
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron-catalyzed three-component 1,2-azidoalkylation of conjugated dienes via activation of aliphatic C–H bonds†
Zhen-Yao Dai,
Chenxi Lin,
Derek B. Hu and
Jennifer M. Schomaker *
*
Department of Chemistry, University of Wisconsin–Madison, Madison, Wisconsin 53706, USA. E-mail: schomakerj@chem.wisc.edu
First published on 5th March 2025
Abstract
Azidoalkyation is an efficient strategy for the conversion of unsaturated precursors into nitrogen-containing structural motifs. Herein, we describe a convenient and highly regioselective iron-catalyzed 1,2-azidoalkylation of 1,3-dienes that employs TMSN3 as a coupling partner with hydrocarbons that bear diverse C–H bonds. This chemistry is achieved through the direct functionalization of strong C(sp3)–H bonds and is facilitated by a combination of hydrogen atom transfer (HAT) and iron catalysis. Notably, the protocol operates with catalyst loadings as low as 0.2 mol% and furnishes access to versatile β-unsaturated azido products with high levels of site-, regio-, and stereoselectivities. Mechanistic studies suggest that the reaction proceeds via a radical pathway; depending on the electronic properties of the diene, the allylic radical intermediate may engage through either group transfer or a single electron oxidation process.
Introduction
The selective transformation of feedstock chemicals into value-added molecules is a key objective of modern synthetic chemistry, where the development of new methods offers significant potential to simplify synthetic routes and modify complex molecules through later-stage functionalization. Conjugated dienes are a readily available class of synthetic precursors that have emerged as a versatile platform for the efficient preparation of more elaborate building blocks.1 Over the past few decades, extensive research has focused on transition metal-catalyzed difunctionalizations of 1,3-dienes.2–5 This process typically involves the generation of a metal–carbon species, followed by migratory insertion to form an allyl metal intermediate (Scheme 1A).6–18 The use of dienes as substrates introduces additional challenges that include the high reactivity of allylic intermediates generated from azidation of 1,3-dienes, as well as control over the site-, regio-, and Z/E-selectivities. Additionally, side reactions such as polymerization can further reduce the efficiency of the reaction. In prior work, we developed iron-catalyzed site- and regioselective 1,2-azidoamidations of 1,3-dienes, utilizing N-fluorobenzenesulfonimide (NFSI) as a dual oxidant and radical precursor to furnish versatile precursors to 1,2-diamines (Scheme 1B).19 | ||
| Scheme 1 Radical azido-difunctionalization of alkenes and 1,3-dienes. | ||
The majority of the current methodologies for the carboazidation of alkenes require pre-functionalized radical precursors, such as alkyl halides, alkyl sulfonyl chlorides, or Togni-type reagents; this need adds additional steps and reduces the overall efficiency of the process (Scheme 1C).20–27 The C(sp3)–H bonds of hydrocarbons are abundant chemical feedstocks, but display relatively high bond dissociation energies (BDEs) and low bond polarity, challenges that must be overcome to use them as radical precursors.28–31 Therefore, the development of carboazidation approaches that can directly activate a wide variety of simple hydrocarbons is highly desirable. In this context, hydrogen atom transfer (HAT) is an appealing strategy for the homolytic cleavage of diverse C(sp3)–H bonds.32–37
Organic azides are an important class of molecules that are present in many natural products and bioactive molecules; in addition, the azide is a highly versatile handle for subsequent organic transformations.38–41 The Luo42 and Nishihara groups43 reported the azidoalkylation of terminal styrenes using Fe or Cu catalysts, respectively; however, the C–H partners were limited to cycloalkanes, chloroalkanes, or cyclic ethers. More recently, Feng and coworkers described the asymmetric azidoalkylation of activated alkenes, but substrates were restricted to α-aryl-substituted aryl enones.44 Although these reports advanced the state-of-the-art,22–24,42–47 carboazidations of 1,3-dienes via direct C–H activation is still underdeveloped, particularly in applications to more complex molecular scaffolds. There are only a few reported examples where the hydrocarbon coupling partner is limited to dichloromethane,45 aldehydes48,49 and acetonitrile.50
We envisioned that combining azidation with an HAT process that is compatible with diverse hydrocarbon coupling partners could provide efficient carboazidation of 1,3-dienes to furnish allylic azides as useful precursors to amine building blocks for syntheses of bioactive and pharmaceutically relevant compounds (Scheme 1D).23–25,39–41 Herein, we describe the 1,2-azidoalkylation of diverse 1,3-dienes employing a broad array of inexpensive hydrocarbon chemical feedstocks. The chemistry proceeds through the direct and selective activation of the aliphatic C–H bonds and merges HAT with iron-catalyzed azidation. The reaction mechanism depends on the electronic features of the 1,3-diene precursor and proceeds via a single-electron transfer/HAT/radical addition through either group transfer or a single-electron oxidation process to construct the new C–C bond.
