
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-index facet-rich quaternary PtCuFeCo octopods as anti-CO poisoning bifunctional electrocatalysts for direct methanol/ethylene glycol fuel cells†
Kaiyu
Dong
and
Qiang
Yuan
 *
*
State Key Laboratory of Green Pesticide, Center for R&D of Fine Chemicals, College of Chemistry and Chemical Engineering, Guizhou University, Guiyang, Guizhou province 550025, P. R. China. E-mail: qyuan@gzu.edu.cn
First published on 29th April 2025
Abstract
High-index facets and doping strategies can generate unanticipated effects for Pt-based nanomaterials, but there is still a tremendous challenge to integrate the two advantages to construct advanced bifunctional electrocatalysts for direct proton/anion exchange membrane alcohol fuel cells. Herein, we successfully synthesized quaternary Pt41.8Cu51.6Fe5.0Co1.6 octopod nanocrystals (ODNs) with high-index facets through a double active auxiliary doping strategy. Electrochemical activity analysis reveals that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C could serve as an alluring bifunctional electrocatalyst for acidic methanol oxidation reaction (MOR) and alkaline ethylene glycol oxidation reaction (EGOR), displaying mass activities of 2.44 and 23.54 A mgPt−1, respectively, which were 6.4 and 8.2 times higher than those of commercial Pt/C. Notably, Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C demonstrated high power densities superior to those of Pt/C in practical direct proton exchange membrane methanol fuel cell (81.4 mW cm−2versus 41.8 mW cm−2 of commercial Pt/C) and direct anion exchange membrane ethylene glycol fuel cell (217.5 mW cm−2versus 93.6 mW cm−2 of commercial Pt/C) devices. Physical characterization studies indicated that the superior activity originated from the exposed surface of the high-index facets and the optimization of the Pt d-band center by alloying; in addition, the near-surface hydrophilic Fe and Co auxiliaries also facilitated the generation of active hydroxyl species, which further boosted the 6e− MOR and 10e− EGOR processes and anti-CO poisoning ability, as confirmed via in situ Fourier transform infrared spectroscopy. This work provides a feasible example for constructing efficient bifunctional low-Pt electrocatalysts for practical direct proton/anion exchange membrane alcohol fuel cell devices by integrating the merits of doping and high-index facets.
Introduction
As an ideal energy conversion device, direct alcohol fuel cells (DAFCs) offer various advantages, including safer and easier storage and higher fuel energy density (e.g., ethylene glycol (EG): 5.9 kW h L−1; methanol: 4.8 kW h L−1) compared to hydrogen fuel cells.1–5 However, the anodic alcohol oxidation reaction (AOR), which involves the transfer of multiple electrons and various carbonaceous intermediates, has very slow kinetics, leading to problems such as oxidation inefficiencies even with commercial Pt/C electrocatalysts.6–9 Therefore, a considerable amount of research has been performed to enhance the AOR activity of Pt catalysts.10–13 The fabrication of Pt-based nanocrystals surrounded by high-index facets (e.g., (311), (511), etc.) is a promising strategy.14–16 These high-index facets are typically composed of numerous step and kink atoms, which can serve as active sites and interact with reactants more efficiently, thus enhancing the electrocatalytic oxidation performance.17,18 For example, Tian et al. observed that Pt-based nanocatalysts with high-index facets delivered excellent electrocatalytic performance for the oxidation reactions of small molecules, such as formic acid and ethanol.19 In addition, the high-index facets of Pt usually have lower coordination numbers and thus higher surface energies, which allow for high-index facets to easily break chemical bonds.20–22 However, single Pt catalysts with high-index facets are susceptible to poisoning by carbonaceous intermediates such as CO, resulting in catalyst deactivation.Doping and alloying strategies have been evidenced as effective approaches to boost the electrocatalytic performance of Pt-based catalysts.23–27 These strategies modulate the electronic structure of the electrocatalyst surface via stimulation of electronic effects (strain and/or ligand effects) to optimize the adsorption energy of the catalyst for reactants and intermediates, thus enhancing the activity and resistance to toxicity of the catalyst.28,29 For example, Gatalo et al. substantially improved the durability of the oxygen reduction reaction by doping a small amount of Au into transition metal-rich Pt3Cu/C catalysts.30 Fang et al. employed compositional engineering to anchor trace Ir in PtCu aerogels as methanol oxidation electrocatalysts, which not only enhanced the electrocatalytic performance for the methanol oxidation reaction (MOR) but also considerably increased the resistance of the catalyst to CO poisoning.31 Currently, most of the anodic electrocatalysts reported can only be applied to the AOR in a single medium (alkaline or acidic).12,32–34 Hence, the following question arises: “Can we achieve bifunctional properties superior to those of commercial Pt/C by exploiting the design of bimetallic-doped low-Pt alloy nanoelectrocatalysts with high-index facet structures?” Accordingly, it would be extremely fascinating to combine the advantages of both doping and alloying with the superiority of high-index facets to investigate AOR electrocatalysts that can be employed in different media (alkaline and acidic).
Herein, we synthesized Pt41.8Cu51.6Fe5.0Co1.6 octopod nanocrystals (ODNs), doped with Fe and Co double-active auxiliaries, as a bifunctional electrocatalyst through a facile solvothermal method. The surface of Pt41.8Cu51.6Fe5.0Co1.6 ODNs is surrounded by abundant high-index facets, which provide substantial active sites for the catalytic reaction. The mass activity (MA) of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C in acidic MOR and alkaline ethylene glycol oxidation reaction (EGOR) was observed to reach 2.44 and 23.54 A mgPt−1, respectively, which was considerably better than those of commercial Pt/C. Moreover, the introduction of Fe and Co active auxiliaries notably enhanced the CO poisoning resistance of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C. In situ Fourier transform infrared (FTIR) spectroscopy revealed that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C achieved complete oxidation of methanol and EG both in acidic MOR and alkaline EGOR with non-CO pathways. What's more, Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C achieved peak power densities (PPD) of 81.4 and 217.5 mW cm−2 in practical direct proton exchange membrane methanol fuel cell (DPEMMFC) and direct anion exchange membrane ethylene glycol fuel cell (DAEMEGFC) devices, which were 1.9 and 2.3 times higher than those of commercial Pt/C, respectively, demonstrating that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C possesses promising potential for practical application as an anode catalyst.
