
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic access to organyl-substituted 1,2,3-benzodiazaborines with turn-on fluorescence activity†
Leonie
Wüst‡
ab,
Johannes
Chorbacher‡
ab,
Tim
Wellnitz
ab,
Samuel
Nees
ab,
Holger
Helten
 *ab and
Holger
Braunschweig
*ab
*ab and
Holger
Braunschweig
*ab
aJulius-Maximilians-Universität Würzburg, Institute of Inorganic Chemistry, Am Hubland, 97074 Würzburg, Germany. E-mail: holger.helten@uni-wuerzburg.de; h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron (ICB), Am Hubland, 97074 Würzburg, Germany
First published on 27th March 2025
Abstract
1,2,3-Benzodiazaborines (DABs), isoelectronic to isoquinoline alkaloids, have attracted considerable interest due to their unique reactivity and promising potential applications, including but not limited to pH sensors, chiral probes or antibacterial agents. Although DABs with hemiboronic acid functionality have been known since the 1960s and were extensively studied since then, a method to convert the borinic acid functionality into a borane moiety with carbon-based substituents has remained elusive and is of interdisciplinary interest. Herein, we present a straightforward and inexpensive two-step synthesis of aryl- and alkyl-substituted DABs starting from established hemiboronic acid derivatives and commercially available reagents. Computational studies on the electronic situation of the aryl-substituted derivatives revealed a more pronounced degree of aromaticity compared to the parent hemiboronic acid compounds. The synthesized DABs proved to be highly sensitive multi-anion fluorescence “turn-on” chemosensors for detection and differentiation of relevant anions such as cyanide (CN−) and fluoride (F−). Overall, the synthetic approach presented herein expands the library of accessible DABs to alkyl and aryl derivatives and opens new possibilities to functionalize these BN-alkaloids for applications in fields like fluorescence sensing, material science, and medicinal chemistry.
Introduction
Alkaloids represent a pivotal group of naturally occurring substances, renowned for their versatile biological activities and extensive utilization in the pharmaceutical, dye production, flavoring, and pesticide industries.1–8 Driven by their manifold applications, synthetic chemists are constantly challenged to devise innovative strategies for the modification of these intricate structures. Traditionally, efforts to alter their chemical and physical properties have been focused on changing the substituent pattern.9,10 However, the systematic tuning of properties such as pharmacological activity or photoluminescence can also be achieved by incorporating an isoelectronic and isosteric B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N unit in place of a CC unit within a given molecular scaffold.11–18 Despite the preservation of the total electron count and overall structure, BN-doped materials usually exhibit distinct photophysical, physicochemical, and biological properties due to the induced dipole moment and their ability to form hydrogen bonds.12,19–23 This BN/CC isosterism renders the field of BN heterocycles increasingly relevant to material science and medicinal chemistry and can be extended from simple hydrocarbons to the more complex alkaloids such as indoles or isoquinolines.24,25 If the CC bond adjacent to the nitrogen atom in isoquinoline is replaced by a BN bond, the resulting heterocycle is the parent 1,2,3-benzodiazaborine (DAB) (Fig. 1a).26,27
N unit in place of a CC unit within a given molecular scaffold.11–18 Despite the preservation of the total electron count and overall structure, BN-doped materials usually exhibit distinct photophysical, physicochemical, and biological properties due to the induced dipole moment and their ability to form hydrogen bonds.12,19–23 This BN/CC isosterism renders the field of BN heterocycles increasingly relevant to material science and medicinal chemistry and can be extended from simple hydrocarbons to the more complex alkaloids such as indoles or isoquinolines.24,25 If the CC bond adjacent to the nitrogen atom in isoquinoline is replaced by a BN bond, the resulting heterocycle is the parent 1,2,3-benzodiazaborine (DAB) (Fig. 1a).26,27
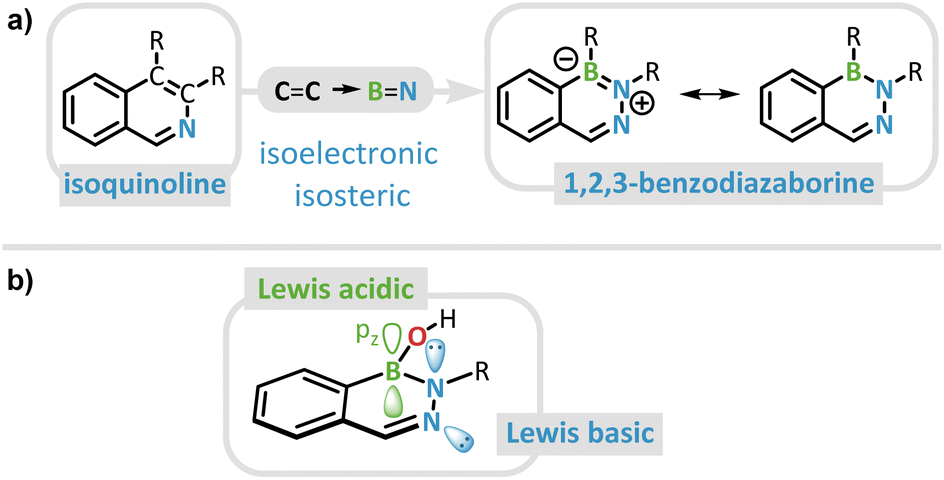 | ||
| Fig. 1 (a) Isoquinoline and the isoelectronic and isosteric parent compound 1,2,3-diazaborine. Note: in the following, we will refrain from drawing the formal charges for all neutral DABs. (b) General DAB scaffold with highlighted Lewis acidic and Lewis basic sides. | ||
Similar to isoquinolines, hemiboronic acid DABs have been studied since the early 1980s for their antibacterial properties.16,28–30 As of today, their pharmaceutical relevance has expanded to bioconjugation reactions due to their rapid, “click”-chemistry-like formation kinetics as well as targeted drug release triggered by reactive oxygen species (ROS).31–34 The latter is facilitated by the semi-intact Lewis acidity of the boron center, resulting from the absence of significant heteroaromatic effects. While the related class of 1,2-azaborines retains a degree of aromaticity in the BN heterocyclic unit that effectively saturates the boron atom, such effects are diminished in 1,2,3-diazaborines due to the presence of an additional nitrogen atom.27,35 This trend can be rationalized by considering the BN/CC isosterism and the reduced aromaticity of pyridine relative to benzene.36 In combination with the isoquinolinic nitrogen atom, DABs possess a bifunctional core structure, having both Lewis basic and Lewis acidic centers at their disposal (Fig. 1b).27,35
In the predominant structural motif of DABs, the Lewis acidity is partially compensated by the donor character of the borinic acid's OH functionality.37 Hence, these DAB borinic acids have been shown by Hall et al. to react as Lewis acids, forming the conjugate bases, only under comparatively harsh pH conditions.27 A few years earlier, Groziak et al. already reported the formation of a zwitterion, in which the tetravalent boron atom of the DAB results in a non-planar central heterocyclic motif.38 Since 2018 these substituted DAB structures have demonstrated significant potential as reversible enzyme inhibitors by binding to cellular nucleophiles due to this coordinative flexibility. They serve as a key example of the broader reactivity spectrum of BN-doped alkaloids in comparison to their CC counterparts.39
To date, numerous examples of hemiboronic acid DABs featuring various N-substituents and different backbone modifications have been reported.40–42 Although recent advancements have expanded to novel, monocyclic derivatives,43 a method for replacing the borinic acid functionality with carbon-based substituents remains elusive. While our group previously described the synthesis of a ferrocenyl-substituted, non-annulated DAB through a ring expansion reaction of an antiaromatic borole precursor, the substrate scope of this approach seemed to be restricted to a single derivative.44 Herein, we present a simple two-step synthesis of aryl- and alkyl-substituted benzenoid DABs, starting from readily available hemiboronic acid derivatives. Furthermore, we investigated the potential of one of these DAB derivatives as a highly selective “turn-on” chemosensor for anions and successfully achieved a ratiometric determination and differentiation of the anions F−, CN−, and NCO− in a laboratory environment. To complement our experimental findings, we conducted comprehensive computational studies on our model derivatives, assessing their aromaticity by utilizing NICS and ACID methodologies.
