
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRIBOsensor for FRET-based, real-time ribose measurements in live cells†
Mina
Ahmadi
 ,
Zhuangyu
Zhao‡
and
Ivan J.
Dmochowski
*
,
Zhuangyu
Zhao‡
and
Ivan J.
Dmochowski
*
Department of Chemistry, University of Pennsylvania, Philadelphia, PA, USA. E-mail: ivandmo@sas.upenn.edu
First published on 8th April 2025
Abstract
D-Ribose is a building block of many essential biomolecules, including all nucleic acids, and its supplementation can enhance energy production, particularly under stress conditions such as ischemia and heart failure. The distribution, biosynthesis, and regulation of ribose in mammalian systems remain poorly understood. To explore intracellular ribose dynamics, we developed a genetically encoded fluorescence resonance energy transfer (FRET) sensor using ribose binding protein (RBP) and enhanced cyan and yellow fluorescent proteins (FPs). The RIBOsensor, which positions one FP near the active site of RBP, achieves the necessary sensitivity for cellular imaging by increasing the FRET signal upon ribose binding, compared to traditional N- and C-terminal FP orientations. This sensor rapidly, reversibly, and selectively detects labile ribose in live cells—enabling longitudinal studies—and can be employed for intracellular ribose quantitation, which provides a valuable tool for investigating ribose transport and metabolism in normal and disease states.
Introduction
D-Ribose is a key constituent of biomolecules such as DNA, RNA, ATP, NADH, and many cofactors, as well as a potential source of energy.1 The uptake of extracellular ribose by mammalian cells has been shown to occur through the glucose transporter, GLUT2, a phenomenon known as “ribose salvage”.2 Subsequently, the imported ribose undergoes phosphorylation by ribokinase, to form ribose-5-phosphate (R5P), an essential intermediate in the pentose phosphate pathway (PPP). R5P, in turn, holds the capacity for conversion into other vital metabolites, including phosphoribosyl pyrophosphate (PRPP) – the precursor for de novo ATP synthesis – and bypasses the rate-limiting enzyme in PPP, glucose-6-phosphate dehydrogenase.3D-Ribose can play multiple roles, depending on the cellular metabolic state. For example, D-ribose supplementation can accelerate ATP production, which supports physiologic demands of the body under special circumstances such as diastolic dysfunction caused by ischemia,4 and fatigue caused by congestive heart failure.5,6 Conversely, urine D-ribose level is elevated in type 2 diabetes mellitus (T2DM) suggesting T2DM is associated with abnormal metabolism of D-ribose in addition to glucose.7,8 Notably, D-ribose reacts with hemoglobin more rapidly than D-glucose, thereby accelerating the glycosylation of hemoglobin in red blood cells, a key marker of hyperglycemia (HbA1C).9 Moreover, the interaction of D-ribose with serum proteins and cellular proteins generates advanced glycation end products, which bind to cell membrane receptors and can lead to high blood pressure, diabetic nephropathy, and neurodegenerative disorders, including Alzheimer's disease.10,11 The complex roles of ribose in both normal human physiology and disease progression underscore the need for more comprehensive investigations of ribose biodistribution, dynamics, biosynthetic pathways, and regulatory mechanisms within mammalian systems. A key challenge is D-ribose quantitation, particularly in living cells where there is an abundance of ribose-containing molecules and structurally related pentose sugars. Direct, robust, and noninvasive methods for real-time monitoring of intracellular ribose are crucial for understanding its role in various physiological and pathological processes, as well as its regulation within cells. Whereas bioanalytical approaches such as mass spectrometry, 129Xe NMR biosensing,12 and small-molecule sensors employing fluorescence13 or 19F NMR spectroscopy14 have been advanced for quantifying ribose, many of these approaches are not compatible with cells (not least, due to the high background levels of RNA and ATP), and the real-time measurement of labile intracellular ribose has remained inaccessible. Whereas, many strategies have been developed and widely implemented for glucose monitoring, both in cells and in vivo, these capabilities are completely lacking for ribose monitoring.15–18Fluorescent proteins are widely employed for live-cell visualization and quantification of biological analytes. Genetically encoded FRET sensors, in particular, serve as valuable ratiometric tools for quantifying biological molecules within different cellular compartments under both resting and stress conditions.19 Periplasmic binding proteins (PBPs) have shown huge potential for the design of genetically encoded FRET sensors,20 and were previously explored—in one example—for ribose sensing.21 PBPs, a ubiquitous protein superfamily extensively distributed in bacteria and archaea, play a pivotal role in the cellular uptake of solutes. Their functionality relies on a distinctive ligand-induced conformational change, which achieves remarkable specificity towards their native ligands. This feature offers a unique avenue for monitoring ligand concentrations in mammalian cells, allowing for the detection of a diverse array of targets, ranging from inorganic ions22 to metalloids,23 vitamins,24 amino acids,25 and sugars,26 with affinities naturally spanning the low nM to low μM range and readily modified via single mutations in the PBP binding pocket.20 The PBP binding pocket can also be tuned to recognize non-native, yet structurally similar ligands.27 PBPs can be introduced into mammalian cells, where there are no competing protein analogs. In this work, we incorporated ribose binding protein (RBP) as the sensor module attached to enhanced cyan fluorescent protein (ECFP) and enhanced yellow fluorescent protein (EYFP) following two strategies: (1) using N- and C-termini of RBP as the two attachment sites (similar to a published prototype,21 which resulted in a small decrease in FRET signal upon ribose binding), or (2) inserting one of the FRET partners internally (Scheme 1). We showed that insertion of one of the FRET partners close to the ribose binding pocket creates a “turn on” sensor with greatly improved FRET signal going from apo- to ligand-bound. The resulting RIBOsensor is capable of selectively, sensitively, and robustly detecting labile ribose in live cells, enabling longitudinal studies and quantitative cellular assays.
