
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluoride-induced redox-switchable behaviour of a palladium(II)/(IV) couple†
Sebastián
Martínez-Vivas
 ,
Macarena
Poyatos
* and
Eduardo
Peris
*
,
Macarena
Poyatos
* and
Eduardo
Peris
*
Institute of Advanced Materials (INAM), Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universitat Jaume I, Av. Vicente Sos Baynat s/n, Castellón, E-12071, Spain. E-mail: poyatosd@uji.es; eperis@uji.es
First published on 7th March 2025
Abstract
We report the preparation of two Pd(II) complexes based on (CCC)-pincer-NHC (NHC = N-heterocyclic carbene) ligands. One of these complexes features an NDI unit (NDI = naphthalene-diimide) attached to the CCC-pincer ligand. Reacting these complexes with bromine results in the formation of the corresponding palladium(IV) complexes [PdBr3(NDI-CCC)] and [PdBr3(CCC)]. The NDI-containing pincer complex exhibits a strong sensitivity to fluoride ions, which can induce a one-electron reduction of the naphthalene-diimide moiety. We demonstrate that the addition of fluoride induces the reduction of the NDI moiety via the formation of hydroxide anions, which are the effective reducing agents of the process. The addition of fluoride significantly affects the reactivity of the NDI-containing palladium complexes. For example, the palladium(IV) complex [PdBr3(NDI-CCC)] can transfer bromide to styrene in a stoichiometric manner, but this reaction is inhibited in the presence of fluoride. Similarly, the palladium(II) complex [PdI(NDI-CCC)] catalyzes the oxidative homocoupling of arylpyridines, but its catalytic activity is quenched when excess fluoride is added. Notably, we demonstrate that this process can be deactivated and reactivated by sequentially introducing an excess of fluoride and NOBF4, revealing a rare instance of a redox-switchable process within a Pd(II)/Pd(IV) catalytic cycle. In contrast, the (CCC)-pincer palladium(II) and (IV) complexes lacking the NDI unit show no sensitivity to fluoride. Our study demonstrates that a simple reagent, such as the fluoride anion, can effectively modulate the reactivity of the Pd(II)/Pd(IV) pair. More broadly, it shows that fluoride serves as a simpler alternative to the metal-based reducing agents commonly used in redox-switchable catalysis.
Introduction
Palladium(IV) complexes have been increasingly recognized as intermediates in many catalytic reactions,1 but these species were initially viewed with skepticism owing to a lack of evidence supporting their viability. This is because high-valent Pd compounds were traditionally considered very unstable under ambient conditions, as they tend to undergo rapid decomposition via reductive elimination processes. Exploiting Pd(II)/Pd(IV) catalysis brings new opportunities in terms of mechanistic information and catalytic applications, because it provides complementary reactivities that are unattainable for Pd(0)/Pd(II) couples.2 Compared to Pd(0)/Pd(II) catalysis, Pd(II)/Pd(IV) processes benefit from the resistance of Pd(IV) species to undergo β-H elimination, and because Pd(IV) compounds undergo facile reductive elimination. This explains why high-valent palladium complexes promote facile carbon–halogen bond-forming reductive elimination, while this process is both kinetically and thermodynamically unfavorable from Pd(II) complexes. The impressive recent advances in Pd(II)/Pd(IV) catalysis related to carbon–halogen bond formation3 and C–H activation1b,4 have stimulated detailed studies to unveil the factors that dictate the energetics of oxidative addition at palladium(II), and reductive elimination from palladium(IV) complexes.1d,2,5It is now widely recognized that Pd(IV) complexes can be stabilized by using rigid multidentate strong electron-donating ligands,2,6 among which bis-chelating3a,b and pincer NHC-based5a,7 ligands (NHC = N-heterocyclic carbene) are gaining increasing attention. We have been recently interested in studying how oxidative addition processes at NHC-based pincer rhodium(I) complexes can be controlled by using supramolecular interactions,8 or by the addition of halides.9 In particular, we observed that pincer-NHC-based ligands functionalized with naphthalene-diimides (NDIs) are highly sensitive to the addition of fluoride or chloride,9,10 and this has a strong influence on the catalytic performance of their related metal complexes. Our studies were based on the fact that NDI-containing compounds are known to engage in strong anion–π interactions,11 thus we initially hypothesized that the combination of pincer ligands with NDIs should constitute an effective way of designing anion-sensitive pincer ligands, which could be further used for approaching new catalytic transformations or for facilitating the stabilization of metal complexes in high oxidation states. In two previous studies, we demonstrated how the addition of halides can be used to modulate the electron-donating strength of NDI-functionalized bis-NHC pincer ligands, thereby influencing the electron richness of the metal centers to which they are bound.9,10 Building on this foundation, we now synthesized two NDI-functionalized (CCC)- and (CNC)-NHC-based pincer complexes of Pd(II), and approached the preparation of their related Pd(IV) complexes by oxidation with molecular bromine. We thought that the stability conferred to the Pd(IV) complex by the rigid NDI-functionalized tridentate pincer ligand could be modulated by increasing the electron donating strength of the ligand by addition of the fluoride anion.9 As we describe herein, the reactivity and catalytic properties of the reported NDI-functionalized pincer complexes of Pd(IV) are highly influenced by the addition of fluoride.