Results and discussion
We initially considered direct substitution at the boron center of well-established and readily accessible DAB borinic acids such as compound 1-OH, using suitable organometallic nucleophiles. However, borinic acids like 1-OH are generally prone to forming the corresponding B–O–B anhydrides through condensation reactions.27,45 Upon reaction with these moisture-sensitive organometallic reagents (e.g. phenyllithium), this was anticipated to lead to the formation of various side products and result in a non-stoichiometric overall reaction. We therefore synthesized 1-OH following established literature procedures and subsequently converted its borinic acid functionality into the corresponding trimethylsilyl (TMS) ether 1-OTMS.27,32 This approach was inspired by a 1972 study by Roques and Florentin, who investigated the in situ preparation of 1-OTMS using the silylating reagent N,O-bis(trimethylsilyl)-acetamide (BSA) and its reactivity towards organometallic reagents.46 Despite the growing interest in DABs in recent years, this approach from over 50 years ago seems to have gone unacknowledged until now. We modified their protocol for the silylation step, enabling us to isolate and fully characterize compound 1-OTMS in double-digit gram quantities, achieving a high yield of 82% (Scheme 1). Compound 1-OTMS is a hydrolysis-sensitive solid and shows a broad singlet at 26.7 ppm in the 11B NMR spectrum. Compared to the starting material 1-OH, which contains a borinic acid functionality (δ(11B) = 28.7 ppm), the 11B NMR resonance of 1-OTMS is slightly shifted upfield due to the β-effect of the TMS group. | ||
| Scheme 1 Silylation of DAB 1-OH with N,O-bis(trimethylsilyl)acetamide (BSA) to the silyl ether 1-OTMS (TMS = trimethylsilyl). Inset: solid-state molecular structure of 1-OTMS. | ||
Roques and Florentin reported the successful boron substitution of their TMS ether derivatives with organolithium reagents (e.g. MeLi, PhLi) to neutral DABs 1-R in high yields of 80–89% after aqueous work-up.46 We attempted to reproduce these results by reacting the isolated compound 1-OTMS with one equivalent of phenyllithium in diethyl ether, monitoring the reaction via11B NMR spectroscopy (see ESI Fig. S83†). While the resonance at 26.6 ppm, corresponding to the starting material 1-OTMS, remained present after the addition of phenyllithium, a minor resonance at 36.3 ppm, within the region of a three-coordinated boron atom, indicated the formation of the desired compound 1-Ph in trace amounts. However, a second resonance at −3.8 ppm, falling within the typical region of a four-coordinate boron atom, was also observed. Subsequent work-up under Schlenk conditions revealed that the four-coordinate species was the diazaborinate 2-Li, formed by the addition of a second equivalent of phenyllithium to 1-Ph. These results contradict the initial reports by Roques and Florentin over fifty years ago, who were limited to melting point determination and lacked access to modern spectroscopic tools.46
While a rational synthesis of the borate 2-Li is feasible in good yields of 75% using two equivalents of phenyllithium (Scheme 2a), we were still interested in a selective synthesis of 1-Ph. Despite attempts using milder reaction conditions, such as lower temperatures and non-polar solvents, no significant improvement was observed. As a result, we switched to Grignard reagents as a comparatively less reactive alternative (Scheme 2b). When phenylmagnesium bromide is used in stoichiometric quantities, compound 1-Ph can be isolated without significant amounts of a borate by-product. While initial substitution experiments in ether solvents yielded satisfactory results, dichloromethane is known to enhance the selectivity of Grignard reagents and provide milder reaction conditions,47 resulting in improved yields of up to 75–93%. This approach is not limited to aryl substituents; we also successfully prepared the alkyl-substituted DAB 1-Me by an analogous reaction with methylmagnesium bromide. While 1-R (R = aryl) is indefinitely stable in solution and when stored as solid exposed to air and moisture for several months, compound 1-Me slowly hydrolyzes in solution to the borinic acid precursor 1-OH (t1/2 ≈ 2 d in CD2Cl2, 25 °C). The second attack on the boron center and the formation of a borate is also prevented if a sterically more demanding organolithium reagent is employed. We found that the reaction of 1-OTMS with equimolar amounts of duryl- or mesityllithium also yields the neutral DABs 1-Dur and 1-Mes, respectively, in sufficient quantities (Scheme 2c).
 | ||
| Scheme 2 Synthesis of the neutral, monosubstituted DABs 1-R and the diazaborinate 2-Li (p-OMe-C6H4 = 4-methoxyphenyl, p-CF3-C6H4 = 4-trifluoromethylphenyl, Mes = 2,4,6-trimethylphenyl, Dur = 2,3,5,6-tetramethylphenyl). (a) Et2O, −78 °C → rt, 16 h; (b) CH2Cl2 or Et2O or THF, −78 °C or 0 °C → rt, 1–2 d; (c) Et2O, 0 °C → rt, 16 h. | ||
All neutral DABs 1-R exhibit characteristic 11B NMR resonances within the expected range of aryl-substituted boron–nitrogen heterocycles (35.8–38.8 ppm).44,48 Upon borate formation, compound 2-Li displays a significant high-field shift, resonating at −3.2 ppm in the typical region of a tetravalent boron compound. This shift is attributed to both the coordinative change from trigonal-planar to tetrahedral and the introduction of a negative charge at the boron atom. Furthermore, the singlet resonance of the aldimine hydrogen atom in the 1H NMR spectrum serves as a valuable spectroscopic probe and provides insights into the electronic situation in the diazaborine heterocycle. Quaternization causes a significant high-field shift of 1.3 ppm for this proton in 2-Li, which can be attributed to both the introduction of the negative charge and the complete disruption of heteroaromatic effects. We also attempted to experimentally assess the Lewis acidity of 1-Ph employing the Gutmann–Beckett method.49–52 However, no discernible shifting of the 11B or 31P NMR resonances was detected upon adding equimolar amounts of triethylphosphine oxide (Et3PO) even at low temperatures.