Results and discussion
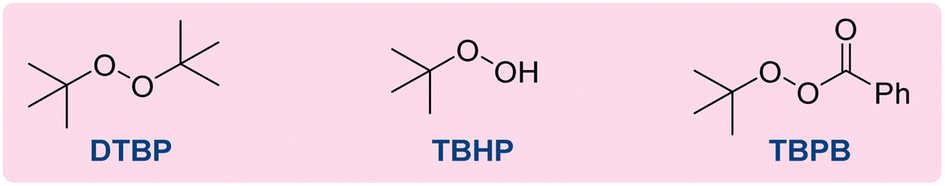
Our study of the three-component azidoalkylation reaction commenced using 1-phenyl-1,3-butadiene 1a as the model substrate, toluene as both the radical precursor and the solvent, di-tert-butyl peroxide (DTBP) as an oxidant and Fe(OTf)2 as the catalyst. We found that the use of only a 0.5 mol% loading of the catalyst, coupled with vigorous stirring of the reaction mixture at 110 °C under nitrogen for 16 h, resulted in the desired adduct 1 in a yield of 70% (Table 1, entry 1). Next, different catalyst loadings were explored; results indicated that increasing the amount of catalyst increased the formation of unwanted diazidation products, while reducing the loading gave diminished reactivity (entries 2–5). A variety of other iron salts were investigated (entries 6–11); however, Fe(OTf)2 proved to be the optimal catalyst. Control experiments underscored the necessity for both the iron salt and heat for the reaction to proceed (entries 12 and 13). Furthermore, substituting DTBP with either tert-butylperoxybenzoate (TBPB) or tert-butylhydroperoxide (TBHP) resulted in no detectable amount of 1,2-azido benzylation product 1 (entries 13, 14).|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldb (1/2)/% |
| a Reaction conditions: 1 (0.10 mmol), TMSN3 (0.25 mmol), DTBP (0.25 mmol), Fe(OTf)2 (0.5 mol%), toluene (2 mL), 20 h, under N2. b Determined by 1H NMR analysis using 1,3,5-triacetylbenzene as an internal standard. | ||
| 1 | None | 70/<5 |
| 2 | 1 mol% of Fe(OTf)2 | 53/10 |
| 3 | 2 mol% of Fe(OTf)2 | 35/30 |
| 4 | 0.2 mol% of Fe(OTf)2 | 62/<5 |
| 5 | 0.1 mol% of Fe(OTf)2 | 24/<5 |
| 6 | FeCl2 instead of Fe(OTf)2 | 21/<5 |
| 7 | FeBr2 instead of Fe(OTf)2 | 41/<5 |
| 8 | FeSO4 instead of Fe(OTf)2 | 20/<5 |
| 9 | FeCl3 instead of Fe(OTf)2 | 44/<5 |
| 10 | Fe(acac)3 instead of Fe(OTf)2 | 45/<5 |
| 11 | Fe(OTf)3 instead of Fe(OTf)2 | 58/12 |
| 12 | w/o Fe(OTf)2 | <5 |
| 13 | Conducted at room temperature | <5 |
| 14 | TBPB instead of DTBP | <5 |
| 15 | TBHP instead of DTBP | <5 |
 |
||
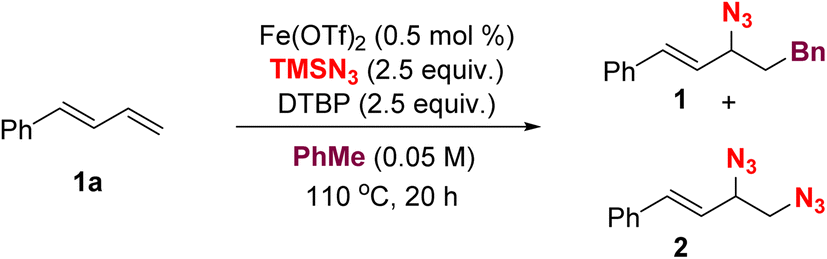
With the optimal reaction conditions in hand, the scope of the radical azidoalkylation reaction was investigated by evaluating an array of 1,3-dienes, as shown in Table 2. First, diverse 1-phenyl-1,3-butadienes, bearing both electron-donating and electron-withdrawing substituents at the para-, meta-, or ortho-positions of the phenyl group, were explored and found to be suitable substrates in this protocol. The corresponding azidoalkylation products 3–19 were obtained in good yields with high levels of regio-, site- and stereocontrol, largely favoring 1,2- over 1,4-addition and selectively installing the azide at the more substituted C3 carbon of the diene. In general, electron-rich and electron-neutral 1-arylsubstituted dienes tended to give higher yields (3–7) as compared to those bearing electron-poor aryl substituents (8–14). Dienes bearing pharmaceutically important heteroaryl groups that include naphthalene, indole, benzofuran and thiophene effectively furnished the target azides 20–23 in moderate yields. Notably, introducing an additional alkyl substituent on the C1, C2 or C3 position of the 1-phenyl-1,3-butadiene gave smooth conversion to the corresponding products 24–26 in moderate-to-good