Results and discussion
Cu(acac)2, Pt(acac)2, Fe(acac)3, and Co(acac)2 were employed as metal precursors, decyltrimethylammonium bromide (DTAB) as a structure-directing agent, trisodium citrate dihydrate (C6H5Na3O7·2H2O) as a reducing agent, and oleylamine (OAm) and N,N-dimethylformamide (DMF) as solvents. PtCuFeCo ODNs with high-index facets were synthesized by a simple solvothermal method (Fig. 1a). Transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images displayed the well-defined edges of the ODNs (size ∼30 nm), including eight symmetrical legs with a length of ∼8 nm (Fig. 1b and c). Aberration-corrected HAADF-STEM (AC-HAADF-STEM) images indicated that the surface of the ODNs featured two different lattice spacings of 0.195 and 0.223 nm, corresponding to the (200) and (111) facets of the face centered cubic (fcc) Pt, respectively, which was confirmed from the corresponding fast Fourier transform (FFT) image (Fig. 1d and S1†). In addition, the lattice spacing of the (200) and (111) facets was significantly smaller than the standard lattice spacing of 0.196 and 0.227 nm for Pt (200) and (111) (PDF #04-0802), respectively, suggesting that the smaller Cu, Fe, and Co atoms entered the Pt lattice and thus led to lattice contraction. More importantly, alternating (511) and (311) high-index facets could be clearly observed in the edge portion of the ODNs (Fig. 1d and S1†), and these results indicated the presence of a large number of exposed high-index facets over the entire surface of the ODNs. Previous studies showed that high-index facets comprising low-coordination unsaturated step atoms can serve as catalytically active sites, facilitating electrocatalytic processes.35,36 Moreover, these step atoms are located in a short-range ordered surface environment and are less susceptible to migration.37 High-resolution elemental mapping images (Fig. 1e) revealed that the elements Pt and Cu were uniformly distributed in the ODN, while a small amount of the elements Fe and Co presented discrete distributions, confirming the successful synthesis of Fe and Co co-doped PtCuFeCo alloy ODNs. Inductively coupled plasma emission spectroscopy (ICP-OES) results demonstrated that the atomic ratios of Pt/Cu/Fe/Co in PtCuFeCo ODNs were 41.8/51.6/5.0/1.6, hence denoting them as Pt41.8Cu51.6Fe5.0Co1.6 ODNs. Furthermore, Pt39.4Cu53.5Fe6.1Co1.0 ODNs, Pt40.9Cu52.9Fe3.4Co2.8 ODNs, and PtCu ODNs were prepared by varying the amounts of Co and Fe (Fig. S2 and Table S1†). | ||
| Fig. 1 (a) Schematic diagram of the synthesis of PtCuFeCo ODNs. (b) TEM image of Pt41.8Cu51.6Fe5.0Co1.6 ODNs. (c) HAADF-STEM image of Pt41.8Cu51.6Fe5.0Co1.6 ODNs. (d) AC-HAADF-STEM image of Pt41.8Cu51.6Fe5.0Co1.6 ODNs, and the inset shows the corresponding FFT image. (e) High-resolution elemental mapping images of a single Pt41.8Cu51.6Fe5.0Co1.6 ODN. (f) PXRD pattern of Pt41.8Cu51.6Fe5.0Co1.6 and PtCu ODNs. | ||
The powder X-ray diffraction (PXRD) pattern of Pt41.8Cu51.6Fe5.0Co1.6 ODNs (Fig. 1f) showed the typical fcc PtCuFeCo alloy structure, with diffraction peaks located at 42.3°, 49.2°, 72.0°, and 87.3° corresponding to the (111), (200), (220), and (311) facets, respectively. Compared with standard peaks of Pt (PDF #04-0802) and the diffraction peaks of PtCu ODNs, the diffraction peaks of Pt41.8Cu51.6Fe5.0Co1.6 ODNs were observed to be clearly shifted to smaller angles, further confirming that Fe and Co atoms were successfully doped into the Pt lattice,38,39 which coincided with the lattice compression results derived from the abovementioned AC-HAADF-STEM images.
Prior to electrocatalytic testing, the catalysts synthesized were loaded onto XC-72 via ultrasonication, following which they were denoted as Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C, Pt39.4Cu53.5Fe6.1Co1.0 ODNs/C, Pt40.9Cu52.9Fe3.4Co2.8 ODNs/C and PtCu ODNs/C (Fig. S3†). First, we investigated the alcohol electrooxidation performance of the following catalysts, each with a different Fe and Co co-doping level: Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C, Pt39.4Cu53.5Fe6.1Co1.0 ODNs/C, and Pt40.9Cu52.9Fe3.4Co2.8 ODNs/C (Fig. S4†). Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C exhibited superior alcohol oxidation properties and stability in comparison with Pt39.4Cu53.5Fe6.1Co1.0 ODNs/C and Pt40.9Cu52.9Fe3.4Co2.8 ODNs/C. Therefore, the subsequent discussion considers Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C as a model to study the electrooxidation properties of alcohols in comparison with Pt/C and PtCu ODNs/C. Cyclic voltammetry (CV) curves of Pt/C, Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C, and PtCu ODNs/C were recorded in N2-saturated 0.1 M HClO4 solution (Fig. S5a†), and their electrochemically active surface areas (ECSAs) were calculated to be 57.5, 77.2, and 65.0 m2 gPt−1, respectively (Fig. S5b†). The ECSA of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C was slightly larger than that of Pt/C, implying higher utilization of the exposed Pt atoms on the surface of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C surrounded by high-index facets. MOR CV curves for the three catalysts are shown in Fig. 2a. The Tafel slope of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C (128.1 mV dec−1) was lower than those of Pt/C (218.6 mV dec−1) and PtCu ODNs/C (130.2 mV dec−1), signifying that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C had faster reaction kinetics in the MOR process (Fig. S6†). Fig. 2b and S7† display the specific activity (SA) of the three catalysts, with the peak SA of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C being 3.16 mA cm−2, which was 4.8 and 1.5 times higher than that of Pt/C (0.66 mA cm−2) and PtCu ODNs/C (2.05 mA cm−2), respectively. Among the three catalysts, Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C reached the highest MA value, 2.44 A mgPt−1, which was 6.4 and 1.8 times higher than that of Pt/C (0.38 A mgPt−1) and PtCu ODNs/C (1.33 A mgPt−1), respectively. The MA of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C exceeded most of the Pt-based MOR electrocatalysts reported in the literature (Fig. 2c and Table S2†). These results illustrated the exceptional effect of complex structural (nanooctopods surrounded by abundant high-index facets) and compositional (fcc PtCuFeCo quaternary alloys) engineering in the enhancement of the MOR.