We investigated the photophysical absorption and emission properties of all neutral DABs 1-R and the borate 2-Li. Additionally, we synthesized compound 2-TBA (Fig. 2) by a salt metathesis reaction with tetrabutylammonium (TBA) chloride, in order to investigate the compounds' photophysical properties independently of any potential solvent interactions with the lithium cation. The variation of the different third substituents at the boron center shows no significant influence on the UV-vis absorption behavior of the neutral DABs 1-R (see ESI Table S2†). The UV-vis spectra of the compounds 1-R all show a maximum of the lowest-energy absorption band between 304–307 nm in THF. In contrast to the 1-R DABs, the diazaborinate salts 2-Li and 2-TBA show a bathochromically shifted band at λmax = 398 nm (Fig. 2 and Table 1). Our calculations of vertical singlet excitation by TD-DFT at the tHCTHhyb53/def2-TZVPP54,55 level of theory revealed that the lowest-energy absorptions of all compounds can be assigned to π → π* transitions, which correspond to HOMO → LUMO excitations (Fig. 7 and Table 4).
 | ||
| Fig. 2 Normalized UV-vis (dashed lines) and emission (solid lines) spectra of 2-Li (left) and 2-TBA (right) in THF, toluene and as a PMMA film. Inset: structures of the compounds. | ||
| No. | λ abs,max [nm] | λ abs,max [nm] | λ abs,max [nm] | λ em,max [nm] | λ em,max [nm] | λ em,max [nm] | Φ fl , [%] | Φ fl , [%] | Φ fl , [%] |
|---|---|---|---|---|---|---|---|---|---|
| a THF. b PMMA film. c Toluene. d Fluorescence quantum yields, determined using an integration sphere. | |||||||||
| 2-Li | 398 | 396 | 391 | 460 | 455 | 510 | 18 | 30 | — |
| 2-TBA | 398 | 399 | 404 | 459 | 463 | 479 | 19 | 27 | 8 |
| 3-F | 369 | 367 | 368 | 468 | 442 | 467 | 6 | 71 | 12 |
| 3-CN | 381 | 385 | 378 | 451 | 446 | 457 | 17 | 80 | 36 |
| 3-NCO | 371 | 371 | 369 | 451 | 440 | 455 | 5 | 74 | 48 |
All 1-R compounds show only little to no light emission after excitation in THF or toluene, which might be due to non-radiative deexcitation processes caused by intramolecular motions.56 The diazaborinate salts 2-Li and 2-TBA however, show pronounced fluorescence properties in solution as well as in the solid state (Fig. 2 and Table 1). For the latter, we prepared PMMA films by slowly evaporating a THF solution of PMMA and the sample to give a homogeneous layer in the cuvette. In THF, both compounds exhibit almost identical photophysical properties. They show a narrow blue emission band with a maximum at 460 nm for 2-Li and at 459 nm for 2-TBA with quantum yields of Φfl = 18–19%. In the films of the rigid PMMA matrix both compounds exhibit similar properties with increased quantum yields of Φfl = 30% for 2-Li and Φfl = 27% for 2-TBA.
In contrast, the emission properties in toluene differ significantly. Both compounds were poorly soluble in toluene. 2-Li showed a very weak, broad, strongly red-shifted emission band (λmax = 510 nm), while 2-TBA displayed a slightly bathochromically shifted narrow emission band (λmax = 479 nm). By exchanging the counterion, a moderate emission at 2-TBA with a quantum yield of Φfl = 8% is observed in toluene, in contrast to the nearly completely quenched fluorescence of 2-Li. This behavior is likely caused by the formation of a contact ion pair between the borate 2 and the lithium cation in toluene.
Since the DAB compounds 1-R are only weakly emissive or non-emissive, while the borate 2 shows pronounced fluorescence emission, we considered exploring this behavior for anion sensing. Many organoboron compounds that feature a three-coordinate boron center have the affinity to bind small, nucleophilic anions, thus enabling detection thereof.57–63 For our studies, we used the bench-stable compound 1-Ph, which is kinetically well stabilized by the attached phenyl group but is not as strongly sterically shielded as 1-Mes and 1-Dur, making it potentially more accessible for nucleophilic attack. Therefore, we tested the interaction of 1-Ph with various anions (F−, Cl−, Br−, I−, CN−, NCO−, SCN−) for its application as potential “turn-on” sensor using the respective TBA salts. We used a stock solution of 1-Ph in THF and added 10 equivalents of each salt solution. Then, we analyzed the mixtures by UV-vis absorption and fluorescence emission spectroscopy in THF. Addition of the anions Cl−, Br−, I−, and SCN− gave no response, but upon addition of F−, CN−, and NCO− a strong bathochromic shift in absorbance and intense blue fluorescence was observed (Fig. 3). Due to the adverse effects of fluorides on the environment,64,65 despite their widespread use,66,67 as well as the toxicity of cyanides,68 it is highly desirable to selectively detect these ions.
 | ||
| Fig. 3 Absorption (left) and emission (right, λex = 300 nm) spectra of 1-Ph in THF (conc. 5 × 10−5 M) upon addition of different anions (10 eq. of Bu4N+ salts). Inset: cuvettes of the samples under UV light. For figures with different color coding, see Fig. S136 and S137.† | ||
To investigate the sensing process in more detail and further determine the properties of the formed products, we targeted the synthesis of the compounds 3-F, 3-CN, and 3-NCO. This was achieved by reacting 1-Ph with a small excess of the corresponding TBA salt at room temperature in THF (Scheme 3).
