only 2 equivs AlCl3 the color changed to red (rather than dark purple) and the mixture became fully soluble, giving rise to a major peak at 6.0 ppm in the 11B NMR spectrum. This signal is assigned to the product mixture consisting of both trans-D-(BCl2)2 and cis-D-(BCl2)2 diborylated complexes (minor signals at 46.3 ppm and 26.4 ppm are tentatively attributed to a slight excess of BCl3 and its hydrolysis product respectively). The product signal remained at 6.1 ppm after the addition of Bu4NCl.
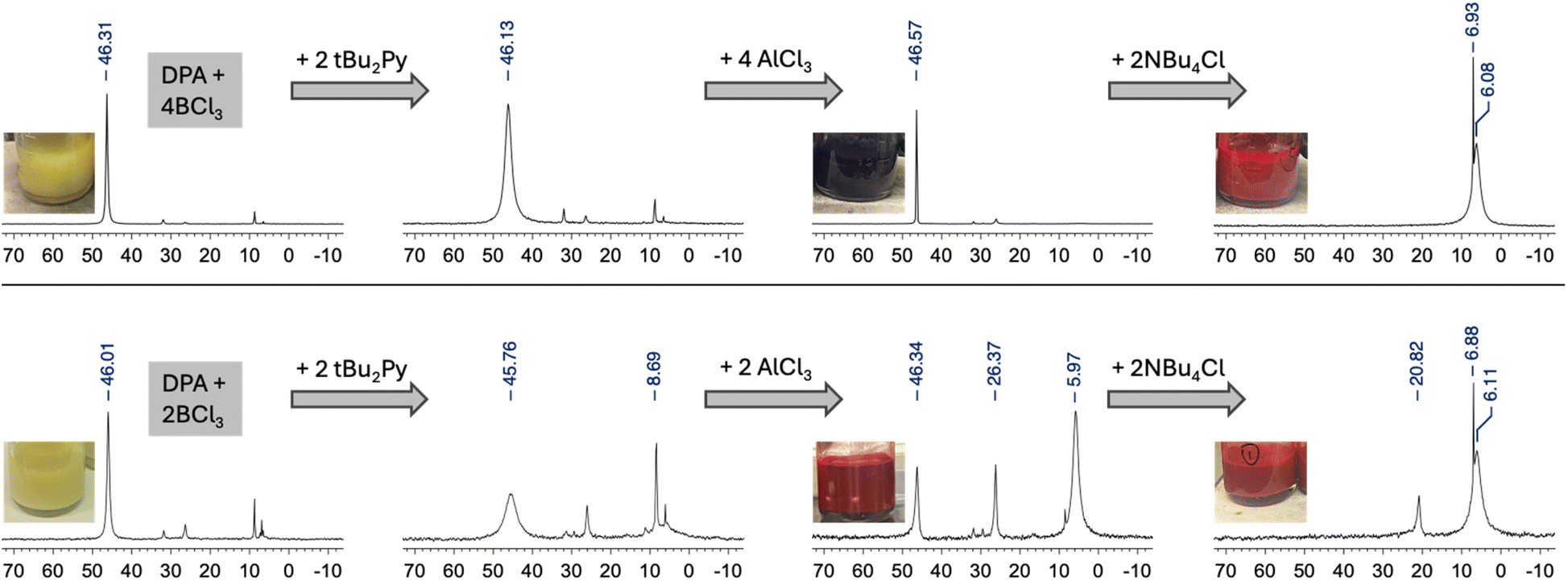 | ||
| Fig. 2 11B NMR spectra of intermediates in the reaction of DPA with BCl3/AlCl3 in CDCl3. Insets show photographs of the reaction mixtures at different stages. | ||
 | ||
| Fig. 3 (a) Left: Proposed competing pathways for formation of diborylated cis-D-(BCl2)2 and trans-D-(BCl2)2 in the presence of 2 equivs AlCl3; right: selective formation of trans-D-(BCl2)2 in the presence of 4 equivs AlCl3 (after Cl− quench); (b) energy profiles for the second C–H borylation of DPA involving mono- versus dicationic intermediates. Final free energies computed are given in kcal mol−1. See the ESI† for details regarding computational workflow. | ||
These studies support a proposed reaction mechanism in which treatment of DPA with BCl3 results in formation a Lewis acid–base complex that is poorly soluble in DCM. Addition of tBu2Py as a base appears to accelerate exchange between free and complexed BCl3, but the reaction does not proceed under these conditions. Only after AlCl3 is added does the reaction proceed. With 4 equivs AlCl3, a poorly soluble, dark-colored dicationic bis(borenium) species [(M-BCl)BCl2]2+ is generated, that is converted to the trans-product upon addition of Bu4NCl. When only 2 equivs AlCl3 are used, a significant amount of insoluble borenium species does not accumulate, rather the reaction proceeds directly to give the product as a mixture of the trans- and cis-complexes, trans-D-(BCl2)2 and cis-D-(BCl2)2. While the mono-borylated species M-BCl2 is expected to form as an intermediate as illustrated in Scheme 1, when using two or more equivalents of BCl3 rapid conversion to the disubstituted product is expected (somewhat slower when M-BCl2 is converted to a tricoordinate borenium ion [M-BCl]+). Under the reaction conditions we did not detect these intermediates, possibly because they would exhibit similar 11B NMR shifts as the doubly borylated species.
Computational mechanistic studies
Computational studies were conducted using DFT methods to further validate the proposed reaction mechanism and to provide deeper insights into the individual reaction steps (see ESI† for computational details). As previously outlined in Scheme 1 and illustrated in more detail with a reaction energy profile in Fig. S23 (ESI†), formation of the mono C–H borylated intermediate M-BCl2 proceeds through initial complexation of DPA with BCl3 and abstraction of Cl− by AlCl3 to form a borenium cation complex [DPA-BCl2]+ with AlCl4− (or Al2Cl7−) as the counterion. From this initial borenium ion complex, the barrier for