 | ||
| Fig. 2 MOR performance of commercial Pt/C, Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C, and PtCu ODNs/C. (a) CV curves of different catalysts. (b) Histograms of MA and SA. (c) Comparison of MOR MA between Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C and other advanced electrocatalysts in acidic media. (d) i–t tests. (e) Long-time i–t test of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C. (f) Performance comparison of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C before and after the long-term i–t test. (g) In situ FTIR spectra of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C during the MOR. (h) CO stripping tests in 0.1 M HClO4. | ||
Chronoamperometry (i–t) tests were performed for 3600 s at 0.75 V vs. RHE potential to assess the MOR stability of the three catalysts (Fig. 2d). The current density of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C was always higher than those of Pt/C and PtCu ODNs/C during the 3600 s i–t test, suggesting that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C had the best MOR stability. After 3600 s, the MA of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C decayed by only 1.5%, which was significantly lower than those of Pt/C (36.9%) and PtCu ODNs/C (21.2%) (Fig. S8†). The current density of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C was consistently higher than that of Pt/C and PtCu ODNs/C even during the i–t test at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 s (Fig. S9†). Additionally, Fig. 2e reveals that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C did not significantly change after five consecutive i–t tests. The MA of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C attenuated only by 8.2% after a long i–t test (Fig. 2f), indicating the outstanding stability of this particular catalyst. The MOR processes on Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C, PtCu ODNs/C, and Pt/C in acidic media were further explored using in situ FTIR spectroscopy (Fig. 2g, S10 and S11†). Characteristic bands of the intermediate species CHO and HCOOH at ∼1450 and ∼1235 cm−1 were observed on both the catalysts.40,41 Both these catalysts seemed to have asymmetric telescopic vibrations of CO2 at ∼2343 cm−1, demonstrating that CO2 was the major product of the MOR.42 Compared with Pt/C and PtCu ODNs/C, Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C exhibited CO2 production at a lower potential of 0.4 V vs. RHE (Fig. S12†), which implied that the doping with Fe and Co atoms facilitated the MOR process on Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C. No CO adsorption was observed on Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C, whereas bridge adsorption (COB) of CO occurred on Pt/C and was also difficult to eliminate at high potentials (Fig. S11b†).43,44 These results illustrated that the non-CO pathway 6e− methanol oxidation process was more accessible on Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C (CH3OH − 6e− + H2O → 6H+ + CO2). Moreover, the resistance of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C to CO poisoning was further verified via CO stripping experiments in 0.1 M HClO4 (Fig. 2h). The CO oxidation onset potential (0.503 V vs. RHE) and CO oxidation potential (0.751 V vs. RHE) of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C were observed to be negatively shifted relative to Pt/C and PtCu ODNs/C, implying that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C possessed more superior resistance to CO poisoning. This may be because the introduction of Fe and Co double active auxiliaries expedites the generation of hydroxyl species at low potentials and facilitates the oxidation of CO intermediates at the neighboring Pt sites.12,45–48
800 s (Fig. S9†). Additionally, Fig. 2e reveals that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C did not significantly change after five consecutive i–t tests. The MA of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C attenuated only by 8.2% after a long i–t test (Fig. 2f), indicating the outstanding stability of this particular catalyst. The MOR processes on Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C, PtCu ODNs/C, and Pt/C in acidic media were further explored using in situ FTIR spectroscopy (Fig. 2g, S10 and S11†). Characteristic bands of the intermediate species CHO and HCOOH at ∼1450 and ∼1235 cm−1 were observed on both the catalysts.40,41 Both these catalysts seemed to have asymmetric telescopic vibrations of CO2 at ∼2343 cm−1, demonstrating that CO2 was the major product of the MOR.42 Compared with Pt/C and PtCu ODNs/C, Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C exhibited CO2 production at a lower potential of 0.4 V vs. RHE (Fig. S12†), which implied that the doping with Fe and Co atoms facilitated the MOR process on Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C. No CO adsorption was observed on Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C, whereas bridge adsorption (COB) of CO occurred on Pt/C and was also difficult to eliminate at high potentials (Fig. S11b†).43,44 These results illustrated that the non-CO pathway 6e− methanol oxidation process was more accessible on Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C (CH3OH − 6e− + H2O → 6H+ + CO2). Moreover, the resistance of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C to CO poisoning was further verified via CO stripping experiments in 0.1 M HClO4 (Fig. 2h). The CO oxidation onset potential (0.503 V vs. RHE) and CO oxidation potential (0.751 V vs. RHE) of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C were observed to be negatively shifted relative to Pt/C and PtCu ODNs/C, implying that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C possessed more superior resistance to CO poisoning. This may be because the introduction of Fe and Co double active auxiliaries expedites the generation of hydroxyl species at low potentials and facilitates the oxidation of CO intermediates at the neighboring Pt sites.12,45–48
