
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBifunctional additive-driven shape transitions of block copolymer particles through synergistic quaternization and protonation†
Zhengping
Tan‡
 a,
Soohyun
Ban‡
b,
Younghyeon
Ahn
a,
Kang Hee
Ku
*b and
Bumjoon J.
Kim
*a
a,
Soohyun
Ban‡
b,
Younghyeon
Ahn
a,
Kang Hee
Ku
*b and
Bumjoon J.
Kim
*a
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 34141, Republic of Korea. E-mail: bumjoonkim@kaist.ac.kr
bSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan, 44919, Republic of Korea. E-mail: kangheeku@unist.ac.kr
First published on 8th March 2025
Abstract
Block copolymer (BCP) particles with tailored shapes and nanostructures hold promise for applications in cell adhesion, photonic system, and energy storage due to their unique optical and rheological properties. Conventional approaches relying on surfactant-mediated self-assembly often limit particle geometries to simple structures. Herein, we present a versatile approach to expand the morphology of poly(styrene-block-2-vinylpyridine) (PS-b-P2VP) BCP particles through the incorporation of 9-bromononanoic acid (BNA), a bifunctional additive that facilitates synergistic quaternization and protonation. Increasing the BNA-to-2VP molar ratio enhances P2VP hydrophilicity and decreases the pH value, driving dramatic shape transitions from onion-like spheres to tulip bulbs, ellipsoids, discs, and Janus cups. This morphological diversity is attributed to synergetic interfacial instability-driven water infiltration and pH-induced repulsion of protonated P2VP chains. Additives with a single functional group, however, yield limited morphologies, such as tulip bulbs or onion-like spheres. Notably, Janus cups fabricated via this strategy exhibit selective cargo-loading capabilities, highlighting the importance of precise control over the internal composition and structure of BCP particles.
Introduction
Polymeric particles with tunable shapes and well-defined internal structures have gained significant attention due to the ability to precisely manipulate optical, mechanical, and interfacial properties, enabling applications in cell adhesion, photonics, and energy storage.1–7 Various fabrication methods, including self-organized precipitation, polymerization-induced self-assembly (PISA), and lithography, have been explored to produce nanostructured polymer particles.8–12 Among these approaches, the confined self-assembly of block copolymers (BCPs) in evaporative emulsions has emerged as a versatile platform for generating highly tunable particle morphologies by leveraging interfacial tension and microphase separation.13–18 Adjusting the interfacial environment, often by selecting surfactants to tune interactions between BCP domains and the surrounding medium, has led to the formation of anisotropic particles, primarily oblate or ellipsoidal structures.19–22 Expanding the range of accessible morphologies, however, requires additional strategies to modify interfacial properties at the molecular level.Recent efforts have focused on functional additives that selectively modify interfacial interactions, thereby reshaping BCP particles. For example, quaternizing additives have been shown to enhance hydrophilicity, promote water uptake, and lower interfacial tension, which in turn transforms ellipsoidal polystyrene-block-poly(2-vinylpyridine) (PS-b-P2VP) particles into accordion- or larva-like structures.23,24 Similarly, light-responsive quaternizing additives have enabled dynamic shape transitions under UV irradiation.25 In addition to quaternization, directional interactions such as hydrogen bonding provide another pathway for morphological control.26–28 A striking example involves hydroxyl-functionalized 3-pentadecylphenol, which forms hydrogen bonds with pyridine-containing blocks, driving polystyrene-block-poly(4-vinylpyridine) (PS-b-P4VP) to assemble into oblate particles with PS cylinders. Subsequent selective removal of small molecules via metal precursor adsorption further refines the structure, offering a strategy for constructing anisotropic architectures for carbon materials.29
Beyond non-covalent interactions, pH modulation offers another effective mechanism for inducing shape transitions by altering domain swelling and interfacial stability.30–32 Inorganic acids, such as hydrochloric acid, have been used to directly modulate pH, destabilize interfaces and generate anisotropic structures.33 Meanwhile, spiropyran photoacid molecules have enabled rapid, reversible shape transitions by leveraging light-driven chemical potential changes to trigger P4VP protonation.34 More recently, hydrolyzed quaternizing agents have been shown to enhance P2VP hydrophilicity while simultaneously inducing protonation under acidic conditions, leading to elongated ellipsoids, buds, or vesicles.35 However, these approaches lack temporal control, as the gradual hydrolysis of quaternizing agents causes slow, uneven acidification, leading to morphological inconsistencies. Thus, a key challenge remains: achieving precise, multi-step morphological evolution within a single system without relying on sequential or independent chemical modifications.