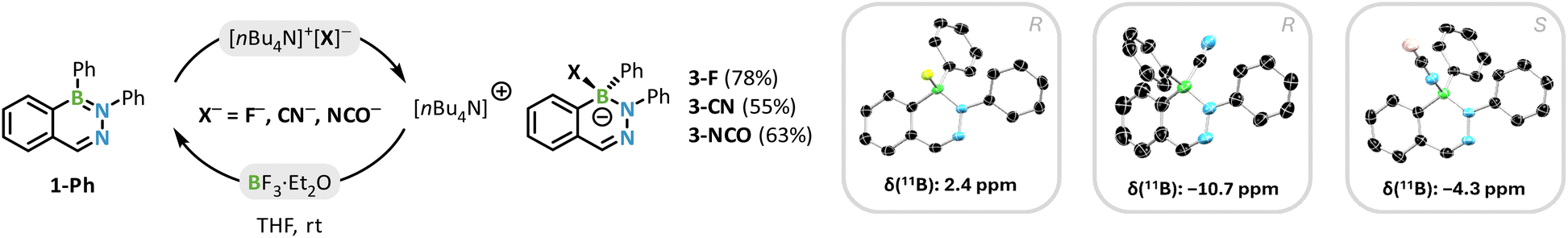 | ||
| Scheme 3 Reaction of 1-Ph with fluoride, cyanide and cyanate tetrabutylammonium salts to the borates 3-F, 3-CN, and 3-NCO. Inset: solid state structures of 3-F, 3-CN, and 3-NCO (nBu4N+ cation omitted for clarity). | ||
The excess salt was then removed by recrystallization in MeCN and/or washing with toluene, giving the compounds 3-X (X = F, CN, NCO) in moderate to good yields (55–78%). The compounds were unambiguously identified by NMR spectroscopy, HRMS, and X-ray diffractometry. By 11B NMR spectroscopy the compounds can be easily distinguished, as there is a notable shift between the compounds: F− (2.4 ppm) > NCO− (−4.3 ppm) > CN− (−10.7 ppm). We propose a straightforward nucleophilic addition (AN) for the formation of 3-X, leading to a racemic product mixture of (R/S)-diazaborinates, and obtained crystallographic proof of both enantiomers (vide infra).§69
When comparing the photophysical properties, it is noticeable that the compounds exhibit a less pronounced bathochromic shift in the absorption (in THF: λmax = 369, 381, and 371 nm for 3-F, 3-CN, and 3-NCO, respectively) compared to 2-TBA. The emission bands in THF, toluene, and as a PMMA film are slightly shifted but show a comparable shape (see ESI Fig. S99, S106 and S113†). The quantum yields in THF are Φfl = 6% (3-F), 17% (3-CN) and 5% (3-NCO), which is also reflected in the intensity of the emission sensing experiments in Fig. 4, and therefore slightly lower than those of the 2-R compounds. In the PMMA film and in toluene, however, there is a clear increase in the quantum yield for the 3-X compounds reaching up to Φfl = 80% (for 3-CN) in the PMMA film, and in toluene up to Φfl = 48% (for 3-NCO) respectively (see Table 1), which, to the best of our knowledge, are currently among the highest quantum yields of DAB compounds without additionally attached fluorophores.
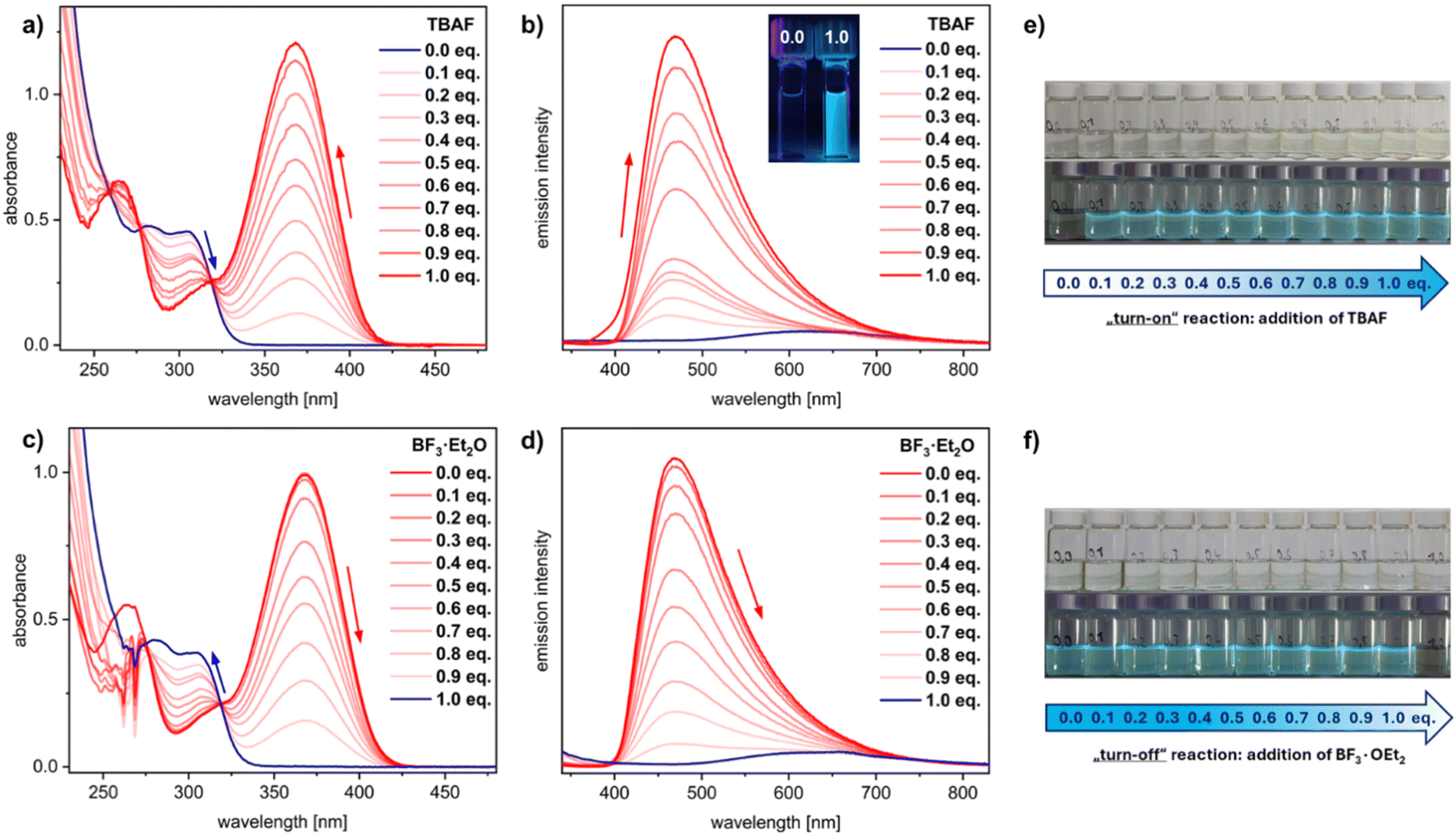 | ||
| Fig. 4 (a) Absorption and (b) emission spectra of 1-Ph upon addition of different equivalents of TBAF (top) as well as (c) absorption and (d) emission spectra of 3-F upon addition of different equivalents of BF3·Et2O (all in THF, conc. 5 × 10−5 M). (e) Solutions of 1-Ph and (f) 3-F in THF after addition of different equivalents of TBAF/BF3·Et2O (top) and under UV light (bottom). | ||
To further investigate the sensitivity of 1-Ph, we assessed its response to the anions F−, CN−, and NCO− by UV-vis and fluorescence titration experiments. For this purpose, we conducted a series of measurements in which varying equivalents of the respective TBA salt solution in THF were added to a stock solution of 1-Ph in THF for each sample. Similar trends were observed for all anions: with increasing equivalents of the TBA salts, the UV-vis band of the parent compound 1-Ph (λmax = 305 nm) decreased while a red-shifted band appeared and increased (for TBAF: λmax = 369 nm) (Fig. 4a).