Notably, Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C delivered a similar excellent EGOR performance in alkaline media. Fig. 3a shows the EGOR CV curves of the three catalysts in alkaline EG solution. Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C (115.9 mV dec−1) displayed the lowest Tafel slope among the three catalysts, indicating its faster reaction kinetics in the EGOR (Fig. S13†). Fig. 3b and S14† present the MA and SA values of the three catalysts for the EGOR, which were as high as 23.54 A mgPt−1 and 30.49 mA cm−2 for Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C. These values were correspondingly 8.2 and 6 times higher than those of Pt/C (2.87 A mgPt−1/4.99 mA cm−2) and 1.7 and 1.4 times higher than those of PtCu ODNs/C (13.88 A mgPt−1/21.35 mA cm−2). The EGOR performance of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C surpassed that of most of the excellent Pt-based EGOR catalysts previously reported in the literature (Fig. 3c and Table S3†). The current density of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C was consistently higher than those of Pt/C and PtCu ODNs/C during the 3600 s i–t test (Fig. 3d). After the i–t test, the MA of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C still remained to be 97.7% of the initial value, which was superior to those of Pt/C (86.0%) and PtCu ODNs/C (87.2%), implying that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C possessed a remarkable EGOR stability (Fig. S15†). Even during the i–t test at 10800 s, the current density of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C remained consistently higher than that of Pt/C and PtCu ODNs/C (Fig. S16†). In addition, Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C was still able to reach 90.5% of the initial MA after five i–t tests (Fig. 3e and S17†), which further demonstrated that Fe and Co co-doped Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C not only exhibited improved EGOR activity but also had a greatly enhanced EGOR stability. Fig. 3f illustrates that the CO oxidation onset potential (0.361 V vs. RHE) and the CO oxidation peak potential (0.698 V vs. RHE) of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C in 1.0 M KOH solution remarkably negatively shifted relative to those of Pt/C (0.445 V vs. RHE/0.791 V vs. RHE) and PtCu ODNs/C (0.371 V vs. RHE/0.722 V vs. RHE), which confirmed that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C possessed an equally excellent anti-CO poisoning ability in alkaline medium. The similarities and differences between Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C, PtCu ODNs/C and Pt/C in the EGOR were investigated by in situ FTIR (Fig. 3g, h, and S18†). Stretching vibrations of the aldehyde group in glyoxal and glycolic acid were observed at ∼1074 cm−1, and the band at ∼1310 cm−1 indicated the generation of oxalate.49,50 The appearance of characteristic bands at ∼1074, ∼1235, ∼1327, ∼1410, and ∼1575 cm−1 implied glycolate production.51,52 Characteristic bands at ∼1360, ∼1412, and 2342 cm−1 were attributed to the C1 species HCO3−, CO32−, and CO2, respectively.53–55 Furthermore, CO formation was not observed from in situ FTIR spectra of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C, revealing that this catalyst not only achieved C–C bond breaking of EG in alkaline medium but also accomplished the EG oxidation process via a 10e− non-CO pathway (CH2OH–CH2OH + 14OH− → 2CO32− + 10H2O + 10e−). Compared with Pt/C and PtCu ODNs/C, the water consumption bands (∼1875 and ∼2660 cm−1) were strengthened during the EGOR process on Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C. Moreover, glycolate formation was observed at a low potential of 0.3 V vs. RHE, and CO2 generation was recorded at 0.6 V vs. RHE (Fig. 3g and S19†), suggesting that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C had a lower EGOR initiation potential and more rapid reaction kinetics. This was facilitated not only by the abundant active sites exposed on the octopod structure surrounded by the high-index facets but also because of the doping with Fe and Co double-active auxiliaries, which substantially enhanced the ability of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C to activate H2O, thus expediting the oxidation process of carbonaceous intermediates adsorbed on the Pt sites.14,45
 | ||
| Fig. 3 EGOR performance of commercial Pt/C, Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C, and PtCu ODNs/C. (a) CV curves of different catalysts. (b) Histograms of MA and SA. (c) Comparison of EGOR MA between Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C and other advanced electrocatalysts. (d) i–t tests at 0.72 V vs. RHE. (e) Long-time i–t test of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C. (f) CO stripping tests in 1.0 M KOH. In situ FTIR spectra of (g) Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C and (h) Pt/C during the EGOR. | ||
In view of the remarkable properties exhibited by Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C in both the MOR and EGOR, we employed it as an anode catalyst and assembled it with Pt/C as the membrane electrode assembly (MEA) to obtain polarization curves in practical DPEMMFC and DAEMEGFC devices (Fig. 4a). The PPD of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C in the DPEMMFC in an O2 atmosphere reached 81.4 mW cm−2, which was 1.9 times higher than that of commercial Pt/C (41.8 mW cm−2) (Fig. 4b). Upon replacing O2 with inexpensive air, Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C still achieved the PPD of 60.6 mW cm−2 in the DPEMMFC, while Pt/C reached the PPD of only 21.7 mW cm−2 (Fig. 4c). Similarly, the PPD of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C in the DAEMEGFC in an O2 atmosphere reached an astonishing 217.5 mW cm−2 at a high current density of 1 A cm−2, which was 2.3 times higher than that of commercial Pt/C (93.6 mW cm−2 at 316.3 mA cm−2) (Fig. 4d). Meanwhile, in the DAEMEGFC in an air atmosphere, the PPD of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C could still be maintained up to 130.2 mW cm−2, which was considerably better than that of Pt/C (64.5 mW cm−2) (Fig. 4e). Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C performed better than Pt/C both in DPEMMFC and DAEMEGFC devices (Fig. 4f), which indicated that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C possessed enormous application potential as an anodic electrocatalyst in practical DPEMMFC and DAEMEGFC devices and could be an effective alternative to Pt/C.
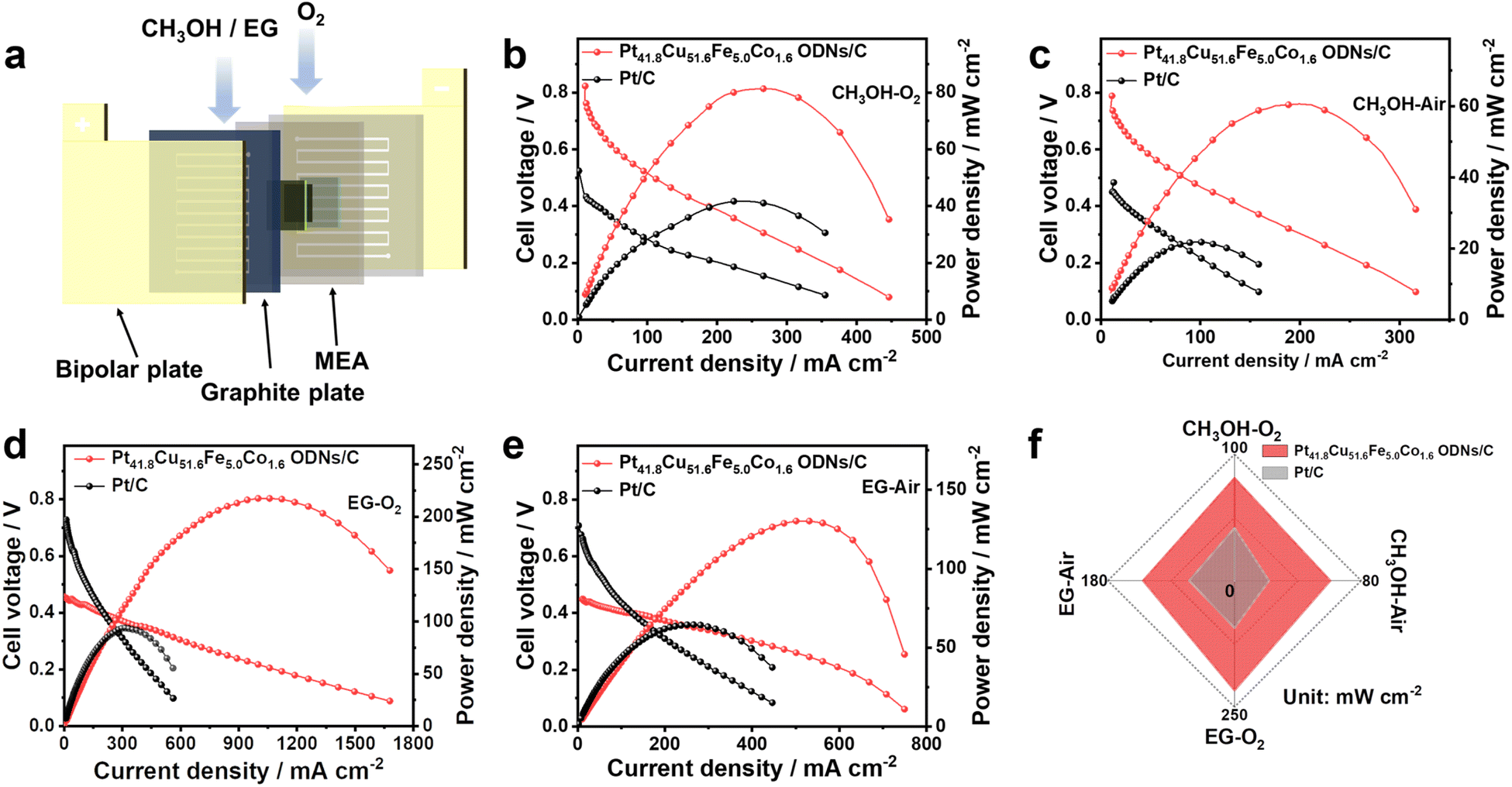 | ||
| Fig. 4 (a) Structure diagram of the DAFC device. Polarization curves of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C and Pt/C in (b) CH3OH–O2 and (c) CH3OH–air. Polarization curves of Pt41.8Cu51.6Fe5.0Co1.6 and Pt/C in (d) EG–O2 and (e) EG–air. (f) Performance comparison diagram. | ||
To further understand the reason behind the enhanced electrocatalytic performance of Pt41.8Cu51.6Fe5.0Co1.6 ODNs, we analyzed their electronic states by X-ray photoelectron spectroscopy (XPS). High-resolution Pt 4f XPS spectra (Fig. 5a) showed that Pt mainly existed in the metallic state, alongside a small amount in the oxidized state on the surface of Pt41.8Cu51.6Fe5.0Co1.6 ODNs. The Pt0 4f7/2 peak of Pt41.8Cu51.6Fe5.0Co1.6 ODNs was observed at 70.7 eV, and the Pt0 4f5/2 peak appeared at 74.1 eV, which was observed to be significantly negatively shifted relative to those of PtCu ODNs and Pt/C. Cu 2p XPS spectra of Pt41.8Cu51.6Fe5.0Co1.6 ODNs (Fig. 5b) indicated the coexistence of the metallic and oxidized states of Cu on the near surface of Pt41.8Cu51.6Fe5.0Co1.6 ODNs. The Cu0 2p3/2 of Pt41.8Cu51.6Fe5.0Co1.6 ODNs underwent a slight positive shift compared to PtCu ODNs. Fe and Co were primarily present in their oxidized states on the near surface of Pt41.8Cu51.6Fe5.0Co1.6 ODNs (Fig. S20†). Additionally, the binding energies of Fe2+ 2p3/2 and Co2+ 2p3/2 of Pt41.8Cu51.6Fe5.0Co1.6 ODNs were observed to be positively shifted by 0.8 and 1.6 eV compared to the standard Fe2+ 2p3/2 (709.6 eV) and Co2+ 2p3/2 (780.4 eV). The electronegativity of Pt (2.28) is slightly higher than those of Cu (1.90), Fe (1.83) and Co (1.88), which facilitates the transfer of electrons from Cu, Fe and Co to Pt. This was corroborated by the XPS results, which demonstrated that Pt became the electron-rich center on the surface of Pt41.8Cu51.6Fe5.0Co1.6 ODNs, thereby moderating the d-band center and accelerating the electrocatalytic reaction process. Moreover, Fe, Cu and Co in the form of oxides were able to activate H2O and generate more OH species, thus expediting the removal of oxygenated intermediates from nearby Pt sites, as evidenced by the above-mentioned in situ FTIR results.56 Valence band spectra (Fig. 5c) revealed that the d-band center of Pt41.8Cu51.6Fe5.0Co1.6 ODNs (−4.26 eV) was significantly shifted downward relative to those of Pt/C (−3.91 eV) and PtCu ODNs (−4.02 eV), which was capable of weakening the bonding strength between Pt and the intermediate, accelerating the reaction kinetics and enhancing the electrocatalytic oxidation performance.57,58 The oxidation states of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C during the MOR and EGOR were further investigated by in situ Raman spectroscopy (Fig. 5d and e). The Raman signals located at 1351 cm−1 and 1590 cm−1 originated from the D and G bands of the carbon carriers. COOH signals (1170 cm−1 and 1475 cm−1) of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C were observed at low potentials regardless of the MOR and EGOR, suggesting that Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C could initiate the MOR and EGOR at low potentials. The Raman signals at 471 cm−1 and 585 cm−1 in Fig. 5d and at 434 cm−1 and 510 cm−1 in Fig. 5e were derived from the vibrations of Cu/Fe/Co–O at hydroxyl species due to the enhanced adsorption affinity of transition metals for hydroxyl species,59–62 which also implied that the doping of Fe and Co facilitated the generation of reactive hydroxyl species, thus accelerating the reaction kinetics of the MOR and EGOR. Combined XPS analysis and in situ spectroscopy revealed that the introduction of Fe and Co atoms optimized the electronic structure of Pt and lowered the d-band center, thus expediting the reaction. The doping with the hydrophilic Fe and Co double active auxiliaries activated more hydroxyl species and facilitated the oxidation of reactants and intermediates,11,33 achieving the complete oxidation of methanol and EG. This resulted in Pt41.8Cu51.6Fe5.0Co1.6 ODNs exhibiting high activity, stability, and resistance to CO poisoning in both the MOR and EGOR and excellent power densities even in practical DAFCs (Fig. 5f).
 | ||
| Fig. 5 (a) Pt 4f XPS spectrum. (b) Cu 2p XPS spectrum. (c) Valence band spectra of Pt/C, PtCu ODNs, and Pt41.8Cu51.6Fe5.0Co1.6 ODNs. In situ Raman spectroscopy during the (d) MOR and (e) EGOR. (f) Mechanism of the reaction. | ||
Conclusion
In summary, we successfully prepared optimal Pt41.8Cu51.6Fe5.0Co1.6 ODNs as an efficient bifunctional electrocatalyst via doping with Fe and Co double active auxiliaries. Benefiting from the octopod structure surrounded by high-index facets and the co-doping of Fe and Co active auxiliaries, Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C exhibited the highest MA/SA for both acidic MOR and alkaline EGOR, which were 2.44 A mgPt−1/3.16 mA cm−2 and 23.54 A mgPt−1/30.49 mA cm−2, respectively. The introduction of Fe and Co double active auxiliaries activated more active OH species and expedited the oxidation of CO intermediate species, thus improving the anti-CO poisoning ability of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C. In situ FTIR spectra revealed the non-CO reaction pathways of Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C for acidic MOR and alkaline EGOR, showing that this catalyst achieved efficient complete oxidation of methanol and EG. In practical DPEMMFC (81.4 mW cm−2/60.6 mW cm−2) and DAEMEGFC (217.5 mW cm−2/130.2 mW cm−2) devices, Pt41.8Cu51.6Fe5.0Co1.6 ODNs/C exhibited PPD that considerably exceeded those of Pt/C in both O2 and air atmospheres. This study offered a new strategy for the fabrication of universal catalysts for use in both acidic and alkaline media and introduced a new perspective for designing Pt-based catalysts for use in practical DAFCs.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22465009), Education Department of Guizhou Province (2021312), Foundation of Guizhou Province (2019-5666), and Science Foundation for Postgraduate Students of Guizhou Province (2024YJSKYJJ025).Notes and references
- M. Li, K. Duanmu, C. Wan, T. Cheng, L. Zhang, S. Dai, W. Chen, Z. Zhao, P. Li, H. Fei, Y. Zhu, R. Yu, J. Luo, K. Zang, Z. Lin, M. Ding, J. Huang, H. Sun, J. Guo, X. Pan, W. A. Goddard, P. Sautet, Y. Huang and X. Duan, Nat. Catal., 2019, 2, 495–503 CrossRef CAS.
- X. Fu, C. Wan, Y. Huang and X. Duan, Adv. Funct. Mater., 2022, 32, 2106401 CrossRef CAS.
- H. Lv, L. Sun, Y. Wang, S. Liu and B. Liu, Adv. Mater., 2022, 34, 2203612 CrossRef CAS.
- A. Kowal, M. Li, M. Shao, K. Sasaki, M. B. Vukmirovic, J. Zhang, N. S. Marinkovic, P. Liu, A. I. Frenkel and R. R. Adzic, Nat. Mater., 2009, 8, 325–330 CrossRef CAS.
- Z. Wang, J. S. Kang, D. Göhl, P. Paciok, D. S. Gonçalves, H.-K. Lim, D. Zanchet, M. Heggen, Y. Shao-Horn, M. Ledendecker and Y. Román-Leshkov, Adv. Energy Mater., 2024, 14, 2304092 CrossRef CAS.
- M. Li, Z. Zhao, W. Zhang, M. Luo, L. Tao, Y. Sun, Z. Xia, Y. Chao, K. Yin, Q. Zhang, L. Gu, W. Yang, Y. Yu, G. Lu and S. Guo, Adv. Mater., 2021, 33, 2103762 CrossRef CAS.
- M. Tang, M. Sun, W. Chen, Y. Ding, X. Fan, X. Wu, X. Z. Fu, B. Huang, S. Luo and J. L. Luo, Adv. Mater., 2024, 36, 2311731 CrossRef CAS PubMed.
- N. Kakati, J. Maiti, S. H. Lee, S. H. Jee, B. Viswanathan and Y. S. Yoon, Chem. Rev., 2014, 114, 12397–12429 CrossRef CAS.
- Z. Zhang, J. Liu, J. Wang, Q. Wang, Y. Wang, K. Wang, Z. Wang, M. Gu, Z. Tang, J. Lim, T. Zhao and F. Ciucci, Nat. Commun., 2021, 12, 5235 CrossRef CAS PubMed.
- Y. Lv, L. Lin, R. Xue, P. Zhang, F. Ma, T. Gan, J. Zhang, D. Gao, X. Zheng, L. Wang, Y. Qin, H. Zhao, Y. Dong, Y. Wang and Y. Zhu, Adv. Energy Mater., 2024, 14, 2304515 CrossRef CAS.
