
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd(μ-L)4Pt vs. Pd(μ-L)4RuCl2: chlorido ancillary ligands as defining factors in the host–guest interactions of M(μ-L)4M′ heterodimetallic supramolecular architectures†
Hayden B.
Gearing
 a,
Monika
Cziferszky
b,
Tilo
Söhnel
ac,
L. James
Wright
a,
James D.
Crowley
*cd and
Christian G.
Hartinger
*a
a,
Monika
Cziferszky
b,
Tilo
Söhnel
ac,
L. James
Wright
a,
James D.
Crowley
*cd and
Christian G.
Hartinger
*a
aSchool of Chemical Sciences, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: c.hartinger@auckland.ac.nz
bInstitute of Pharmacy/Pharmaceutical Chemistry, University of Innsbruck, Centre for Chemistry and Biomedicine, Innrain 80–82/IV, 6020 Innsbruck, Austria
cMacDiarmid Institute for Advanced Materials and Nanotechnology, Victoria University of Wellington, PO Box 600, Wellington 6140, New Zealand
dDepartment of Chemistry, University of Otago, PO Box 56, Dunedin 9054, NewZealand. E-mail: jcrowley@chemistry.otago.ac.nz
First published on 24th March 2025
Abstract
In supramolecular architectures, the interactions between host and guest molecules are governed by non-covalent forces such as hydrogen (H) bonding, hydrophobic and electrostatic interactions. We alter here the cavity microenvironment to control the interactions between guest and host molecules and study the effects of introducing axial chlorido ligands through the use of an octahedral building block in M(μ-L)4M′ architectures. We prepared the heterodimetallic Pd(μ-L)4Pt C4Pt and Pd(μ-L)4RuCl2C4Ru architectures and demonstrated the role of ‘classic’ non-covalent forces in their host–guest chemistry with anionic and neutral molecules, while the cages also underwent disassembly and reassembly upon addition of external stimuli. This culminated in the isolation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest complex between C4Pt and the dianionic 1,5-naphthalenedisulfonate which was characterized by single crystal X-ray diffraction studies. These showed the guest occupied the central cavity and was held in place by H bonding. The endo-chlorido ligand in C4Ru played an important role in the capture of neutral guest molecules. In particular, it allowed for finetuning of the cavity properties of the supramolecular architectures by limiting the formation of H bonds and restricting the cavity size while offering alternative interactions.
:1 host–guest complex between C4Pt and the dianionic 1,5-naphthalenedisulfonate which was characterized by single crystal X-ray diffraction studies. These showed the guest occupied the central cavity and was held in place by H bonding. The endo-chlorido ligand in C4Ru played an important role in the capture of neutral guest molecules. In particular, it allowed for finetuning of the cavity properties of the supramolecular architectures by limiting the formation of H bonds and restricting the cavity size while offering alternative interactions.
Introduction
In recent decades, self-assembled metallosupramolecular architectures (MSAs), such as M2L4-type cages, have been designed and studied for a wide range of applications.1–5 For example, [Pd2L4]4+ (or [Pd(μ-L)4Pd]4+) MSAs have been examined for their use as catalysts,6–8 as luminescent materials,9,10 in medical applications,11,12 for molecular recognition and, of particular interest, for their ability to encapsulate drugs and other molecules in host–guest chemistry.5,13–15 In most cases for these cage-like hosts, binding of guest molecules is facilitated by non-covalent forces such as hydrogen (H) bonding, hydrophobic and coulombic interactions.16–19The synthetic principles that direct the construction of high-symmetry architectures are well-understood.3,20 These same strategies have also been used to great effect in installing secondary metal centers, which serve as structural and/or behavioral components.21–26 However, recently lower-symmetry MSAs have been attractive targets as they may facilitate the introduction of different chemical properties into a single cage molecule. To this end many heteroditopic ligand scaffolds have been employed to generate lower symmetry architectures such as heteroleptic,27–31 pseudo-heteroleptic,32–34 and multi-cavity Pd(II)-based architectures.18,29,35–39
Another strategy, however, is to introduce non-symmetry by installation of different metal centers at either end of cage architectures to yield, for example, heterodimetallic MM′L4 compounds. Such a strategy has been implemented to incorporate metal centers such as Fe,40 Cu,41 Ru,42 Pt43–46 and others, into [PdML4] systems. The first syntheses of heterodimetallic [Pd(μ-L)4Pt] cages were reported by Lisboa et al. using subcomponent self-assembly.43 In a different approach, Pearcy et al. established a stepwise approach by sequential coordination of initially the more kinetically inert Pt(II) center followed by the more labile Pd(II) center to heterotopic imidazole and pyridyl N-donor scaffolds.46 Preston and co-workers recently introduced an alternative Pt-based building block for the construction of kinetically robust Pt2L4 cages.47 In such MSAs, the different coordination properties of two metal centers or of different coordination motifs in ditopic ligands can be exploited for stimulus-induced partial (and reversible) disassembly of the host molecule, and release of a molecular guest.17,40,43 For example, a [Pd(μ-L)4Pt]4+ cage was found to bind benzoquinone and 2,6-diaminoanthraquinone guests strongly,43 and also showed interactions with cisplatin and 5-fluorouracil. Only a few other examples of such MSAs have been reported since.44,46–49
Based on a preliminary study, in which we explored the preparation and sulfonate binding ability of [Pd(μ-L)4Pt]4+ in comparison to cages featuring an octahedral trans-Ru(II)Cl2 moiety in place of the Pt center ([Pd(μ-L)4RuCl2]2+; Fig. 1), we develop here a broad understanding of how the introduction of such a moiety to the cavity affects the guest binding properties of heterodimetallic architectures. Furthermore, we assessed the stimulus-responsive abilities of these cages for disassembly and reassembly, and the effect on binding of anionic guests.