To address this limitation, we introduce a bifunctional additive, 9-bromononanoic acid (BNA), which integrates both bromo and carboxyl functional groups (Scheme 1). This molecular design enables a dual mode of action: (1) quaternization of the P2VP block, which enhances hydrophilicity, and (2) sequential protonation of the P2VP block, further inducing disassembly under acidic conditions. By adjusting the amount of BNA, we can finely tune the pH of the aqueous environment from neutral to acidic, as well as control the degree of P2VP modification. Through this synergistic mechanism, we investigate how the enhanced hydrophilicity of P2VP affects particle morphologies, driving shape transitions from onion-like spheres to tulip bulbs, ellipsoids, discs, and Janus cups in comparison to the effects of single-functional additives. Additionally, we examine the selective cargo uptake capabilities of Janus cups, emphasizing their ability to encapsulate homopolymers or polymer-grafted nanoparticles, which underscores the structural diversity achievable with these architectures.
 | ||
| Scheme 1 Schematic illustration for the co-assembly of PS-b-P2VP and BNA additive within the evaporative oil-in-water emulsion. The bromo functional group induces quaternization of nitrogen in P2VP, while the carboxylic acid functional group lowers the system's pH, promoting the protonation of residual nitrogen in unquaternized P2VP. | ||
Experimental section
Materials
PS102k-b-P2VP97k (number-average molecular weights (Mn) = 199 kg mol−1, dispersity (Đ) = 1.12), P2VP8k (Mn = 8 kg mol−1, Đ = 1.15) and PS8k (Mn = 8 kg mol−1, Đ = 1.12) were purchased from Polymer Source, Inc. Poly(vinyl alcohol) (PVA, 87–89% hydrolyzed, Mn = 13–23 kg mol−1), cetyltrimethylammonium bromide (CTAB), 9-bromononanoic acid (BNA), 9-nonanoic acid (NA), 9-bromononane (BN), 1,4-dibromobutane (DBB, purity 99%) were purchased from Sigma-Aldrich and used without further purification.Preparation of PS-b-P2VP(BNA)x particles
A chloroform solution containing PS-b-P2VP BCPs and BNA additive was prepared and stirred at room temperature for 12 h. The feed molar ratios of BNA to the 2VP unit (x) varied from 0 to 1.5. Then, the polymer solution (0.1 mL, 10 mg mL−1) was emulsified with 1 mL of surfactant-containing aqueous solution (CTAB, 10 mg mL−1) by vortexing at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 1 min. Subsequently, chloroform was evaporated at room temperature for 24 h. The residual surfactants were removed by repeating centrifugation at 12000 rpm and finally re-dispersed in water for further characterization.
000 rpm for 1 min. Subsequently, chloroform was evaporated at room temperature for 24 h. The residual surfactants were removed by repeating centrifugation at 12000 rpm and finally re-dispersed in water for further characterization.
Crosslink of janus cups and selective loading with polymers
Dibrobutane (DBB) was used to selectively crosslink the P2VP block. DBB was dissolved in ethanol at a concentration of 50 mg mL−1 and then added to the aqueous suspension of Janus cups (1 mL, 5 mg mL−1) (DBB:2VP unit = 5:1). After being stirred at 40 °C for 24 h, the crosslinked Janus cups were separated by centrifugation (12000 rpm, 3 min) and washed in ethanol for 2 times. Then, 1 mg crosslinked Janus cups and different amounts of hP2VP8k or hPS8k (weight fraction of homopolymers are 25 wt%, 75 wt%, 86 wt%) or polymer-grafted gold nanoparticles (P2VP@AuNPs or PS@AuNPs, weight fraction is 30 wt%) were placed in 2 mL tetrahydrofuran (THF) in a vial. After that, the above solution was dialyzed into water. The Janus cups loaded with hP2VP8k, hPS8k, P2VP@AuNPs, or PS@AuNPs were collected after dialysis.