Intense blue fluorescence was already observed upon addition of 0.1 equivalents of the ions, indicating the compounds' applicability as efficient “turn-on” sensors (see ESI Fig. S118, S125 and S132†). When adding from 0.1 to 1.0 equivalents, an almost linear increase in the intensities of absorption and emission was observed, enabling ratiometric sensing of the ions (Fig. 4a and b). In the case of fluoride, full saturation was achieved with one equivalent added, while cyanide and cyanate required 1.1–1.2 equivalents for complete conversion (see ESI Fig. S122 and S129†), which could also be due to deviations from ideal concentrations. By isolating the anion-binding products 3-X it was also possible to monitor the reverse reaction. This was achieved by adding BF3·Et2O, which allowed us to fully reverse the reaction ratiometrically by titrating 0.1–1.0 equivalents of the Lewis acid, similar to the anion sensing (Fig. 4c, d, S126 and S133†). This characteristic could enable a sustainable use of the sensor, as it could be recycled after use.
Single crystal X-ray diffraction experiments were performed on the neutral DABs 1-R as well as all the diazaborinates 2-Li, 2-TBA and 3-X. Fig. 5 depicts the solid-state molecular structures of 1-Ph, 2-Li, and 2-TBA. Selected bond lengths and angles are summarized in Table 2. Compound 2-Li features a complexation of the lithium cation via the Lewis basic N2 atom (N2–Li1 2.015(7) Å). The steric constraints imposed by Li(OEt2)2+ result in a heavily twisted DAB ring with a B1–N1–N2–C1 torsion angle of 22.2(4)°. Due to the position of the diazaborinate moiety in 2-TBA in the crystal, located on a 2-fold symmetry axis, only a qualitative discussion of its solid-state structure is permissible. In both 1-Ph and 2-TBA, the central DAB ring maintains an approximately planar conformation, with a B1–N1–N2–C1 torsion angle of 1.71(19)° in 1-Ph (for a side view, see ESI Fig. S85†).
 | ||
| Fig. 5 Molecular structures of 1-Ph, 2-Li, and 2-TBA in the solid state (ellipsoids drawn at 50% probability; [nBu4N]+ counterion for 2-TBA omitted). The complexing ether is rendered as wireframe for clarity. For selected bond lengths (Å) and angles (°) see Table 2. For side views, see ESI Fig. S85.† | ||
| No. | δ 1H (H−1) [ppm] | δ 11B [ppm] | B1–N1 [Å] | B1–N1–N2–C1 [°] DAB torsion | N1–N2–C8–C9 [°] coplanarity |
|---|---|---|---|---|---|
| 1-Ph | 8.58 | 36.2 | 1.4210(19) | 1.71(19) | 67.79(16) |
| 2-Li | 7.13–7.20 | −3.2 | 1.582(4) | 22.2(4) | 36.5(4) |
| 2-TBA | 7.08 | −4.8 | Whole molecule disorder | ||
| 3-F | 7.27 | 2.4 | Whole molecule disorder | ||
| 3-CN | 7.07 | −10.7 | 1.558(3) | 14.9(4) | 21.3(3) |
| 3-NCO | 7.17–7.20 | −4.3 | 1.562(4) | 4.0(5) | 5.0(4) |
The B1–N1 bond lengths in 1-OTMS and 1-Ph of 1.4452(15) Å and 1.4210(19) Å, respectively, fall within the expected range for neutral boron–nitrogen heterocycles with partial BN double bond character.70,71 However, the corresponding B1–N1 bond length in 2-Li of 1.582(4) Å is significantly elongated, indicating the loss of π character and thus a reduced bond order. A NBO analysis72 (ωB97X-D73/def2-SVP55) supports this interpretation and revealed a strong ionic character of 58% for the B1–N1 bond of the diazaborinate and a bond order of 0.75, considerably reduced compared to 1-Ph (1.75).
Similarly to 2-TBA, the other diazaborinates 3-X reported herein exhibit distinctive 2-fold whole molecule disorders, likely caused by the central chirality of these compounds. For example, compound 3-F crystallized in the centrosymmetric space group P21/n, resulting in the superimposition of the R and S enantiomers. For 3-CN and 3-NCO, two crystallographically identical molecules crystallized in the asymmetric unit, with only one molecule exhibiting a whole molecule disorder in each case. Notably, and unlike 2-Li, 3-NCO features an approximately planar diazaborinate heterocycle with a small B1–N1–N2–C1 torsion angle of 4.0(4)°. The naphthalinoid DAB unit and the phenyl substituent at the N1 atom in 3-NCO are nearly co-planar with a N2–N1–C10–C11 torsion angle of 5.0(4)°, which may be correlated to the observed luminescent properties of these compounds. However, further investigations are required to fully understand this phenomenon, which are still subject of ongoing projects. Compound 3-CN is overall more twisted than 3-NCO, but less severe compared to 2-Li.
Density functional theory (DFT) calculations were performed on the neutral DABs 1-R (R = OH, OTMS, Ph), all diazaborinates 2 and 3-X, as well as the parent isoquinoline 4, using the Gaussian 16 program (see ESI for computational studies of all compounds†).74 All calculations were carried out for the free diazaborinates without the inclusion of the corresponding cations (ωB97X-D73/def2-SVP,55 see ESI†). Recently, Hall et al. assessed the aromatic character of 1-OH by nucleus-independent chemical shift (NICS) calculations. They reported a small degree of aromaticity for the central DAB ring motive based on isotropic shielding tensors (resp. NICSiso(0) and NICSiso(1)).27 To provide further insights into the degree and type of aromaticity in the neutral DABs 1-R, the NICS values of the zz tensor perpendicular to the XY ring plane (NICSzz-XY) were scanned (Table 3).