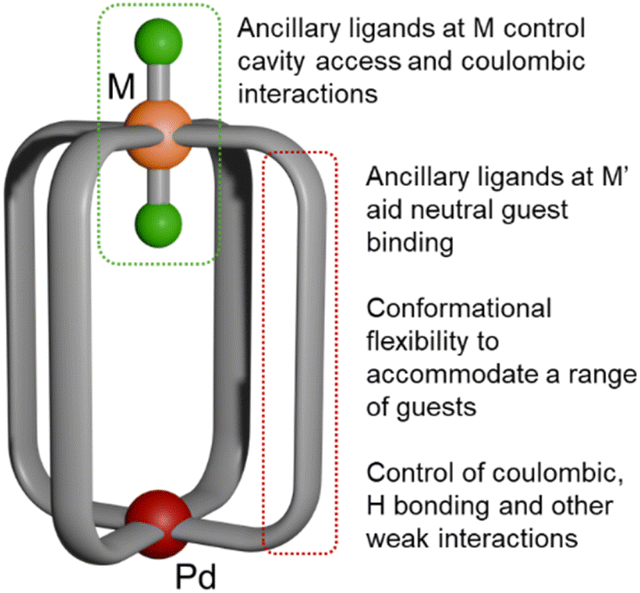 | ||
| Fig. 1 Schematic structure of Pd(μ-L)4RuCl2 cages. Pd(II), red; Ru(II), orange; Cl, green; bridging ligand, grey. | ||
Results and discussion
Making use of a modular approach in the subcomponent self-assembly of heterobimetallic MSAs,43,44,46–49 we aimed to introduce rational changes to PdML4 MSAs by altering the Pd⋯M distances through variation of the bridging ligands, exploring the impact of ancillary Cl ligands on M and the effects of coulombic, H bonding and hydrophobic interactions. We previously reported Pd(μ-L)4RuCl2 MSAs C1Ru–C3Ru (Fig. 2) that allowed discrimination between small, similarly sized, anionic guest molecules by restricting the cavity size with axially-coordinated ligands to the Ru center while maintaining the stimulus-responsive reversible opening and closing of PdPt analogs.49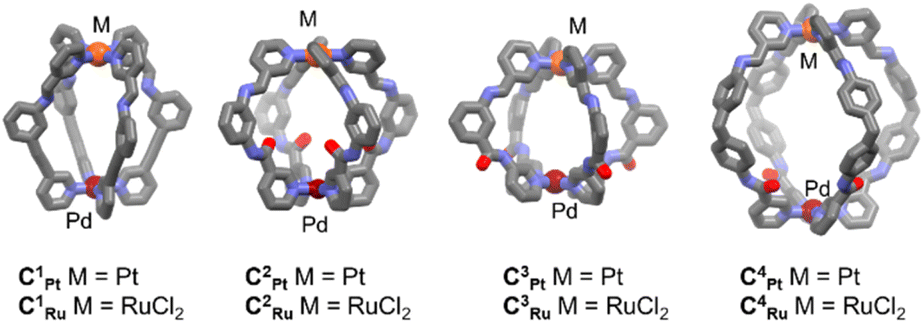 | ||
| Fig. 2 Model structures for the systematic variation of Pd(μ-L)4M MSAs C1Pt–C4Pt and C1Ru–C4Ru which feature different functionalities to control guest binding. M = Pt or RuCl2. | ||
Extension of the Pd⋯M distances compared to previously reported heterodimetallic MSAs C1Pt–C3Pt and C1Ru–C3Ru, and therefore enlargement of the cage cavity, was achieved by non-symmetric functionalization of 4,4′-methylenedianiline (MDA). MDA has been used to generate homometallic MSA with large accessible cavities.50,51 While the MDA unit can undergo hydrophobic interactions with guest molecules to accommodate a wider array of guests, we introduced an amide motif at the Pd end of the cage instead of the alkynyl linker that was used previously.49 The amide function can in principle act as either an H bond donor or acceptor. The second metal coordination site was built using an imine formation reaction between the free amino groups of the MDA units and the 3-pyridinecarboxaldehyde (3-PA) complex precursors [Pt(3-PA)4](BF4)2 (Ptpyald)43 or trans-[Ru(3-PA)4Cl2] (Rupyald).49 The synthesis of the non-symmetrically functionalized precursor 1 was achieved via amide coupling with nicotinic acid and N-Boc-4,4′-methylenedianiline in the presence of triethylamine (TEA) and O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) under reflux. Removal of the protecting Boc group was achieved in a 1:5 trifluoroacetic acid:chloroform mixture to ultimately give pyridylamine derivative 2 (Scheme 1). Preparation of the cages C4Pt and C4Ru was accomplished through self-assembly between 2, [Pd(CH3CN)4](BF4)2, and Ptpyald or Rupyald in stoichiometric ratios of 4:1:1 at room temperature in dry DMSO, with a reaction time of ca. 24 h. The longer reaction time required relative to previously reported systems is likely due to the higher degree of flexibility of the linker in this case and the need to self-correct.51 This helps to overcome the initial formation of the kinetically favored [Pd2(2)4]4+ complex where ligand 2 is coordinated through the pyridyl and aniline N atoms, which has been previously observed by Chand and co-workers (Fig. S31–S33†).32
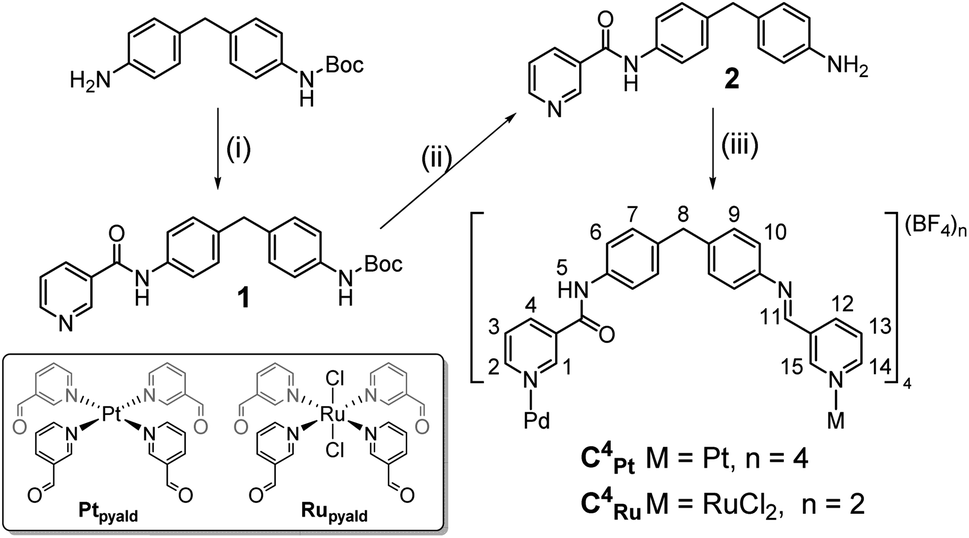 | ||
| Scheme 1 Synthetic route to low-symmetry PdPt C4Pt and PdRu C4Ru cages. Reaction conditions: (i) nicotinic acid, HATU, TEA, CH3CN, reflux, 16 h. (ii) TFA:CHCl3 (1:5), 3 h. (iii) [Pd(CH3CN)4](BF4)2, Ptpyald or Rupyald, DMSO, 24 h. | ||
The formation of C4Pt and C4Ru was tracked by 1H NMR spectroscopy, with each cage showing quantitative conversion after 24 h. The 1H NMR spectra showed the steady diminution of the aldehyde and amine peaks from 2 and Ptpyald or Rupyald, respectively, with the appearance of imine signals at ca. δ 8.5–8.6 ppm. In addition, the pyridyl protons (H-1–H-4) shifted downfield upon coordination of the pyridyl-amide motifs to the Pd(II) center. Electrospray ionization-mass spectrometry (ESI-MS) data obtained showed a major peak for [C4Pt(–4BF4)]4+ at m/z 467.6310 (m/zcalc 467.6311) with an isotopic pattern consistent with a PdPt system (Fig. S22†). For C4Ru a major peak at m/z 924.2015 was observed (m/zcalc 924.2011) corresponding to [C4Ru(–2BF4)]2+ (Fig. S30†). 