Results and discussion
Scheme 1 shows the synthetic procedure for the preparation of the three Pd(II) bis-NHC complexes 4–6, which are described in the present study. The NDI-functionalized (CCC)-pincer palladium complex 4 was obtained by reaction of the NDI-substituted phenyl bisimidazolium salt [1](I)2 with palladium acetate in the presence of sodium acetate in DMSO at 140 °C (40% yield). By using the same synthetic procedure, but using the methyl-substituted phenyl-bisimidazolium salt [2](I)2, (CCC)-pincer complex 5 was obtained in 45% yield. The (CNC) pincer complex 6 was prepared by reacting the pyridyl-bisimidazolium salt [3](I)2 with silver oxide in CH2Cl2, and then by transmetalating the in situ preformed silver-NHC complex with [PdCl2(COD)] in the presence of Na(BArF4). All three Pd(II) complexes were characterized by NMR spectroscopy and mass spectrometry (see the ESI† file for full details). All three complexes are very stable in solution and in the solid state for long periods of time.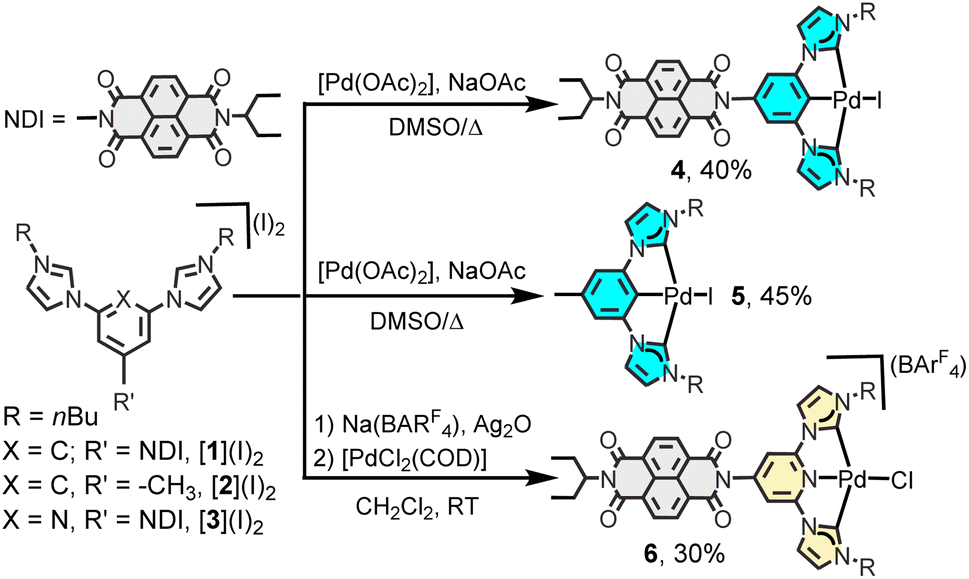 | ||
| Scheme 1 Synthesis of pincer-bis(NHC) Pd(II) complexes. | ||
The structure of the (CNC)-pincer complex 6 was further verified by single crystal X-ray diffraction studies (Fig. 1). The structure shows the pincer coordination of the pyridyl-bis-NHC ligand, which establishes a Pd–N distance of 1.995(6) Å, and Pd–C distances of 2.044(8) and 2.061(7) Å. The angle between the plane formed by the pincer ligand and the NDI moiety is 67.75°. The longer axis of the NDI moiety and the Cl–Pd–N axis establish an angle of 163.54°, thus slightly deviating from linearity.
 | ||
| Fig. 1 Molecular structure of complex 6. Hydrogen atoms and counter anion (BArF4) are omitted for clarity. Carbon atoms in grey, palladium in blue, nitrogen atoms in red, chlorine atom in yellow and oxygen atoms in green. | ||
In order to prepare the related pincer complexes of Pd(IV), three equivalents of Br2 were added to solutions of complexes 4–6 in CH2Cl2, and the resulting mixtures were allowed to react for five minutes at room temperature (see Scheme 2). Under these mild reaction conditions, both 4 and 5 evolved to their related Pd(IV) tribromide complexes 7 and 8, which were obtained in very high yield (>80%). These tribromide complexes were obtained regardless of the presence of the iodo ligands in 4 and 5. The NDI-containing pincer complex of Pd(IV) 7 was also obtained from the preformed pincer Pd–Br complex 9, which was obtained at room temperature by reaction of 4 with an excess of NaBr in acetone. Complexes 7 and 8 were characterized by NMR spectroscopy and mass spectrometry (see the ESI† file for full details). Complex 7 showed great stability in the solid state and in solution, while 8 tended to decompose at room temperature in hours in a solution of chlorinated solvents such as CHCl3 or CH2Cl2. The (CNC)-pincer palladium(II) complex 6 did not produce the desired Pd(IV) complex under these reaction conditions, or even when the reaction was allowed to proceed for a longer reaction time or at higher temperatures. While all Pd(II) complexes 4–6 contain rigid strong electron-donating pincer bis-NHC ligands, the unattainability of the targeted Pd(IV) from 6 is most likely related to the lower electron richness of the metal center due to the cationic nature of the complex. This may also be the reason why Pd(IV) with (CNC)-pincer bis-NHC complexes have not been reported so far.