 | ||
| Scheme 1 Design of the genetically encoded FRET-based RIBOsensor (A) representation of two approaches to attach fluorescence protein pair (ECFP and EYFP) to RBP (PDB ID: 2DRI and 1URP, closed and open conformations respectively) to create RBP-FRET sensor: (1) attach the pair to the N- and C-termini of RBP and (2) keep one of the FPs at either N- or C-terminus of RBP and fuse the other FP internally. The values reported in orange are α-carbon distances of RBP amino acids at the connection sites. The changes in the distance (ΔDistance) from open to closed conformations are reported on the double arrows. (B) Representation of RIBOsensor linear peptide sequence. | ||
Results and discussion
Design and in vitro characterization of a working RIBOsensor
PBP-FRET sensors are typically constructed by inserting the PBP between two fluorescent proteins (FPs) using short peptide linkers.19 Although this approach is straightforward, and the FRET response can potentially be tuned by adjusting the length and amino acid content of the linkers,19 the resulting signal change, either loss or gain of FRET upon ligand binding, is limited by the protein conformation and dynamics at the N- and C-termini of the PBP. We first tried to improve upon the published ribose sensor,21 by making modifications to the linkers within the linear fusion protein sequence ECFP-linker-RBP-linker-EYFP. By eliminating the linker and deleting 9 amino acids from the C-terminus of ECFP and 2 and 5 amino acids from the N-terminus of RBP and EYFP, respectively, we developed the ECFP-RBP-EYFP ribose sensor. This design, by restricting FP movements at their attachment sites, slightly improved the ΔFRET signal (change in the ratio of emission intensity at 529 nm (EYFP) and 476 nm (ECFP), I529/I476, excited at 433 nm, from saturation to apo) compared to the published version, from −0.19 (ref. 21) to −0.24. However, this small, negative change in FRET signal was not amenable to quantitative measurements of cellular ribose in fluorescence microscopy studies. It was shown previously for a glucose–galactose binding protein that inserting one of the FPs at an internal allosteric site, if tolerated by the PBP, can lead to a more significant FRET change than placing the FPs at the N- and C-termini and optimizing the linkers.28 Internal insertion sites were also explored in PBPs for the design of single-wavelength indicators, often using circularly permuted or split fluorescent proteins.29 To leverage this in the design of a cellular ribose sensor, we used molecular dynamics (MD) simulations to sample the available RBP conformations and calculated the change in distance of the α-carbons of each amino acid of RBP to either the N- or C-terminus when transitioning from the apo- (open) to the ligand-bound (closed) conformation. In addition to monitoring the distance changes between the FP attachment points, a set of candidate insertion sites within RBP was identified that remain solvent-exposed and are not directly involved in ribose binding (Fig. 1). | ||
| Fig. 1 Determination of internal site of insertion on RBP. Using molecular dynamics simulations, the average α-carbon-to-α-carbon distance between each amino acid of RBP to the reference (either N- or C-terminus) over 50-ns free movement in water is calculated for both open and closed conformations and the changes in the distances, open minus closed are reported (ΔDistance). The three investigated loops containing A133, A162, and N13 for the internal fusion are highlighted on the closed conformation of RBP (PBD ID: 2DRI) in blue, red, and green colors, respectively. | ||
Based on these criteria, we identified three promising sites within the loops near the ribose binding pocket, A133, A162, and N13, for the insertion of one FP through short linkers, while the other FP remained at the N- or C-terminus of RBP, also attached by a short linker. Internally oriented constructs are named using parentheses to include the internally fused FP and its site of attachment while “–” is used to show the FP attachment to either N- or C-terminus. For example, ECFP-RBP (A133.EYFP) was created with ECFP located at the N-terminus and EYFP inserted between Ala 133 and Gly 134 (Scheme 1). All constructs were generated using gBlocks™ gene fragments and cloned via restriction enzyme digestion into pET28 and pCDNA3.1 vectors for bacterial and mammalian expression, respectively. For in vitro characterization, sensors were overexpressed in the BL21(DE3) E. coli strain, purified using HisTrap nickel affinity chromatography, and subjected to on-column denaturation with 8 M urea buffer to release endogenous ribose bound to RBP, followed by on-column renaturation, elution, and size-exclusion chromatography. SDS-PAGE and MALDI-TOF MS confirmed the purity and correct mass of the protein sensors (Fig. S1–S3†). As hypothesized from MD simulations, internal fusion of one of the FPs resulted in switching the sensor response from a loss to a gain of FRET signal upon ribose binding for all three versions. Although two of the internally fused sensors, RBP(N13.ECFP)-EYFP and ECFP-RBP(A162.EYFP), produced similar or smaller ΔFRET signal compared to ECFP-RBP-EYFP (Fig. 2A and S4†), the ECFP-RBP(A133.EYFP) sensor resulted in a significant improvement in ΔFRET, +0.68 (Fig. 2B and Table 1). Consequently, we selected the ECFP-RBP(A133.EYFP) sensor, from now on referred to as the RIBOsensor, for further characterization and cell studies. Direct titration of ribose to purified apo RIBOsensor revealed a dissociation constant of 30.8 ± 7.0 μM, while the sensor remained selective for ribose compared to other relevant biological analytes (Fig. 2C and D and S5†). It is important to note that the Hill1 fitting used here to determine Kd is based on calibrating the ratio signal for borderline “ratio” values, the FRET ratio of the fully unbound and bound sensor. In a method developed by Pomorski et al.,30 calibrating for borderline values of emission intensities at both wavelengths—in this case, 529 nm (EYFP) and 476 nm (ECFP)—was shown to result in a more accurate Kd determination. For the RIBOsensor, the two data fitting methods produced similar in vitro Kd values (Fig. S6†), and using the “ratio” approach allowed direct comparison to in situ measurements of Kd gleaned from cell population studies.
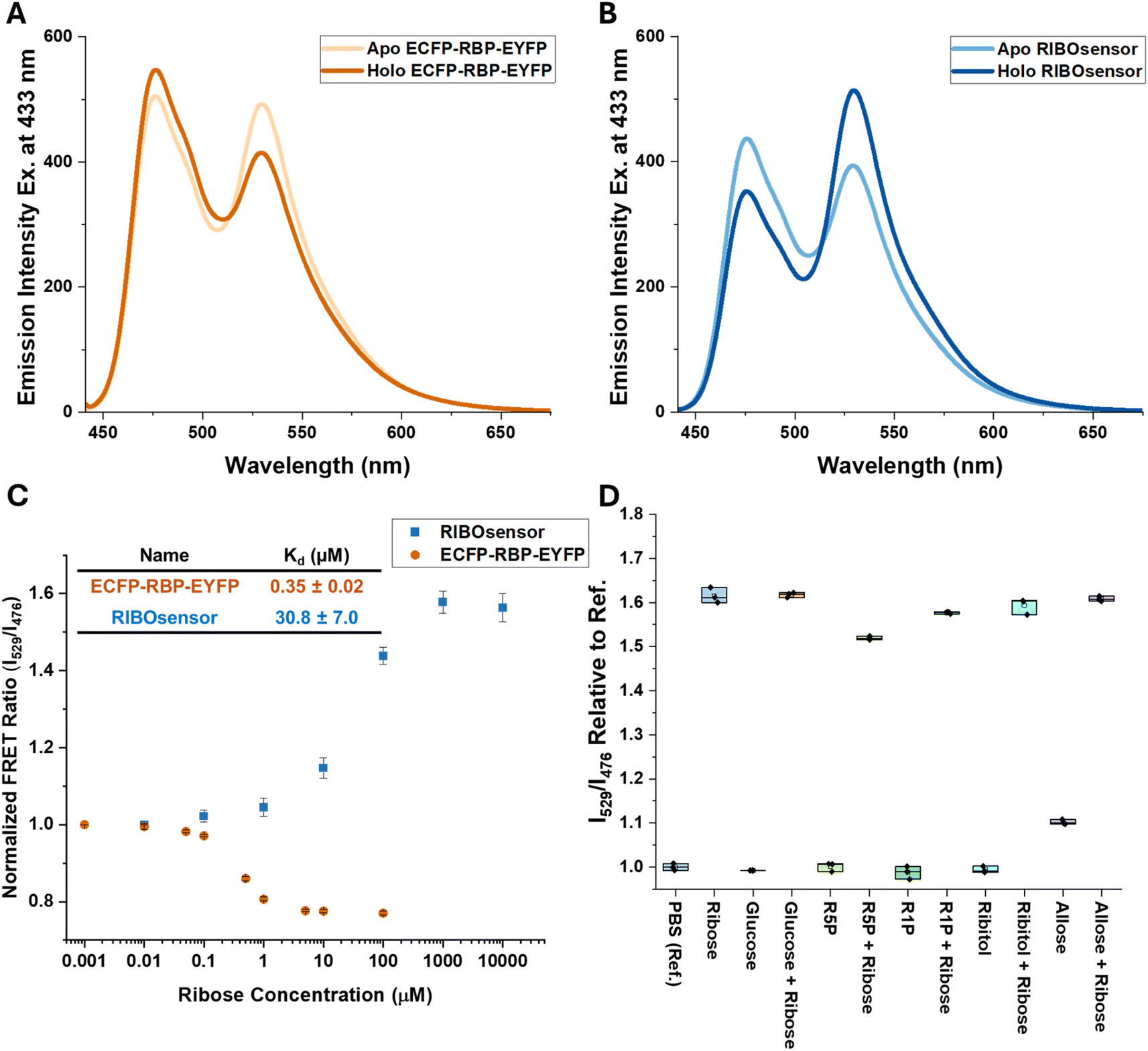 | ||
| Fig. 2 Development of RBP-FRET sensor. FRET response of 1 μM ECFP-RBP-EYFP (A) and RIBOsensor (B) in the absence and presence of a saturating concentration of D-ribose monitored by fluorescence spectroscopy. (C) Titration of 0.7 μM pure sensor with 0.01 μM to 10 mM D-ribose in PBS. With 433 nm excitation wavelength, emission of EYFP at 529 nm over ECFP at 476 nm is graphed vs. ribose concentration. Kd values of ECFP-RBP-EYFP and RIBOsensor are calculated using the Hill equation fitting on titration data points. The data represent the means of 6 and 4 independent replicates for RIBOsensor and ECFP-RBP-EYFP, respectively, with vertical bars indicating standard deviation. (D) In vitro selectivity and competition assay using purified RIBOsensor and relevant biological analytes. Measurements were done in PBS using 10 mM of each analyte in the presence or absence of 10 mM D-ribose. The data represent 3 independent replicates for each condition. | ||