1H DOSY NMR spectroscopy confirmed the resonances observed for C4Pt and C4Ru in each case corresponded to a single species, with diffusion coefficients of 1.16 × 10−10 and 2.78 × 10−10 m2 s−1, respectively. These values are consistent with larger assemblies compared to those of previously reported hetero- and homodimetallic cages.43,44,46,48,50,51C4Pt was isolated in high yield (92%) after the addition of ethyl acetate to the DMSO solution, whereas C4Ru could not be isolated in pure form by precipitation. However, NMR studies showed quantitative formation of this compound in solution and therefore solutions of C4Ru in DMSO or DMSO-d6 were prepared as stock solutions for further studies. The molecular structure of C4Pt (Fig. 3) was determined by single crystal X-ray diffraction analysis. The structure featured three of the amide carbonyl moieties facing outwards, allowing the amide-NH groups to serve as H bond donors to encapsulated BF4− within the cavity, an orientation which is essential for binding of guests with H bond accepting properties (vide infra). Near the Pt center, three of the imine lone pairs are not available as H bond acceptors for guests due to their exo orientation. The structure is the largest heterodimetallic PdPtL4 MSA reported so far with the Pd⋯Pt distance measuring at 15.48 Å, while C1Pt–C3Pt (as well as C2Ru and C3Ru) were in the range of 9.5–11.5 Å.49 In contrast, the molecular structure of C4Ru (Fig. 3) features all four carbonyl oxygen atoms oriented towards the cavity and no classical H bond donors available for host–guest binding, while ‘non-classic’ short C–H⋯O bonds were observed to a co-crystallized guest DMSO molecule. In solution, the cages are more flexible, as shown by cross-peaks for the amide NH and imine CH protons to the neighboring endocyclic pyridyl protons in the NOESY spectra of both C4Pt and C4Ru (Fig. S18 and S26†). Although the Pd⋯M distances for C4Ru and C4Pt, are similar (15.21 and 15.48 Å respectively) the presence of the endo-chlorido ligand in C4Ru reduces its cavity size considerably (Fig. 3). A significant difference was found in the packing for the two cages. They both stack linearly along the Pd⋯M axis, however, the intermolecular Pd⋯Ru distance is significantly shorter at 5.76 Å compared to the corresponding Pd⋯Pt spacing of 8.36 Å. This appears driven by electrostatic interactions between the exo-chlorido ligand of one C4Ru cage and the secondary coordination sphere of the Pd center of a neighboring molecule of C4Ru (Ru–Cl⋯Pd 3.34 Å).
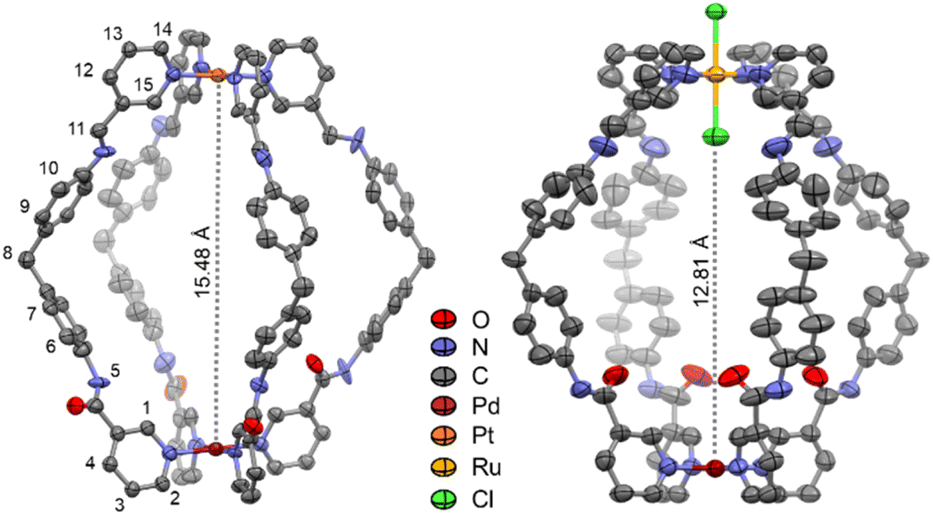 | ||
| Fig. 3 Molecular structures of C4Pt (left) and C4Ru (right). The Pd⋯Pt and Pd⋯Cl intra-cage distances are highlighted by dashed lines. Hydrogen atoms, solvents and counteranions have been omitted for clarity. Due to the space group for C4Pt, the representation is a combination of the Pt-imine and Pd-amide ends, superimposed at the bridging methylene groups (see Fig. S60† for clarity). | ||
Disassembly and reassembly reactions
Similar to the previously reported Pd(μ-L)4Pt and Pd(μ-L)4RuCl2 cage assemblies,49 the larger cavity cages C4Pt and C4Ru showed selective disassembly at the palladium center upon addition of 4-dimethylaminopyridine (DMAP). Addition of DMAP to C4Pt or C4Ru in DMSO-d6 resulted in decreasing intensities of the signals assigned to the heterodimetallic architectures in the 1H NMR spectra. This was accompanied by the appearance of resonances attributed to the monometallic ‘open-form’ [PtL4]2+ and [RuCl2L4] complexes, as well as a new set of peaks corresponding to [Pd(DMAP)4]2+ (Fig. 4). | ||
Fig. 4 Stacked 1H NMR spectra (DMSO-d6, 298 K, 400 MHz) of the disassembly of C4Pt upon addition of DMAP, and subsequent reassembly on addition of p-TsOH. Shifts in peaks are indicated with dashed lines. Spartan ′24®52 MMFF models of the closed and open forms of C4Pt have been included for visualization.  [Pd(DMAP)4](BF4)2, [Pd(DMAP)4](BF4)2,  (HDMAP)(TsO). (HDMAP)(TsO). | ||
Upon addition of p-toluenesulfonic acid (p-TsOH), the DMAP in [Pd(DMAP)4]2+ is protonated and the Pd ion re-coordinates to the accessible pyridyl nitrogen donors to reassemble the respective MSA. This process was slower than observed previously for [PdPtL4]4+ cages (1 day compared to several hours for the smaller MSAs), most likely due to the greater length and higher flexibility of the bridging ligands.43,46,49
Host–guest chemistry