 | ||
| Scheme 2 Oxidation of Pd(II) complexes with bromine. | ||
The molecular structure of 7 was confirmed by means of single crystal X-ray diffraction studies. The structure (Fig. 2) consists of a phenylene-bis-imidazolylidene ligand with an appended NDI moiety coordinated to palladium in a pincer fashion. Three bromo ligands complete the pseudo-octahedral coordination sphere about the palladium center. The NDI moiety is disposed in a quasiperpendicular orientation with respect to the equatorial plane of the complex, as shown by the angle established between these two planes, which measures 82.5°. The Pd–Ccarbene distances measure 1.966(9) and 2.074(8) Å, while the Pd–Br distances are 2.4694(11), 2.4619(12) and 2.5649(12) Å, the latter one being the one assigned to the bromo ligand trans to the phenylene ring, whose longer distance is attributed to the trans influence exerted by the aryl ligand.
 | ||
| Fig. 2 Molecular structure of complex 7. Hydrogen atoms and solvent molecules (CH2Cl2) were omitted for clarity. Carbon atoms in grey, palladium in blue, nitrogen atoms in red, oxygen atoms in green and bromine atoms in brown. | ||
Given that NDIs are known to be sensitive to the addition of halides, we decided to compare the reactivity of 7 and 8 towards the addition of tetrabutyl ammonium fluoride (TBAF). It is well known that the addition of fluoride to NDI induces the reduction of NDI to the radical anion NDI˙− and sometimes to the dianion NDI2−.12 During some time the ability of fluorides to reduce NDIs was a matter of controversy, but some detailed studies by Gabbai and co-workers proposed that, in the presence of solvents with protons (e.g. acetonitrile, DMSO, DMF), the deprotonated solvent is the actual electron donor for the reduction of NDI.12b More recently, two independent studies by Ghosh12d and Tam and Xu12e proved that, in the absence of protic solvents, fluoride reacts with water (coming from commercial TBAF·3H2O) forming the very stable difluoride anion HF2−. The OH− anion thus produced forms an anion–π complex with the NDI unit, and then the electron transfer from OH− to NDI is produced facilitating the formation of NDI˙− and finally NDI2−.12d
Fig. 3 shows the series of spectra obtained from the reaction of complex 7 upon addition of increasing amounts of TBAF in DMSO-d6. As can be observed from these spectra, the addition of TBAF produces the gradual formation of Pd(II) complex 9. Four equivalents of TBAF are needed for the complete transformation of 7 into 9. This stoichiometry is consistent with previous studies that suggest that two equivalents of fluoride are needed to deprotonate water to form OH− and HF2−, with OH− performing the electron transfer process to NDI.12b,d,e As two electrons are needed in the reduction of Pd(IV) to Pd(II), the whole reduction process requires four equivalents of fluoride. Further addition of TBAF (five equivalents) results in the disappearance of the proton resonances due to the formation of a paramagnetic species, which can be attributed to the reduction of the NDI unit to NDI˙− radical in 9˙−. Subsequent addition of two equivalents of NOBF4 allowed the reoxidation of 9˙− to 9 (Fig. 3, bottom spectrum), thus showcasing the reversibility of the process.
 | ||
| Fig. 3 Selected region of the 1H NMR spectra of complex 7 in DMSO-d6 upon the addition of increasing amounts of TBAF·3H2O showing the formation of the Pd(II) complex 9, and finally the paramagnetic species 9˙−. Subsequent addition of NOBF4 reoxidizes 9˙− to 9 (bottom spectrum). | ||
Under the same conditions, the addition of increasing amounts of TBAF over a DMSO-d6 solution of Pd(IV) complex 8 showed that this Pd(IV) is perfectly stable in the presence of fluoride (no changes were observed in the resulting 1H NMR spectra, see Fig. S41 in the ESI† for details), thus indicating that the presence of the NDI unit is needed to induce the reduction of Pd(IV) to Pd(II). To determine whether a direct connection between the NDI unit and the metal center is required for the reduction of Pd(IV), we conducted an experiment in which an equimolar mixture of compound 8 and N,N′-di-(1-ethylpropyl)-naphthalene-diimide was reacted with four equivalents of TBAF in DMSO-d6. The 1H NMR spectra revealed that the resonances of the NDI protons disappeared, indicating the formation of NDI˙−. However, the signals corresponding to compound 8 remained unchanged (see Fig. S42 in the ESI† for details). This control experiment demonstrates that a direct connection between the NDI unit and the metal is essential for Pd(IV) reduction, and that the electron transfer between the NDI unit and the metal is intramolecular.