| Name | Sites of FP connection | ECFP Linker(s) | EYFP Linker(s) | ΔDistancec (Å) (open-close) | ΔFRET responsed |
|---|---|---|---|---|---|
| a RIBOsensor. b Other than linkers, 9 amino acids from the C-terminus of ECFP and 2 and 5 amino acids from the N-terminus of RBP and EYFP, respectively, are also deleted. c Between α-carbon of RBP amino acids at the site of FP connections. d Change in the ratio of I529/I476, excited at 433 nm, from apo to ribose saturation. | |||||
| ECFP-RBP-EYFP | N-terminus/C-terminusb | — | — | −0.24 | −0.238 |
| ECFP-RBP(A133.EYFP)a | N-terminus/A133 | GGTGGA | AG/GS | +2.79 | 0.677 |
| ECFP-RBP(A162.EYFP) | N-terminus/D162 | GGTGGA | AG/GS | +4.10 | 0.243 |
| RBP(N13.ECFP)-EYFP | N13/C-terminus | AG/GS | GAGTGG | +0.95 | 0.060 |
Evaluating RIBOsensor dynamics in temporal ribose measurements in HEK293T cells
Based on the promising in vitro data, we expressed the RIBOsensor in HEK293T cells to characterize its in situ response and develop an assay for estimating labile ribose concentrations. Confocal microscopy confirmed the cytosolic localization, uniform distribution, and intact nature of the sensor upon donor fluorophore excitation with a 458 nm laser or direct excitation of the acceptor fluorophore at 515 nm (Fig. S7†). The response of the pure apo-sensor to 10% fetal bovine serum (FBS) indicated a ∼20 μM ribose concentration in this standard growth media (Fig. S8†). To minimize ribose salvage prior to our cell measurements, we experimented with the effect of lowering the FBS and glucose concentrations in the growth media on the FRET signal of HEK293T cells expressing RIBOsensor. HEK293T cells tolerated growth media with 1% FBS and no glucose, which significantly decreased the FRET signal compared to cells grown in normal media with 10% FBS and 25 mM glucose (Fig. S9 and S10†). Because HEK293T cells grow very slowly in 1% FBS and no glucose, the FBS and glucose concentration were adjusted to 2% and 5 or 25 mM, respectively, to maintain cell viability, except in cases of full starvation, where no glucose was used to achieve minimal FRET readouts. To examine the temporal response of HEK293T cells to ribose, cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) containing 2% FBS and 25 mM glucose and transfected with the RIBOsensor. At the time of initial measurement (t0), the growth media was switched to DMEM supplemented with 1 mM ribose, and the ECFP and EYFP emission channels were recorded simultaneously every 10 min while the cells were excited with the 458 nm laser (Fig. 3A and B and S11†). As the starved cells salvaged ribose from the media, a gradual increase in the FRET ratio (IEYFP/IECFP) was observed, with a maximum around 60 min followed by a decrease in FRET ratio, suggesting a regulatory mechanism for ribose uptake coupled with metabolism (Fig. 3B). It is noteworthy that PBPs are rapid binders,31 and RIBOsensor also binds rapidly to D-ribose (Fig. S12†). To confirm that the observed peak in FRET ratio was due to ribose regulation rather than sensor saturation, in another experiment cells were exposed to DMEM containing 1 mM ribose and 20 μM digitonin to permeabilize the cell membrane and facilitate ribose diffusion. In this condition, the FRET ratio reached a higher range faster compared to intact cells (Fig. 3A and B). | ||
| Fig. 3 Temporal measurements of ribose in HEK293T cells using confocal microscopy and flow cytometry (A) spatial–temporal imaging of RIBOsensor response in live HEK293T cells upon addition of 1 mM D-ribose extracellularly in the absence or presence of 20 μM digitonin to permeabilize the cells. Pseudocolored ratiometric images were generated from confocal images of cells excited by 458 nm laser and simultaneously recording ECFP (470–510 nm) and EYFP (527–567 nm) channels. Cells were grown in 2% FBS and 25 mM glucose. Scale bar is 10 μm (B) quantified FRET response across the cells represented in (a). (C) Flow cytometry data on HEK293T cells expressing RIBOsensor and grown in 2% FBS and 25 mM glucose. FRET response (Ex: 405 nm; Em: 450/50 nm for ECFP, 525/50 nm for EYFP) was measured post addition of 1 mM D-ribose up to 16 h. Saturated signal, shown in gray, is measured from cells treated with 15 μM digitonin and 50 mM D-ribose. (D) Growth of the FRET value, corresponds to the high FRET peak (labeled as purple in c) vs. time. | ||
Our confocal laser scanning microscopy measurements on single cells indicate that photobleaching occurs in both ECFP and EYFP channels. However, the overall FRET ratio is less significantly affected (Fig. S13†). In addition to photobleaching, confocal microscopy presents other challenges: cells may not survive for the entire duration of the measurements, the field of view can drift out of focus, and despite closing the chamber between measurements and using a very low laser power—limiting laser exposure to the measurement periods—these factors make single-cell measurements challenging to reproduce precisely. Nonetheless, there is value in being able to obtain spatial and temporal data for ribose localization and concentration within living cells. In flow cytometry, cells are exposed to light only briefly, minimizing photobleaching. Additionally, flow cytometry allows for the analysis of a large number of cells, enabling conclusions to be drawn with greater statistical significance. To assess ribose transport dynamics in a population of cells with intact membrane, we examined the response of starved HEK293T cells expressing the RIBOsensor to the addition of 1 mM ribose using flow cytometry. Cells were cultured in DMEM with 2% FBS and 25 mM glucose, and at the flow cytometer were irradiated at 405 nm to excite ECFP. Ribose salvage was observed for different cell populations up to 16 h after ribose addition (Fig. 3C and S14†). The FRET response (EmEYFP(525/50nm)/EmECFP(450/50nm) initially increased, peaked, and then declined (Fig. 3D), mirroring observations from single-cell confocal microscopy measurements. Notably, even after 8 h, while the majority of cells exhibited higher FRET values (peak indicated by the purple circle in Fig. 3C), a significant portion of cells still displayed lower FRET values (peak indicated by the green circle in Fig. 3C). Both single-cell confocal microscopy and cell-population flow cytometry studies confirm the RIBOsensor's effectiveness for detecting both increases and decreases in intracellular ribose levels that reflect labile ribose dynamics. Importantly, the RIBOsensor remains intact and fluorescent in cells for up to 56 h post-induction, as confirmed by a nondenaturing gel (Fig. S15†).