Characterization
Scanning electron microscopy (SEM, Magellan 400), and transmission electron microscopy (TEM, Tecnai F20, 200 kV) were used to observe the surface and internal structure of various BCP particles. The BCP particle solution was drop-cast on the silicon wafer for SEM samples or on the carbon-supported grid for TEM samples. The TEM grids were exposed to iodine vapor for 1 min to selectively stain P2VP domains before TEM analysis. Fourier-transform infrared spectroscopy (FT-IR) measurement was performed on a Nicolet iS500 FT-IR spectrometer (Thermo Fisher Scientific Instrument). FT-IR samples were prepared by concentrating the particles by centrifugation, followed by drying in a vacuum at 40 °C. 1H nuclear magnetic resonance (NMR) spectra of reacted PS-b-P2VP with BNA were obtained from a Bruker AVANCE III HD instrument at 400 MHz using DMF-d7 as solvent. Note that the DMF-d7 was used for NMR solvent due to the existence of a polar carboxyl group in BNA. The degree of reaction (α) was determined by the integral ratios between the proton adjacent to non-reacted nitrogen (δ 8.1 to 8.5 ppm) and the proton adjacent to reacted nitrogen (δ 8.8 to 9.5 ppm). The pH values of particle suspension were measured by an Ohaus ST3100 pH Bench. Note that the pH values of particle suspensions were measured before the removal of residual surfactants to prevent any loss of the free additive molecules.Results and discussion
A series of polymer particles composed of PS102k-b-P2VP97k and BNA were prepared using a solvent-evaporative emulsion method, with the feed molar ratios of BNA to 2VP units (x) varied from 0 to 1.5. CTAB was employed as a surfactant in all cases to selectively interact with the PS domains, stabilizing the particles in the aqueous phase.22,36–38 Upon solvent evaporation, the particles exhibited diverse morphologies depending on the value of x. Fig. 1 presents the morphological evolution of PS-b-P2VP(BNA)x particles at different x of 0.1, 0.5, 0.8, 1.2 and 1.5. In the absence of BNA (x = 0), the pristine PS-b-P2VP particles exhibit a concentric onion-like shape, with domain spacing of 26 nm for PS (DPS) and 22 nm for P2VP (DP2VP) (Fig. S1a and b†). As x increased from 0 to 1.5, the overall shape of the PS-b-P2VP(BNA)x particles changed from concentric spheres to tulip bulbs, ellipsoids, discs, and Janus cups. The preferential interaction between BNA and pyridyl units in the PS-b-P2VP was confirmed by the 1H NMR spectra (Fig. S2†) as a new proton peak from the quaternized pyridinium ring appeared between 8.8 and 9.5 ppm.35,39 | ||
| Fig. 1 SEM and TEM images of PS-b-P2VP(BNA)x particles. (a–e) SEM and (f–j) TEM images of PS-b-P2VP(BNA)x particles at different x values of (a and f) 0.1, (b and g) 0.5, (c and h) 0.8, (d and i) 1.2 and (e and j) 1.5. Scale bars in (a–e) are 1 μm while 100 nm in (f–j). Inset in (i) is the cartoon of disc particles. | ||
When x values increased from 0 to 0.3, the PS-b-P2VP(BNA)x particles maintained onion-like spherical structures with concentric lamellae (Fig. 1a, f, S1c and d). At x = 0.5, tulip bulb-like particles having a mixture of axial and radial lamellae emerged (Fig. 1b and g), driven by the enhanced hydrophilicity of P2VP blocks from the interaction with BNA, resulting in weaker selective surroundings.35,40 At x = 0.8, well-defined ellipsoidal particles with alternating lamellar domains (DPS = 36 nm, DP2VP = 32 nm) were formed (Fig. 1c and h). This formation corresponds to the interfacial tension between the P2VP(BNA) solution in chloroform and the aqueous surfactant solution (γP2VP(BNA)) closely matching that of PS (γPS), indicating neutral interfacial conditions (γPS = 3.99 mN m−1, γP2VP(BNA) = 3.93 mN m−1 in Table S1†). A control experiment using mixed surfactants (PVA/CTAB) also produced similar ellipsoidal particles with identical domain spacings, confirming that BNA induces interfacial neutralization (Fig. S3†). When x was increased to 1.2, disc-like particles with sandwich-like structures were observed, where P2VP domains formed the outer surfaces, and the PS domain was positioned in the center (Fig. 1d and i). Further increasing x to 1.5 resulted in Janus cups, characterized by P2VP inner layer and PS outer layer, as shown in Fig. 1e and j.
The formation of disc-like particles proceeds through the following steps (Fig. 2a). During the evaporation process, BNA reacts with P2VP chains, increasing their hydrophilicity and lowering interfacial tension (3.48 mN m−1), which promotes the formation of ellipsoidal particles with alternating layers. Water molecules then penetrate into the P2VP domains, increasing incompatibility with the PS blocks and resulting in a more pronounced undulated surface. Finally, electrostatic repulsion induced by quaternization and protonation leads to the disassembly of ellipsoidal particles into single disc-like particles, with P2VP domains on the surfaces and PS domains at the core (thickness: 59 ± 5 nm, Fig. 2b–d).41,42 This one-pot strategy for nanodisc formation advances the confined self-assembly of anisotropic BCP particles, addressing the inherent challenge of stabilizing disc-like structures. Previous studies required sequential assembly and disassembly steps to generate similar morphologies.43–45 In contrast, our findings show that precisely tuning BNA concentrations induces direct disc formation through a synergistic mechanism of enhanced hydrophilicity, water infiltration, and electrostatic repulsion. This approach simplifies nanodisc fabrication and offers an alternative pathway within confined self-assembly. The nanodiscs obtained in this study are slightly thicker than those formed via conventional ellipsoidal disassembly (56 ± 8 nm).32 This difference suggests that BNA-mediated protonation enhances P2VP swelling under acidic conditions, further contributing to their distinct structural characteristics (Fig. S4†).