|
|
||||||
|---|---|---|---|---|---|---|
| Ring A | Ring B | |||||
| 4 | 1-OH | 1-Ph | 4 | 1-OH | 1-Ph | |
| NICSiso(0) | −6.5 | −1.3 | −1.8 | −8.5 | −8.7 | −8.5 |
| NICSiso(±1) | −9.8 | −4.4 | −5.1 | −11.1 | −11.5 | −11.6 |
| NICSzz(0) | −6.6 | 11.8 | 7.2 | −9.8 | −11.1 | −11.4 |
| NICSzz(±1) | −25.1 | −8.3 | −12.2 | −27.9 | −29.2 | −30.0 |

For all three compounds, the NICSzz-XY scan confirms diamagnetic ring currents in both rings A and B. The scan profiles exhibit the expected π aromatic behavior, with maxima at ca. 1.0 to 1.5 Å above and below the ring plane (see ESI Fig. S139†). In 4 and 1-Ph, the aromatic contributions from the phenyl substituents result in slightly asymmetric scan curves. The BN-doping in 1-OH and 1-Ph leads to a significant loss of aromaticity in ring A (compare NICSzz(−1/1) of −12.2 ppm for 1-Ph and −8.3 ppm for 1-OHvs. −25.1 ppm for 4), while the annulated ring B experiences a minor aromatic stabilization, as evidenced by the lower NICSzz(−1/1) value of 4 compared to 1-Ph (−27.9 ppm for 4vs. −30.0 ppm for 1-Ph). As expected, the exchange of the boronic acid functionality in 1-OH for the phenyl substituent in 1-Ph results in lower NICSzz(−1/1) values, indicating increased aromaticity of the DAB ring A. This is likely due to the absence of the borinic acids' +M effect, which leaves the pz-orbital of the boron atom less saturated and more available for electron delocalization. The increased aromaticity of 1-Ph is substantial for the reversiblity of the sensing reaction, as rearomatiziation offers a sufficient driving force.
To visualize diatropic ring currents, the anisotropy of the induced current density (ACID) isosurfaces of 4, 1-Ph, and 1-OH were calculated, considering both σ and π contributions, with the magnetic field orthogonal to the molecular ring plane (Fig. 6). The ACID plots support the picture derived from the NICSzz scans. At identical isovalues, the isosurface break in 1-Ph, particularly around the BN bond, is less prominent than in the borinic acid precursor 1-OH, indicating stronger conjugative effects in 1-Ph. Overall, the NICS and ACID calculations show a more pronounced degree of aromaticity of the aryl substituted DAB 1-Ph compared to the well-established borinic acid DAB 1-OH. However, compared to the parent isoquinoline 4, the aromaticity of the BN-doped diazaborine ring A of 1-Ph is still diminished.
 | ||
| Fig. 6 ACID isosurfaces of 4, 1-OH, and 1-Ph (σ and π contributions at isovalue 0.05 a.u.). The magnetic field is orthogonal with respect to the molecular ring plane of the annulated ring system. Current density vectors are plotted onto the ACID isosurface to indicate diatropic (aromatic) ring currents. | ||
In addition, we investigated the photoabsorption properties of 1-R, 2, and 3-X using time-dependent DFT (TD-DFT) methods (tHCTHhyb53/def2-TZVPP54,55) in a model mimicking tetrahydrofuran solvation (Table 4). The primary absorption band was selected based on the criterion of maximum oscillator strength, and the orbital contributions were analyzed. All transitions correspond to S0 → S1 HOMO(π) → LUMO(π*) transitions and are in good agreement with the experimentally determined absorption maxima (vide supra).
| No. | Transition | λ exp [nm] | λ calc [nm] | Oscillator strength | Main excitation |
|---|---|---|---|---|---|
| 1-OH | S0 → S1 | 307 | 314 | 0.47 | HOMO → LUMO |
| 1-OTMS | S0 → S1 | 304 | 307 | 0.43 | HOMO → LUMO |
| 1-Ph | S0 → S1 | 305 | 307 | 0.22 | HOMO → LUMO |
| 2 | S0 → S1 | 398 | 401 | 0.37 | HOMO → LUMO |
| 3-F | S0 → S1 | 369 | 374 | 0.50 | HOMO → LUMO |
| 3-CN | S0 → S1 | 381 | 394 | 0.38 | HOMO → LUMO |
| 3-NCO | S0 → S1 | 371 | 380 | 0.45 | HOMO → LUMO |
Fig. 7 depicts the calculated frontier molecular orbitals (FMOs) of 4, 1-OH, 1-Ph, and 2, which all display π symmetry. The neutral, BN-doped DABs 1-OH and 1-Ph exhibit remarkably similar orbital symmetries, differing from the parent CC-isoquinoline 4 in the position of the nodal planes. In 1-OH, the highest occupied molecular orbital (HOMO) coefficients extend continuously over both rings A and B. This effect is less pronounced in compound 1-Ph, where the orbital lobes exhibit some discontinuity at the C–B bond. Consequently, the contribution of the boron center to the HOMO of 1-Ph is lower. A reverse situation is observed in the lowest unoccupied molecular orbitals (LUMO) of 1-OH and 1-Ph.
 | ||
| Fig. 7 Frontier molecular orbitals of 4, 1-OH, 1-Ph, and 2 (ωB97X-D/def2-SVP/isovalue 0.06 eÅ−3). | ||
The LUMO of 1-Ph shows a substantial contribution from the boron center, conferring a weak Lewis acidic character to the boron atom. This facilitates the addition of nucleophiles like phenyllithium (to 2-Li) or the anions CN−, NCO− and F− (to 3-X). In contrast, 1-OH lacks such contributions. Within the central DAB ring system of the neutral DAB 1-Ph the HOMO exhibits the most significant contributions from the BN and CN bonds. Although the HOMO of 2 also features contributions from the CN bond, a nodal plane exists between the boron and the adjacent nitrogen atom, and the orbital coefficient at the nitrogen atom displays pz character. This exclusion of the boron center from the π system in 2-R might be responsible for the substantially different optical properties of the emissive diazaborinates relative to their non-emissive neutral counterparts 1-R. An in-depth experimental and computational study of the fluorescence process is currently under investigation and is not yet thoroughly understood. FMOs of all anion-bound diazaborinates resemble the FMO situation in 2 (see ESI†).