In contrast, when either compound 8 or compound 7 was reacted with two equivalents of cobaltocene (Cp2Co), both complexes were readily reduced to their corresponding Pd(II) forms, the bromo-analogue of 5 and 9, respectively (see Fig. S43 in the ESI for details†). Further addition of two equivalents of Cp2Co to compound 9 led to the reduction of the NDI moiety, forming 9˙−. These experiments highlight that, while Cp2Co promotes the indiscriminate reduction of both Pd(IV) complexes, TBAF can be used as a more selective additive to reduce only those complexes containing the NDI unit.
In order to shed some light into the mechanism of the reduction of the NDI-containing Pd(IV) complex 7 into the Pd(II) complex 9, we performed a series of electrochemical and spectroelectrochemical studies. The cyclic voltammetry of complex 7 shows one irreversible cathodic peak at −0.502 V (vs. Fc+/Fc), and two reversible waves at −1.00 and −1.44 V (see Fig. S31 and S32 in the ESI† for details). The first reduction event at −0.502 V is attributed to the one electron reduction of the palladium(IV) center, while the two reversible waves at more negative potentials are due to the one- and two electron reductions of the NDI unit. This experiment indicates that, as the HOMO level of NDI˙− lies above the LUMO of Pd(IV), once the fluoride anion induces the reduction of NDI, then an electron transfer occurs between NDI˙− and the metal center facilitating the reduction of Pd(IV). In the absence of the NDI unit, this process does not occur, and this explains the absence of reactivity between 8 and fluoride.
In order to confirm that the addition of fluoride induces the reduction of the NDI unit, we compared the series of spectra resulting from the UV-Vis spectroelectrochemical (SEC) experiments, and from the UV-Vis titration of 4 with TBAF in DMSO. The spectra resulting from both experiments are shown in Fig. 4. As can be observed from Fig. 4a, the UV-vis spectrum of complex 4 shows a vibronically-resolved band centered at 380 nm, which is attributed to transitions centered at the NDI unit. Upon applying a negative potential of −1200 mV, this band reduces its intensity and new featureless bands centered at 470 and 620 nm appear, which are consistent with the appearance of the one-electron reduced species [PdI(CCC-NDI˙−)] (4˙−). When more negative potentials are applied (Vap = −1600 mV), a further decrease of the intensity of the band at 380 nm is observed, with the concomitant appearance of two bands at 710 and 770 nm, thus confirming the complete formation of 4˙−. A very similar situation is produced when fluoride is added over a solution of 4 in DMSO. As can be observed in Fig. 4b, the addition of five equivalents of TBAF produced a species whose UV-Vis spectrum is quasi-identical to that obtained from the electrochemical one-electron reduction of 4, thus strongly suggesting that [PdI(CCC-NDI˙−)] (4˙−) was formed. Further addition of TBAF (up to 10 equivalents) results in the quantitative formation of (4˙−). This experiment shows that the NDI unit at 4 can be reduced by one electron upon addition of a large excess of fluoride, in agreement with previous studies.12d,e In order to check if the addition of OH− anions would produce a similar effect on the reduction of the NDI unit, we also carried out a reaction between 4 and TBAOH. As can be observed in Fig. S38 of the ESI,† the addition of two equivalents of TBAOH to a DMSO-d6 solution of 4 results in the disappearance of the proton resonances due to the formation of a paramagnetic species, whose UV-Vis spectrum matches perfectly with that of 4˙−. This experiment aligns with all previous studies suggesting that the reduction of the NDI unit by the fluoride anion proceeds through the OH−–π complex with the NDI moiety.12b,d,e
 | ||
| Fig. 4 (a) UV-Vis-SEC monitoring reduction of 4 in dry DMSO (0.25 M [N(nBu)4][PF6]). The solid lines represent the UV-Vis spectra of complex 4 (grey), the partial formation (red) and the complete formation (blue) of the one-electron reduced species 4˙−. (b) UV-Vis spectroscopic changes in the spectrum of 4 observed upon the addition of different amounts of TBAF·3H2O [0 equivalents of TBAF (grey); 5 equivalents of TBAF (red); 10 equivalents of TBAF (blue)]. | ||
We next decided to evaluate the effect of the addition of fluoride on the bromine transfer reaction to styrene. The stoichiometric halogen transfer reactions from Pd(IV) halide-containing complexes to olefins and alkynes have been previously reported by Kraft3a,b and Guo,5a and constitute a process that exemplifies the high tendency of Pd(IV) complexes to form carbon–halogen bonds. The reactions were carried out in a sealed NMR tube at 80 °C in CDCl3, using an excess of 5 equivalents of styrene, and the conversion was determined by 1HNMR spectroscopy based on the formation of the corresponding Pd(II) complexes and in the formation of the related amount of 1,2-dibromoethylbenzene. As can be observed from the time-dependent reaction profiles shown in Fig. 5, after 10 h of reaction the bromine transfer process is produced in 70% for complex 7, while complex 8 only reaches 50% conversion. More interesting is the fact that, in the presence of five equivalents of TBAF, the NDI-containing Pd(IV) complex 7 does not cause the formation of 1,2-dibromoethylbenzene, while under the same reaction conditions 8 shows exactly the same bromination capacity as in the absence of TBAF. This result shows that the bromination abilities of 7 can be quenched in the presence of the fluoride anion, as a consequence of its reduction from Pd(IV) to Pd(II).