yields and E/Z selectivity. The non-terminal 1,3-diene 27a was also tolerated, delivering the corresponding adduct 27 in 36% yield and in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1 diastereomeric ratio (dr). Additionally, alkyl-substituted 1,3-butadienes were effective in the azidoalkylation, affording the desired products 28 in 63% yield and 29 in 39% yield with 1:1.2 site-selectivity, respectively. Finally, 1,3-dienes tethered to bioactive molecules, including menthol, borneol, ibuprofen, citronellol, gemfibrozil, cholesterol and tocopherol, all reacted smoothly in the three-component coupling to deliver the target products 30–36 in moderate-to-good yield and excellent Z/E selectivities, showcasing the utility of this chemistry in more complex molecule settings.
:1.1 diastereomeric ratio (dr). Additionally, alkyl-substituted 1,3-butadienes were effective in the azidoalkylation, affording the desired products 28 in 63% yield and 29 in 39% yield with 1:1.2 site-selectivity, respectively. Finally, 1,3-dienes tethered to bioactive molecules, including menthol, borneol, ibuprofen, citronellol, gemfibrozil, cholesterol and tocopherol, all reacted smoothly in the three-component coupling to deliver the target products 30–36 in moderate-to-good yield and excellent Z/E selectivities, showcasing the utility of this chemistry in more complex molecule settings.

The scope of aliphatic C–H coupling partners was examined next with a series of substituted 1-methylarenes (Table 3). Electron-donating and electron-withdrawing substituents at the para position of the phenyl group were well-tolerated to deliver the azides 37–46 with moderate-to-good yields. Compared with the C–H bond located α to oxygen, the benzylic position of 4-methylanisole proved more reactive to furnish 39 in 46% yield. In the case of 4-methylcumene, a 31% yield and 1:1.2 rr of 46 was observed. When substituents on the phenyl group were moved from the p- to m- or o-positions, the corresponding products 47–52 were afforded in good yield. A 3-methyl thiophene served as an alkylating agent to afford the heteroarene product 53. Moreover, 2,3-dimethylbutane preferentially reacted at the tertiary C–H bonds to furnish 54 in 54% yield with a 6:1 rr and in 9:1 site-selectivity. Cycloalkanes of varying ring sizes (5-to-8 carbons) proceeded smoothly to provide the corresponding products 55–58 in moderate-to-good yields. Additionally, isopropyl ether, anisole and methyl tert-butyl ether displayed a preference for the α-oxy alkylated products 59–61. N,N-dimethylamide gave a low yield of 62, while a thiol ether 63a and an acetal 64a gave 63–64 in moderate yields. Unfortunately, the use of methanol as the source of the carbon-centered radical gave 65 in low yield. Both acetone and acetonitrile were capable of serving as the radical coupling partner to furnish the corresponding azides 66 and 67. The tertiary C–H bonds of methyl isobutyrate engaged the diene to furnish azide 68 in 66% yield, with a 6:1 rr and in 20:1 site-selectivity. Notably, the C–H bond of an aldehyde group in 69a and 70a could be cleaved and added to the terminal carbon of the 1,3-diene to give the azidocarbonylation products 69 and 70 in moderate yields. However, alkyl aldehydes or formate resulted in decarbonylation and decarboxylation to yield 71 and 72.