 | ||
| Fig. 2 (a) Schematics illustrating the proposed mechanism of the formation of disc-like particles when x = 1.2. (b–d) Representative (b) top-view SEM, (c) TEM, and (d) side-view SEM images of disc-like particles. (e) Schematics illustrating the proposed mechanism of the formation of Janus cups when x = 1.5. (f and g) TEM and SEM images of particles captured after evaporation for 4 h and 8 h, respectively. (h) TEM images of Janus cups tilted at angles of −30 to 30°. | ||
Increasing the BNA/2VP ratio further alters the assembly, eventually leading to the formation of Janus cups (Fig. 2e). Similar to the disc formation, PS-b-P2VP chains first migrate to the emulsion interface due to the enhanced hydrophilicity of P2VP upon reacting with BNA (γP2VP(BNA) = 3.32 mN m−1). At higher BNA content, enhanced water infiltration destabilizes the emulsion, causing it to break into smaller droplets with water cores. During this process, polymer chain rearrangement occurs asymmetrically, as quaternized P2VP alters interfacial tension gradients, leading to preferential bending rather than symmetric bilayer stabilization.33,35 As protonation proceeds under acidic conditions, strong electrostatic repulsion within the swollen P2VP domains increases the internal surface area of the hollow structures. The combination of interfacial instability and electrostatic expansion drives asymmetric deformation, ultimately leading to Janus cup morphology. Additionally, the aqueous surrounding, composed of CTAB with a strong affinity for PS, further contributes to asymmetric stabilization by preferentially interacting with the PS domains. Freeze-drying experiments on PS-b-P2VP(BNA)1.5 emulsions at different evaporation times support this mechanism. After 4 hours of evaporation, hollow cores were clearly observed, confirming that water infiltration and solvent removal are essential in the formation of Janus cups (Fig. 2f and g). The Janus cups exhibit a constant thickness of 24 nm ± 2 nm and a narrow size distribution (406 ± 11 nm < length (L) < 715 ± 8 nm) (Fig. S5†). Tilting TEM images (Fig. 2h, −30° to 30°) confirm the open hollow structure, with the P2VP domain inside and the PS domain on the exterior. We note that Janus cups obtained in this study differ from those reported previously, which were formed through crosslinking and disassembly of particles.40,46,47 In those systems, the resulting structures either exhibited a sandwich-like arrangement from bud-like particles or a mixed inner and outer composition. In contrast, our system follows a distinct pathway to asymmetry, where interfacial instability and swelling-driven electrostatic repulsion consistently yield Janus cups with a P2VP-inner and PS-outer configuration.
BNA molecules, with their bromo and carboxyl groups, exhibit complementary dual functionalities. They quaternize the nitrogen in P2VP, reducing interfacial tension, while also enabling protonation of the nitrogen under acidic conditions. Once the pH drops below the pKa of P2VP, protonation is triggered, significantly enhancing the hydrophilicity of the P2VP blocks.48,49 To elucidate the role of quaternization and protonation in driving morphological changes, pH values and reaction degree (α, reacted n2VP/(reacted n2VP + n2VP)) were systematically measured and correlated with the resulting particle shapes (Fig. 3 and Table S2†). As x increased from 0.1 to 1.5, α values increased from 10.5% to 21.2%, while the pH values decreased from 6.96 to 3.25 (Table S3†). Notably, when x was higher than 0.6, the pH dropped below 4.7, initiating significant protonation of the P2VP blocks. This protonation was confirmed through the FT-IR spectrum of obtained PS-b-P2VP(BNA)x particles as shown in Fig. S6.† The peak was broadened near 1000 cm−1 and a new peak at 1637 cm−1 corresponding to N–H bending vibration emerged, indicating the formation of ionized 2VP units.23,33,41 A significant morphological transition occurs when x reaches 0.9, marked by the appearance of a mixture of ellipsoidal and disc-like particles (purple box in Fig. 3). This shift results from increased repulsion between the P2VP blocks at a pH of 4.45 and an α value of 18%, leading to the partial disassembly of ellipsoidal particles into discs. As x increases to 1.2 and 1.5, stronger repulsion and swelling of the P2VP blocks further promote the complete transformation into disc-like particles, followed by a morphological evolution into Janus cups.