 | ||
| Fig. 5 Time-dependent reaction profiles of the bromine transfer reaction from (a) complex 7 and (b) complex 8, to styrene. The reactions were monitored by 1H NMR spectroscopy, by recording the corresponding spectra every 5 minutes. The red empty dots represent evolution of the reaction carried out in the absence of fluoride. The blue triangles in (b) represent the evolution of the reaction in the presence of 5 equivalents of TBAF for the reaction carried out with 8. Addition of 5 equivalents of TBAF to the reaction performed with 7 shows no formation 1,2-dibromoethylbenzene. | ||
We also wanted to test if the addition of fluoride could have any effect on the catalytic properties of the two Pd(II) complexes under study. As a model reaction, we studied the oxidative homocoupling of 2-arylpyridines. This reaction has been studied for almost two decades using Pd,13 Cu,14 Ru15 and Co16-based homogeneous catalysts in combination with various oxidants. The process is considered to be the first evidence of a Pd(IV) mediated C–H bond activation.13a Based on the well-accepted mechanism for the Pd-catalysed dehydrogenative homocoupling of aryl-pyridines, the reaction proceeds through a Pd(II)/Pd(IV) mechanism which involves two distinct C–H activation steps [one at Pd(II) and the other at Pd(IV)] followed by the reductive elimination of the final homocoupled product.13a We first performed the reactions at room temperature using catalysts 4 and 5 (5 mol%) using a 0.11 M solution of 2-phenylpyridine in CD3CN, and using NO+BF4− and I2 as oxidants to promote the Pd(II)–Pd(IV) step. Neither of these two oxidants facilitated the homocoupling process under these reaction conditions. Then, we performed the reactions using complexes 4 and 5 as catalysts under the same reaction conditions using selectfluor (0.66 mmol) as oxidant. We decided to use selectfluor because this reagent has been used widely as an oxidant of transition metals, especially for facilitating catalytic processes involving selective oxidative C–H functionalization,17 and it has also been used to facilitate the oxidative homocoupling of arenes using palladium(II) complexes with pincer-type ligands.18 As mentioned before, all reactions were performed at room temperature and their progress was monitored by 1H NMR spectroscopy. Under these conditions, the reactions followed zeroth order kinetics with respect to phenyl-pyridine (see Fig. S47 in the ESI† for details). This most likely indicates that the substrate is coordinated to the metal in the resting state of the process, and that the product is not released from the coordination sphere of the complex in the rate-determining step (RDS) of the cycle. With this data in hand, the oxidation from Pd(II) to Pd(IV) is most likely the RDS of the catalytic cycle. Analysis of the reaction rates at three different catalyst concentrations allowed us to determine that the reaction follows first order kinetics with respect to the catalyst (see Fig. S46 in the ESI† for details). Complexes 4 and 5 show quasi-identical reaction rates for the dimerization of 2-phenyl pyridine [(2.8 ± 0.1) × 10−6 and (3.0 ± 0.1) × 10−6 M s−1, for 4 and 5 respectively]. We next wanted to test the effect of the addition of TBAF on the reaction rate of this process. The reactions were performed using both catalysts 4 and 5, under the same standard reaction conditions (5 mol% of catalyst loading, room temperature in CD3CN), but with the addition of five equivalents of TBAF with respect to the catalyst. As can be observed in the reaction profiles shown in Fig. 6a and b, the addition of TBAF on the reaction carried out with 5 did not have any effect on the reaction rate, while the rate for the reaction carried out with the NDI-appended catalyst 4 was reduced by a factor of 0.5 when 5 equivalents of fluoride were added. This partial inhibition of the catalytic activity of 4 could be due to the partial reduction of the NDI moiety of the catalyst, in agreement with the UV-Vis spectroscopic changes observed upon addition of fluoride as discussed above (Fig. 4b), which showed that the addition of 5 equivalents of TBAF was not enough to produce the complete reduction of NDI. With this result in hand, we performed another reaction using catalyst 4 and added ten equivalents of TBAF. Under these reaction conditions, the catalytic activity of 4 was almost completely quenched, as the reaction rate is five times slower than in the absence of fluoride, as can be observed in Fig. 6b.