Quantifying labile ribose concentration in HEK293T cells
Lastly, we aimed to develop a method for estimating the labile ribose concentration in HEK293T cells. Using the apparent dissociation constant and also the minimum readout (Rmin) and maximum readout (Rmax) for the FRET ratio signal, the concentration of the target analyte can be estimated in live cells using formula (1) and the FRET ratio in the experimental condition (denoted as Rexp in formula (1)).32 | (1) |
The critical parameters to establish are Rmin and the dissociation constant. To determine the most accurate value for Rmin, we explored two strategies: (1) starving the cells in media containing 2% FBS and no glucose for several passages, and (2) generating a non-responsive control version of the sensor by mutating key amino acids in the binding pocket of RBP, following the methodology developed for a FRET heme sensor.33 As mentioned, to reduce salvaged ribose, HEK293T cells were grown initially in 2% FBS with either 5 or 25 mM glucose. To further deplete the ribose pool, cells were cultured in 2% FBS and glucose-free media, which likely forced the cells to utilize endogenous ribose for energy. Additionally, co-transfection of the RIBOsensor with wild-type RBP, which has a ribose dissociation constant of 0.13 μM,21 further reduced the FRET signal due to competition (Fig. S16†). To pursue the first strategy and create a non-responsive RBP sensor, we mutated Phe 16 to Ala, a change previously shown to shift the dissociation constant to the millimolar range,21 resulting in the ECFP-RBP(A133.EYFP)-F16A sensor. Although this sensor did not respond to saturated ribose concentrations (Fig. S17†), lower FRET values were observed using the RIBOsensor in starvation conditions (Fig. S16†). This discrepancy may arise from the effect of the F16A mutation on RBP folding, which likely differs from the wild type and alters the FRET ratio at the apo state of the RIBOsensor and ECFP-RBP(A133.EYFP)-F16A. This limits the utility of the non-responsive control sensor for determining Rmin. Consequently, the starvation method was selected for Rmin determination. Lastly, establishing the in situ dissociation constant is crucial. Various microenvironmental factors, including pH, buffer composition, ionic strength, temperature, and intracellular metabolites and cellular crowding, have been shown to significantly influence FRET sensor readouts and, consequently, the apparent binding constant. While in vitro measurements can be conducted under conditions that closely resemble the cellular environment, the most accurate determination of the apparent Kd requires measurements within living cells. Research indicates that in situ Kd values may be slightly or significantly higher than those obtained in vitro.18,34–37
To determine this, HEK293T cells were grown in 2% FBS and 5 mM glucose for multiple passages. Waiting 48 h after transfection with the RIBOsensor, cells were collected, washed in PBS, and divided equally. They were then treated independently with 15 μM digitonin and varying concentrations of ribose in PBS buffer for 30 min. The cells were collected by centrifugation, resuspended in PBS buffer, and analyzed by flow cytometry. The FRET signal shifted towards higher values with increasing ribose concentration (Fig. S18b†). The range of FRET ratios for each cell population treated with different ribose concentrations was determined by subtracting the lowest FRET value from the highest in that population and labeled as ΔFRET (Fig. S18c†). This in situ titration yielded an apparent Kd value of 680 ± 85 μM (Fig. 4A and S18d†). Furthermore, in situ competition experiments confirmed that the RIBOsensor in live cells retains selectivity for ribose over glucose and ribose-5-phosphate despite the higher Kd (Fig. 4B). Using the in situ Kd value, the FRET ratio of starved HEK293T cells co-transfected with RIBOsensor and wild-type RBP as Rmin, with peak value of 1.50, and the FRET ratio of cells treated with a saturating ribose concentration in the presence of digitonin as Rmax, with peak value of 2.32, the endogenous labile ribose concentration in HEK293T cells grown in common growth media (10% FBS and 25 mM glucose) was determined to be 130 ± 20 μM (Fig. 4C).
 | ||
| Fig. 4 In situ characterization of RIBOsensor to measure endogenous ribose concentration in HEK293T cells (A) in situ titration using HEK293T cells grown in 2% FBS, 5 mM glucose and expressing RIBOsensor. 48 h after transfection cells were collected and washed in PBS buffer. Cells were then treated with 15 μM digitonin and increasing concentration of D-ribose from 0.01 μM to 100 mM for 30 min, collected, resuspended in PBS and subjected to flow cytometry. ΔFRET was defined as the range of FRET (max value − min value) for each condition. Corresponding Kd value was determined by Hill equation fitting. The data represent the means of 4 independent replicates, with vertical bars indicating standard deviation. (B) In situ competition using HEK293T cells grown in 2% FBS, 5 mM glucose and expressing RIBOsensor. Cells were treated with 15 μM digitonin and 10 mM of each analyte in the presence or absence of 10 mM D-ribose. The data represent at least 3 independent replicates for each condition. (C) Flow cytometry data of the FRET values of HEK293T cells. Rmin corresponds to the cells grown in 2% FBS, no glucose and co-transfected with RIBOsensor and wild-type RBP. Rmax corresponds to the cells grown in 2% FBS, no glucose and treated for 30 min with 15 μM digitonin and 50 mM D-ribose. Experimental media corresponds to cells grown in 10% FBS and 25 mM glucose. FRET values of the peak of the cells in each condition are reported on the graph by arrows and used to calculate ribose concentration in HEK293T cells using formula (1). | ||
Conclusions
In this study, we developed the first genetically encoded FRET ribose sensor with the demonstrated capability of real-time, live-cell ribose measurements, for both imaging ribose in single cells by fluorescence microscopy and assessing ribose dynamics in cell populations using flow cytometry. We followed two strategies to attach donor and acceptor FPs to RBP. The first linker-modification strategy slightly improved FRET response upon ribose binding, likely by restricting the movements of FPs at the N- and C-termini of RBP. A more successful design strategy was guided by MD simulations, which identified several interior attachment sites for one of the fluorescent proteins; the A133 site for EYFP insertion resulted in the RIBOsensor. This novel attachment site not only improved the magnitude of the ΔFRET response more than 3-fold relative to standard FP attachment to RBP at N- and C-termini, but also generated a ‘turn-on’ sensor, which has advantages for many quantitative fluorescence assays. These features make it possible, finally, to achieve quantitative measurements of intracellular ribose, and its reversible dynamics. It is noteworthy that RBP consists of N- and C-terminal lobes (Scheme 1). Both lobes are Rossmann-like fold domains containing a beta-sheet and, respectively, four and five alpha helices. The C-terminal lobe has fewer loops and more alpha helices compared to the N-terminal lobe, resulting in a more rigid environment for the amino acids near the RBP binding pocket in the C-terminal lobe. Consequently, amino acids in the C-terminal lobe are more significantly affected by structural changes in RBP upon ligand binding. Additionally, the N-terminus is located on the opposite side of the C-terminal lobe. As a result, a greater change in the distances of each amino acid from the N- or C-terminus is observed for the amino acids located in the C-terminal lobe when transitioning from open to closed conformation (Fig. 1). The FRET response in a given FP pair depends first on separating the FPs with the intervening RBP by roughly the Fӧrster distance, 4.9 nm for ECFP/EYFP pair,38 and then eliciting a change in the distance between the chromophores upon ribose binding/RBP closure. MD simulations helped to identify promising attachment sites on RBP to maximize the impact of conformational changes (from open to closed conformation) on the FPs. However, these predictions focus on the FP attachment sites, and the actual chromophore behavior is influenced by the flexibility at the attachment sites and the relative orientation of the FPs with respect to RBP and each other. This could explain