| a Unless indicated, reactions were run with 1,3-diene (0.1 mmol), TMSN3 (0.25 mmol), DTBP (0.25 mmol), Fe(OTf)2 (0.0002 mmol), R–H (2 mL), 110 °C, under N2, 16 h. b R–H/PhH = 1/1 (2 mL) as solvent. c R–H/MeCN = 1/1 (2 mL) as solvent. d 10 equiv. of R–H, PhH (2 mL) as solvent. e Ratio of 1,2- and 1,4-addition. |
|---|
 |
The synthetic utility of the azidoalkylation chemistry was demonstrated by carrying out selected post-functionalizations of azide 3 (Scheme 2). The reaction of 3a was conducted on a larger scale employing slightly adjusted conditions to furnish 3 in 76% yield. Treatment of the azide 3 with P(OEt)3 yielded the phosphoramide 73. Compound 3 was readily reduced using PPh3/H2O and subjected to amidation to give 74, followed by subsequent ring-closing metathesis with Grubbs II at low concentrations to generate the target lactam 75. The azide was readily transformed to the primary amine 76via reduction of 3 with Pd(OH)2/C and H2, while a copper-catalyzed Huisgen cycloaddition between azide 3 and phenylacetylene delivered the corresponding triazole 77 in a 93% yield. Treatment of the azides 66 and 68 with PPh3/H2O furnished the subsequent imidization product 78 and lactam 79 in excellent yields of 92% and 98%, respectively.
 | ||
| Scheme 2 Derivatizations of the azidoalkylation product 3. | ||
To gain additional insights into the mechanism of this diene azidoalkylation reaction, we conducted a series of mechanistic probe experiments. The addition of 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO) to the reaction of 3a under the standard conditions completely inhibited the formation of the desired product 3. The TEMPO-trapped adduct 81 was detected by high-resolution mass spectrometry, supporting the intermediacy of a benzylic radical likely generated from HAT from toluene by the tert-butoxyl radical (Scheme 3a, reaction 1). Notably, when an electron-donating group (EDG) was present on the C1 phenyl ring in 3a, the allylic radical-trapped product 80a was not observed (Scheme 3a, reaction 1). However, with the introduction of an electron-withdrawing group (EWG) to the phenyl ring of 10a, 80b was detected (Scheme 3a, reaction 2). As suggested by these experiments, the more electron-deficient allylic radical has a longer lifetime as compared to the more electron-rich allylic radical, indicating that a dual mechanistic pathway might exist in this reaction. The reaction of the radical clock 82 also supported the existence of a radical pathway, as the ring-opened 83 was obtained in 34% yield in a 16:1 E:Z ratio, suggesting that the in situ-generated cyclopropylcarbinyl radical undergoes fast ring-opening to give an allylic radical, which is then rapidly trapped by the azide radical (Scheme 3b). In addition, a kinetic isotope effect experiment clearly showed that such an effect was present, as a KIE of 3.03 (kH/kD) was observed for the reaction of diene 1a in toluene and toluene-d8 (Scheme 3c), suggesting that the aliphatic C–H bond cleavage might be involved in the rate-determining step of the reaction. We also conducted parallel KIE experiments and observed a KIE of 2.22 (kH/kD) (see the ESI† for details), also supporting the likelihood that C–H bond cleavage is involved in the rate-determining step of the reaction. Finally, we conducted a series of Hammett studies (Scheme 3d). The negative slope of the Hammett plot indicated the possibility that allylic cation intermediates might be generated during the reaction. However, when electron-withdrawing groups (OCF3, CO2Et, SCF3, CF3) were introduced on the aryl group of the substrates, the expected negative trend was not observed (Scheme 3d), indicating that electron-deficient 1,3-dienes likely proceed through allylic radical intermediates, followed by a group transfer process to generate the final products.