Conclusions
In conclusion, we herein present functionalized 1,2,3-benzodiazaborines (DABs), which are BN-substituted isoelectronic analogs of isoquinoline alkaloids, as well as a series of emissive borates derived therefrom. Through a modular approach using organometallic reagents we were able to synthesize a series of boron-functionalized DABs starting from an easily accessible hemiboronic acid. Using this approach, a borate was synthesized that showed emissive properties in solution and enhanced emission in the solid state. Based on this observation, we tested a suitable phenyl-functionalized DAB for its ability to bind small anions, thus operating as a fluorescence “turn-on” sensor. We succeeded in selective ratiometrically detecting and differentiating the anions F−, CN− and NCO−, as well as in reversing the process with the Lewis acid BF3·Et2O. By isolating the resulting products, we were able to characterize further highly fluorescent borates of this compound class, which set a new benchmark for fluorescent DABs. While the herein presented diazaborinate-system serves as a fundamental proof of concept for the anion sensing process and nesessitates futher modifications to enhance its stability in non-organic media, this characteristic is very promising for the future design of air-stable and sustainable sensors that can potentially be recycled after use. Structural studies revealed a pronounced planarity in the backbone of all neutral compounds, especially the phenyl-functionalized DAB, demonstrating their isosteric relationship to isoquinoline. Furthermore, NICS and ACID calculations revealed a more pronounced degree of aromaticity of the phenyl-substituted DAB compared to the precursor hemiboronic acid. We expect our synthetical approach to aryl-substituted DABs to be highly relevant for interdisciplinary fields, such as material science and medicinal chemistry.75 In future studies we plan to explore the full potential of this novel class of DABs and derived borates and aim at extending our investigations to further modify their photophysical properties.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for all compounds have been deposited at the CCDC under 2394196–2394208.Author contributions
L. W., J. C., H. H. and H. B. conceived the project. L. W. performed all experiments on DAB synthesis and boron substitution and performed the quantum chemical calculations. J. C. performed all photophysical experiments and synthesized the anion sensing compounds. T. W. and S. N. performed the XRD experiments. L. W., J. C., H. H. and H. B. discussed the results, wrote and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Ivo Krummenacher for useful comments on and for editing an earlier draft of this manuscript. We also thank Dr Krzysztof Radacki for help with X-ray diffraction experiments and with quantum chemical calculations.References
- B. Debnath, W. S. Singh, M. Das, S. Goswami, M. K. Singh, D. Maiti and K. Manna, Mater. Today Chem., 2018, 9, 56–72 CrossRef CAS.
- S. Qiu, H. Sun, A. H. Zhang, H. Y. Xu, G. L. Yan, Y. Han and X. J. Wang, Chin. J. Nat. Med., 2014, 12, 401–406 CAS.
- N. Li, Q. Wang, J. Zhou, S. Li, J. Liu and H. Chen, Molecules, 2022, 27, 3291 CrossRef CAS PubMed.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS PubMed.
- A. Farah, T. de Paulis, D. P. Moreira, L. C. Trugo and P. R. Martin, J. Agric. Food Chem., 2006, 54, 374–381 CrossRef CAS PubMed.
- M. T. Ayseli and Y. İpek Ayseli, Trends Food Sci. Technol., 2016, 48, 69–77 CrossRef CAS.
- Y. Wu, D. Ren, C. Gao, J. Li, B. Du, Z. Wang and S. Qian, Int. J. Pest Manag., 2021, 69, 288–298 CrossRef.
- M. Jacobson, Insecticides of Plant Origin, ACS Symposium Series, Am. Chem. Soc., 1989, 387, 1–10 Search PubMed.
- A. E. Wahba and M. T. Hamann, Mar. Drugs, 2010, 8, 2395–2416 CrossRef CAS PubMed.
- L. Zhai, Y. Tang, Y. Zhang, S.-H. Huang, L. Zhu and R. Hong, Chem. Rec., 2022, 22, e202100197 CrossRef CAS PubMed.
- B. C. Das, M. Adil Shareef, S. Das, N. K. Nandwana, Y. Das, M. Saito and L. M. Weiss, Bioorg. Med. Chem., 2022, 63, 116748 CrossRef CAS PubMed.
- P. Zhao, D. O. Nettleton, R. G. Karki, F. J. Zécri and S.-Y. Liu, Chem. Med. Chem., 2017, 12, 358–361 CrossRef CAS PubMed.
- C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS.
- C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS PubMed.
- M. Kawaguchi, Adv. Mater., 1997, 9, 615–625 CrossRef CAS.
- C. Baldock, G. J. de Boer, J. B. Rafferty, A. R. Stuitje and D. W. Rice, Biochem. Pharmacol., 1998, 55, 1541–1549 CrossRef CAS PubMed.
- H. Helten, Chem.–Eur. J., 2016, 22, 12972–12982 CrossRef CAS PubMed.
- H. Helten, Organoboron and Related Group 13 Polymers, in Comprehensive Organometallic Chemistry IV, ed. D. O’Hare, K. Meyer and G. Parkin, Elsevier, New York, 2022, pp. 71–134 Search PubMed.
- D. H. Knack, J. L. Marshall, G. P. Harlow, A. Dudzik, M. Szaleniec, S.-Y. Liu and J. Heider, Angew. Chem., Int. Ed., 2013, 52, 2599–2601 CrossRef CAS PubMed.
- J. Chorbacher, M. Maier, J. Klopf, M. Fest and H. Helten, Macromol. Rapid Commun., 2023, 44, 2300278 CrossRef CAS PubMed.
- M. Maier, J. Chorbacher, A. Hellinger, J. Klopf, J. Günther and H. Helten, Chem.–Eur. J., 2023, 29, e202302767 CrossRef CAS PubMed.
- Z. Liu and T. B. Marder, Angew. Chem., Int. Ed., 2008, 47, 242–244 CrossRef CAS PubMed.
- M. d. R. Merino-García, L. A. Soriano-Agueda, J. d. D. Guzmán-Hernández, D. Martínez-Otero, B. Landeros Rivera, F. Cortés-Guzmán, J. E. Barquera-Lozada and V. Jancik, Inorg. Chem., 2022, 61, 6785–6798 CrossRef PubMed.
- M. Shigeno, M. Imamatsu, Y. Kai, M. Kiriyama, S. Ishida, K. Nozawa-Kumada and Y. Kondo, Org. Lett., 2021, 23, 8023–8027 CrossRef CAS PubMed.
- E. R. Abbey and S.-Y. Liu, Org. Biomol. Chem., 2013, 11, 2060–2069 RSC.
- M. J. S. Dewar and R. C. Dougherty, J. Am. Chem. Soc., 1962, 84, 2648–2649 CrossRef CAS.
- M. Z. H. Kazmi, J. P. G. Rygus, H. T. Ang, M. Paladino, M. A. Johnson, M. J. Ferguson and D. G. Hall, J. Am. Chem. Soc., 2021, 143, 10143–10156 CrossRef CAS PubMed.
- C. Baldock, J. B. Rafferty, S. E. Sedelnikova, P. J. Baker, A. R. Stuitje, A. R. Slabas, T. R. Hawkes and D. W. Rice, Science, 1996, 274, 2107–2110 CrossRef CAS PubMed.
- G. Högenauer and M. Woisetschläger, Nature, 1981, 293, 662–664 CrossRef PubMed.
- M. A. Grassberger, F. Turnowsky and J. Hildebrandt, J. Med. Chem., 1984, 27, 947–953 CrossRef CAS PubMed.
- A. Chowdhury, S. Chatterjee, A. Kushwaha, S. Nanda, T. J. Dhilip Kumar and A. Bandyopadhyay, Chem.–Eur. J., 2023, 29, e202300393 CrossRef CAS PubMed.