why the N13 position resulted in a suboptimal ΔFRET response and why the A162 position, despite having a greater Δdistance compared to A133, produced a smaller ΔFRET response. While the proximity of EYFP to the ribose binding pocket in RIBOsensor influenced the dissociation constant, RBP remained selective for ribose over other relevant metabolites, including R5P and glucose. The affinity of PBPs for their ligand can be modulated, as needed, by single point mutations in their binding pocket.20 In this study, the only mutation applied to RIBOsensor was F16A to remove ribose binding and generate a non-responsive control sensor. Otherwise, the as-measured intracellular Kd of ∼650 μM is well poised for physiologic studies, particularly for assessing “hyper-ribose” metabolic conditions. Additionally, we established a methodology to determine endogenous labile ribose concentrations in HEK293T cells by measuring in situ dissociation constants and minimum sensor readouts. We note that incomplete depletion of the intracellular ribose pool can produce an Rmin value that is still too high. In this scenario, the measured concentration of labile ribose may exceed 130 μM. Nevertheless, this measured value, averaged over a large population of HEK293T cells grown in nutrient-rich conditions, is the first, to the best of our knowledge, measurement of endogenous ribose in live cells. A previous estimate of ribose concentration (without experimental validation)39 is more than 10-fold lower than our measurement, which highlights the gap in current understanding about free ribose in cells. The RIBOsensor will enable a survey of labile ribose in many cell types grown under different nutrient conditions, in both healthy and disease states. Various studies indicate that glucose concentrations in mammalian cells range from a few hundred micromolar to millimolar levels.40 Our current findings in HEK293T cells suggest that the intracellular free ribose concentration may be comparable to the lower end of the glucose range. This highlights the need to examine a broader range of cells, both healthy and diseased, at different growth stages to better understand the ‘normal’ concentration range of free ribose for its multiple roles in RNA function and energy metabolism. Our cell studies demonstrate a robust RIBOsensor readout, highlighting the potential for investigating ribose metabolism, regulation, and distribution in mammalian cells and tissues under normal or stressed conditions. The RIBOsensor enables real-time, dynamic monitoring of ribose metabolism, regulation, and distribution in mammalian cells and tissues, both under normal and stressed conditions. It can monitor variations in ribose levels across cell types, as has been extensively studied for glucose, and provide insights into key regulatory steps of the PPP pathway, potentially enhancing our understanding of ribose dysregulation in diseases like type 2 diabetes. The RIBOsensor's robust, selective, and reliable readout even in the complex cytosolic microenvironment makes it a promising diagnostic tool for ribose measurements in biofluids without the need for extraction of free ribose and extensive sample preparation. Additionally, simple fluorescent measurements and minimal instrumentation requirements make the RIBOsensor well-suited for clinical applications compared to other ribose quantification techniques.Experimental
Molecular dynamics simulations
MD simulations were performed with NAMD software41 using the Bridges-2 Regular Memory system at the Pittsburgh Supercomputing Center (PSC).42,43 Initial structures were obtained from the X-ray structure of RBP in the ribose-bound conformations (PDB: 1DRJ).44 Each protein was solvated in a TIP3P water box,45 and 150 mM NaCl was used to neutralize total charge. Each system was minimized with 1000 steps, after which the system was equilibrated to a temperature of 300 K and a pressure of 1 atm using the Langevin thermostat and barostat methods. The CHARMM36 force field was used to perform all MD simulations.46 Periodic boundary conditions were employed, and the SHAKE algorithm47 was applied to constrain the lengths of all bonds that involve a hydrogen.For the ribose-bound structure, a 100-ns MD trajectory was obtained. The snapshots of the last 50 ns were used to calculate residue distances, which were measured as the average α-C distances between the N-terminal or C-terminal residue and each of the other residues using VMD 1.9.3.48 For the ribose-free MD simulation, the ribose molecule was omitted from the same initial structure, and the system was allowed to equilibrate in a 70-ns simulation. The protein relaxed to the open conformation within the first 20 ns, and the last 50 ns trajectory was analyzed in the same way as the ribose-bound RBP. Subsequently, the average distance change of each residue relative to the N-terminus or C-terminus was determined by taking the difference between the two simulations.
Generation of ribose sensor genetic constructs
Ribose sensor constructs were created using gBlocks™ gene fragments (Integrated DNA Technologies, IDT) with appropriate restriction sites at their 5′ and 3′ ends (BamHI, CsiI, HindIII). These fragments were used to modify Addgene plasmid 17860,21 which served as the template. After constructing the desired ribose sensor gene, it was inserted into the pET28a(+) vector using XbaI and HindIII restriction sites. To insert the ribose sensor-encoding gene into the pCDNA3.1 vector, BamHI and HindIII restriction sites were used. All constructs were verified by Sanger sequencing (Penn Genomic and Sequencing Core).Expression of ribose sensors in E. coli and purification
Plasmids encoding different ribose sensors were transformed into BL21(DE3) competent cells (New England Biolabs), which were then grown in 1 L of LB Miller broth containing 50 μg mL−1 kanamycin at 37 °C until a final OD600 of 0.6–0.8 was reached. Protein expression was induced by adding 1 mM IPTG, followed by overnight incubation at 18 °C. The cells were then pelleted by centrifugation and frozen at −20 °C if needed. Cell lysis was performed using chicken egg white lysozyme (Thermo Scientific, Cat. J60701.14) in 20 mM Tris base buffer (pH 7.9) with benzonase nuclease (Sigma-Aldrich, Burlington, MA, USA) and protease inhibitor cocktail tablets (cOmplete Mini, EDTA free, Roche Diagnostics). After stirring the lysate at rt for 30 min, NaCl (0.5 M) and imidazole (20 mM) were added. The lysate was clarified by centrifugation, and the supernatant was loaded onto HisTrap™ FF nickel affinity columns (Cytiva) pre-equilibrated with 20 mM Tris base (pH 7.9) and 0.5 M NaCl. Each ribose sensor bound to the column was unfolded using 15 column volumes of 20 mM Tris base (pH 7.9), 0.5 M NaCl, 20 mM imidazole, and 8 M urea to remove any endogenous ribose bound to the RBP. The sensor was then refolded on-column with 20 column volumes of 20 mM Tris base (pH 7.9), 0.5 M NaCl, and 20 mM imidazole, and subsequently eluted with 20 mM Tris base (pH 7.9), 0.5 M NaCl, and 500 mM imidazole. The eluate was concentrated and further purified by size exclusion chromatography in phosphate-buffered saline (PBS) using a HiLoad™ 16/600 Superdex™ 200 pg column (GE Life Sciences). Protein concentrations were determined from the absorbance at 280 nm using the extinction coefficient 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 490 M−1 cm−1 (for RIBOsensor), as calculated by PROTPARAM.49 Protein purity and size were verified by native gel, SDS-PAGE, and MALDI-TOF mass spectrometry, respectively. For native gel analysis protein sample was mixed with 5× loading dye (250 mM Tris.HCl, pH 6.8, 30% glycerol, 0.05% bromophenol blue). In-gel fluorescence images of pure protein were captured using 405 nm and 546 nm lasers on a G:Box mini 6/9 gel imager. Afterwards, gel was stained with Coomassie blue and destained in methanol/acetic acid/water. For SDS-PAGE analysis, protein sample was mixed with 5× loading dye (10% SDS, 500 mM dithiothreitol, 50% glycerol, 250 mM Tris, pH 6.8, and 0.05% bromophenol blue) and boiled for at least 5 min before loading onto a 4–15% Mini-PROTEAN TGX precast protein gel (BioRad). Gel was stained with Coomassie blue and destained in methanol/acetic acid/water. The MS spectra were collected by Bruker rapifleX MALDI-TOF instrument. The matrix was a saturated solution of sinapinic acid in 70% acetonitrile with 0.1% trifluoroacetic acid.