For the PdPt and PdRu MSAs C1Pt–C3Pt and C1Ru–C3Ru, the anionic guests mesylate (MsO) and tosylate (TsO) were found to bind in the cavities of C3Pt and/or C3Ru, and interactions with the neutral guests 5-fluorouracil and, to a minor extent, cisplatin were also observed.49 The binding of guest molecules was influenced by the presence of H bond accepting and H bond donating groups, by coulombic and hydrophobic interactions, as well as steric constraints. In particular, the presence of the axial, endo-Cl ligand in C3Ru restricted the accessibility of the cage and partially compensated for the positive charge at the Ru center.With the design of C4Pt and C4Ru, it was anticipated that the steric constraints associated with guest binding would be relaxed and this would allow for easier access of guests into the cavities. Therefore, C4Pt and C4Ru were studied for their ability to interact with the MsO (CH3SO3−) and TsO (C7H7SO3−) mono-anions as well as the larger di-anion 1,5-naphthalene disulfonate (NDS; C10H6S2O62−) by 1H NMR spectroscopy and host–guest modelling. Larger disulfonates have been found by others to bind into the cavities of Pd2L4 cages.53–57 For the mono-anionic guests, the modelling studies suggested that two guests would fit into the cavities of C4Pt and C4Ru, whereas for the larger di-anionic NDS a single molecule would fill the space (Fig. S63 and S64†). In contrast, it was found that NDS could not bind effectively in the cavity of C4Ru and the smaller C3Pt (Fig. S65†). The binding constants (Table 1) were calculated from the NMR studies using the stoichiometries inferred by the Spartan ′24® docking models (ESI†).
| MSA | Binding constants M−1 | ||
|---|---|---|---|
| MsO | TsO | NDS | |
| C1Ru | 75 ± 2 | 297 ± 66 | — |
| C2Pt | 279 ± 11 | 237 ± 13 | — |
| C2Ru | 209 ± 4 | 189 ± 7 | — |
| C3Pt | (1.30 ± 0.18) × 104 | (3.50 ± 1.48) × 104 | — |
| C3Ru | 314 ± 5 | 93 ± 3 | — |
| C4Pt | 453 ± 5 | 581 ± 10 | > 105 |
| C4Ru | 283 ± 32 | 370 ± 44 | 1053 ± 168 |
Titrations of MsO into a solution of C4Pt in DMSO-d6 monitored by 1H NMR spectroscopy showed interactions both inside and outside of the cage (Fig. S36 and S37†). For the interior protons H-1 and H-15, a plateau in the change of chemical shifts was reached at [G]:[H] ratios of ca. 6, whereas the exterior protons H-2 and H-14 did not plateau up to a [G]:[H] ratio of ca. 10. For C4Ru, only very minor shifts for the interior protons were observed (Fig. S46 and S47†), indicating the role the endo-chlorido ligand plays in blocking H-15 from forming H bonds to guest molecules. In general, the binding constants observed for C4Pt and C4Ru were very small in comparison to C3Ru and, in particular, to C3Pt. The weaker binding may be due to a mismatch between cavity and guest size.49 The larger TsO guest interacted with protons inside and outside the cavity of C4Pt without reaching a plateau within 10 eq. of guest added (Fig. S38 and S39†), while it mainly interacted with the exo-cavity proton H-2 of C4Ru (Fig. S48 and S49†). The binding constants were slightly larger for both MSAs than observed for MsO which may be explained by favorable hydrophobic interactions, as also seen between two molecules of TsO in the cavity of C4Pt in modelling studies (Fig. S64†). As for MsO, the interaction with C3Pt was significantly more pronounced than for C4Pt (Table 1). The much larger cavity of C4Pt may not favor the encapsulation of MsO and TsO. The endo-chlorido ligand of the RuCl2 motif of C4Ru reduces the cavity size compared to C4Pt and this is reflected by the smaller binding constant observed for TsO.
Significant shifts of signals in the 1H NMR spectra were observed when titrating NDS into solutions of C4Pt (e.g., H-15 shifted by ca. 0.4 ppm downfield, the amide proton H-5 by 0.1 ppm upfield), which can be explained by a combination of the sulfonate groups being excellent H bond acceptors, and also having a doubly negative charge compared to the single negative charges of MsO and TsO. The binding constant K obtained for NDS encapsulation by C4Pt was overall the highest at > 105 M−1 and demonstrates selectivity for this type of guest (Fig. 5). The binding of NDS in the cavity of C4Pt is likely driven by a combination of coulombic interactions of the sulfonate groups with both Pd(II) and Pt(II) metal centers as well as H bonding to the endo-α-pyridyl protons H-1 and H-15, similar to the interactions observed for quinones and other guests with related structures in previously reported homometallic MSAs.17,43 These interactions result in a pseudo-chelate effect where the guest interacts with both “ends” of the cage cavity. Binding of NDS to C4Ru was observed to be much weaker (K = 1053 ± 168 M−1) which reflects the effective blockage of the imine proton H-15 by the endo chlorido ligand (with loss of the chelate effect), the reduced effective charge at the Ru center and steric constraints, which consequently places the opposite sulfonate moiety into a non-ideal position or orientation for effective hydrogen bonding to protons H-1 and H-5 (Fig. S65†).
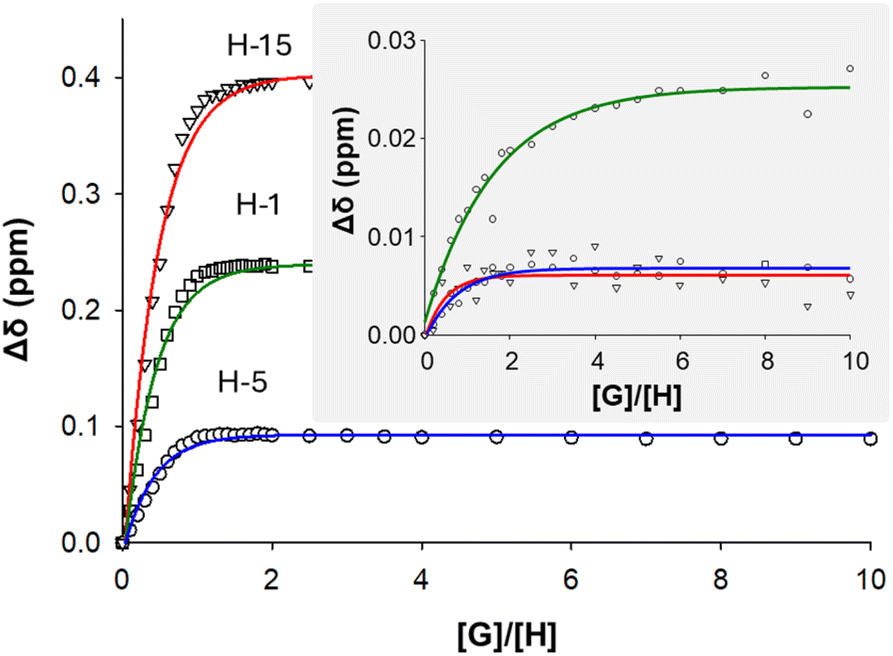 | ||
| Fig. 5 Absolute changes in chemical shifts of H-1, H-5, and H-15 in the 1H NMR spectra of C4Pt and C4Ru (inset) upon titration with up to 10 eq. NDS. | ||
The interactions observed by NMR spectroscopy were supported by X-ray diffraction analysis of a crystal in which C4Pt was found to have encapsulated NDS (C4Pt⊂NDS; Fig. 6). Each flexible arm adopted a unique orientation in space, as also seen in the molecular structure of C4Pt, to allow for the accommodation of a single NDS molecule in the cavity. The sulfonate motifs form hydrogen bonds to two of the amide groups and two of the α-pyridyl protons at the Pd end of C4Pt. At the Pt center, the second sulfonate motif formed H bonds to the endo-facing α-pyridyl protons. Host–guest complex formation was supported by coulombic interactions of the sulfonate groups of NDS with both the Pd and Pt centers, as seen for 1,8-naphthalimide sulfonates in Pd2L4 cages.56 However, the observed solid state structure is a snapshot of one possible host–guest conformation, and the solution NMR data is consistent with dynamic interconversion between different conformations (Fig. S40†).
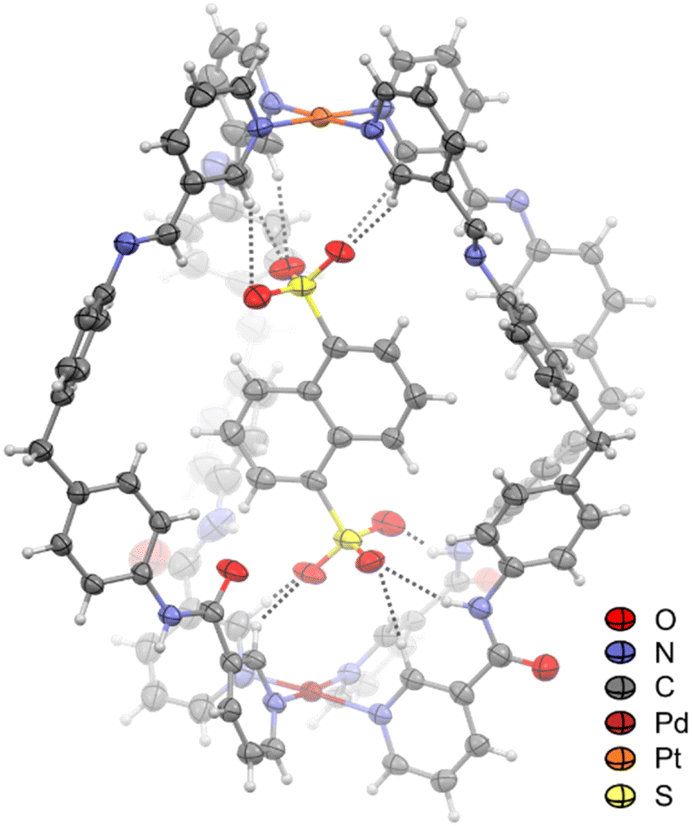 | ||
| Fig. 6 Molecular structure of C4Pt⊂NDS drawn at 50% probability level. Solvents and counteranions have been omitted for clarity. The dashed lines indicate H bonds between the NDS oxygen atoms and protons of C4Pt. | ||
Investigation of the host guest properties of C4Pt and C4Ru conducted by ESI-MS confirmed the NMR spectroscopic data. Mass spectra of 1:1 mixtures of C4Pt and MsO, TsO or NDS gave peaks at m/z 655.1681, 680.5120, and 1079.2435 which were assigned to [C4Pt(–4BF4) + MsO]3+, [C4Pt(–4BF4) + TsO],3+, and [C4Pt(–4BF4) + NDS]2+, respectively (Fig. 7 and S42–S44†), demonstrating interactions between the host and guest molecules. In competitive studies with 1:1:1:1 mixture of the anionic guests and C4Pt, the stronger binding of NDS to C4Pt was evidenced by the appearance of only the C4Pt⊂NDS peak, with no evidence of peaks corresponding to the C4Pt, C4Pt⊂MsO, or C4Pt⊂TsO species (Fig. 7 and S45†). This may be likened to properties similar to the chelate effect found for multidentate ligands in metal complexes. Furthermore, the lower binding constants observed for C4Ru⊂MsO, C4Ru⊂TsO and C4Ru⊂NDS were consistent with the absence of host–guest complexes detected by ESI-MS.
 | ||
| Fig. 7 Stacked mass spectra of C4Pt, C4Pt incubated with MsO, TsO, or NDS (1:1) as well as with a mixture of guests (1:1:1:1). | ||
Modelling (Spartan ′24®, MMFF) of the host–guest chemistry of C4Pt and C4Ru with the neutral molecules cisplatin (cis), oxaliplatin (ox) and 5-fluorouracil (5-FU) within the cavities of C4Pt and C4Ru suggested 1:1, 1:2, and 1:3 adducts were possible (Fig. S66–S70†). Mixtures of these guests with C4Pt or C4Ru at molar ratios of 5:1 in DMSO-d6 caused no shifts of proton resonances in the 1H NMR spectra, most likely due to competition with DMSO for the binding site(s) within the cavities. However, qualitative guest-binding studies17 of cis, ox, and 5-FU with saturated solutions of C4Pt or C4Ru in CD3CN or 10% DMSO/CD3CN, respectively, could be carried out using 1H NMR spectroscopy (Fig. 8). The 1H NMR spectra of C4Pt⊂cis and C4Pt⊂5-FU showed significant shifts of internally facing resonances (H-1, H-5 and H-15) while no effects on the externally facing protons H-2 and H-14 were observed (Fig. 8, S52 and S56†). Incubation with ox resulted in less pronounced changes but shifts of the endo-facing protons were still visible. These data point towards interactions of the drugs within the cage cavity, rather than externally around the Pd and Pt centers, as was seen with the anionic guests in addition to endo-bonding (vide supra). The overall changes in chemical shifts, however, were less pronounced than those of previously reported MM′L4 MSAs with smaller cavities.49 Based on the relative change in chemical shift of these internal protons, we can conclude that the guest species are bound closer to the Pd center, likely due to H bond formation between the carbonyl oxygen atoms and the platinum guests and the NH protons in the case of 5-FU. Analogous studies with C4Ru revealed prominent shifts of the amide NH (H-5) and imine CH (H-11; Fig. S56†) with only minor shifts of exo-facing protons, also suggesting interaction within the cavity of C4Ru. Broadening of peaks H-12, H-14, and H-15 was observed in CD3CN (Fig. S56; † and to a lesser extent in DMSO-d6), which may be explained at least in part by C–H⋯Cl interactions with the exo- and endo-chlorido ligands at the Ru center.
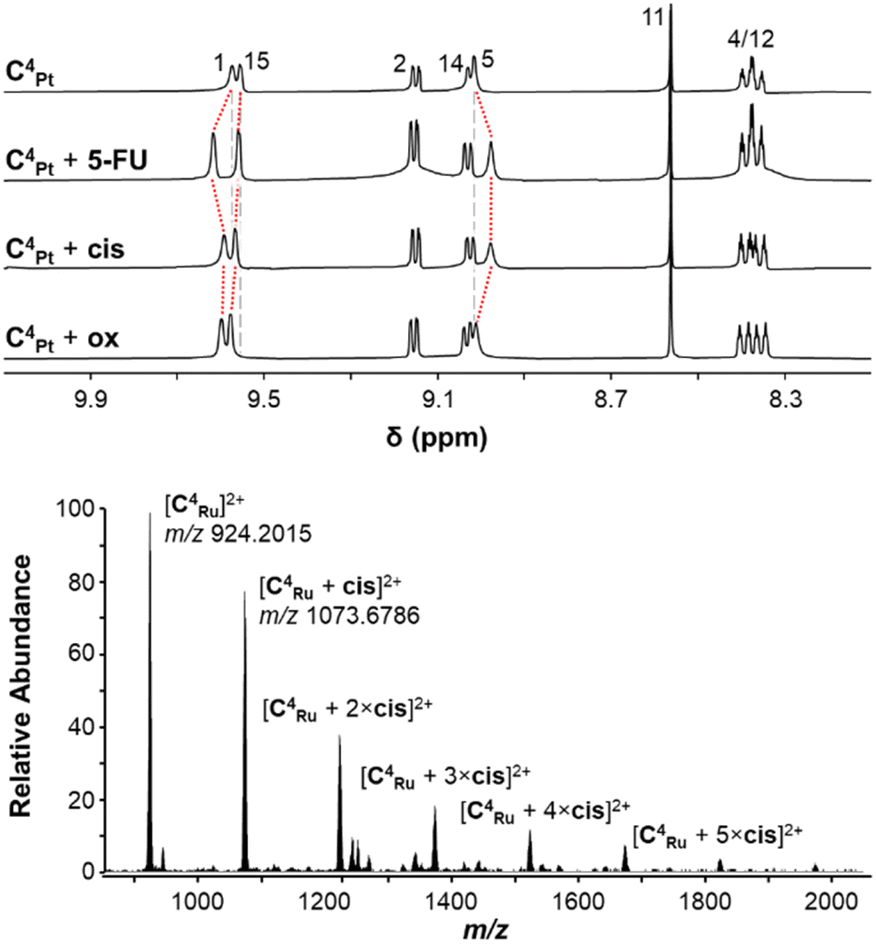 | ||
| Fig. 8 (top) 1H NMR spectra (CD3CN, 298 K, 400 MHz) of C4Pt and mixtures of C4Pt with 5-FU, cis, or ox (1:5). Shifts of endo-facing protons H-1, H-5, and H-15 are shown with dashed red lines. Vertical grey lines indicate the same chemical shift for the corresponding peak of C4Pt (top spectrum). (bottom) ESI-mass spectrum of a 1:5 mixture of C4Ru and cis. | ||
The mass spectra of 1:5 mixtures of C4Pt or C4Ru and 5-FU indicated only weak interactions, with the 5-FU adducts being detected at <5% abundances relative to the MSAs (Table S1, Fig. S53 and S57†). Mixtures of both MSAs with the platinum complexes however revealed for C4Pt host–guest complexes with up to two molecules of cis and 3 eq. of ox and even higher-level interaction with C4Ru with up to six molecules of cis or ox attached to the cage (Fig. 8, S54, S55, S58, S59 and Table S1†). Some of these interactions will occur on the outside of the cavity, facilitated by the chlorido ligands, as molecular modelling suggested a maximum of three molecules of cis or ox are able to be bound within C4Pt or C4Ru (Fig. S66–S70†). The higher number of adducts observed for C4Ru may be due to interactions between the exo-chlorido ligands and the Pt centers of the guests as found in the molecular structure of C4Ru (vide supra). The differences between cis/ox and 5-FU to act as guest molecules can be explained by the H bond donating properties of the former. HCD (higher energy collisional dissociation) fragmentation of the isolated host–guest ions of [C4Pt]4+ and [C4Ru]4+ showed release of the guest molecules at NCE 10 (normalised collision energy) with the parent cage ions [C4Pt]4+ and [C4Ru]4+ remaining intact at m/z 467.6302 and m/z 924.2015, respectively (see Fig. S53–S55 and S57–S59†). The relative intensities of the host:guest ions versus the host ions at NCE 10 reflect differences in the gas phase stability of the inclusion complexes with the neutral guest molecules and decrease in the order ox > cis > 5-FU for C4Pt and 5-FU > ox > cis for C4Ru.
Conclusions
Heterodimetallic M(μ-L)4M′ MSAs based on non-symmetric bridging ligands have been shown to disassemble and reassemble upon the addition of external stimuli as well as host a variety of guest molecules in their cavities. We obtained the Pd(μ-L)4Pt C4Pt and Pd(μ-L)4RuCl2C4Ru MSAs via subcomponent self-assembly using a 4,4′-methylenedianiline-based linker. This approach resulted in the largest Pd(μ-L)4M cages known so far. With inter-metal distances of 15.48 and 15.21 Å for C4Pt and C4Ru, respectively, they are much larger than the closely related C1Pt–C3Pt and C1Ru–C3Ru MSAs. C4Pt and C4Ru underwent rapid selective opening at the Pd center upon addition of four eq. of DMAP to liberate palladium(II) as [Pd(DMAP)4]2+, however the kinetics of reassembly with addition of p-TsOH were much slower than found for the equivalent smaller cages, which can be explained by the increased flexibility and length of the bridging ligands. The sulfonate-containing guests MsO and TsO were bound only weakly in the cavities of C4Pt and C4Ru. However, the 2− charged guest NDS interacted strongly with C4Pt although the binding constant to C4Ru was 2 orders of magnitude lower. This lower binding constant can be attributed to the endo-chlorido ligand at the Ru center preventing H bonding to the internal α-pyridyl protons as well as reducing the coulombic interactions between the cage and the guest. Single crystal X-ray crystallographic studies demonstrated that the host–guest complex C4Pt⊂NDS formed with one NDS guest occupying the cavity. The smaller MSAs C3Pt and C3Ru allowed effective discrimination between MsO and TsO based on size and coulombic interactions. However, these cages were too small to bind NDS effectively. The binding constants observed for C4Pt and C4Ru with MsO and TsO were significantly smaller when compared to C3Pt which we explain by a mismatch between cavity and guest size.1H NMR spectroscopy and ESI-MS also demonstrated binding with the neutral guests cis, ox, and 5-FU within the cavity of both cages, with the number of guest molecules incorporated determined by the sizes and H bond donor properties of the guest molecules.
This study presents the second example of a M(μ-L)4M′ cage with an octahedral metal center and demonstrates that the chlorido ligands coordinated to the Ru center have a larger role to play in tuning the host–guest interaction than anticipated from studies on smaller cages. Introduction of chlorido co-ligands expands the range of possible interactions that can occur between the host and guest molecules but also changes the overall charge of the cage and reduces direct accessibility to host metal centers, which are both important areas to further explore in the search for new applications of supramolecular architectures.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Hayden B. Gearing: synthesis, binding studies, original draft; Monika Cziferszky: mass spectrometry studies; Tilo Söhnel: crystallography; L. James Wright: conceptualization, funding, writing – review and editing; James D. Crowley: supervision, conceptualization, funding, writing – review and editing; Christian G. Hartinger: supervision, conceptualization, funding, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. B. G. thanks the University of Auckland for a PhD scholarship. The work was supported by the Marsden Fund Council from Government funding (UOA1726), administered by the Royal Society of New Zealand. We are grateful to Dr Timothy Christopher and Dr Githal Arachchige for collecting the X-ray diffraction and mass spectrometry data.References
- S. J. Pike and P. J. Lusby, Chem. Commun., 2010, 46, 8338–8340 RSC
.
- A. Schmidt, A. Casini and F. E. Kühn, Coord. Chem. Rev., 2014, 275, 19–36 CrossRef CAS
- N. B. Debata, D. Tripathy and H. S. Sahoo, Coord. Chem. Rev., 2019, 387, 273–298 CrossRef CAS
- A. Pöthig and A. Casini, Theranostics, 2019, 9, 3150–3169 CrossRef PubMed
- C. J. T. Cox, J. Hale, P. Molinska and J. E. M. Lewis, Chem. Soc. Rev., 2024, 53, 10380–10408 RSC
- V. Martí-Centelles, A. L. Lawrence and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 2862–2868 CrossRef PubMed
- R. L. Spicer, A. D. Stergiou, T. A. Young, F. Duarte, M. D. Symes and P. J. Lusby, J. Am. Chem. Soc., 2020, 142, 2134–2139 CrossRef CAS PubMed
- X.-C. Zhou, L.-X. Wu, X.-Z. Wang, Y.-L. Lai, Y.-Y. Ge, J. Su, X.-P. Zhou and D. Li, Inorg. Chem., 2022, 61, 5196–5200 CrossRef CAS PubMed
- J. E. M. Lewis, A. B. S. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Chem. Sci., 2014, 5, 1833–1843 RSC
- A. B. S. Elliott, J. E. M. Lewis, H. van der Salm, C. J. McAdam, J. D. Crowley and K. C. Gordon, Inorg. Chem., 2016, 55, 3440–3447 CrossRef CAS PubMed
- T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Acc. Chem. Res., 2013, 46, 2464–2474 CrossRef CAS PubMed
- A. Casini, B. Woods and M. Wenzel, Inorg. Chem., 2017, 56, 14715–14729 CrossRef CAS PubMed
- D. Preston, K. F. White, J. E. M. Lewis, R. A. S. Vasdev, B. F. Abrahams and J. D. Crowley, Chem.–Eur. J., 2017, 23, 10559–10567 CrossRef CAS PubMed
- C. T. McTernan, J. A. Davies and J. R. Nitschke, Chem. Rev., 2022, 122, 10393–10437 CrossRef CAS PubMed
- W. M. Bloch, S. Horiuchi, J. J. Holstein, C. Drechsler, A. Wuttke, W. Hiller, R. A. Mata and G. H. Clever, Chem. Sci., 2023, 14, 1524–1531 RSC
- D. A. McMorran and P. J. Steel, Angew. Chem., Int. Ed. Engl., 1998, 37, 3295–3297 CrossRef CAS
- J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC
- S. Sharma, M. Sarkar and D. K. Chand, Chem. Commun., 2023, 59, 535–554 RSC
- N. Kishida, H. Sasafuchi, T. Sawada and M. Yoshizawa, Chem. Sci., 2024, 15, 13234–13239 RSC
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed
- X. Sun, D. W. Johnson, D. L. Caulder, K. N. Raymond and E. H. Wong, J. Am. Chem. Soc., 2001, 123, 2752–2763 CrossRef CAS PubMed
- J. Yang, M. Bhadbhade, W. A. Donald, H. Iranmanesh, E. G. Moore, H. Yan and J. E. Beves, Chem. Commun., 2015, 51, 4465–4468 CAS
- C. Shen, A. D. W. Kennedy, W. A. Donald, A. M. Torres, W. S. Price and J. E. Beves, Inorg. Chim. Acta, 2017, 458, 122–128 CAS
- D. Rota Martir, D. B. Cordes, A. M. Z. Slawin, D. Escudero, D. Jacquemin, S. L. Warriner and E. Zysman-Colman, Chem. Commun., 2018, 54, 6016–6019 CAS
- D. Rota Martir and E. Zysman-Colman, Chem. Commun., 2019, 55, 139–158 CAS
- M. Hardy, N. Struch, J. J. Holstein, G. Schnakenburg, N. Wagner, M. Engeser, J. Beck, G. H. Clever and A. Lützen, Angew. Chem., Int. Ed., 2020, 59, 3195–3200 CAS
- W. M. Bloch, Y. Abe, J. J. Holstein, C. M. Wandtke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2016, 138, 13750–13755 CAS
- W. M. Bloch, J. J. Holstein, W. Hiller and G. H. Clever, Angew. Chem., Int. Ed. Engl., 2017, 56, 8285–8289 CAS
- S. Sudan, R.-J. Li, S. M. Jansze, A. Platzek, R. Rudolf, G. H. Clever, F. Fadaei-Tirani, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2021, 143, 1773–1778 CAS
- D. Preston and J. D. Evans, Angew. Chem., Int. Ed. Engl., 2023, 62, e202314378 CAS
- E. Benchimol, I. Regeni, B. Zhang, M. Kabiri, J. J. Holstein and G. H. Clever, J. Am. Chem. Soc., 2024, 146, 6905–6911 CAS
- S. S. Mishra, S. V. K. Kompella, S. Krishnaswamy, S. Balasubramanian and D. K. Chand, Inorg. Chem., 2020, 59, 12884–12894 CAS
- P. Molinska, A. Tarzia, L. Male, K. E. Jelfs and J. E. M. Lewis, Angew. Chem., Int. Ed. Engl., 2023, 62, e202315451 CrossRef CAS PubMed
- A. Tarzia, W. Shan, V. Posligua, C. Cox, L. Male, B. Egleston, R. Greenaway, K. Jelfs and J. E. M. Lewis, Chem.–Eur. J., 2024, e202403336 Search PubMed
- D. Preston, J. E. M. Lewis and J. D. Crowley, J. Am. Chem. Soc., 2017, 139, 2379–2386 CrossRef CAS PubMed
- J. A. Findlay, K. M. Patil, M. G. Gardiner, H. I. MacDermott-Opeskin, M. L. O'Mara, P. E. Kruger and D. Preston, Chem.–Asian J., 2022, 17, e202200093 CrossRef CAS PubMed
- J. E. M. Lewis, Angew. Chem., Int. Ed., 2022, 61, e202212392 CrossRef CAS PubMed
- A. Platzek, S. Juber, C. Yurtseven, S. Hasegawa, L. Schneider, C. Drechsler, K. E. Ebbert, R. Rudolf, Q.-Q. Yan, J. J. Holstein, L. V. Schäfer and G. H. Clever, Angew. Chem., Int. Ed., 2022, 61, e202209305 CrossRef CAS
- A. Kumar, S. Krishnaswamy and D. K. Chand, Angew. Chem., Int. Ed., 2024, e202416332 Search PubMed
- W. D. J. Tremlett, T. Söhnel, J. D. Crowley, L. J. Wright and C. G. Hartinger, Inorg. Chem., 2023, 62, 3616–3628 CrossRef CAS PubMed
- S. Thoonen, S. E. Walker, S. Brandon, A. I. McKay, M. J. Paterson, J. D. Crowley, K. L. Tuck, D. R. Turner, ChemRxiv, 2025, DOI:10.26434/chemrxiv-2024-lfgkf-v2.
- H. B. Gearing, T. Söhnel, P. Young, L. Lisboa, L. J. Wright, J. D. Crowley and C. G. Hartinger, Chem. Commun., 2024, 60, 10950–10953 RSC
- L. S. Lisboa, J. A. Findlay, L. J. Wright, C. G. Hartinger and J. D. Crowley, Angew. Chem., Int. Ed., 2020, 59, 11101–11107 CrossRef CAS PubMed
- L. S. Lisboa, M. Riisom, H. J. Dunne, D. Preston, S. M. F. Jamieson, L. J. Wright, C. G. Hartinger and J. D. Crowley, Dalton Trans., 2022, 51, 18438–18445 RSC
- L. Lisboa, D. Preston, J. McAdam, L. Wright, C. Hartinger and J. Crowley, Angew. Chem., Int. Ed., 2022, 61, e202201700 CrossRef CAS PubMed
- A. C. Pearcy, L. S. Lisboa, D. Preston, N. B. Page, T. Lawrence, L. J. Wright, C. G. Hartinger and J. D. Crowley, Chem. Sci., 2023, 14, 8615–8623 RSC
- Z. T. Avery, M. G. Gardiner and D. Preston, Angew. Chem., Int. Ed., 2024, e202418079 Search PubMed
- L. Lisboa, D. Preston, J. McAdam, L. Wright, C. Hartinger and J. Crowley, Angew. Chem., 2022, 61, e202201700 CrossRef CAS PubMed
- H. Gearing, T. Soehnel, P. G. Young, L. Lisboa, L. J. Wright, J. D. Crowley and C. Hartinger, Chem. Commun., 2024, 60, 10950–10953 RSC
- S. Bandi and D. K. Chand, Chem.–Eur. J., 2016, 22, 10330–10335 CrossRef CAS PubMed
- Q. V. C. van Hilst, A. C. Pearcy, D. Preston, L. J. Wright, C. G. Hartinger, H. J. L. Brooks and J. D. Crowley, Chem. Commun., 2024, 60, 4302–4305 RSC
-
Spartan '24, Wavefunction, Inc Search PubMed
- G. H. Clever, S. Tashiro and M. Shionoya, Angew. Chem., Int. Ed., 2009, 48, 7010–7012 CrossRef CAS PubMed
- G. H. Clever, S. Tashiro and M. Shionoya, J. Am. Chem. Soc., 2010, 132, 9973–9975 CrossRef CAS
- T. R. Schulte, J. J. Holstein and G. H. Clever, Angew. Chem., Int. Ed., 2019, 58, 5562–5566 CrossRef CAS PubMed
- D. Preston, K. M. Patil, A. T. O'Neil, R. A. S. Vasdev, J. A. Kitchen and P. E. Kruger, Inorg. Chem. Front., 2020, 7, 2990–3001 RSC
- H. Lee, J. Tessarolo, D. Langbehn, A. Baksi, R. Herges and G. H. Clever, J. Am. Chem. Soc., 2022, 144, 3099–3105 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: The syntheses of the MSAs, characterization data, additional figures of the molecular structures, experimental data for the disassembly and reassembly reactions, as well as host guest binding studies by NMR, ESI-MS and molecular modelling. CCDC 2389021–2389023. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00209e |
| This journal is © The Royal Society of Chemistry 2025 |