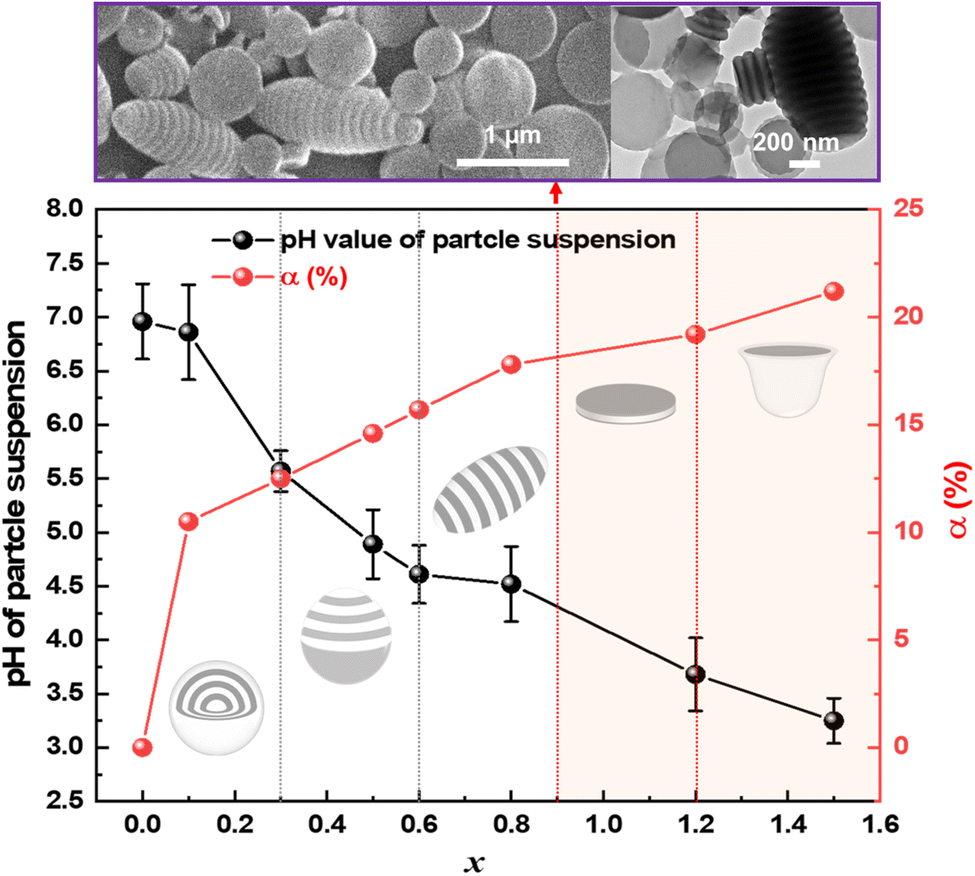 | ||
| Fig. 3 The α of P2VP in PS-b-P2VP(BNA)x particles and the pH values of particle suspensions as a function of x of BNA molecules. The dashed lines indicate the transition points of particle shapes. We define these morphologies as spheres, tulip bulbs, ellipsoids, discs, and Janus cups in the order of increasing x. Cartoons in the inset show the particle shape transitions. Representative SEM and TEM images on the top of the graph showed the mixture of ellipsoids and disc-like particles at x = 0.9. | ||
To elucidate the critical role of bifunctional groups in driving the shape transition of BCP particles, control experiments were conducted using additives containing either a bromo group (9-bromononane, BN) or a carboxyl group (nonanoic acid, NA) alone. In the presence of BN, the particle suspension maintained a nearly constant neutral pH value (∼6.90, Fig. 4 and Table S3†). Under these conditions, PS-b-P2VP(BN)x particles primarily adopt tulip bulb-like morphologies, with no further shape changes but increased domain spacings (DPS and DP2VP) as shown in Fig. 4 and S7.† The persistence of these morphologies results from the limited hydrophilicity enhancement of P2VP, caused by the lower reactivity between BN and PS-b-P2VP. This behavior is similar to our previous work, where ellipsoidal particles were stabilized in a broad range when molecular additives exhibited low quaternization with PS-b-P2VP.35 Specifically, DP2VP increased from 24 nm to 54 nm and DPS increased from 29 nm to 64 nm as x increased from 0.1 to 1.2. This domain expansion is attributed to the swelling of P2VP domains by surroundings due to quaternization by BN.35 In a parallel experiment, NA was introduced to the emulsions, resulting in a decrease in suspension pH from 6.96 to 3.45 (Fig. 4 and Table S3†). Despite the significant pH reduction, PS-b-P2VP(NA)x particles exhibited only limited morphological transitions, evolving from spheres to tulip bulb-like structures and eventually to onion-like spheres as x increased from 0.1 to 1.2 (Fig. 4 and S8†). The lack of pronounced shape anisotropy suggests that NA alone can't drive the complex morphological transformations observed with bifunctional additives due to the limited enhanced hydrophilicity of P2VP from protonation by a single carboxyl group in NA. These findings highlight the pivotal role of bi-functional additives in promoting anisotropic shape transformations in BCP particles. The combination of bromo and carboxyl groups enables simultaneous quaternization and protonation of the P2VP domains, which is essential for achieving the diverse and well-defined anisotropic structures observed in this system.