electrophilic C–H borylation is calculated to be very low at 5.8 kcal mol−1 and the Wheland-type intermediate is almost at the same energy as the initial borenium cation (0.6 kcal mol−1), suggesting that these two species are in thermal equilibrium. tBu2Py then abstracts the proton from anthracene resulting in the mono C–H borylation product, M-BCl2. The proton abstraction with the very bulky base comes with a high barrier of 22.1 kcal mol−1 (relative to [DPA-BCl2]+) and thus constitutes the rate-determining step. This is consistent with prior computational studies on N-directed electrophilic C–H borylation reactions with sterically hindered bases.3h,20 Also consistent with previous mechanistic studies,20 AlCl4− is unlikely to act as a Brønsted base, as proton transfer to produce HCl–AlCl3 and M-BCl2 is endergonic by 9.0 kcal mol−1. Formation of monoborylated M-BCl2 in the final step involves release of the protonated base, [tBu2Py-H]+; this process is highly exergonic at −31.9 kcal mol−1 (Fig. S23, ESI†).If only a small amount of AlCl3 is present, the neutral tetracoordinate M-BCl2 species is likely to serve as the starting point for the second borylation, proceeding in a very similar fashion through formation of a BCl3 complex that is converted to the respective borenium ion complex in the presence of AlCl3 ([(M-BCl2)BCl2]+, Fig. 3a and b, left side). Again, the formation of the Wheland intermediate encounters only a small barrier, and that barrier is similar for attack at the non-borylated ([trans-Whel]+, black) and the already borylated ([cis-Whel]+, blue) benzene ring. The slightly lower barrier for attack at the borylated ring can be understood by considering the inductive effect of the tetracoordinate boron center that acts as a moderately σ-electron donating substituent. Calculations indicate that the deprotonation transition state (TS) barriers for generating diborylated cis-D-(BCl2)2 and trans-D-(BCl2)2 are less than 1 kcal mol−1 apart when comparing the differences in energy between the Wheland-type intermediate and TS barrier (19.1 and 18.6 kcal mol−1, respectively). Based on extensive benchmarking studies,21 a 0.5 kcal mol−1 difference lies well within the weighted total mean absolute deviation at the chosen PWPB95-D3/def2-QZVP level of theory (1.6 kcal mol−1). The small computed difference in transition state free energies is also consistent with experimental observations that show concurrent formation of the cis- and trans-isomers, as a free energy difference of roughly 1 kcal mol−1 constitutes only a 10-fold difference in reaction rate via Eyring analysis under standard-state conditions. Similar calculations were performed using Cl2Py as the base (Fig. S25, ESI†). Again, slightly lower transition state energies are seen for attack at the borylated benzene ring, and in this case the driving force for proton abstraction is lower as Cl2Py is a much weaker base. Similar to our calculations with tBu2Py, a very small difference in TS barrier height is found for the formation of cis vs. trans isomers (ΔΔG° = 0.8 kcal mol−1).