 | ||
| Scheme 3 Mechanistic studies. | ||
Based on our mechanistic studies, a plausible mechanism for the Fe-catalyzed azidoalkylation of 1,3-dienes is proposed in Scheme 4. First, a single-electron oxidation process occurs through the reaction of the iron(II) trifluoromethanesulfonate complex with DTBP to produce an oxidized iron species A and a tert-butoxyl radical. Intermediate A then reacts with TMSN3 to produce the iron(III) azide species B. The tert-butoxyl radical then selectively abstracts an H-atom from the coupling partner to generate the alkyl radical R˙. This carbon-centered radical may add to the terminal carbon atom of the 1,3-diene to selectively generate the allylic radical C. This allylic radical intermediate is subsequently captured by B. In pathway a, electron-deficient dienes are more likely to be favored. A group transfer process leads to the formation of the final product and regenerates the active iron catalyst. In contrast, electron-rich dienes are expected to proceed via pathway b, where a single-electron transfer (SET) occurs first between the iron(III) azide B and C, which is then followed by nucleophilic attack by the azide species. In addition, the tert-butoxyl radical can potentially abstract the TMS group from TMSN3, which leads to the generation of a free azido radical (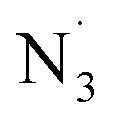 ). A similar process may occur to furnish the undesired diazidation product from the 1,3-diene precursor, thereby reducing the overall conversion to the desired azidoalkylation product.
). A similar process may occur to furnish the undesired diazidation product from the 1,3-diene precursor, thereby reducing the overall conversion to the desired azidoalkylation product.
 | ||
| Scheme 4 Proposed catalytic cycle. | ||
Conclusion
In conclusion, we have described a practical iron-catalyzed azidoalkylation of 1,3-dienes based on the direct and selective abstraction of aliphatic C–H bonds enabled by a HAT process. A wide array of commercially available hydrocarbons bearing diverse C–H bonds were employed in the coupling to provide access to synthetically useful amines. Mechanistic studies indicated that the electronic properties of the precursor 1,3-dienes influence the mechanistic pathway of the transformation, with reaction proceeding via either group transfer for electron-deficient 1,3-dienes or sequential single-electron transfer and nucleophilic attack for electron-rich 1,3-dienes to furnish the azidoalkylation products.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. M. S. thanks the NSF CHE-1954325 and the American Chemical Society Pharmaceutical Roundtable Green Chemistry Institute Panel for funding. NMR facilities at UW-Madison are funded by the National Science Foundation (NSF; CHE-9208463, CHE-9629688) and National Institutes of Health (NIH; RR08389-01). The Q-Exactive mass spectrometer was acquired from an NIH-S10 award (NIH-1S10OD020022-1). The Bruker D8 VENTURE Photon III X-ray diffractometer was partially funded by NSF Award #CHE-1919350 to the UW–Madison. The Bruker Quazar APEX2 was purchased by University of Wisconsin–Madison. Dr Charles G. Fry, Dr Heike Hofstetter, and Dr Cathy Clewett at UW–Madison are thanked for helping with NMR techniques. Dr Martha M. Vestling at UW–Madison is thanked for mass spectrometry characterization.References
- X. Li, C. Pei and J. Gong, Shale Gas Revolution: Catalytic Conversion of C1–C3 Light Alkanes to Value-Added Chemicals, Chem, 2021, 7, 1755–1801 CAS.
- X. Wu and L.-Z. Gong, Palladium(0)-Catalyzed Difunctionalization of 1,3-Dienes: From Racemic to Enantioselective, Synthesis, 2019, 51, 122–134 CrossRef CAS.
- Y. Xiong, Y. Sun and G. Zhang, Recent Advances on Catalytic Asymmetric Difunctionalization of 1,3-Dienes, Tetrahedron Lett., 2018, 59, 347–355 CrossRef CAS.
- P.-Z. Wang, W.-J. Xiao and J.-R. Chen, Recent Advances in Radical-Mediated Transformations of 1,3-Dienes, Chin. J. Catal., 2022, 43, 548–557 CrossRef CAS.
- E. McNeill and T. Ritter, 1,4-Functionalization of 1,3-Dienes With Low-Valent Iron Catalysts, Acc. Chem. Res., 2015, 48, 2330–2343 CrossRef CAS PubMed.