 | ||
| Fig. 6 Time-dependent reaction profiles of the catalytic dehydrogenative homocoupling of 2-phenylpyridine using 5 (a) and 4 (b) as catalysts. Red empty dots represent the profiles for the reactions using the catalysts (5 mol%) in the absence of additives. Blue solid dots and green squares represent the profiles for the reactions carried out with the catalyst +5 and +10 equivalents of TBAF, respectively. All reactions were performed in CD3CN at room temperature, and the evolution was monitored by 1H NMR spectroscopy. | ||
Finally, we wanted to see if the influence of fluoride addition on the activity of 4 was reversible. In order to do this, we performed an experiment in which we followed the homocoupling of 2-phenylpyridine using catalyst 4, and then we sequentially added ten equivalents of TBAF and NO+BF4−. As can be observed in the reaction profile shown in Fig. 7, the addition of TBAF induced the complete inhibition of the activity of the catalyst, which was fully recovered after addition of ten equivalents of NO+BF4−. With these results in hand, a question that remains is whether the catalytic switchable behavior is facilitated by the NDI˙−/NDI redox couple or the Pd(II)/Pd(IV) couple, as the process is facilitated by sequentially adding a reducing agent (or in this case fluoride, which acts as a proxy of a reducing reagent), and an oxidant (NO+) which, in principle, could have an effect on both redox pairs. The answer to this question can be found in the following experimental results: (i) the inhibition of the activity of the catalyst is explained as a consequence of the one-electron reduction of the NDI moiety at the catalyst, which avoids the formation of the Pd(IV) species (catalyst 5, lacking the NDI unit, is not affected by the addition of fluoride); (ii) the recovery of the activity of the catalyst after the addition of NO+BF4− is due to the reoxidation of the NDI˙− radical formed after addition of fluoride, because NO+ is not a strong enough oxidant to facilitate the Pd(II)–Pd(IV) step (as we mentioned before, the homocoupling reaction does not work when NO+ alone is used as an oxidant); (iii) selectfluor, while being an oxidant, does not seem to react with the one-electron reduced NDI unit, as it is not consumed after addition of ten equivalents of TBAF; and (iv) after reactivating the catalyst upon addition of NO+BF4−, the Pd(II)–Pd(IV) step of the catalytic cycle is facilitated by selectfluor, which remains the active transition metal oxidant. All this means that all the fluoride-induced redox-switchable properties of 4 are centered at the NDI unit rather than at the Pd center.
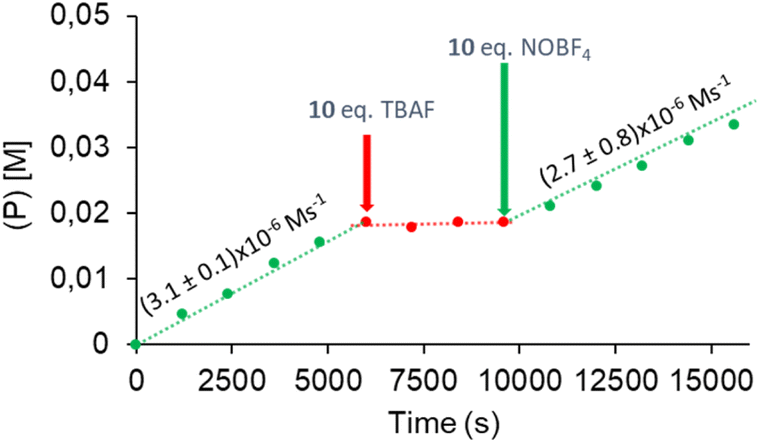 | ||
| Fig. 7 Time-dependent reaction profile of the catalytic dehydrogenative homocoupling of 2-phenylpyridine initiated with 4 as catalyst (5 mol%), and then deactivated and re-activated by sequentially adding TBAF (10 equivalents with respect to the catalyst) and NO+BF4− (10 equivalents with respect to the catalyst). The reaction was performed in CD3CN at room temperature. Product yields determined by 1H NMR spectroscopy. | ||
In order to further explore the scope of 4 in the coupling of aryl-pyridines, we also tested its activity in the homocoupling of substituted phenyl pyridines, such as 2-(p-tolyl)-pyridine and 2-(p-methoxyphenyl)-pyridine and also phenanthroline, where it produced the expected homocoupling products with yields ranging from 80–90% after 12 h of reaction, except for 2-(p-tolyl)-pyridine, which was converted into its dimer in only 52% yield.