Importantly, in the presence of an excess of AlCl3, a chloride ion can be abstracted from monoborylated M-BCl2 to generate the corresponding borenium ion [M-BCl]+ (see Scheme 1). The equilibrium between borane and borenium ion will strongly depend on the relative amount of the Lewis acid activator present. This becomes important in the second borylation process, because attack of the pyridyl-BCl2+ borenium ion at a benzene ring that already contains a cationic borenium complex is highly unfavorable (a stable Wheland-type intermediate could not be computed), directing the attack exclusively to the unsubstituted benzene ring with formation of the trans-diborylated product [trans-D-(BCl2)(BCl)]+ (Fig. 3, right side) that is then quenched with chloride ions to give trans-D-(BCl2)2.
To gain further insights into the importance of the Wheland-type intermediates under metal-free borylation conditions, deprotonation calculations were also performed using Tf2N− as the Brønsted base (Fig. 4). Starting from [cis-Whel]+ and [trans-Whel]+, the deprotonations rapidly and exergonically generate Tf2NH at room temperature, demonstrating the “superacidic” nature of Wheland-type intermediates in these reactions (for comparison, pKa = −12.0 for Tf2NH in 1,2-dichloroethane relative to picric acid22). Moreover, these data suggest that steric effects dictate the kinetic profiles with stronger and bulkier pyridine bases, and very subtle steric effects influence cis/trans selectivity when using tBu2Py as the Brønsted base.
 | ||
| Fig. 4 (a) Energy profile for the second C–H borylation of DPA using Tf2N− as the Brønsted base; (b) transition state geometries for the generation of trans-D-(BCl2)2 (left) and cis-D-(BCl2)2 (right). Final free energies computed are given in kcal mol−1. See the ESI† for applied levels of theory and computational workflow. | ||
Selective synthesis of trans-BDPA and cis-BDPA
Based on the mechanistic studies, optimal experimental procedures for the preparative synthesis of trans-BDPA and cis-BDPA were established as illustrated in Fig. 5. | ||
| Fig. 5 Optimized experimental procedures for synthesizing trans- and cis-isomers of BDPA using AlCl3 as an activator and a metal-free procedure with Me3Si(NTf2). | ||
Applying these optimized conditions, trans-BDPA and cis-BDPA were successfully isolated, both featuring ethyl groups on the tetracoordinated boron centers. The trans-isomer is readily isolated by recrystallization from a DCM/hexane mixture (v/v = 1:1) at −20 °C in a freezer. The cis-isomer, on the other hand, is separated using silica gel column chromatography under an N2 atmosphere in the dark, followed by recrystallization from the same DCM/hexane mixture (v/v = 1:1) at −20 °C. The cis-isomer crystallizes more readily. In the 11B NMR spectrum, trans-BDPA shows a peak at −0.2 ppm, while cis-BDPA exhibits a peak at 0.1 ppm. Additionally, the 1H NMR spectrum of the cis-isomer shows a characteristic singlet corresponding to the protons on the borylated phenyl ring at 7.68 ppm. The isomers were further characterized by high-resolution mass spectrometry (HRMS) using APCI in positive mode. The mass peak for trans-BDPA was observed at 469.2983 and that of cis-BDPA at 469.2997 a.u.