- K. Chen, H. Zhu, S. Liu, J. Bai, Y. Guo, K. Ding, Q. Peng and X. Wang, Switch in Selectivities by Dinuclear Nickel Catalysis: 1,4-Hydroarylation of 1,3-Dienes to Z-Olefins, J. Am. Chem. Soc., 2023, 145, 24877–24888 CAS.
- H. Wang, R. Zhang and W. Zi, Synergistic Palladium/Copper-Catalyzed 1,4-Difunctionalization of 1,3-Dienes for Stereodivergent Construction of 1,5-Nonadjacent Stereocenters, Angew. Chem., Int. Ed., 2024, 63, e202402843 CrossRef CAS.
- X.-Y. Ruan, D.-X. Wu, W.-A. Li, Z. Lin, M. Sayed, Z.-Y. Han and L.-Z. Gong, Photoinduced Pd-Catalyzed Enantioselective Carboamination of Dienes via Aliphatic C–H Bond Elaboration, J. Am. Chem. Soc., 2024, 146, 12053–12062 CrossRef CAS PubMed.
- Y. Xiong and G. Zhang, Enantioselective 1,2-Difunctionalization of 1,3-Butadiene by Sequential Alkylation and Carbonyl Allylation, J. Am. Chem. Soc., 2018, 140, 2735–2738 CrossRef CAS PubMed.
- Y. Li, Y. Dong, X. Wang, G. Li, H. Xue, W. Xin, Q. Zhang, W. Guan and J. Fu, Regio-, Site-, and Stereoselective Three-Component Aminofluorination of 1,3-Dienes via Cooperative Silver Salt and Copper Catalysis, ACS Catal., 2023, 13, 2410–2421 CrossRef CAS.
- L.-F. Fan, R. Liu, X.-Y. Ruan, P.-S. Wang and L.-Z. Gong, Asymmetric 1,2-Oxidative Alkylation of Conjugated Dienes via Aliphatic C–H Bond Activation, Nat. Synth., 2022, 1, 946–955 CrossRef CAS.
- N. H. Watkins, Y. Kwon and Q. Wang, Copper-Catalyzed 1,4-Aminohydroxylation and Aminothiolation of 1,3-Dienes by Carbonyl-Assisted Migration, Chem Catal., 2024, 4, 101147 CrossRef CAS.
- S.-S. Chen, M.-S. Wu and Z.-Y. Han, Palladium-Catalyzed Cascade Sp2 C−H Functionalization/Intramolecular Asymmetric Allylation: From Aryl Ureas and 1,3-Dienes to Chiral Indolines, Angew. Chem., Int. Ed., 2017, 56, 6641–6645 CrossRef CAS.
- M. M. Parsutkar and T. V. RajanBabu, α- and β-Functionalized Ketones from 1,3-Dienes and Aldehydes: Control of Regio- and Enantioselectivity in Hydroacylation of 1,3-Dienes, J. Am. Chem. Soc., 2021, 143, 12825–12835 CrossRef CAS PubMed.
- X. Pang, Z.-Z. Zhao, X.-X. Wei, L. Qi, G.-L. Xu, J. Duan, X.-Y. Liu and X.-Z. Shu, Regiocontrolled Reductive Vinylation of Aliphatic 1,3-Dienes with Vinyl Triflates by Nickel Catalysis, J. Am. Chem. Soc., 2021, 143, 4536–4542 CrossRef CAS PubMed.
- X.-H. Yang and V. M. Dong, Rhodium-Catalyzed Hydrofunctionalization: Enantioselective Coupling of Indolines and 1,3-Dienes, J. Am. Chem. Soc., 2017, 139, 1774–1777 CrossRef CAS.
- S. M. Thullen and T. Rovis, A Mild Hydroaminoalkylation of Conjugated Dienes Using a Unified Cobalt and Photoredox Catalytic System, J. Am. Chem. Soc., 2017, 139, 15504–15508 CrossRef CAS.
- F. Burg and T. Rovis, Diastereoselective Three-Component 3,4-Amino Oxygenation of 1,3-Dienes Catalyzed by a Cationic Heptamethylindenyl Rhodium(III) Complex, J. Am. Chem. Soc., 2021, 143, 17964–17969 CrossRef CAS.