Conclusions
In summary, we synthesized three different bis-NHC-based pincer complexes of Pd(II) and demonstrated how oxidation to their corresponding Pd(IV) complexes was strongly influenced by the type of pincer ligand coordinated to the metal. The neutral Pd(II) complexes with a phenylene-bis-NHC pincer ligand were easily oxidized to their related [PdBr3(CCC)] Pd(IV) complexes upon reaction with Br2. In contrast, the Pd(II) complex with a pyridyl-bis-NHC pincer ligand showed limited oxidation to Pd(IV), likely due to the lower electron richness of the metal in the cationic complex.We also investigated the reactivity of the [PdBr3(CCC)] Pd(IV) complexes, which varied significantly depending on whether the (CCC)-pincer ligand was attached to an NDI unit or not. Upon the addition of an excess of fluoride, the NDI-containing complex 7 was rapidly reduced to its corresponding Pd(II) complex 9, with the formula [PdBr(NDI-CCC)]. In contrast, compound 8, which lacks the NDI unit, remained stable even in the presence of an excess of fluoride. The sensitivity of 7 to fluoride was further demonstrated in two model reactions with organic substrates. The bromine transfer from 7 to styrene was completely inhibited by an excess of fluoride, and while both 4 and 5 were active catalysts in the dehydrogenative homocoupling of 2-arylpyridines, only the activity of 4 was inhibited by an excess of fluoride.
These observations suggest that fluoride induces the reduction of the NDI unit in the catalyst, which is followed by two successive electron transfers from the NDI anion (NDI˙−) to Pd(IV), leading to the reduction of Pd(IV) to Pd(II). It is also important to note that, consistent with previous studies,12b,d,12e our work provides additional experimental evidence supporting the idea that fluoride anions reduce NDI through the formation of hydroxide anions. These hydroxide anions then form an OH−–π complex with the NDI moiety, facilitating electron transfer from OH− to NDI and promoting the formation of the NDI˙− radical.
In practical terms, our findings demonstrate that the redox transition between the Pd(II)/Pd(IV) couple can be easily modulated by adding fluoride to NDI-containing complexes, providing a novel approach to control this critical chemical step. Additionally, our results highlight how combining NDI units with metal complexes can reveal new reactivity patterns that have yet to be explored. This method offers a simpler alternative to traditional metal-based reducing agents and may pave the way for applications in adaptive catalysis and sensor development.
Finally, we demonstrated that the catalytic activity of the NDI-containing catalyst 4 could be switched on and off by sequentially adding excess fluoride and NO+BF4−. To the best of our knowledge, this is the first example of switchable catalysis based on a Pd(II)/Pd(IV) cycle. Furthermore, this sequence represents one of the rare instances of a halide-induced redox-switchable process, a phenomenon previously reported only once.10
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
S. Martínez-Vivas prepared the complexes and carried out all experimental work. M. Poyatos and E. Peris designed and supervised the study. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Ministerio de Ciencia y Universidades (PID2021-127862NB-I00) and the Universitat Jaume I (UJI-B2021-39). We are grateful to the Serveis Centrals d'Instrumentació Científica (SCIC-UJI) for providing spectroscopic facilities.Notes and references
- (a) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177 CrossRef CAS PubMed; (b) J. J. Topczewski and M. S. Sanford, Chem. Sci., 2015, 6, 70 RSC; (c) P. Sehnal, R. J. K. Taylor and I. J. S. Fairlamb, Chem. Rev., 2010, 110, 824 CrossRef CAS PubMed; (d) L. M. Xu, B. J. Li, Z. Yang and Z. J. Shi, Chem. Soc. Rev., 2010, 39, 712 RSC.
- B. Zhang, X. C. Yan and S. Guo, Chem.–Eur. J., 2020, 26, 9430 CrossRef CAS PubMed.
- (a) A. S. McCall, H. W. Wang, J. M. Desper and S. Kraft, J. Am. Chem. Soc., 2011, 133, 1832 CrossRef CAS PubMed; (b) A. S. McCall and S. Kraft, Organometallics, 2012, 31, 3527 CrossRef CAS; (c) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (d) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (e) T. Furuya, H. M. Kaiser and T. Ritter, Angew. Chem., Int. Ed., 2008, 47, 5993 CrossRef CAS PubMed; (f) T. Furuya and T. Ritter, J. Am. Chem. Soc., 2008, 130, 10060 CrossRef CAS PubMed; (g) T. Furuya, D. Benitez, E. Tkatchouk, A. E. Strom, P. P. Tang, W. A. Goddard and T. Ritter, J. Am. Chem. Soc., 2010, 132, 3793 CrossRef CAS PubMed.
- (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (b) K. Yamamoto, J. K. Li, J. A. O. Garber, J. D. Rolfes, G. B. Boursalian, J. C. Borghs, C. Genicot, J. Jacq, M. van Gastel, F. Neese and T. Ritter, Nature, 2018, 554, 511 CrossRef CAS PubMed; (c) D. C. Powers, D. Y. Xiao, M. A. L. Geibel and T. Ritter, J. Am. Chem. Soc., 2010, 132, 14530 CrossRef CAS PubMed.
- (a) X. Yan, H. Wang and S. Guo, Angew. Chem., Int. Ed., 2019, 58, 16907 CrossRef CAS PubMed; (b) M. Nappi and M. J. Gaunt, Organometallics, 2019, 38, 143 CrossRef CAS.