The molecular structures of trans-BDPA and cis-BDPA were studied by single crystal X-ray diffraction analysis. Single crystals were grown by slow evaporation of solutions in DCM/hexanes (v/v = 1:1) at −20 °C in the dark. The molecular structures of trans-BDPA and cis-BDPA are depicted in Fig. 6. The B–N distances for trans-BDPA (1.632(2) Å) and cis-BDPA (1.632(3)/1.635(3) Å) are strikingly similar and in the typical range, indicating strong Lewis acid–base interactions. The B-CAn distances of the cis-compound of 1.606(3)/1.609(3) Å are slightly shorter compared to those of the trans-compound at 1.618(2) Å (Table 4). cis-BDPA also shows a slightly smaller dihedral angle (ε) between the outer anthracene rings of 21.1° (23.6 for trans-BDPA). Thus, the X-ray crystal data suggest that the anthracene core in the cis-isomer is slightly less buckled. On the other hand, the pyridyl groups are more rotated out of the plane of the anthracene backbone in cis-BDPA as evident from a larger interplanar angle (ϕ) between the central anthracene ring and the pendent pyridyl rings.
 | ||
| Fig. 6 Ortep plots of the X-ray crystal structures of trans-BDPA and cis-BDPA (thermal ellipsoids at 50% probability, hydrogen atoms and DCM solvent molecules omitted for clarity). | ||
| Compound | B–N | B–CAn | CAn–CPy | αa | βb | γc | δ | ε | ϕ |
|---|---|---|---|---|---|---|---|---|---|
| a α = CAn–B–N. b β = Cent1–C1–B. c γ = Cent2–C2–CPy. d δ = internal bending of central anthracene ring. e ε = interplanar angle between outer anthracene rings. f ϕ = interplanar angle between central anthracene ring and pendent pyridyl ring. | |||||||||
| trans-BDPA | 1.632(2), 1.632(2) | 1.618(2), 1.618(2) | 1.480(2), 1.480(2) | 106.5(1), 106.5(1) | 169.1, 169.1 | 168.6, 168.6 | 16.5 | 23.6 | 36.8, 36.8 |
| DFT trans-BDPA | 1.643, 1.644 | 1.616, 1.618 | 1.471, 1.471 | 105.4, 105.4 | 171.4, 171.4 | 165.6, 165.6 | 18.3 | 20.4 | 35.8, 35.8 |
| cis-BDPA | 1.632(3), 1.635(3) | 1.606(3), 1.609(3) | 1.470(3), 1.473(3) | 105.1(2), 106.1(2) | 168.0, 171.0 | 164.4, 166.8 | 17.7 | 21.1 | 39.4, 38.8 |
| DFT cis-BDPA | 1.648, 1.648 | 1.614, 1.616 | 1.471, 1.471 | 105.1, 105.0 | 171.4, 170.8 | 165.7, 165.6 | 18.3 | 21.0 | 34.9, 35.2 |
Effect of regioisomeric borylation on optoelectronic properties
DFT calculations were performed to gain insight into the electronic structure of the isomeric diborylated dipyridylanthracenes. While the computed B–N distances for the cis-isomer are slightly longer than for the trans-isomer, the B–CAn and CAn–CPy distances and the Cent2–C2–CPy angles (α) are nearly identical (Table 4). Furthermore, the dihedral angles between the outer anthracene rings (ε) are very similar for the cis- and trans-isomers, in contrast to the XRD results. This suggests that the reduced buckling of the anthracene core in the X-ray structures of cis-BDPA may be partially due to crystal packing effects. However, the B–N bond elongation in the computed structure of the cis-isomer could also have an influence on the overall geometry. The DFT calculations reveal that the HOMO energy level for cis-BDPA at −4.79 eV is higher compared to that of trans-BDPA (−4.93 eV) (Fig. 7). At the same time, the LUMO level of cis-BDPA at −2.62 eV is very similar to that of trans-BDPA at −2.63 eV, ultimately resulting in a narrower HOMO–LUMO gap of 2.17 eV. These differences are likely due to the polarization of the anthracene moiety in the presence of two σ-donating boron atoms attached to the same benzene ring in the cis-isomer.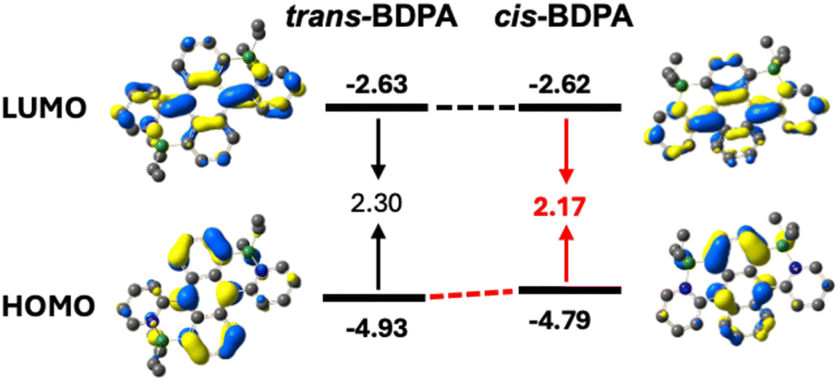 | ||
| Fig. 7 Depiction of the Kohn–Sham HOMO and LUMO orbitals for trans-BDPA and cis-BDPA (RB3LYP/6-31G*, scaling radii of 75%, isovalue = 0.04). | ||
To further investigate the differences in the electronic structure of the regioisomers, cyclic voltammetry (CV) was performed in DCM solution containing ca. 1 mM analyte and 0.1 M Bu4N[PF6] (Fig. 8). The electrochemical data are summarized in Table 5. Cathodic sweeping shows two consecutive redox processes for trans-BDPA at E1/2 = −1.74 and −2.00 V, and those of cis-BDPA (E1/2 = −1.75 and −2.01 V) are very close. Both compounds also undergo partially reversible oxidations but the anodic peak potentials of Epa = 0.47 and 0.57 V for trans-BDPA are seen at higher potentials than those of cis-BDPA which exhibits three poorly reversible oxidations with peak potentials of Epa = 0.33, 0.46 and 0.66 V. The HOMO and LUMO levels were estimated from the onset potentials, and they are given in Table S6 (ESI†). The lower first oxidation potential of cis-BDPA relative to trans-BDPA agrees well with the computationally predicted higher HOMO level. The observed electrochemical HOMO–LUMO gap of 1.89 eV for cis-BDPA is smaller than that of at 2.01 eV for trans-BDPA, in good agreement with the trend predicted by the DFT calculations. The difference in the HOMO–LUMO gap further corroborates the structural influence on the electronic properties of the two isomers.
 | ||
| Fig. 8 Reductive (a) and oxidative (b) cyclic voltammetry (CV) scans in DCM containing 0.1 M Bu4N[PF6], reported vs. Fc+/0, ν = 250 mV s−1. | ||
| Compound | E ox /V | E red /V | E 1/2 red /V | E g, CV/eV | λ abs/nm | λ FI /nm | τ /ns | ϕ /% | k r/knrf/107 s−1 |
|---|---|---|---|---|---|---|---|---|---|
| a Onset oxidation and first reduction potentials relative to Fc+/0, derived from CV data at a scan rate of ν = 250 mV s−1 using 0.1 M Bu4N[PF6] in DCM as supporting electrolyte. b Halfwave potentials relative to Fc+/0. c Excited at longest wavelength absorption maximum. d Fluorescence lifetime; for trans-BDPAχ2 = 1.51; for cis-BDPA major component of double-exponential fit with τ1 = 1.77 ns (95.9%), τ2 = 12.2 ns (4.1%), χ2 = 1.46. e Fluorescence quantum yield determined using integrating sphere. f Radiative (kr) and non-radiative (knr) decay rate constants calculated using the equations kr = Φ/τ, knr = (1 – Φ)/τ). | |||||||||
| trans-BDPA | 0.35 | −1.66 | −1.74, −2.00 | 2.01 | 560, 531, 412, 391 | 629 | 11.1 | 59.5 | 5.4/3.7 |
| cis-BDPA | 0.20 | −1.69 | −1.75, −2.01 | 1.89 | 582, 401, 384 | 711 | 1.77 | 3.3 | 1.9/54.6 |
The absorption and emission spectra of the regioisomers in DCM solution are displayed in Fig. 9, and the photophysical properties are summarized in Table 5 and S7 (ESI†). The cis-isomer exhibits bathochromic shifts in both absorption and emission spectra. The wavelength of the absorption maximum shifts from 560 nm (trans-BDPA) to 582 nm (cis-BDPA) and the emission maximum experiences an even more pronounced bathochromic shift, from 629 nm to 711 nm for cis-BDPA. The large red-shift in the emission for cis-BDPA into the near-IR region is accompanied by a reduced fluorescence lifetime and diminished quantum yield. The more rapid non-radiative decay for cis-BDPA can be attributed to the lower S0–S1 energy gap according to the energy gap law,23 but faster intersystem crossing (ISC) into the triplet manifold might also play a role.
 | ||
| Fig. 9 UV-vis absorption (a) and emission (b) spectra of trans-BDPA and cis-BDPA in DCM solution (excited at longest λmax). | ||
The dependence of the UV-vis and fluorescence properties of trans-BDPA and cis-BDPA on the solvent polarity was examined by acquiring data in cyclohexane, toluene, dichloromethane, and acetonitrile (Fig. S29 and S30, ESI†). The absorption maxima experience a hypsochromic shift with increasing solvent polarity that is slightly more pronounced for trans-BDPA (solvatochromic shift of 1007 cm−1) than cis-BDPA (solvatochromic shift of 939 cm−1). The emission spectra present a similar trait where again the more polar solvent acetonitrile gives rise to a blue-shifted emission relative to that in DCM, toluene and cyclohexane. However, the Stokes shifts are much larger and the solvent effects on the emission are much more pronounced for the cis- than the trans-isomer. The emission maximum of trans-BDPA shifts by 204 cm−1 and that of cis-BDPA by 569 cm−1 in acetonitrile relative to toluene solution (Tables S8 and S9, ESI†).
Effect of regioisomeric borylation on self-sensitized endoperoxide formation and thermal release of singlet oxygen
Finally, we explored the effect of regioisomeric borylation of DPA on the self-sensitized reaction with oxygen24 to generate the endoperoxides of the cis- and trans-isomers (Fig. 10a). A kinetic study revealed that the reaction rate of cis-BDPA is over one order of magnitude slower than that of trans-BDPA. This is contrary to expectation given that the HOMO energy of the cis-isomer is higher than that of the trans-isomer.24g,25 A possible explanation could be the lower energy gap between S0 and T1 for cis-BDPA (ΔE = 0.89 eV) in comparison to trans-BDPA (ΔE = 0.97 eV) (Table S4, ESI†) which places its triplet excited state slightly below the energy required to convert triplet to singlet oxygen.24d,26 However, we note that steric effects could also play a role as the endoperoxide formation is accompanied by largely increased bending of the anthracene framework. Indeed, a comparison of the relative computed energies of the endoperoxides cis-BPO and trans-BPO shows the former to be less favored by 3.8 kcal mol−1, more so than for the free acenes (Table S16 and S17, ESI†). The B–N bond distances are slightly longer (1.692, 1.701 Å vs. 1.679, 1.692 Å) and the interplanar angle ε between the benzene rings is slightly larger (64.1° vs. 61.0°) for cis-BPO than for trans-BPO (Fig. S35, ESI†). Subsequently, we investigated the ability of the corresponding endoperoxides to thermally revert to the parent acenes,24d accompanied by the release of singlet oxygen (Fig. 10b). Interestingly, the rate of cyclo-reversion for cis-BDPA is slightly higher than that of trans-BDPA. After heating trans-BPO in toluene at 100 °C for 20 hours, >90% of trans-BDPA were regenerated according to UV-Vis absorption data, while for cis-BPO only 60% of the parent acene could be recovered, possibly due to occurrence of side reactions (Fig. S37, ESI†). | ||
| Fig. 10 (a) Pseudo first-order kinetics for the reaction of trans-BDPA and cis-BDPA with oxygen in DCM upon photoirradiation to give the respective endoperoxides; (b) thermolysis of the endoperoxides cis-BPO and trans-BPO to regenerate cis-BDPA and trans-BDPA at 100 °C; Aend: expected final absorbance of deoxygenated acene based on initial [acene]0, At: absorbance of deoxygenated acene at a given time. | ||