- Z.-Y. Dai, I. A. Guzei and J. M. Schomaker, Iron-Catalyzed Site- and Regioselective 1,2-Azidoamidations of 1,3-Dienes, Org. Lett., 2024, 26, 269–273 CrossRef CAS PubMed.
- F. Wang, X. Qi, Z. Liang, P. Chen and G. Liu, Copper-Catalyzed Intermolecular Trifluoromethylazidation of Alkenes: Convenient Access to CF3-Containing Alkyl Azides, Angew. Chem., 2014, 126, 1912–1917 CrossRef.
- L. Xu, X.-Q. Mou, Z.-M. Chen and S.-H. Wang, Copper-Catalyzed Intermolecular Azidocyanation of Aryl Alkenes, Chem. Commun., 2014, 50, 10676–10679 RSC.
- L. Ge, H. Zhou, M.-F. Chiou, H. Jiang, W. Jian, C. Ye, X. Li, X. Zhu, H. Xiong, Y. Li, L. Song, X. Zhang and H. Bao, Iron-Catalysed Asymmetric Carboazidation of Styrenes, Nature Catal., 2020, 4, 28–35 CrossRef.
- W. Liu, M. Pu, J. He, T. Zhang, S. Dong, X. Liu, Y.-D. Wu and X. Feng, Iron-Catalyzed Enantioselective Radical Carboazidation and Diazidation of α,β-Unsaturated Carbonyl Compounds, J. Am. Chem. Soc., 2021, 143, 11856–11863 CrossRef CAS PubMed.
- Y.-A. Yuan, D.-F. Lu, Y.-R. Chen and H. Xu, Iron-Catalyzed Direct Diazidation for a Broad Range of Olefins, Angew. Chem., 2016, 128, 544–548 CrossRef.
- L. Wu, Z. Zhang, D. Wu, F. Wang, P. Chen, Z. Lin and G. Liu, Anionic Bisoxazoline Ligands Enable Copper-Catalyzed Asymmetric Radical Azidation of Acrylamides, Angew. Chem., Int. Ed., 2021, 60, 6997–7001 CrossRef CAS.
- H. Xiong, N. Ramkumar, M.-F. Chiou, W. Jian, Y. Li, J.-H. Su, X. Zhang and H. Bao, Iron-Catalyzed Carboazidation of Alkenes and Alkynes, Nat. Commun., 2019, 10, 122 CrossRef.
- F. Wang, X. Qi, Z. Liang, P. Chen and G. Liu, Copper-Catalyzed Intermolecular Trifluoromethylazidation of Alkenes: Convenient Access to CF3-Containing Alkyl Azides, Angew. Chem., Int. Ed., 2014, 53, 1881–1886 CrossRef CAS PubMed.
- C. S. Day, A. Fawcett, R. Chatterjee and J. F. Hartwig, Mechanistic Investigation of the Iron-Catalyzed Azidation of Alkyl C(Sp3)–H Bonds with Zhdankin's Λ3-Azidoiodane, J. Am. Chem. Soc., 2021, 143, 16184–16196 CrossRef CAS PubMed.
- Z.-Y. Dai, S.-Q. Zhang, X. Hong, P.-S. Wang and L.-Z. Gong, A Practical FeCl3/HCl Photocatalyst for Versatile Aliphatic C–H Functionalization, Chem Catal., 2022, 2, 1211–1222 CrossRef CAS.
- Y. Wang, P. Hu, J. Yang, Y.-A. Zhu and D. Chen, C–H Bond Activation in Light Alkanes: A Theoretical Perspective, Chem. Soc. Rev., 2021, 50, 4299–4358 RSC.
- O. Baudoin, Multiple Catalytic C−H Bond Functionalization for Natural Product Synthesis, Angew. Chem., Int. Ed., 2020, 59, 17798–17809 CrossRef CAS.
- M. Rivas, V. Palchykov, X. Jia and V. Gevorgyan, Recent Advances in Visible Light-Induced C(Sp3)–N Bond Formation, Nat. Rev. Chem, 2022, 6, 544–561 CrossRef.
- L. Capaldo, D. Ravelli and M. Fagnoni, Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C–H Bonds Elaboration, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS.