- J. Vicente, A. Arcas, F. Julia-Hernandez and D. Bautista, Inorg. Chem., 2011, 50, 5339 CrossRef CAS PubMed.
- (a) H. B. Li, B. Zhang, R. Feng and S. Guo, Dalton Trans., 2024, 53, 11470 RSC; (b) D. C. Najera, G. E. Martinez and A. R. Fout, Organometallics, 2023, 42, 832 CrossRef CAS PubMed.
- S. Martinez-Vivas, M. Poyatos and E. Peris, Angew. Chem., Int. Ed., 2023, 62, e202307198 CrossRef CAS PubMed.
- S. Martínez-Vivas, D. G. Gusev, M. Poyatos and E. Peris, Angew. Chem., Int. Ed., 2023, 62, e202313899 CrossRef PubMed.
- S. Martínez-Vivas, S. Gonell, M. Poyatos and E. Peris, ACS Catal., 2024, 7600 CrossRef.
- (a) M. R. Ajayakumar, P. Mukhopadhyay, S. Yadav and S. Ghosh, Org. Lett., 2010, 12, 2646 CrossRef CAS PubMed; (b) M. R. Ajayakumar, D. Asthana and P. Mukhopadhyay, Org. Lett., 2012, 14, 4822 CrossRef CAS PubMed; (c) J. F. Zhao, G. Li, C. Y. Wang, W. Q. Chen, S. Chye, J. Loo and Q. C. Zhang, RSC Adv., 2013, 3, 9653 RSC; (d) A. Mitra, R. J. Clark, C. T. Hubley and S. Saha, Supramol. Chem., 2014, 26, 296 CrossRef CAS; (e) M. Giese, M. Albrecht and K. Rissanen, Chem. Commun., 2016, 52, 1778 RSC; (f) Y. J. Zhao, Y. Cotelle, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 4270 CrossRef CAS PubMed.
- (a) S. Guha, F. S. Goodson, L. J. Corson and S. Saha, J. Am. Chem. Soc., 2012, 134, 13679 CrossRef CAS PubMed; (b) G. Belanger-Chabot, A. Ali and F. P. Gabbai, Angew. Chem., Int. Ed., 2017, 56, 9958 CrossRef CAS PubMed; (c) S. Saha, Acc. Chem. Res., 2018, 51, 2225 CrossRef CAS PubMed; (d) B. Chowdhury, S. Sinha, R. Dutta, S. Mondal, S. Karmakar and P. Ghosh, Inorg. Chem., 2020, 59, 13371 CrossRef CAS PubMed; (e) T. L. D. Tam and J. W. Xu, Chem. Commun., 2019, 55, 6225 RSC.
- (a) K. L. Hull, E. L. Lanni and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 14047 CrossRef CAS PubMed; (b) J. M. Racowski, A. R. Dick and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 10974 CrossRef CAS PubMed; (c) F. Saito, H. Aiso, T. Kochi and F. Kakiuchi, Organometallics, 2014, 33, 6704 CrossRef CAS; (d) W. B. Liu, Y. Q. Zhu and C. J. Li, Synthesis, 2016, 48, 1616 CrossRef CAS; (e) J. Vána, J. Bartácek, J. Hanusek, J. Roithová and M. Sedlák, J. Org. Chem., 2019, 84, 12746 CrossRef PubMed; (f) S. R. Whitfield and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 15142 CrossRef CAS PubMed.
- (a) X. Chen, G. Dobereiner, X. S. Hao, R. Giri, N. Maugel and J. Q. Yu, Tetrahedron, 2009, 65, 3085 CrossRef CAS; (b) S. Strekalova, A. Kononov, I. Rizvanov and Y. Budnikova, RSC Adv., 2021, 11, 37540 RSC.
- (a) X. Y. Guo, G. J. Deng and C. J. Li, Adv. Synth. Catal., 2009, 351, 2071 CrossRef CAS; (b) T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 15640 CrossRef CAS PubMed; (c) M. Muntzeck, F. Pippert and R. Wilhelm, Tetrahedron, 2020, 76, 131314 CrossRef CAS.
- Y. L. Xie, D. D. Xu, W. W. Sun, S. J. Zhang, X. P. Dong, B. Liu, Y. B. Zhou and B. Wu, Asian J. Org. Chem., 2016, 5, 961 CrossRef CAS.
- K. Yang, M. Song, A. I. M. Ali, S. M. Mudassir and H. Ge, Chem.–Asian J., 2020, 15, 729 CrossRef CAS PubMed.
- L. A. Wright, E. G. Hope, G. A. Solan, W. B. Cross and K. Singh, Dalton Trans., 2015, 44, 7230 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details dealing with the characterization, electrochemical and spectroelectrochemical measurements, reactivity studies and catalytic experiments. The data related to the crystallographic studies. CCDC 2387767, 2387768 and 2387900. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08203f |
| This journal is © The Royal Society of Chemistry 2025 |