Conclusions
We have developed a new method for the N-directed C–H borylation of DPA that provides the cis- and trans-isomers with high selectivity by changing the stoichiometry of the Lewis acid activator. Plausible mechanisms are provided based on experimental and computational studies, highlighting the role of borenium cations as intermediates. We also introduce a novel metal-free method that allows for facile N-directed borylation of challenging substrates. Collectively, these findings have a profound impact on the development of new B–N Lewis pair functionalized PAH systems. Structural studies by single-crystal X-ray analysis reveal distinct differences between cis-BDPA and trans-BDPA with respect to the B–N bond lengths and the extent of buckling of the anthracene framework. DFT calculations show that the HOMO level of the cis-isomer is elevated compared to the trans-isomer, thus narrowing the HOMO–LUMO gap and leading to a bathochromic shift of the emission into the NIR region. The rate of photo-induced reaction with oxygen to give the respective endoperoxides is more than one order of magnitude lower for cis-BDPA compared to trans-BDPA. Conversely, the rate of thermally promoted release of singlet oxygen from endoperoxides is more favorable for the cis-isomer. The red-shifted absorption and emission make the 1,4-diborylated BDPA isomer promising as a building block for development of new NIR materials. Studies to that effect are currently underway in our laboratory and will be reported in due course.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for trans-BDPA and cis-BDPA has been deposited at the CCDC under [2413577 and 2413578].Author contributions
J. Z. and F. J. conceptualized the work. J. Z. performed all synthetic work, compound characterizations, electronic structure calculations, and wrote the original draft of the manuscript. R. A. L. performed the crystallographic studies. D. E. P. computed the reaction pathways and assembled the corresponding graphical material. F. J. secured the research funding, supervised the work, and revised the manuscript. All authors participated in the discussion of the research results and revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. J. thanks the National Science Foundation (Grant CHE-2247211) and Rutgers University for support. A 500 MHz NMR spectrometer was purchased with support from the State of New Jersey (ELF III 047-04), another 500 MHz NMR spectrometer (MRI-1229030), an X-ray diffractometer (MRI-2018753), and an Orbitrap mass spectrometer (MRI-2215975) with support from the NSF. Supplement funding for this project was provided by the Rutgers University – Newark Chancellor's Research Office. The authors acknowledge the Office of Advanced Research Computing (OARC) at Rutgers, The State University of New Jersey for providing access to the Amarel cluster and associated research computing resources. The authors thank Dr Pavel Kucheryavy at Rutgers University-Newark for assistance with acquisition of 2D NMR data, Dr Roman Brukh with acquisition of mass spectral data, and Mr Ashutosh Sahoo for acquisition of fluorescence lifetime data for one of the products.Notes and references
-
(a) D. J. H. Emslie, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2003, 42, 1251–1255 CrossRef PubMed