- X. Zhang, X. Lin, X. Hu, S. Geng, F. Xiao, K. Jiang, S. Li, Z. Zheng and L. Bu, ACS Catal., 2024, 14, 84–93 CrossRef CAS.
- Q. Yang, S. Zhang, F. Wu, L. Zhu, G. Li, M. Chen, A. Pei and Y. Feng, J. Energy Chem., 2024, 90, 327–336 CrossRef CAS.
- Y. Nie, Z. Li, Y. Wang, X. Zheng, L. Luo, X. Xia, S. Yang, C. Du, Y. Huang and Y. Wang, Appl. Catal., B, 2024, 343, 123494 CrossRef CAS.
- X. Zhang, X. Liu, D. Wu, L. Hu, H. Zhang, Z. Sun, S. Qian, Z. Xia, Q. Luo, L. Cao, J. Yang and T. Yao, Nano Lett., 2024, 24, 3213–3220 CrossRef CAS.
- Q. Wang, S. Wang, X. Han, X. Guo, H. Huang, K. Kang, P. Zhao and S. Xie, Inorg. Chem., 2024, 63, 11424–11430 CrossRef CAS PubMed.
- L. Ji, M. Lin, W. Yan, Y. Cai, G. Li, J. Zheng, S. Zhou, Z. Huang, H. Liao, H. Lin, C. Xiao, B. Zhang, Y. Jiang and S. Sun, ACS Catal., 2023, 13, 13846–13855 CrossRef CAS.
- C. Xiao, B. A. Lu, P. Xue, N. Tian, Z. Y. Zhou, X. Lin, W. F. Lin and S. G. Sun, Joule, 2020, 4, 2562–2598 CrossRef CAS.
- M. Luo, Y. Sun, X. Zhang, Y. Qin, M. Li, Y. Li, C. Li, Y. Yang, L. Wang, P. Gao, G. Lu and S. Guo, Adv. Mater., 2018, 30, 1705515 CrossRef.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- J. X. Tang, N. Tian, L. P. Xiao, Q. S. Chen, Q. Wang, Z. Y. Zhou and S. G. Sun, J. Mater. Chem. A, 2022, 10, 10902–10908 RSC.
- Z. Y. Zhou, Z. Z. Huang, D. J. Chen, Q. Wang, N. Tian and S. G. Sun, Angew. Chem., Int. Ed., 2010, 49, 411–414 CrossRef CAS.
- S. Liu, N. Tian, A. Y. Xie, J. H. Du, J. Xiao, L. Liu, H. Y. Sun, Z. Y. Cheng, Z. Y. Zhou and S. G. Sun, J. Am. Chem. Soc., 2016, 138, 5753–5756 CrossRef CAS PubMed.
- X. Tian, X. Zhao, Y. Q. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. J. M. Hensen, X. W. Lou and B. Y. Xia, Science, 2019, 366, 850–856 CrossRef CAS PubMed.
- F. Xiao, Q. Wang, G. L. Xu, X. Qin, I. Hwang, C. J. Sun, M. Liu, W. Hua, H. W. Wu, S. Zhu, J. C. Li, J. G. Wang, Y. Zhu, D. Wu, Z. Wei, M. Gu, K. Amine and M. Shao, Nat. Catal., 2022, 5, 503–512 CrossRef CAS.
- H. Huang, K. Li, Z. Chen, L. Luo, Y. Gu, D. Zhang, C. Ma, R. Si, J. Yang, Z. Peng and J. Zeng, J. Am. Chem. Soc., 2017, 139, 8152–8159 CrossRef CAS.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef CAS.
- Y. F. Wang, S. L. Zhang, Y. X. Deng, S. H. Luan, C. K. Wang, L. F. Ding, X. Jiang, D. M. Sun and Y. W. Tang, Rare Met., 2025, 44, 300–310 CrossRef CAS.
- Z. P. Wu, D. T. Caracciolo, Y. Maswadeh, J. Wen, Z. Kong, S. Shan, J. A. Vargas, S. Yan, E. Hopkins, K. Park, A. Sharma, Y. Ren, V. Petkov, L. Wang and C. J. Zhong, Nat. Commun., 2021, 12, 859 CrossRef CAS PubMed.
- T. He, W. Wang, F. Shi, X. Yang, X. Li, J. Wu, Y. Yin and M. Jin, Nature, 2021, 598, 76–81 CrossRef CAS PubMed.
- M. Gatalo, P. Jovanovič, G. Polymeros, J. P. Grote, A. Pavlišič, F. Ruiz-Zepeda, V. S. Šelih, M. Šala, S. Hočevar, M. Bele, K. J. J. Mayrhofer, N. Hodnik and M. Gaberšček, ACS Catal., 2016, 6, 1630–1634 CrossRef CAS.
- Q. Fang, H. Wang, X. Lv, X. Wei, X. Luo, J. Huang, L. Jiao, W. Gu, W. Song and C. Zhu, ACS Sustainable Chem. Eng., 2021, 9, 13039–13046 CrossRef CAS.
- S. Li, G. Wang, H. Lv, Z. Lin, J. Liang, X. Liu, Y. G. Wang, Y. Huang, G. Wang and Q. Li, J. Am. Chem. Soc., 2024, 146, 17659–17668 CrossRef CAS PubMed.
- H. Lei, N. Ma, K. Li, Y. Wang, Q. Yuan, J. Fan, J. Shui and Y. Huang, Energy Environ. Sci., 2024, 17, 7792–7802 RSC.
- K. Xu, L. Liang, T. Li, M. Bao, Z. Yu, J. Wang, S. M. Thalluri, F. Lin, Q. Liu, Z. Cui, S. Song and L. Liu, Adv. Mater., 2024, 36, 2403792 CrossRef CAS PubMed.
- J. Wu and H. Yang, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef CAS.
- C. Zhang, Z. Chen, H. Yang, Y. Luo, Z. Qun Tian and P. Kang Shen, J. Colloid Interface Sci., 2023, 652, 1597–1608 CrossRef CAS PubMed.
- Z. Y. Zhou, N. Tian, J. T. Li, I. Broadwell and S. G. Sun, Chem. Soc. Rev., 2011, 40, 4167–4185 RSC.
- L. Huang, Y. Q. Su, R. Qi, D. Dang, Y. Qin, S. Xi, S. Zaman, B. You, S. Ding and B. Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 25530–25537 CrossRef CAS.
- L. Bu, J. Liang, F. Ning, J. Huang, B. Huang, M. Sun, C. Zhan, Y. Ma, X. Zhou, Q. Li and X. Huang, Adv. Mater., 2023, 35, 2208672 CrossRef CAS.
- X. M. Qu, L. X. You, X. C. Tian, B. W. Zhang, G. D. Mahadevan, Y. X. Jiang and S. G. Sun, Electrochim. Acta, 2015, 182, 1078–1084 CrossRef CAS.
- A. Fan, C. Qin, R. Zhao, H. Sun, H. Sun, X. Dai, J. Y. Ye, S. G. Sun, Y. Lu and X. Zhang, Nano Res., 2022, 15, 6961–6968 CrossRef CAS.
- K. Liu, W. Wang, P. Guo, J. Ye, Y. Wang, P. Li, Z. Lyu, Y. Geng, M. Liu and S. Xie, Adv. Funct. Mater., 2019, 29, 1806300 CrossRef.
- Y. W. Zhou, Y. F. Chen, K. Jiang, Z. Liu, Z. J. Mao, W. Y. Zhang, W. F. Lin and W. B. Cai, Appl. Catal., B, 2021, 280, 119393 CrossRef CAS.
- T. Xia, K. Zhao, Y. Zhu, X. Bai, H. Gao, Z. Wang, Y. Gong, M. Feng, S. Li, Q. Zheng, S. Wang, R. Wang and H. Guo, Adv. Mater., 2023, 35, 2206508 CrossRef CAS.
- H. Liu, R. Jia, C. Qin, Q. Yang, Z. Tang, M. Li and Z. Ma, Adv. Funct. Mater., 2023, 33, 2210626 CrossRef CAS.
- K. Zhan, C. Feng, X. Feng, D. Zhao, S. Yue, Y. Li, Q. Jiao, H. Li and Y. Zhao, ACS Sustainable Chem. Eng., 2020, 8, 6273–6281 CrossRef CAS.
- C. Lee, C. K. Hwang, J. W. An, J. S. Jang, B. Koo, J. M. Kim, K. Shin, C. S. Lee and K. R. Yoon, Appl. Catal., B, 2024, 358, 124371 CrossRef CAS.
- H. Li, Y. Han, H. Zhao, W. Qi, D. Zhang, Y. Yu, W. Cai, S. Li, J. Lai, B. Huang and L. Wang, Nat. Commun., 2020, 11, 5437 CrossRef CAS.
- X. Yang, Q. Yuan, T. Sheng and X. Wang, Chem. Sci., 2024, 15, 4349–4357 RSC.
- J. X. Tang, Q. S. Chen, L. X. You, H. G. Liao, S. G. Sun, S. G. Zhou, Z. N. Xu, Y. M. Chen and G. C. Guo, J. Mater. Chem. A, 2018, 6, 2327–2336 RSC.
- J. Li, L. Li, X. Ma, X. Han, C. Xing, X. Qi, R. He, J. Arbiol, H. Pan, J. Zhao, J. Deng, Y. Zhang, Y. Yang and A. Cabot, Adv. Sci., 2023, 10, 2300841 CrossRef CAS PubMed.
- B. W. Zhang, W. H. Lai, T. Sheng, X. M. Qu, Y. X. Wang, L. Ren, L. Zhang, Y. Du, Y. X. Jiang, S. G. Sun and S. X. Dou, J. Mater. Chem. A, 2019, 7, 5214–5220 RSC.
- X. Yang, Q. Yuan, J. Li, T. Sheng, K. X. Yao and X. Wang, Nano Lett., 2023, 23, 3467–3475 CrossRef CAS.
- Y. Qin, W. Zhang, F. Wang, J. Li, J. Ye, X. Sheng, C. Li, X. Liang, P. Liu, X. Wang, X. Zheng, Y. Ren, C. Xu and Z. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202200899 CrossRef CAS.
- L. Xin, Z. Zhang, J. Qi, D. Chadderdon and W. Li, Appl. Catal., B, 2012, 125, 85–94 CrossRef CAS.
- J. Huang, Y. Liu, M. Xu, C. Wan, H. Liu, M. Li, Z. Huang, X. Duan, X. Pan and Y. Huang, Nano Lett., 2019, 19, 5431–5436 CrossRef CAS PubMed.
- X. Li, Y. Huang, Z. Chen, S. Hu, J. Zhu, P. Tsiakaras and P. Kang Shen, Chem. Eng. J., 2023, 454, 140131 CrossRef CAS.
- Z. Ma, Z. P. Cano, A. Yu, Z. Chen, G. Jiang, X. Fu, L. Yang, T. Wu, Z. Bai and J. Lu, Angew. Chem., Int. Ed., 2020, 59, 18334–18348 CrossRef CAS PubMed.
- Y. Wang, H. Meng, R. Yu, J. Hong, Y. Zhang, Z. Xia and Y. Wang, Angew. Chem., Int. Ed., 2025, 64, e202420752 CrossRef CAS PubMed.
- Y. Ye, J. Xu, X. Li, Y. Jian, F. Xie, J. Chen, Y. Jin, X. Yu, M.-H. Lee, N. Wang, S. Sun and H. Meng, Adv. Mater., 2024, 36, 2312618 CrossRef CAS PubMed.
- H. Wang, A. Guan, J. Zhang, Y. Mi, S. Li, T. Yuan, C. Jing, L. Zhang, L. Zhang and G. Zheng, Chin. J. Catal., 2022, 43, 1478–1484 CrossRef CAS.
- Z. Wang, Y. Li, X. Zhao, S. Chen, Q. Nian, X. Luo, J. Fan, D. Ruan, B.-Q. Xiong and X. Ren, J. Am. Chem. Soc., 2023, 145, 6339–6348 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization studies (TEM, HAADF-STEM, and SEM), CV curves, in situ FTIR spectra, and tables. See DOI: https://doi.org/10.1039/d5sc00525f |
| This journal is © The Royal Society of Chemistry 2025 |