 | ||
| Fig. 4 A plot of pH values of particle suspensions and representative shape of particles as a function of x by using BNA, BN, and NA additives. Scale bars in SEM images are 1 μm. | ||
Janus materials, characterized by their distinct dual properties, offer unique functionalities such as directional interactions, selective responsiveness, and asymmetric assembly, making them valuable for a wide range of applications.50–53 Here, the Janus cups with a cavity and open cap were explored for selective cargo loading, leveraging their unique structural properties. The Janus cups consist of an inner P2VP domain and an outer PS domain, offering distinct interaction potentials for selective encapsulation. To load the P2VP8k homopolymer (hP2VP8k) into the Janus cups, the hP2VP8k was mixed with the crosslinked Janus cups in THF. The solvent quality for P2VP was then adjusted by dialyzing the mixture into water for 12 h. Notably, the Janus cups retained their structure after crosslinking with DBB for 24 h, as shown in Fig. S9.† By varying the weight ratio of hP2VP8k to the Janus cups, the degree of homopolymer incorporation was precisely tuned. For instance, increasing the amount of hP2VP8k (e.g., 25 wt%, 75 wt%, and 86 wt%) resulted in greater protrusion of homopolymer within the cavity (Fig. 5a). This effect is attributed to the reduced solubility and aggregation of hP2VP8k chains in polar environments during solvent exchange, promoting their preferential interaction with the P2VP cavity. In contrast, hPS8k exhibited selective adsorption onto the exterior PS domain of the Janus cups, rather than filling the inner cavity, as shown in Fig. 5b. With increasing hPS8k content (e.g., 25 wt%, 75 wt%, and 86 wt%), the external PS area grew, indicating the preferential interaction of hPS8k with the PS outer surface, consistent with previous findings on triblock copolymer-based Janus cups.40 Furthermore, to explore the potential of Janus cups beyond synthetic homopolymers, we investigated their selective loading behavior using polymer-modified gold nanoparticles (e.g., P2VP@AuNPs and PS@AuNPs) as shown in Fig. 5c and d. These nanoparticles were grafted with P2VP8k and PS8k, respectively, to assess their preferential localization within the Janus cups. Our results revealed that P2VP8k@AuNPs selectively loaded into the cavity of the Janus cups, demonstrating a strong affinity toward the inner P2VP domain (Fig. 5c). In contrast, PS8k@AuNPs preferentially attached to the exterior PS domain rather than infiltrating the inner cavity (Fig. 5d). This finding highlights how polymer composition governs nanoparticle localization within Janus structures, enhancing their selective encapsulation capabilities. This selective behavior makes them promising for applications in catalysis, sensing, and targeted delivery, offering a strategy for designing anisotropic nanostructures with tunable functionalities.
 | ||
| Fig. 5 Selective loading of homopolymers (hPS or hP2VP) and polymer-grafted gold NPs into PS-b-P2VP Janus cups. (a and b) TEM images of Janus cups loaded with different weight ratios of (a) P2VP8k and (b) PS8k homopolymers. (c and d) TEM images of Janus cups loaded with (c) P2VP8k@AuNPs and (d) PS8k@AuNPs. Scale bars are 50 nm. | ||
Conclusions
In summary, we introduced a bifunctional approach to achieve diverse morphologies in PS-b-P2VP BCP particles using an additive with both bromo and carboxyl groups. The bromo group of BNA facilitated the quaternization of nitrogen in P2VP, while its carboxylic acid group decreased the system's pH, promoting the protonation of residual nitrogen in unquaternized P2VP. This dual functionalities induced synergistic effects that drove distinct morphological transitions, ranging from onion-like spheres with concentric layers (x < 0.5) to ellipsoids with stacked lamellae (0.5 < x ≤ 0.8), discs with P2VP outer layers (0.8 < x ≤ 1.2), and Janus cups (1.2 < x ≤ 1.5). Control experiments with single-function additives validated the necessity of this dual functionalities for achieving such structural diversity. Notably, the Janus cups fabricated in this study exhibited selective cargo-loading capabilities, with distinct P2VP and PS domains facilitating targeted encapsulation of homopolymers and polymer-grafted gold nanoparticles. This work demonstrates the unique potential of bi-functional additives as a precise tool for developing anisotropic BCP structures, offering new avenues in tailored particle design.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Z. T. and S. B. contributed equally. Conceptualization: Z. T., B. J. K., K. H. K.; funding acquisition: B. J. K., K. H. K.; investigation: Z. T., S. B., Y. A.; formal analysis: Z. T.; validation: S. B.; visualization: Z. T., S. B., K. H. K.; writing-original draft preparation: Z. T.; writing-review and editing: S. B., Y. A., B. J. K., K. H. K.; supervision: B. J. K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Korea Research Foundation Grant, funded by the Korean Government (2022R1C1C1006324) and Korea Toray Fellowship funded by Korea Toray Science Foundation. This work was also supported by Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea government (MOTIE) (20214000000650).References
- J. A. Champion and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4930–4934 CrossRef CAS PubMed.