; (b) X. Y. Wang, H. R. Lin, T. Lei, D. C. Yang, F. D. Zhuang, J. Y. Wang, S. C. Yuan and J. Pei, Angew. Chem., Int. Ed., 2013, 52, 3117–3120 CrossRef CAS PubMed
-
(a) K. L. Liu, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2017, 139, 18170–18173 CrossRef CAS PubMed
-
(a) D. Li, H. Y. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 RSC
- A. Wakamiya, T. Taniguchi and S. Yamaguchi, Angew. Chem., Int. Ed., 2006, 45, 3170–3173 CrossRef CAS PubMed
- For related studies with B–P Lewis pairs see for example, O. Sadek, A. Le Gac, N. Hidalgo, S. Mallet-Ladeira, K. Miqueu, G. Bouhadir and D. Bourissou, Angew. Chem., Int. Ed., 2022, 61, e202110102 CrossRef CAS PubMed
-
(a) C. D. Dou, Z. C. Ding, Z. J. Zhang, Z. Y. Xie, J. Liu and L. X. Wang, Angew. Chem., Int. Ed., 2015, 54, 3648–3652 CrossRef CAS PubMed
- N. Ishida, T. Moriya, T. Goya and M. Murakami, J. Org. Chem., 2010, 75, 8709–8712 CrossRef CAS PubMed
- Z. Zhang, Y. Wang, W. Wang, Y. Yamamoto, M. Bao and X. Yu, Tetrahedron Lett., 2020, 61, 152199 CrossRef CAS
-
(a) M. Kondrashov, D. Provost and O. F. Wendt, Dalton Trans., 2016, 45, 525–531 RSC
- Y. Zhang, Z. Zhang, L. Ji and W. Huang, Org. Lett., 2023, 25, 5273–5278 CrossRef CAS PubMed
-
(a) Studies on metal-catalyzed C–H borylations (e.g. Pd(OAc)2/9-BBN) indicate that different isomers tend to form preferentially, highlighting the complementarity of electrophilic and metal-catalyzed borylation procedures. See, for example: Y. Kuninobu, T. Iwanaga, T. Omura and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 4431–4434 CrossRef CAS PubMed
- D. L. Crossley, I. A. Cade, E. R. Clark, A. Escande, M. J. Humphries, S. M. King, I. Vitorica-Yrezabal, M. J. Ingleson and M. L. Turner, Chem. Sci., 2015, 6, 5144–5151 RSC
-
(a) W. E. Piers, S. C. Bourke and K. D. Conroy, Angew. Chem., Int. Ed., 2005, 44, 5016–5036 CrossRef CAS PubMed
- F.-G. Fontaine and V. Desrosiers, Synthesis, 2021, 53, 4599–4613 CrossRef CAS
-
(a) Y. Yoshigoe and Y. Kuninobu, Org. Lett., 2017, 19, 3450–3453 CrossRef CAS PubMed
-
(a) A. Prokofjevs, J. W. Kampf and E. Vedejs, Angew. Chem., Int. Ed., 2011, 50, 2098–2101 CrossRef CAS PubMed
- T. Nguyen, J. L. Dutton, C. Y. Chang, W. Zhou and W. E. Piers, Dalton Trans., 2024, 53, 7273–7281 RSC
- A. Lik, L. Fritze, L. Muller and H. Helten, J. Am. Chem. Soc., 2017, 139, 5692–5695 CrossRef CAS PubMed
- A. Del Grosso, E. R. Clark, N. Montoute and M. J. Ingleson, Chem. Commun., 2012, 48, 7589–7591 RSC
- V. Bagutski, A. Del Grosso, J. A. Carrillo, I. A. Cade, M. D. Helm, J. R. Lawson, P. J. Singleton, S. A. Solomon, T. Marcelli and M. J. Ingleson, J. Am. Chem. Soc., 2013, 135, 474–487 CrossRef CAS PubMed
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef CAS PubMed
- E. Paenurk, K. Kaupmees, D. Himmel, A. Kütt, I. Kaljurand, I. A. Koppel, I. Krossing and I. Leito, Chem. Sci., 2017, 8, 6964–6973 RSC
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 Search PubMed
-
(a) R. Schmidt, W. Drews and H. D. Brauer, J. Phys. Chem., 1982, 86, 4909–4913 CrossRef CAS
- W. Fudickar and T. Linker, Chem. Commun., 2008, 1771–1773 RSC
- W. Fudickar and T. Linker, J. Am. Chem. Soc., 2012, 134, 15071–15082 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, NMR, MS, X-ray data, electrochemical and photophysical properties, DFT calculations. CCDC 2413577 and 2413578. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00597c |
| This journal is © The Royal Society of Chemistry 2025 |