- J. P. M. António, J. I. Carvalho, A. S. André, J. N. R. Dias, S. I. Aguiar, H. Faustino, R. M. R. M. Lopes, L. F. Veiros, G. J. L. Bernardes, F. A. da Silva and P. M. P. Gois, Angew. Chem., Int. Ed., 2021, 60, 25914–25921 CrossRef PubMed.
- O. Dilek, Z. Lei, K. Mukherjee and S. Bane, Chem. Commun., 2015, 51, 16992–16995 RSC.
- S. Cambray, A. Bandyopadhyay and J. Gao, Chem. Commun., 2017, 53, 12532–12535 RSC.
- M. Stojanović and M. Baranac-Stojanović, J. Org. Chem., 2016, 81, 197–205 CrossRef PubMed.
- J. Pedersen and K. V. Mikkelsen, RSC Adv., 2022, 12, 2830–2842 RSC.
- D. G. Hall, in Boronic Acids, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 1–99 Search PubMed.
- E. A. Sarina, M. M. Olmstead, D. Kanichar and M. P. Groziak, Acta Cryst., 2015, 71, 1085–1088 CrossRef CAS PubMed.
- J. P. M. António, L. M. Gonçalves, R. C. Guedes, R. Moreira and P. M. P. Gois, ACS Omega, 2018, 3, 7418–7423 CrossRef PubMed.
- H. Cho, S. Lee and M. S. Han, Org. Biomol. Chem., 2022, 20, 4986–4992 RSC.
- S. Shimo, T. Nakamura, K. Takahashi, N. Toriumi and N. Iwasawa, ChemPhotoChem, 2022, 6, e202100195 CrossRef CAS.
- S. Shimo, K. Takahashi and N. Iwasawa, Chem.–Eur. J., 2019, 25, 3790–3794 CrossRef CAS PubMed.
- J. J. Blackner, O. M. Schneider, W. O. Wong and D. G. Hall, J. Am. Chem. Soc., 2024, 146, 19499–19508 CrossRef CAS PubMed.
- F. Lindl, S. Lin, I. Krummenacher, C. Lenczyk, A. Stoy, M. Müller, Z. Lin and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 338–342 CrossRef CAS PubMed.
- K. D. M. Harris, B. M. Kariuki, C. Lambropoulos, D. Philp and J. M. Robinson, Tetrahedron, 1997, 53, 8599–8612 CrossRef CAS.
- B. Roques and D. Florentin, J. Organomet. Chem., 1972, 46, C38–C40 CrossRef CAS.
- H. G. Viehe and M. Reinstein, Chem. Ber., 1962, 95, 2557–2562 CrossRef CAS.
- H. Braunschweig, C. Hörl, L. Mailänder, K. Radacki and J. Wahler, Chem.–Eur. J., 2014, 20, 9858–9861 CrossRef CAS PubMed.
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. Sukumar Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- V. Gutmann, Coord. Chem. Rev., 1976, 18, 225–255 CrossRef CAS.
- P. Erdmann and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202114550 CrossRef CAS PubMed.
- A. E. Ashley, T. J. Herrington, G. G. Wildgoose, H. Zaher, A. L. Thompson, N. H. Rees, T. Krämer and D. O'Hare, J. Am. Chem. Soc., 2011, 133, 14727–14740 CrossRef CAS PubMed.
- A. D. Boese and N. C. Handy, J. Chem. Phys., 2002, 116, 9559–9569 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Cappello, D. A. B. Therien, V. N. Staroverov, F. Lagugne-Labarthet and J. B. Gilroy, Chem.–Eur. J., 2019, 25, 5994–6006 CrossRef CAS PubMed.
- E. Galbraith and T. D. James, Chem. Soc. Rev., 2010, 39, 3831–3842 RSC.
- C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed.
- W. Zhang, G. Li, L. Xu, Y. Zhuo, W. Wan, N. Yan and G. He, Chem. Sci., 2018, 9, 4444–4450 RSC.
- Y. Han, W. Yuan, H. Wang, M. Li, W. Zhang and Y. Chen, J. Mater. Chem. C, 2018, 6, 10456–10463 RSC.
- Y. Chen, W. Chen, Y. Qiao, X. Lu and G. Zhou, Angew. Chem., Int. Ed., 2020, 59, 7122–7130 CrossRef CAS PubMed.
- P. Li, D. Shimoyama, N. Zhang, Y. Jia, G. Hu, C. Li, X. Yin, N. Wang, F. Jäkle and P. Chen, Angew. Chem., Int. Ed., 2022, 61, e202200612 CrossRef CAS PubMed.
- For sensing experiments of a molecule with a DAB structure see: Y. Satta, R. Nishiyabu, T. D. James and Y. Kubo, Tetrahedron, 2017, 73, 2053–2061 CrossRef CAS.
- B. D. Gessner, M. Beller, J. P. Middaugh and G. M. Whitford, N. Engl. J. Med., 1994, 330, 95–99 CrossRef CAS PubMed.
- P. P. Singh, M. K. Barjatiya, S. Dhing, R. Bhatnagar, S. Kothari and V. Dhar, Urol. Res., 2001, 29, 238–244 CrossRef CAS PubMed.
- D. Kanduti, P. Sterbenk and B. Artnik, Mater. Socio Med., 2016, 28, 133–137 CrossRef PubMed.
- L. Cai, S. Lu and V. W. Pike, Eur. J. Org. Chem., 2008, 2853–2873 CrossRef CAS.
- J. O. Egekeze and F. W. Oehme, Vet. Q., 1980, 2, 104–114 CrossRef CAS PubMed.
- M. Axt, J. Alifantes and V. Emílio Uberti Costa, J. Chem. Soc., Perkin Trans. 2, 1999, 2783–2788 RSC.
- P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed.
- E. R. Abbey, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2008, 130, 7250–7252 CrossRef CAS PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0., Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2018 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, M. C. X. Li, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. L. F. Ding, J. G. F. Egidi, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- L. Wüst, O. Steinlein, T. Keim, H. Braunschweig and L. Meinel, EP Pat., 24207621.4, 2024.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and procedures, analytical, computational. CCDC 2394196–2394208. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01500f |
| ‡ These authors contributed equally to this work. |
| § While we obtained crystallographic proof of both enantiomers, attempts to quantify their ratio via1H NMR spectroscopy using the chiral lanthanoid shift reagent Eu(hfc)3 (hfc = (1R)-3-(hep-tafluorbutanoyl)camphorate) were unsuccessful. In case of 3-CN, we observed an abstraction of the cyanide anion due to its excellent ligand properties, along with the simultaneous recovery of 1-Ph, whereas no significant shifting was detected for 3-F (see ESI Appendix).† |
| This journal is © The Royal Society of Chemistry 2025 |