- J. Lee, S. Ban, K. Jo, H. S. Oh, J. Cho and K. H. Ku, ACS Nano, 2024, 18, 5196–5205 CrossRef CAS PubMed.
- D. P. Song, T. H. Zhao, G. Guidetti, S. Vignolini and R. M. Parker, ACS Nano, 2019, 13, 1764–1771 CAS.
- Y. J. Lee, H.-E. Kim, H. Oh, H. Yun, J. Lee, S. Shin, H. Lee and B. J. Kim, ACS Nano, 2022, 16, 2988–2996 CrossRef CAS PubMed.
- S. Kim, M. Ju, J. Lee, J. Hwang and J. Lee, J. Am. Chem. Soc., 2020, 142, 9250–9257 CrossRef CAS PubMed.
- S. Kim, J. Hwang, J. Lee and J. Lee, Sci. Adv., 2020, 6, eabb3814 CrossRef CAS PubMed.
- Q. He, H. Vijayamohanan, J. Li and T. M. Swager, J. Am. Chem. Soc., 2022, 144, 5661–5667 CrossRef CAS PubMed.
- T. Higuchi, A. Tajima, K. Motoyoshi, H. Yabu and M. Shimomura, Angew. Chem., Int. Ed., 2009, 48, 5125–5128 CrossRef CAS PubMed.
- J. H. Moon, A. J. Kim, J. C. Crocker and S. Yang, Adv. Mater., 2007, 19, 2508–2512 CrossRef CAS.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS PubMed.
- C. Tang, C. Zhang, J. Liu, X. Qu, J. Li and Z. Yang, Macromolecules, 2010, 43, 5114–5120 CrossRef CAS.
- D. Li, X. Chen, M. Zeng, J. Ji, Y. Wang, Z. Yang and J. Yuan, Chem. Sci., 2020, 11, 2855–2860 RSC.
- K. H. Ku, J. M. Shin, H. Yun, G.-R. Yi, S. G. Jang and B. J. Kim, Adv. Funct. Mater., 2018, 28, 1802961 CrossRef.
- Q. Guo, Y. Li, Q. Liu, Y. Li and D.-P. Song, Angew. Chem., Int. Ed., 2022, 61, e202113759 CrossRef CAS PubMed.
- J. J. Shin, E. J. Kim, K. H. Ku, Y. J. Lee, C. J. Hawker and B. J. Kim, ACS Macro Lett., 2020, 9, 306–317 CrossRef CAS PubMed.
- Y. Wang, D. Hu, X. Chang and Y. Zhu, Macromolecules, 2022, 55, 6211–6219 CrossRef CAS.
- L. Navarro, A. F. Thünemann and D. Klinger, ACS Macro Lett., 2022, 11, 329–335 CrossRef CAS PubMed.
- C. K. Wong, X. Qiang, A. H. E. Müller and A. H. Gröschel, Prog. Polym. Sci., 2020, 102, 101211 CrossRef CAS.
- K. H. Ku, Y. J. Lee, Y. Kim and B. J. Kim, Macromolecules, 2019, 52, 1150–1157 CrossRef CAS.
- L. Guo, D. Xia, Y. Wang, S. Ding, J. Xu, Y. Zhu and B. Du, Polym. Chem., 2024, 15, 30–39 RSC.
- J. Li, X. Yu, J. Zhang, J. Jin, Y. Pan, X. Ji and W. Jiang, ACS Appl. Mater. Interfaces, 2024, 16, 41332–41340 CrossRef CAS PubMed.
- S. G. Jang, D. J. Audus, D. Klinger, D. V. Krogstad, B. J. Kim, A. Cameron, S.-W. Kim, K. T. Delaney, S.-M. Hur, K. L. Killops, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, J. Am. Chem. Soc., 2013, 135, 6649–6657 CrossRef CAS PubMed.
- S. Lee, J. J. Shin, K. H. Ku, Y. J. Lee, S. G. Jang, H. Yun and B. J. Kim, Macromolecules, 2020, 53, 7198–7206 CrossRef CAS.
- M. Ren, Z. Hou, X. Zheng, J. Xu and J. Zhu, Macromolecules, 2021, 54, 5728–5736 CrossRef CAS.
- D. Hu, Y. Wang, J. Liu, Y. Mao, X. Chang and Y. Zhu, Nanoscale, 2022, 14, 6291–6298 RSC.
- B. V. K. J. Schmidt, C. X. Wang, S. Kraemer, L. A. Connal and D. Klinger, Polym. Chem., 2018, 9, 1638–1649 RSC.
- J. Xu, Y. Yang, K. Wang, Y. Wu and J. Zhu, Mater. Chem. Front., 2017, 1, 507–511 RSC.
- J. G. Kennemur, Macromolecules, 2019, 52, 1354–1370 CrossRef CAS.