- J. Zhang and M. Rueping, Metallaphotoredox Catalysis for Sp3 C–H Functionalizations through Hydrogen Atom Transfer (HAT), Chem. Soc. Rev., 2023, 52, 4099–4120 RSC.
- S. Zhang and M. Findlater, Electrochemically Driven Hydrogen Atom Transfer Catalysis: A Tool for C(Sp3)/Si–H Functionalization and Hydrofunctionalization of Alkenes, ACS Catal., 2023, 13, 8731–8751 CrossRef CAS PubMed.
- L.-C. Wang, Y. Yuan, Y. Zhang and X.-F. Wu, Cobalt-Catalyzed Aminoalkylative Carbonylation of Alkenes toward Direct Synthesis of γ-Amino Acid Derivatives and Peptides, Nat. Commun., 2023, 14, 7439 CrossRef.
- D. L. Golden, S.-E. Suh and S. S. Stahl, Radical C(Sp3)–H Functionalization and Cross-Coupling Reactions, Nat. Rev. Chem, 2022, 6, 405–427 CrossRef CAS PubMed.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Organic Azides: An Exploding Diversity of a Unique Class of Compounds, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- M. Shee and N. D. Pradeep Singh, Chemical Versatility of Azide Radical: Journey from a Transient Species to Synthetic Accessibility in Organic Transformations, Chem. Soc. Rev., 2022, 51, 2255–2312 RSC.
- K. Wu, Y. Liang and N. Jiao, Azidation in the Difunctionalization of Olefins, Molecules, 2016, 21, 352 CrossRef.
- M. Goswami and B. de Bruin, Metal-Catalysed Azidation of Organic Molecules, Eur. J. Org Chem., 2017, 8, 1152–1176 CrossRef.
- W.-Y. Li, C.-S. Wu, Z. Wang and Y. Luo, Fe-Catalyzed Three-Component Carboazidation of Alkenes with Alkanes and Trimethylsilyl Azide, Chem. Commun., 2018, 54, 11013–11016 RSC.
- Y. Ikemoto, S. Chiba, Z. Li, Q. Chen, H. Mori and Y. Nishihara, Carboazidation of Terminal Alkenes with Trimethylsilyl Azide and Cyclic Ethers Catalyzed by Copper Powder under Oxidative Conditions, J. Org. Chem., 2023, 88, 4472–4480 CrossRef CAS PubMed.
- L. Ge, H. Wang, Y. Liu and X. Feng, Asymmetric Three-Component Radical Alkene Carboazidation by Direct Activation of Aliphatic C–H Bonds, J. Am. Chem. Soc., 2024, 146, 13347–13355 CrossRef CAS.
- L. Xu, J. Chen and L. Chu, Solvent-Tuned Chemoselective Carboazidation and Diazidation of Alkenes via Iron Catalysis, Org. Chem. Front., 2019, 6, 512–516 RSC.
- P. Sivaguru, Y. Ning and X. Bi, New Strategies for the Synthesis of Aliphatic Azides, Chem. Rev., 2021, 121, 4253–4307 CrossRef CAS PubMed.
- P. Palamini, E. M. D. Allouche and J. Waser, Iron-Catalyzed Synthesis of α-Azido α-Amino Esters via the Alkylazidation of Alkenes, Org. Lett., 2023, 25, 6791–6795 CrossRef CAS.
- L. Ge, Y. Li and H. Bao, Iron-Catalyzed Radical Acyl-Azidation of Alkenes with Aldehydes: Synthesis of Unsymmetrical β-Azido Ketones, Org. Lett., 2019, 21, 256–260 CrossRef CAS.
- W.-Y. Li, Q.-Q. Wang and L. Yang, Fe-Catalyzed Radical-Type Difunctionalization of Styrenes with Aliphatic Aldehydes and Trimethylsilyl Azide via a Decarbonylative Alkylation–Azidation Cascade, Org. Biomol. Chem., 2017, 15, 9987–9991 RSC.
- J.-W. Hu, Y. Zhong and R.-J. Song, Copper/Iron Controlled Regioselective 1,2-Carboazidation of 1,3-Dienes with Acetonitrile and Azidotrimethylsilane, Org. Biomol. Chem., 2025, 23, 1437–1442 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00307e |
| This journal is © The Royal Society of Chemistry 2025 |