490 M−1 cm−1 (for RIBOsensor), as calculated by PROTPARAM.49 Protein purity and size were verified by native gel, SDS-PAGE, and MALDI-TOF mass spectrometry, respectively. For native gel analysis protein sample was mixed with 5× loading dye (250 mM Tris.HCl, pH 6.8, 30% glycerol, 0.05% bromophenol blue). In-gel fluorescence images of pure protein were captured using 405 nm and 546 nm lasers on a G:Box mini 6/9 gel imager. Afterwards, gel was stained with Coomassie blue and destained in methanol/acetic acid/water. For SDS-PAGE analysis, protein sample was mixed with 5× loading dye (10% SDS, 500 mM dithiothreitol, 50% glycerol, 250 mM Tris, pH 6.8, and 0.05% bromophenol blue) and boiled for at least 5 min before loading onto a 4–15% Mini-PROTEAN TGX precast protein gel (BioRad). Gel was stained with Coomassie blue and destained in methanol/acetic acid/water. The MS spectra were collected by Bruker rapifleX MALDI-TOF instrument. The matrix was a saturated solution of sinapinic acid in 70% acetonitrile with 0.1% trifluoroacetic acid.
In vitro characterization of ribose sensors
Fluorescence spectra of apo- and holo-sensors were recorded for various ribose sensors in three independent experiments, using 1 μM sensor concentration in PBS buffer at rt, with a Jasco FP8300 spectrofluorometer. The excitation wavelength was set to 433 nm, and emission spectra were recorded from 400 to 700 nm. The FRET ratio in vitro was defined as I529nm/I476nm based on fluorescence intensity. To determine the ribose Kd of ECFP-RBP-EYFP and RIBOsensor, each sensor at 0.7 μM was incubated for 15–30 min with increasing concentrations of D-ribose (Alfa Aesar, Cat. A17894) ranging from 0.01 μM to 1 mM and 10 mM, respectively. Fluorescence spectra for each mixture were recorded at rt. The calculated FRET ratios and ribose concentrations from three independent experiments were fitted using the Hill equation (n = 1, non-cooperative binding interaction) in OriginPro 2023:where y represents the FRET ratio and x denotes the ribose concentration. The parameter “k” corresponds to the dissociation constant. To determine the Kd following the Pomorski et al.30 method, the following fitting equations were set and used in OriginPro 2023.
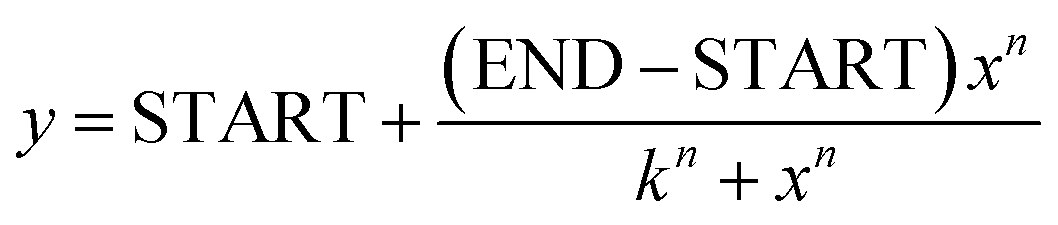

where Ib and Iu are the intensities of the bound and unbound sensors, respectively. x is the concentration of the unbound ligand, K is the dissociation constant, and n is the Hill coefficient. The details of Ib and Iu values are provided in the ESI.† To test the selectivity of the RIBOsensor for ribose over other relevant compounds, 0.7 μM of the protein was incubated with 10 mM of each analyte in the presence and absence of 10 mM ribose. The FRET ratios from three independent experiments for each analyte were normalized to the FRET ratio of the apo-sensor. To test how quickly RIBOsensor responds to D-ribose, FRET ratios of 0.7 μM purified protein incubated with 1 mM D-ribose in PBS were measured either immediately after mixing with D-ribose or after more than 15 min of incubation. For each condition, three independent measurements were conducted.

Mammalian cell culture and transfection
HEK293T cells (ATCC ID: CRL-3216) were maintained in Gibco DMEM (Cat. 11995065) supplemented with 10 μg mL−1 penicillin-streptomycin (Gibco, Cat. 15140122) and 10% FBS (Gibco, Cat. 16000044) at 37 °C with 5% CO2 only for a few passages and then transferred to starved conditions. Except for these few passages, for all other growth media, Gibco DMEM without D-glucose, L-glutamine, Phenol Red, and sodium pyruvate (Cat. A1443001) was used. To prepare the growth media, 10 μg mL−1 penicillin-streptomycin, 6 mM L-glutamine (SAFC, Cat. 59202C), and, depending on the level of starvation, FBS and D-glucose (Sigma, Cat. G7528) were added. Cells were passaged for at least 5 days in starved media before transfection. Plasmids were transiently transfected using polyethyleneimine (PEI) “MAX” (MW 40000 Da) (Polysciences, Inc. Cat. 24765) as the transfection reagent under standard transfection conditions with a 3:1 (w/w) vector/PEI ratio. For a 6 cm dish, 4 μg of plasmid was used, and the amount was adjusted according to the surface area of other plates. Cells were replated on a μ-Slide 8 Well high ibiTreat chamber (ibidi) for confocal microscopy fluorescence imaging or on a 6-well plate for flow cytometric analysis 16 h before use. HEK293T cells grown in starved media are less adherent compared to cells grown in normal media. To prevent cell detachment by the addition of ribose or changing the media for temporal measurements, chambers and plates were coated with 50 μg mL−1 poly-D-lysine (Gibco, Cat. A3890401).
Characterization of RIBOsensor expressed in HEK293T cells
The pellets of HEK293T cells expressing RIBOsensor, collected from a 10 cm dish for each condition, were stored at −80 °C until ready for lysis. Cells were lysed in 300 μL PBS supplemented with benzonase nuclease and a protease inhibitor cocktail. Cells were lysed using a Branson Sonifier probe sonicator (three sets of 6–10 pulses, 15% duty cycle, output setting 3–5), followed by centrifugation at 13000 rpm at 4 °C for 30 min. The supernatant, soluble fraction, was collected for native gel analysis or FRET ratio measurements. For native gel analysis, 24 μL of the soluble fraction were mixed with 5× loading dye (250 mM Tris·HCl, pH 6.8, 30% glycerol, 0.05% bromophenol blue). In-gel fluorescence images were captured using 405 nm and 546 nm lasers on a G:Box mini 6/9 gel imager. Subsequently, the gel was stained with Coomassie blue and destained in methanol/acetic acid/water. For FRET ratio measurements, soluble fractions were incubated with 50 mM D-ribose in PBS and measured immediately after mixing with D-ribose. Same samples were measured again after more than 15 min of incubation. For each condition, three independent measurements were conducted.
Confocal microscopy
HEK293T cells transfected with the RIBOsensor were cultured in the ibiTreat 8-well chamber for 16 h before measurements. Fluorescence images were captured using an Olympus FV1000 laser scanning confocal microscope equipped with UPLFLN 10× or 60× air objectives. For temporal measurements, the ibiTreat chamber was maintained in a stage-top incubator to keep the atmosphere at 5% CO2 and the temperature at 37 °C. For direct excitation of EYFP, HEK293T cells were excited with a 515 nm argon ion laser. For FRET measurements, HEK293T cells were excited with a 458 nm laser, and two individual fluorescence micrographs were simultaneously collected at various spots and time points: the emission of ECFP was collected from 470 nm to 510 nm, and EYFP was collected from 527 nm to 567 nm to minimize bleed-through artifacts. To account for variability in sensor expression levels, cells with medium fluorescence were selected rather than those with the brightest or dimmest fluorescence. Each image was acquired in a 512 × 512 or 1600 × 1600 pixel format with a scan rate of 40.0 μs per pixel. For temporal measurements, cells were grown and transfected in the presence of 2% FBS and 25 mM glucose. D-ribose and digitonin (MilliporeSigma, Cat. CHR103MI) solutions were prepared in DMEM (without D-glucose, L-glutamine, Phenol Red, and sodium pyruvate), which replaced the growth media at time zero. All other measurements were conducted in the growth media unless otherwise noted in the caption. Single-cell images (acquired using the 60× air objective) were processed using ImageJ software. To quantify the average and maximum ECFP or EYFP signal, after subtracting the background, using the “measure” tool in ImageJ the mean and max of signal with a threshold of 10 was calculated for each channel separately. FRET ratio images were generated following the protocol developed by Raz et al.50 To quantify the FRET ratio across the cells, the mean FRET ratio was calculated using the “measure” tool in ImageJ. For images acquired with the 10× objective, 1600 × 1600 pixel, a MATLAB script was used to generate the FRET image across the entire field of view and to calculate the average FRET value. For MATLAB processing, the TIFF files of ECFP and EYFP channels were converted using ImageJ, and the average background for each channel was calculated using the “measure” tool in ImageJ, considering only the cell-free areas of the recorded images for each channel.Flow cytometry and in situ characterization
For each condition, HEK293T cells transfected with the RIBOsensor were cultured in a single 6-well plate 16 h before measurements. For temporal measurements, D-ribose was directly added to the growth media (2 mL volume for a 6-well plate). Cells were collected and washed twice with DPBS (without Ca2+ or Mg2+) before being analyzed on the flow cytometer. The cells were resuspended in 1 mL DPBS and analyzed on an LSRFortessa using a 405 nm laser for donor excitation (ECFP Em. 450/50 nm, EYFP Em. 525/50 nm) and a 488 nm laser for direct excitation of EYFP (Em. 530/30 nm), with a minimum of 10000 cells recorded per condition. Flow cytometry data were analyzed using FlowJo (v10.9.0) software. Cells are gated to remove background signals in ECFP and EYFP channel coming from non-transfected cells, exclude data from debris and aggregated cells, and to remove low FRET cells even in the presence of a saturated concentration of D-ribose. For in situ titration and competition assays, after collecting and washing the cells twice with DPBS (without Ca2+ or Mg2+), they were resuspended in 500 μL of DPBS (without Ca2+ or Mg2+) containing 15 μM digitonin, Benzonase nuclease, and the desired concentrations of D-ribose, D-glucose, or R5P for 30 min. Cells were then collected by centrifugation and resuspended in 500 μL of DPBS (without Ca2+ or Mg2+) before being analyzed on the flow cytometer.
Data availability
Supplementary data characterizing sensors, HEK293T cells confocal microscopy, and flow cytometry are provided in the ESI.† Raw characterization data are provided in an Excel file.Author contributions
Conceptualization, M. A. and I. J. D.; formal analysis, M. A.; funding acquisition, I. J. D.; investigation, M. A.; methodology, M. A. and Z.Z.; project administration, I. J. D.; software, M. A. and Z. Z.; supervision, I. J. D.; validation, M. A.; visualization, M. A.; writing – original draft, M. A.; writing – review & editing, M. A., Z. Z., and I. J. D.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Institutes of Health (R35-GM131907) and the University of Pennsylvania, Department of Chemistry for supporting this work. M. A. received financial support from the NIH Predoctoral Fellowship at the Chemistry-Biology Interface (CBI) at UPenn. The flow cytometry data were generated in the UPenn Cytomics and Cell Sorting Shared Resource Laboratory and is partially supported by the Abramson Cancer Center NCI Grant (P30 016520). The research identifier number is RRid:SCR_022376. MD simulations in this work used Bridges-2 at Pittsburgh Supercomputing Center through allocation CHE180082 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296. We thank Stephen Crane for help in developing MATLAB scripts and Jiayi He for help with MD simulations.References
- P. Jargiello, Somatic Cell Genet., 1978, 4, 647–660 CrossRef CAS PubMed.
- P. M. Clark, G. Flores, N. M. Evdokimov, M. N. McCracken, T. Chai, E. Nair-Gill, F. O'Mahony, S. W. Beaven, K. F. Faull, M. E. Phelps, M. E. Jung and O. N. Witte, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E2866–E2874 CrossRef CAS PubMed.
- A. Stincone, A. Prigione, T. Cramer, M. M. C. Wamelink, K. Campbell, E. Cheung, V. Olin-Sandoval, N.-M. Grüning, A. Krüger, M. Tauqeer Alam, M. A. Keller, M. Breitenbach, K. M. Brindle, J. D. Rabinowitz and M. Ralser, Biol. Rev. Camb. Philos. Soc., 2015, 90, 927–963 CrossRef PubMed.
- L. M. Shecterle, K. R. Terry and J. A. St Cyr, Cureus, 2018, 10, e2291 Search PubMed.
- J. D. Pierce, D. E. Mahoney, J. B. Hiebert, A. R. Thimmesch, F. J. Diaz, C. Smith, Q. Shen, D. P. Mudaranthakam and R. L. Clancy, BMC Cardiovasc. Disord., 2018, 18, 57 CrossRef PubMed.
- S. Li, J. Wang, Y. Xiao, L. Zhang, J. Fang, N. Yang, Z. Zhang, M. I. Nasser and H. Qin, Exp. Ther. Med., 2021, 21, 496 CrossRef CAS PubMed.
- L. Yu, Y. Chen, Y. Xu, T. He, Y. Wei and R. He, Aging, 2019, 11, 4943–4969 CrossRef CAS PubMed.
- Y. Tai, Z. Zhang, Z. Liu, X. Li, Z. Yang, Z. Wang, L. An, Q. Ma and Y. Su, Mol. Biol. Rep., 2024, 51, 220 CrossRef CAS PubMed.
- Z. Zhang, Y. Tai, Z. Liu, Y. Pu, L. An, X. Li, L. Li, Y. Wang, Z. Yang, C. Duan, K. Hou, Q. Zhang, F. Ren, Q. Ma and Y. Su, Blood Cells Mol. Dis., 2023, 99, 102725 CrossRef CAS PubMed.
- C.-Y. Shen, C.-H. Lu, C.-F. Cheng, K.-J. Li, Y.-M. Kuo, C.-H. Wu, C.-H. Liu, S.-C. Hsieh, C.-Y. Tsai and C.-L. Yu, Biomedicines, 2024, 12, 1699 CrossRef CAS PubMed.
- M. Javed, M. I. Ahmad, H. Javed and S. Naseem, Mol. Biol. Rep., 2020, 47, 2289–2299 CrossRef CAS PubMed.
- S. D. Zemerov, B. W. Roose, K. L. Farenhem, Z. Zhao, M. A. Stringer, A. R. Goldman, D. W. Speicher and I. J. Dmochowski, Anal. Chem., 2020, 92, 12817–12824 CrossRef CAS PubMed.
- N. Dey and S. Bhattacharya, Chemistry, 2017, 23, 16547–16554 CrossRef CAS PubMed.
- X.-D. Gao, Y. Hu, W.-F. Wang, X.-B. Zhao, X.-Z. Du and Y.-P. Shi, Anal. Chem., 2022, 94, 11564–11572 CrossRef CAS PubMed.
- S. Walker-Samuel, R. Ramasawmy, F. Torrealdea, M. Rega, V. Rajkumar, S. P. Johnson, S. Richardson, M. Gonçalves, H. G. Parkes, E. Årstad, D. L. Thomas, R. B. Pedley, M. F. Lythgoe and X. Golay, Nat. Med., 2013, 19, 1067–1072 CrossRef CAS PubMed.
- M. Mita, I. Sugawara, K. Harada, M. Ito, M. Takizawa, K. Ishida, H. Ueda, T. Kitaguchi and T. Tsuboi, Cell Chem. Biol., 2022, 29, 98–108 CrossRef CAS PubMed.
- T. Maric, G. Mikhaylov, P. Khodakivskyi, A. Bazhin, R. Sinisi, N. Bonhoure, A. Yevtodiyenko, A. Jones, V. Muhunthan, G. Abdelhady, D. Shackelford and E. Goun, Nat. Methods, 2019, 16, 526–532 CrossRef CAS PubMed.
- C. M. Díaz-García, C. Lahmann, J. R. Martínez-François, B. Li, D. Koveal, N. Nathwani, M. Rahman, J. P. Keller, J. S. Marvin, L. L. Looger and G. Yellen, J. Neurosci. Res., 2019, 97, 946–960 CrossRef PubMed.
- L. Lindenburg and M. Merkx, Sensors, 2014, 14, 11691–11713 CrossRef CAS PubMed.
- K. A. Edwards, Talanta Open, 2021, 3, 100038 CrossRef.
- I. Lager, M. Fehr, W. B. Frommer and S. Lalonde, FEBS Lett., 2003, 553, 85–89 CrossRef CAS PubMed.
- N. Agrawal, N. Soleja, R. Bano, R. Nazir, T. O. Siddiqi and M. Mohsin, ACS Omega, 2021, 6, 14164–14173 CrossRef CAS PubMed.
- N. Soleja, O. Manzoor, P. Khan and M. Mohsin, Sci. Rep., 2019, 9, 11240 CrossRef PubMed.
- N. K. Karpowich, H. H. Huang, P. C. Smith and J. F. Hunt, J. Biol. Chem., 2003, 278, 8429–8434 CrossRef CAS PubMed.
- W. Ko, S. Kim and H. S. Lee, Org. Biomol. Chem., 2017, 15, 8761–8769 RSC.
- M. Fehr, H. Takanaga, D. W. Ehrhardt and W. B. Frommer, Mol. Cell. Biol., 2005, 25, 11102–11112 CrossRef CAS PubMed.
- D. Tavares, A. Reimer, S. Roy, A. Joublin, V. Sentchilo and J. R. van der Meer, Sci. Rep., 2019, 9, 16940–16914 CrossRef PubMed.
- K. Deuschle, S. Okumoto, M. Fehr, L. L. Looger, L. Kozhukh and W. B. Frommer, Protein Sci., 2005, 14, 2304–2314 CrossRef CAS PubMed.
- J. S. Marvin, B. G. Borghuis, L. Tian, J. Cichon, M. T. Harnett, J. Akerboom, A. Gordus, S. L. Renninger, T.-W. Chen, C. I. Bargmann, M. B. Orger, E. R. Schreiter, J. B. Demb, W.-B. Gan, S. A. Hires and L. L. Looger, Nat. Methods, 2013, 10, 162–170 CrossRef CAS PubMed.
- A. Pomorski, T. Kochańczyk, A. Miłoch and A. Kręzel, Anal. Chem., 2013, 85, 11479–11486 CrossRef CAS PubMed.
- D. M. Miller, J. S. Olson, J. W. Pflugrath and F. A. Quiocho, J. Biol. Chem., 1983, 258, 13665–13672 CrossRef CAS.
- A. Miyawaki, J. Llopis, R. Heim, J. M. McCaffery, J. A. Adams, M. Ikura and R. Y. Tsien, Nature, 1997, 388, 882–887 CrossRef CAS PubMed.
- Y. Song, M. Yang, S. V. Wegner, J. Zhao, R. Zhu, Y. Wu, C. He and P. R. Chen, ACS Chem. Biol., 2015, 10, 1610–1615 CrossRef CAS PubMed.
- R. Moussa, A. Baierl, V. Steffen, T. Kubitzki, W. Wiechert and M. Pohl, J. Biotechnol., 2014, 191, 250–259 CrossRef CAS PubMed.
- P. J. Dittmer, J. G. Miranda, J. A. Gorski and A. E. Palmer, J. Biol. Chem., 2009, 284, 16289–16297 CrossRef CAS PubMed.
- R. Lerchundi, N. Huang and C. R. Rose, Front. Cell. Neurosci., 2020, 14, 80 CrossRef CAS PubMed.
- A. J. Boersma, I. S. Zuhorn and B. Poolman, Nat. Methods, 2015, 12, 227–229 CrossRef CAS PubMed.
- G. H. Patterson, D. W. Piston and B. G. Barisas, Anal. Biochem., 2000, 284, 438–440 CrossRef CAS PubMed.
- J. Fortpied, R. Gemayel, D. Vertommen and E. Van Schaftingen, Biochem. J., 2007, 406, 139–145 CrossRef CAS PubMed.
- A. Behjousiar, C. Kontoravdi and K. M. Polizzi, PLoS One, 2012, 7, e34512 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- S. T. Brown, P. Buitrago, E. Hanna, S. Sanielevici, R. Scibek and N. A. Nystrom, presented in part at the Practice and Experience in Advanced Research Computing, New York, NY, USA, 2021 Search PubMed.
- T. J. Boerner, S. Deems, T. R. Furlani, S. L. Knuth and J. Towns, presented in part at the Practice and Experience in Advanced Research Computing, New York, NY, USA, 2023 Search PubMed.
- A. J. Björkman, R. A. Binnie, H. Zhang, L. B. Cole, M. A. Hermodson and S. L. Mowbray, J. Biol. Chem., 1994, 269, 30206–30211 CrossRef.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- R. B. Best, X. Zhu, J. Shim, P. E. M. Lopes, J. Mittal, M. Feig and J. A. D. Mackerell, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- E. Gasteiger, C. Hoogland, A. Gattiker, S. e. Duvaud, M. R. Wilkins, R. D. Appel and A. Bairoch, in The Proteomics Protocols Handbook, ed. J. M. Walker, Humana Press, United States, 2005, pp. 571–607 Search PubMed.
- E. Kardash, J. Bandemer and E. Raz, Nat. Protoc., 2011, 6, 1835–1846 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00244c |
| ‡ Current address: Department of Biology, Massachusetts Institute of Technology, Cambridge, MA, USA. |
| This journal is © The Royal Society of Chemistry 2025 |