- M. Wang, X. Mao, J. Liu, B. Deng, S. Deng, S. Jin, W. Li, J. Gong, R. Deng and J. Zhu, Adv. Sci., 2022, 9, 2202394 CrossRef CAS PubMed.
- J. Lee, K. H. Ku, C. H. Park, Y. J. Lee, H. Yun and B. J. Kim, ACS Nano, 2019, 13, 4230–4237 CrossRef CAS PubMed.
- Z. Hou, M. Ren, K. Wang, Y. Yang, J. Xu and J. Zhu, Macromolecules, 2020, 53, 473–481 CrossRef CAS.
- D. Klinger, C. X. Wang, L. A. Connal, D. J. Audus, S. G. Jang, S. Kraemer, K. L. Killops, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Angew. Chem., Int. Ed., 2014, 53, 7018–7022 CrossRef CAS PubMed.
- Y. Wu, K. Wang, H. Tan, J. Xu and J. Zhu, Langmuir, 2017, 33, 9889–9896 CrossRef CAS PubMed.
- J. Kim, J. Park, K. Jung, E. J. Kim, Z. Tan, M. Xu, Y. J. Lee, K. H. Ku and B. J. Kim, ACS Nano, 2024, 18, 8180–8189 CrossRef CAS PubMed.
- Z. Tan, E. J. Kim, T. N.-L. Phan, J. Kim, J. J. Shin, K. H. Ku and B. J. Kim, Macromolecules, 2022, 55, 9972–9979 CrossRef CAS.
- N. Yan, X. Liu, J. Zhu, Y. Zhu and W. Jiang, ACS Nano, 2019, 13, 6638–6646 CrossRef CAS PubMed.
- D. Hu, X. Chang, Y. Xu, Q. Yu and Y. Zhu, ACS Macro Lett., 2021, 10, 914–920 CrossRef CAS PubMed.
- C. Chen, R. A. L. Wylie, D. Klinger and L. A. Connal, Chem. Mater., 2017, 29, 1918–1945 CrossRef CAS.
- F. Jiménez-Ángeles, H.-K. Kwon, K. Sadman, T. Wu, K. R. Shull and M. Olvera de la Cruz, ACS Cent. Sci., 2019, 5, 688–699 CrossRef PubMed.
- X. Qiang, A. Steinhaus, C. Chen, R. Chakroun and A. H. Gröschel, Angew. Chem., Int. Ed., 2019, 58, 7122–7126 CrossRef CAS PubMed.
- Q. Zhang, H. Fan, L. Zhang and Z. Jin, Macromolecules, 2020, 53, 7025–7033 CrossRef CAS.
- X. Wang, S. Liu, S. Cao, F. Han, H. Wang and H. Chen, Macromolecules, 2019, 52, 6698–6703 CrossRef CAS.
- X. Qiang, S. Franzka, G. Quintieri, X. Dai, C. K. Wong and A. H. Gröschel, Angew. Chem., Int. Ed., 2021, 60, 21668–21672 CrossRef CAS PubMed.
- A. Steinhaus, D. Srivastva, X. Qiang, S. Franzka, A. Nikoubashman and A. H. Gröschel, Macromolecules, 2021, 54, 1224–1233 CrossRef CAS.
- R. Deng, F. Liang, P. Zhou, C. Zhang, X. Qu, Q. Wang, J. Li, J. Zhu and Z. Yang, Adv. Mater., 2014, 26, 4469–4472 CrossRef CAS PubMed.
- X. Qiang, S. Franzka, X. Dai and A. H. Gröschel, Macromolecules, 2020, 53, 4224–4233 CrossRef CAS.
- R. Deng, F. Liang, W. Li, S. Liu, R. Liang, M. Cai, Z. Yang and J. Zhu, Small, 2013, 9, 4099–4103 CrossRef CAS PubMed.
- S. Fujii, S. Kameyama, S. P. Armes, D. Dupin, M. Suzaki and Y. Nakamura, Soft Matter, 2010, 6, 635–640 RSC.
- Q. Yuan, T. P. Russell and D. Wang, Macromolecules, 2020, 53, 10981–10987 CrossRef CAS.
- F. Wurm and A. F. M. Kilbinger, Angew. Chem., Int. Ed., 2009, 48, 8412–8421 CrossRef CAS PubMed.
- F. Tu and D. Lee, J. Am. Chem. Soc., 2014, 136, 9999–10006 CrossRef CAS PubMed.
- A. Walther and A. H. E. Müller, Soft Matter, 2008, 4, 663–668 RSC.
- J. Hu, S. Zhou, Y. Sun, X. Fang and L. Wu, Chem. Soc. Rev., 2012, 41, 4356–4378 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: SEM and TEM images of the particles, NMR data, FT-IR data, degree of reaction, interfacial tension values, and pH values of particle suspension. See DOI: https://doi.org/10.1039/d5sc00259a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |