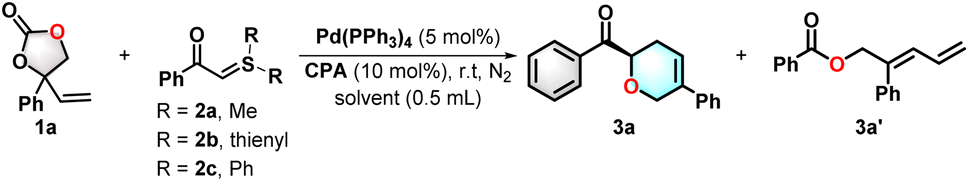DOI:
10.1039/D5SC01050K
(Edge Article)
Chem. Sci., 2025,
16, 8108-8113
Enantioselective [5 + 1] cycloaddition of sulfur ylides and vinylethylene carbonates via synergistic palladium/chiral phosphonic acid catalysis†
Received
10th February 2025
, Accepted 24th March 2025
First published on 8th April 2025
Abstract
An effective method for the synthesis of dihydropyrans through synergistic palladium and chiral phosphonic acid catalysis was reported. This protocol proceeded under mild reactions and provided dihydropyrans in up to 87% yield and up to 97% ee. Meanwhile, various derivations such as oxidation, Wittig-reaction, reductions, nucleophilic substitution, and Baeyer–Villiger were accomplished to furnish interesting compounds. To gain insight into the reaction mechanism, nonlinear relationship experiments and Hammett plot experiments were carried out. In addition, a range of products (3i, 4b, 4f, 4g, and 4j) accessible from this method exhibit various anti-inflammatory activities on NO and ROS inhibition.
Introduction
Chiral di- or tetrahydropyrans are privileged scaffolds in natural product chemistry and pharmaceuticals (Fig. 1).1–6 For example, lovastatin is a lipid-regulating drug that can reduce blood cholesterol, low-density lipoprotein (LDL), and triglyceride concentrations, while increasing high-density lipoprotein (HDL) concentrations.7 Aspergillide C exhibits significant cytotoxicity against mouse lymphocytic leukemia cells (L1210) with LD50 values of 2.0 μg mL−1.3 Ambruticin exhibits potent antifungal activity against a range of fungal pathogens including Coccidioides immitis and Blastomyces dermatitidis.1 Due to the importance of di- or tetrahydropyrans in natural products, human medicines, and other fields, many synthetic strategies and methodologies, including hetero-Diels–Alder reactions, Prins cyclizations, and intramolecular nucleophilic conjugate additions, have been developed for the synthesis of di- or tetrahydropyrans.8–13 The intramolecular asymmetric oxa-Michael reactions were also popular for the preparation of substituted tetrahydropyrans. Zhu and co-workers reported palladium(II)-catalyzed, Selectfluor-mediated formal 6-endo-trig fluorocycloetherification of γ-hydroxyalkenes for the synthesis of functionalized tetrahydropyrans.14 Quintard and Kochem developed stereo-controlled oxa-Michael additions to generate a wide array of chiral tetrahydropyrans via borrowing hydrogen.8 Then, Scheidt reported an enantioselective cross-dehydrogenative coupling to furnish tetrahydropyrans via Lewis acid catalysis.10 Despite the recent progress, the attainment of high stereoselectivity through a ring-forming process in the synthesis of chiral di- or tetrahydropyrans continues to pose a significant synthetic hurdle.
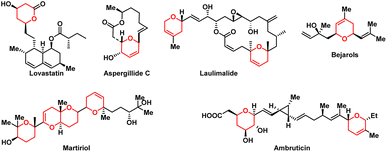 |
| | Fig. 1 Selected bioactive natural products and drugs bearing di- or tetrahydropyrans. | |
The combination of two or more unsaturated structural units to form cyclic organic compounds is among the most useful synthetic constructions in organic chemistry.15 Vinylethylene carbonates (VECs), an important type of zwitterionic allylpalladium species precursor, not only acted as 1,3-dipoles, but also as 1,5-dipoles.16–19 The synthesis of diverse O-heterocycles from vinylethylene carbonates through [3 + 2],20–27 [5 + 2],28–31 [5 + 3],32,33 and [5 + 4]34–40 cycloadditions with different synthons is well known via metal-catalyzed ring-opening strategies. Despite the progress, utilizing vinylethylene carbonates as 1,5-dipoles to participate in the enantioselective [5 + 1] cycloaddition is yet to be disclosed, probably due to other competitive reactions, thus making enantiocontrol difficult. To make TM-catalyzed dipolar [5 + 1] cycloadditions more powerful and general in the synthesis of six-membered cyclic compounds, new strategies for achieving these transformations in high efficiency and selectivity are required. On the other hand, development of suitable C1 partners in the assembly with vinylethylene carbonates is also crucial for the creation of some unique frameworks. Sulfur ylides have gained popularity for use as potential synthetic building blocks in transition-metal-catalyzed reactions as they could be easily used as carbene precursors, which are commonly effective C1 partners.41 In continuation of our research work on synergistic double metal-catalyzed organic transformations,42–45 herein, we disclose a novel [5 + 1] annulation reaction of vinylethylene carbonates and sulfur ylides via synergistic palladium/chiral phosphonic acid catalysis (Scheme 1).
 |
| | Scheme 1 The construction of di- or tetrahydropyran. | |
Results and discussion
Reaction discovery and optimization
Our study initiated the investigation of the model reaction between vinylethylene carbonate 1a and sulfur ylide 2a catalyzed by synergistic Pd(PPh3)4/chiral phosphonic acid to validate the feasibility of the above hypothesis. Sulfur ylide 2a was initially chosen to avoid the complex stereoselectivity control caused by the simultaneous formation of one point. Regrettably, the targeted dihydropyran product was not observed under the Pd/CPA catalytic conditions. Instead, the rearrangement product 3a′ was formed (Table 1, entry 1). When substrate 2b was employed, the desired product 3a was still not achieved under the existing reaction conditions (entry 2). In an attempt to enhance the steric hindrance of the sulfur ylide, the desired product 3a was obtained in 59% yield, albeit with poor enantioselectivity (entry 3). Afterward, diversely substituted and other scaffold-based CPA catalysts were investigated (entries 4–11). When 2,4,6-trimethylphenyl CPA C2 was used, the product was obtained in 66% yield with 16% ee (entry 4). 9-Anthracenyl CPA C3 gave an acceptable result (entry 5). Other tetrahydro BINOL-based CPAs C4–C6 did not give satisfactory results (entries 6–8). The less sterically hindered spiro CPA C7 afforded the tetrahydropyran in 71% yield and 6% ee (entry 9). The use of spiro skeleton-based CPAs C8–C9 bearing bulky groups increased the enantioselectivity (entries 10 and 11), and the best result was obtained in 57% yield with 92% ee. Further screening of the solvents indicated that DCE was still the best choice compared to the other solvents (entries 12–14). The addition of 5 Å molecular sieves dramatically improved the yield to 88% with 93% ee (entry 15). The optimal conditions involved the use of 1a (1.5 mmol, 1.5 equiv.), Pd (PPh3)4 (3 mol%), and C9 (10 mol%) as the catalyst and 5 Å molecular sieves (100 mg) as the additive in DCE (1.0 mL) at 25 °C for 18 hours (entry 16).
Table 1 The screening of reaction conditionsa
|

|
| Entry |
2
|
CPA |
Solvent |
Yield 3a/3a′b (%) |
ee of 3ac |
|
Reaction conditions: 1a (0.06 mmol, 1.2 equiv.), 2 (0.05 mmol, 1.0 equiv.), Pd(PPh3)4 (5 mol%), CPA (10 mol%), solvent (0.5 mL), N2, 24 h.
Determined by 1HNMR using CH2Br2 as the internal standard.
Determined by HPLC.
50 mg of 5 Å molecular sieves were added. Isolated yield is listed in parentheses.
1a (0.15 mmol, 1.5 equiv.), 2c (0.1 mmol, 1.0 equiv.), Pd (PPh3)4 (3 mol%), DCE (1.0 mL), 18 h.
|
| 1 |
2a
|
C1
|
DCE |
ND/30 |
— |
| 2 |
2b
|
C1
|
DCE |
ND/33 |
— |
| 3 |
2c
|
C1
|
DCE |
59/— |
2 |
| 4 |
2c
|
C2
|
DCE |
66/— |
16 |
| 5 |
2c
|
C3
|
DCE |
64/— |
64 |
| 6 |
2c
|
C4
|
DCE |
72/— |
12 |
| 7 |
2c
|
C5
|
DCE |
60/— |
18 |
| 8 |
2c
|
C6
|
DCE |
58/— |
31 |
| 9 |
2c
|
C7
|
DCE |
31/ |
6 |
| 10 |
2c
|
C8
|
DCE |
62/ |
86 |
| 11 |
2c
|
C9
|
DCE |
76/ |
92 |
| 12 |
2c
|
C9
|
THF |
46/— |
89 |
| 13 |
2c
|
C9
|
DCM |
57/— |
93 |
| 14 |
2c
|
C9
|
1,4-Dioxane |
65/— |
94 |
| 15d |
2c
|
C9
|
DCE |
82 (78)/— |
93 |
| 16e |
2c
|
C9
|
DCE |
88 (80)/— |
92 |

|
Substrate scope
With the optimized reaction conditions established, we turned to investigate the scope and limitations of this new [5 + 1] annulation reaction. Initially, we investigated the substitution patterns of vinylethylene carbonates 1 in their reactions with sulfur ylide 2c. As depicted in Scheme 2, we probed the incorporation of various groups onto the para-position of the phenyl ring in vinylethylene carbonates, and the corresponding products 3b–3i were obtained in good yields and high enantioselectivities. Notably, substrates bearing electron-withdrawing groups exhibited enhanced reactivity, yielding the corresponding products in 60–76% yield with enantioselectivity ranging from 86% to 92%. In contrast, substrates with substituents at the meta- and ortho-positions also underwent the reaction successfully, producing dihydropyrans 3j–3q with good yields and enantioselectivity. We were pleased to discover that disubstituted vinylethylene carbonates were well tolerated, affording the desired products 3r–3v in yields of 29–76% with enantioselectivities of 81–90% ee. Interestingly, heteroaryl (thienyl-, furyl-) substituted vinylethylene carbonates proved to be good substrates, yielding products in 21–80% yield with commendable enantioselectivities. Additionally, fluorenyl- and naphthyl-substituted vinylethylene carbonates were also competent in the reaction, providing the targeted products in 26–60% yield with good enantioselectivities. The reaction conditions were also compatible for alkyl-substituted substrates, but a low yield was obtained, offering the dihydropyran 3bb in 35% yield with 93% ee.
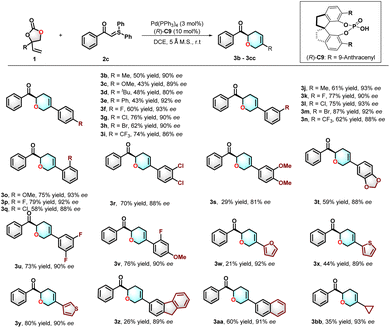 |
| | Scheme 2 Substitution scope of VMCCS.a aReaction conditions: 1 (0.15 mmol), 2c (0.1 mmol), Pd(PPh3)4 (3 mol%), (R)-C9 (10 mol%), DCE (1 mL), 5 Å MS (100 mg), r.t., N2, 18 h. | |
After examining the vinylethylene carbonate scope, we examined a range of sulfur ylides under the optimized conditions (Scheme 3). Gratifyingly, the sulfur ylides bearing various functional groups were compatible with our reaction conditions, including some sensitive groups (bromide, and iodide). Substitution patterns on the para position of sulfur ylides, including electron-donating groups (4a–4c), halides (4d–4g), and electron-withdrawing groups (4h–4j), are well tolerated to give the corresponding chiral dihydropyrans with 46–97% ee. Notably, the para-CN substituent resulted in slightly lower enantiomeric excesses (46% ee) and reduced yields (15%). Additionally, sulfur ylides with meta substituents were also investigated, leading to the formation of enantioenriched dihydropyrans 4k–4l in moderate yields of 57–70% with outstanding enantioselectivity. Ortho-substituted substrates could transform the target product 4m in 24% yield with 84% ee. Furthermore, the enantioselective [5 + 1] annulation of hetero-substituted sulfur ylides was successfully accomplished, yielding the corresponding dihydropyrans 4n–4o in moderate yields of 51–72% and with good enantiomeric excesses of 79–87%. The reaction also accommodated a naphthyl-substituted sulfur ylide, affording tetrahydropyran 4p in 54% yield with 91% ee. The catalytic system also proved effective for disubstituted sulfur ylides, producing dihydropyrans 4q–4t with high enantiomeric excesses. Alkyl-substituted sulfur ylide could tolerate standard conditions to provide the corresponding product 4w in 45% yield with 53% ee. The absolute configuration of 4b was assigned to be R configuration, as unambiguously confirmed by X-ray diffraction.
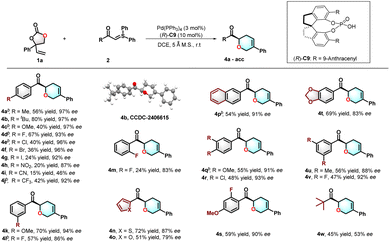 |
| | Scheme 3 Substitution scope of sulfur ylides.a aReaction conditions: 1a (0.15 mmol), 2 (0.1 mmol), Pd(PPh3)4 (3 mol%), (R)-C9 (10 mol%), DCE (1 mL), 5 Å MS (100 mg), r.t., N2, 18–36 h. b1a (0.12 mmol), Pd(PPh3)4 (5 mol%), 36 h. | |
Diverse synthetic application
A 1 mmol scale reaction was conducted to provide product 3a in 81% yield with 97% ee, which demonstrates the practical applicability of this asymmetric catalysis (Scheme 4). Then, we carried out several transformations of the tetrahydropyran 4b. First, addition of organometallic reagents afforded the corresponding α-hydroxyl dihydropyrans 5a and 5b with a high efficiency, good enantioselectivities and diastereoselectivites. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond could be reduced to alcohol 5c in 84% yield with 96% ee and 10
O double bond could be reduced to alcohol 5c in 84% yield with 96% ee and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. Also, the CC double bond of 4b could be efficiently reduced to tetrahydropyran 5d without erosion of enantioselectivity in the presence of Pd/C, albeit with good diastereoselectivities (>20:1). The Wittig reaction of 4b using commercial methyltriphenylphosphonium bromide could proceed to deliver the product 5e in 44% yield with 96% ee. Finally, Baeyer–Villiger oxidation could smoothly convert 4b to the corresponding ester 5f in 84% yield with 96% ee in the presence of m-CPBA as an oxidant. These transformations bearing diverse functional groups while maintaining the optical purity established the potential application and versatility of the methodology in organic synthesis.
:1 dr. Also, the CC double bond of 4b could be efficiently reduced to tetrahydropyran 5d without erosion of enantioselectivity in the presence of Pd/C, albeit with good diastereoselectivities (>20:1). The Wittig reaction of 4b using commercial methyltriphenylphosphonium bromide could proceed to deliver the product 5e in 44% yield with 96% ee. Finally, Baeyer–Villiger oxidation could smoothly convert 4b to the corresponding ester 5f in 84% yield with 96% ee in the presence of m-CPBA as an oxidant. These transformations bearing diverse functional groups while maintaining the optical purity established the potential application and versatility of the methodology in organic synthesis.
 |
| | Scheme 4 Derivation of the compound 4b. | |
Biological studies
Anti-inflammatory assay.
In the evaluation of the anti-inflammatory activity of the synthesized compounds, the levels of inflammatory factors were assessed in RAW264.7 cells. The concentrations of two key pro-inflammatory mediators, nitric oxide (NO) and reactive oxygen species (ROS), are critical for the organism's response to inflammatory stimuli. Elevated levels of NO and ROS have been shown to significantly influence the pathogenesis of various inflammatory diseases. Therefore, the concentrations of these two key pro-inflammatory mediators were detected in RAW264.7 cells. The collected data revealed that compounds 3i, 4b, 4f, 4g, and 4j exhibit moderate concentration-dependent inhibitory activity against NO production (Fig. 2A). In the subsequent anti-inflammatory assay, compounds 3i and 4g demonstrated weak inhibition of ROS secretion in a dose-dependent manner, indicating their effectiveness in suppressing ROS (Fig. 2B).
 |
| | Fig. 2 (A) Chemical structures of tested compounds 3i, 4b, 4f, 4g, and 4j. Effects of compound 3i at 2 μM, 10 μM, and 30 μM on the production of (B) nitric oxide (NO) and (C) reactive oxygen species (ROS) in LPS-induced RAW264.7 macrophages. The cells were pretreated with different concentrations of compounds for 1 h and then treated with LPS. NO concentration (LPS, 25 ng mL−1, 24 h) in the medium was determined by the Griess method. The fluorescence intensity of ROS (LPS, 1 μg mL−1, 24 h) was quantified by using a fluorescence microplate reader. Dexamethasone (DEX.) was employed as a positive control (10 μM). The results shown are representative of three independent experiments. | |
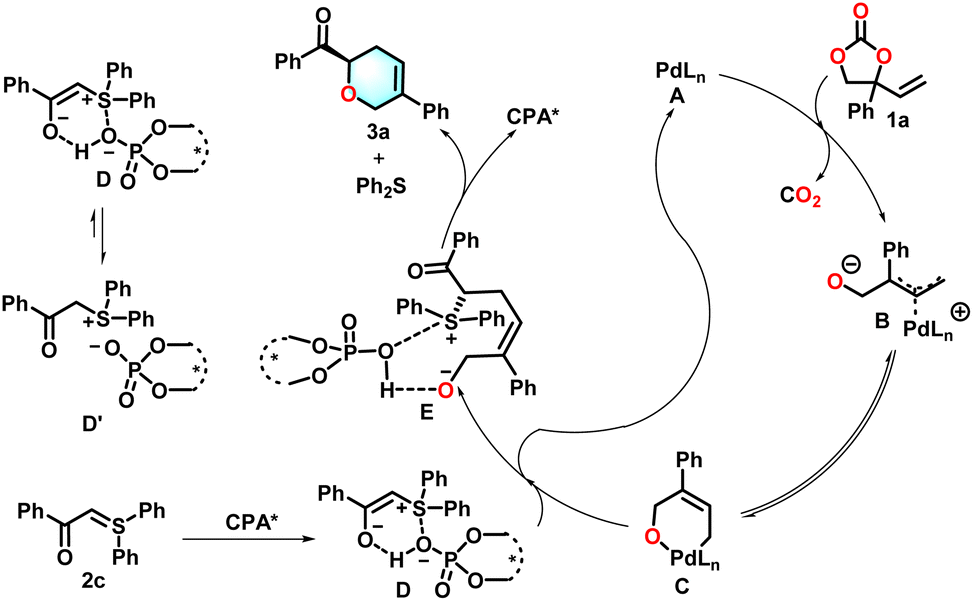
Next, we carried out some additional studies to gain insights into the mechanism, particularly on the mode of catalyst function. We were curious about whether the chiral phosphonic acid activated the vinylethylene carbonates or the sulfur ylide nucleophile. Experiments to probe the relationship between the enantio-purity of C9 and that of product 3a showed that the relationship fell within the experimental error of a linear association. To investigate the chiral phosphonic acid speciation, nonlinear effect (NLE) studies were carried out by variation of the enantiomeric excess of the chiral phosphonic acid catalyst. The ee of the product 3a was linearly related to the ee of chiral phosphonic acid employed (Scheme 5). The linear relationship implies that a mono-chiral phosphonic acid molecule might participate in the stereo-determining step. The slope of a Hammett plot in Scheme 5 that reveals the effect of the electronic properties of the aryl group in the alkene electrophile on the reaction was negative (ρ = −1.38), indicating that a positive charge accumulates in the allyl group in the intermediate.
 |
| | Scheme 5 Nonlinear relationship experiment and Hammett plot. | |
Control experiments were conducted as shown in Scheme 6. The absence of chiral phosphonic acid (eqn (a)) resulted in no detectable product formation. Furthermore, when chiral phosphonic acid was introduced into the reaction mixture after 12 hours of palladium catalysis, the desired product was obtained in 59% yield with 92% ee (eqn (b)). These findings suggest that the reaction is a step-wise cycloaddition. Sulfur ylide 2c (0.02 mmol) was treated with excess chiral phosphoric acid (CPA, 0.04 mmol) in dichloroethane (DCE) at ambient temperature (eqn (c)). Comprehensive characterization by 1H NMR and 13C NMR spectroscopy unambiguously confirmed the formation of intermediate D′ through tautomerization of the initially generated unstable intermediate D. Subsequently, vinyl ethylene carbonate 1a (0.03 mmol) and the Pd(PPh3)4 catalyst (0.0006 mmol, 3 mol%) were introduced into the pre-equilibrated reaction system. Notably, the reaction failed to proceed even after 12 hours of stirring. This observation strongly suggests that excess CPA coordinates with the Pd(PPh3)4 catalyst, thereby preventing the critical transformation of intermediate C. To verify this hypothesis, we reintroduced fresh sulfur ylide 2c (0.02 mmol) into the stagnant reaction mixture. Remarkably, this intervention successfully liberated the Pd catalyst from the CPA–Pd complex, ultimately yielding the desired product 3a in 24% yield with 90% ee. These controlled experimental results show that the CPA does not engage in permanent coordination with the Pd(PPh3)4 catalyst under productive catalytic conditions. To unambiguously establish the catalytic role of intermediate D, we conducted rigorously controlled mechanistic investigations. Initial treatment of sulfur ylide (0.04 mmol) with chiral phosphoric acid (CPA, 0.02 mmol) in anhydrous dichloroethane (DCE) at room temperature under an argon atmosphere led to the clean formation of intermediate D′, as evidenced by 1H NMR spectroscopic analysis (eqn (d)). Crucially, the subsequent addition of vinyl ethylene carbonate 1a (0.06 mmol) and Pd(PPh3)4 catalyst (0.0012 mmol, 3 mol%) to this preformed intermediate system for 12 hours afforded the desired product 3a in 61% isolated yield with 92% ee. These results suggested that intermediate D could be the key transition state. To further ascertain whether Pd(PPh3)4 cooperated with the CPA, a mixture containing Pd(PPh3)4 (0.0015 mmol) and chiral phosphoric acid (CPA, 0.005 mmol) in anhydrous dichloroethane (DCE, 0.5 mL) was stirred for 30 minutes at room temperature under an argon atmosphere (eqn (e)). Subsequently, substrates 1a (0.075 mmol) and 2c (0.05 mmol) were added to this mixture. However, no desired product 3a was observed. In a separate experiment, a mixture of Pd(PPh3)4 (0.0015 mmol), substrate 1a (0.075 mmol), and CPA (0.05 mmol) in anhydrous DCE (0.5 mL) was similarly stirred for 30 minutes under argon at room temperature (eqn (f)), followed by the addition of substrate 2c (0.05 mmol). The desired product 3a was obtained in trace amounts. These findings indicate that CPA does not coordinate with the Pd(PPh3)4 catalyst under the catalytic process.
 |
| | Scheme 6 Control experiments. | |
Based on our experimental results and the literature report, a plausible mechanism is listed in Scheme 7. Initially, the ring-opening of vinylethylene carbonate 1a occurs in the presence of a palladium catalyst, yielding intermediate B.17–19 Concurrently, intermediate B may undergo isomerization to form intermediate C. This intermediate C is then subjected to a reaction with a chiral complex D, which is derived from sulfur ylides and chiral phosphonic acid,46 to generate intermediate E and release species A. Subsequently, an intra-molecular nucleophilic substitution reaction proceeds smoothly, resulting in the formation of the desired product 3a, while simultaneously regenerating chiral CPA.
 |
| | Scheme 7 A plausible mechanism. | |
Conclusions
In summary, we have devised a highly efficient protocol for the enantioselective synthesis of dihydropyrans, achieving high yields and exceptional enantioselectivities through the catalysis of synergistic palladium/chiral phosphonic acid. Mechanistic insights revealed that the pivotal step of the reaction entails the activation of challenging cyclic, aliphatic oxocarbenium ions. It is noteworthy that this reaction can be executed on one mmol scale, and the resultant product can be readily transformed into diverse and valuable chiral building blocks for the synthesis of enantio-enriched compounds. To assess the anti-inflammatory effectiveness of the synthesized compounds, we conducted evaluations of NO and ROS production levels in RAW264.7 cells. Notably, compounds 3i, 4b, 4f, 4g, and 4j exhibited favorable anti-inflammatory activity by inhibiting NO production. In particular, compounds 3i and 4g also demonstrated mild inhibitory effects on ROS production. Moreover, we anticipate that our findings will stimulate further innovation among pharmaceutical chemists in adorning pharmacophores with enantio-enriched di- or tetrahydropyran motifs.
Data availability
All the data supporting this article have been uploaded as part of the ESI.†
Author contributions
M. Ke and F. Chen designed the work. All the syntheses and characterization studies have been done by M. Ke, J. Zheng, J. Zong, J. Wang, G. Zheng, and B. Zhang. Bioactive experiments have been done by K. Tang, and Z. Ju and D. Cheng helped polish the article. The manuscript was written by M. Ke, Z. Ju and F. Chen. The overall work was supervised by Z. Ju and F. Chen. All authors have approved the final version of the article before submission.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded by the National Key Research and Development Program (2021YFF0600704) and the Educational Foundation of Zhejiang University of Technology (no. KYY-HX-20220471).
Notes and references
- J. I. Bowen, X. Zhong, K. Gao, B. Reed, M. P. Crump, L. Wang and C. L. Willis, Chem. Sci., 2024, 15, 5319–5326 RSC
 .
.
- A. Diez-Torrubia, S. Cabrera, S. de Castro, C. García-Aparicio, G. Mulder, I. De Meester, M.-J. Camarasa, J. Balzarini and S. Velázquez, Eur. J. Med. Chem., 2013, 70, 456–468 CrossRef CAS PubMed .
- T. Nagasawa, Org. Lett., 2009, 11, 761–764 CrossRef CAS PubMed .
- S. Pajk, M. Živec, R. Šink, I. Sosič, M. Neu, C.-w. Chung, M. Martínez-Hoyos, E. Pérez-Herrán, D. Álvarez-Gómez, E. Álvarez-Ruíz, A. Mendoza-Losana, J. Castro-Pichel, D. Barros, L. Ballell-Pages, R. J. Young, M. A. Convery, L. Encinas and S. Gobec, Eur. J. Med. Chem., 2016, 112, 252–257 CrossRef CAS PubMed .
- M. D. Shultz, A. K. Cheung, C. A. Kirby, B. Firestone, J. Fan, C. H.-T. Chen, Z. Chen, D. N. Chin, L. DiPietro, A. Fazal, Y. Feng, P. D. Fortin, T. Gould, B. Lagu, H. Lei, F. Lenoir, D. Majumdar, E. Ochala, M. G. Palermo, L. Pham, M. Pu, T. Smith, T. Stams, R. C. Tomlinson, B. B. Touré, M. Visser, R. M. Wang, N. J. Waters and W. Shao, J. Med. Chem., 2013, 56, 6495–6511 CrossRef CAS PubMed .
- C.-Y. Tang, J. Wu, F.-T. Ji, F. Tian, L. Peng and L.-L. Wang, Org. Chem. Front., 2024, 11, 211–216 RSC .
- G. Guan, P. Dai and I. Shechter, Arch. Biochem. Biophys., 2003, 417, 251–259 CrossRef CAS PubMed .
- A. Alexandridis, T. Rancon, A. Halliday, A. Kochem and A. Quintard, Org. Lett., 2024, 26, 5788–5793 CrossRef CAS PubMed .
- P. Kramer, J. Grimmer, M. Bolte and G. Manolikakes, Angew. Chem., Int. Ed., 2019, 58, 13056–13059 CrossRef CAS PubMed .
- A. Lee, R. C. Betori, E. A. Crane and K. A. Scheidt, J. Am. Chem. Soc., 2018, 140, 6212–6216 CrossRef CAS PubMed .
- L.-L. Li, Y.-L. Su, Z.-Y. Han and L.-Z. Gong, Chem.–Eur. J., 2018, 24, 7626–7630 CrossRef CAS PubMed .
- B. Srinivas, D. S. Reddy, N. A. Mallampudi and D. K. Mohapatra, Org. Lett., 2018, 20, 6910–6914 CAS .
- Y.-H. Wang, B.-J. He, S. J. Kalita, Z.-N. Zhao, Y.-C. Li, D.-H. Zhang, Z.-Q. Yi, U. Schneider and Y.-Y. Huang, Org. Chem. Front., 2025, 12, 838–848 CAS .
- J. Gong, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2023, 145, 15735–15741 CAS .
- X. Xu and M. P. Doyle, Acc. Chem. Res., 2014, 47, 1396–1405 CrossRef CAS PubMed .
- W. Guo, J. E. Gómez, À. Cristòfol, J. Xie and A. W. Kleij, Angew. Chem., Int. Ed., 2018, 57, 13735–13747 CAS .
- L. Hu, A. Cai, Z. Wu, A. W. Kleij and G. Huang, Angew. Chem., Int. Ed., 2019, 58, 14694–14702 CAS .
- L. Zuo, T. Liu, X. Chang and W. Guo, Molecules, 2019, 24, 3930–3935 CrossRef CAS PubMed .
- S. Khan, T. Ahmad, T. Rasheed and N. Ullah, Coord. Chem. Rev., 2022, 462, 214526 CAS .
- A. Khan, L. Yang, J. Xu, L. Y. Jin and Y. J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 11257–11260 CrossRef CAS PubMed .
- A. Khan, R. Zheng, Y. Kan, J. Ye, J. Xing and Y. J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 6439–6442 CrossRef CAS PubMed .
- I. Khan, C. Zhao and Y. J. Zhang, Chem. Commun., 2018, 54, 4708–4711 RSC .
- K. Liu, I. Khan, J. Cheng, Y. J. Hsueh and Y. J. Zhang, ACS Catal., 2018, 8, 11600–11604 CrossRef CAS .
- L.-C. Yang, Z. Y. Tan, Z.-Q. Rong, R. Liu, Y.-N. Wang and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 7860–7864 CrossRef CAS PubMed .
- Y. Yang and W. Yang, Chem. Commun., 2018, 54, 12182–12185 RSC .
- S. Singha, E. Serrano, S. Mondal, C. G. Daniliuc and F. Glorius, Nat. Catal., 2020, 3, 48–54 CrossRef CAS .
- J. Wang, L. Zhao, Q. Rong, C. Lv, Y. Lu, X. Pan, L. Zhao and L. Hu, Org. Lett., 2020, 22, 5833–5838 CrossRef CAS PubMed .
- S. Singha, T. Patra, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2018, 140, 3551–3554 CrossRef CAS PubMed .
- Y. Wei, S. Liu, M.-M. Li, Y. Li, Y. Lan, L.-Q. Lu and W.-J. Xiao, J. Am. Chem. Soc., 2019, 141, 133–137 CrossRef CAS PubMed .
- H.-I. Ahn, J.-U. Park, Z. Xuan and J. H. Kim, Org. Biomol. Chem., 2020, 18, 9826–9830 CAS .
- X. Gao, D. Zhu, Y. Chen, H. Deng, F. Jiang, W. Wang, Y. Wu and H. Guo, Org. Lett., 2020, 22, 7158–7163 CrossRef PubMed .
- C. Yuan, Y. Wu, D. Wang, Z. Zhang, C. Wang, L. Zhou, C. Zhang, B. Song and H. Guo, Adv. Synth. Catal., 2018, 360, 652–658 CrossRef CAS .
- B. Niu, X.-Y. Wu, Y. Wei and M. Shi, Org. Lett., 2019, 21, 4859–4863 CrossRef CAS PubMed .
- Z.-Q. Rong, L.-C. Yang, S. Liu, Z. Yu, Y.-N. Wang, Z. Y. Tan, R.-Z. Huang, Y. Lan and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 15304–15307 CrossRef CAS PubMed .
- L.-C. Yang, Z.-Q. Rong, Y.-N. Wang, Z. Y. Tan, M. Wang and Y. Zhao, Angew. Chem., Int. Ed., 2017, 56, 2927–2931 CrossRef CAS PubMed .
- P. Das, S. Gondo, P. Nagender, H. Uno, E. Tokunaga and N. Shibata, Chem. Sci., 2018, 9, 3276–3281 RSC .
- X. Gao, M. Xia, C. Yuan, L. Zhou, W. Sun, C. Li, B. Wu, D. Zhu, C. Zhang, B. Zheng, D. Wang and H. Guo, ACS Catal., 2019, 9, 1645–1654 CrossRef CAS .
- H. Uno, N. Punna, E. Tokunaga, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2020, 59, 8187 CrossRef CAS PubMed .
- C. Xia, D.-C. Wang, G.-R. Qu and H.-M. Guo, Org. Chem. Front., 2020, 7, 1474–1480 RSC .
- G. Yang, Y.-M. Ke and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 12775–12780 CrossRef CAS PubMed .
- L.-Q. Lu, T.-R. Li, Q. Wang and W.-J. Xiao, Chem. Soc. Rev., 2017, 46, 4135–4149 RSC .
- M. Ke, Z. Liu, K. Zhang, S. Zuo and F. Chen, Green Synth. Catal., 2021, 2, 228–232 CrossRef .
- M. Ke, Y. Yu, K. Zhang, S. Zuo, Z. Liu, X. Xiao and F. Chen, Adv. Synth. Catal., 2022, 364, 1849–1854 CrossRef CAS .
- M. Ke, Y. Yu, L. Sun, X. Li, Q. Cao, X. Xiao and F. Chen, Chem. Commun., 2023, 59, 2632–2635 RSC .
- M. Ke, X. Li, J. Zong, B. Wang, J. Zheng, S. Zhang, J.-A. Chen and F. Chen, Org. Lett., 2024, 26, 1201–1206 CrossRef CAS PubMed .
- W. Guo, Y. Luo, H. H. Y. Sung, I. D. Williams, P. Li and J. Sun, J. Am. Chem. Soc., 2020, 142, 14384–14390 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2406615. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01050k |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jinying
Zheng‡
b,
Jiayi
Zong
a,
Keshuang
Tang
a,
Jiahao
Wang
a,
Guohui
Zheng
a,
Boxuan
Zhang
a,
Dang
Cheng
a,
Jinying
Zheng‡
b,
Jiayi
Zong
a,
Keshuang
Tang
a,
Jiahao
Wang
a,
Guohui
Zheng
a,
Boxuan
Zhang
a,
Dang
Cheng