
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing fluorescent probe design through multilayer interaction convolutional networks: advancing biosensing and bioimaging precision†
Gongcheng
Ma‡
a,
Qihang
Ding‡
 *c,
Yuding
Zhang‡
d,
Xiaodong
Zeng‡
gh,
Kai
Zhu
b,
Hongli
Chen
a,
Wenxuan
Zhang
e,
Qingzhi
Wang
a,
Shuman
Huang
b,
Ping
Gong
*d,
Zhengwei
Xu
*b and
Xuechuan
Hong
*fgh
*c,
Yuding
Zhang‡
d,
Xiaodong
Zeng‡
gh,
Kai
Zhu
b,
Hongli
Chen
a,
Wenxuan
Zhang
e,
Qingzhi
Wang
a,
Shuman
Huang
b,
Ping
Gong
*d,
Zhengwei
Xu
*b and
Xuechuan
Hong
*fgh
aSchool of Life Science and Technology, Xinxiang Medical University, Xinxiang, 453003, China
bKey Laboratory of Artificial Intelligence and Personalized Learning in Education of Henan Province, School of Computer and Information Engineering, Henan Normal University, Xinxiang, 453007, China. E-mail: xuzhengweihhu@outlook.com
cDepartment of Chemistry, Korea University, Seoul, 02841, Korea. E-mail: dingqihang@whu.edu.cn
dGuangdong Key Laboratory of Nanomedicine, CAS-HK Joint Lab of Biomaterials, CAS Key Laboratory of Biomedical Imaging Science and System, Shenzhen Engineering Laboratory of Nanomedicine and Nanoformulations, CAS Key Lab for Health Informatics, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, China. E-mail: ping.gong@siat.ac.cn
eInstitute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100050, China
fDepartment of Cardiology, Zhongnan Hospital of Wuhan University, School of Pharmaceutical Sciences, Wuhan University, Wuhan, 430071, China. E-mail: xhy78@whu.edu.cn
gState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China
hShandong Laboratory of Yantai Drug Discovery, Bohai Rim Advanced Research Institute for Drug Discovery, Yantai, 264117, China
First published on 22nd April 2025
Abstract
Fluorescent probes are pivotal in biosensing and bioimaging, necessitating precise spectral tailoring for high-performance applications. Despite their importance, probe design remains largely empirical, a process that is both time-consuming and laborious. To streamline this, we created a comprehensive dataset of over 600 rhodamine fluorescent probes and employed a multilayer interaction convolutional model (MICNet) trained on molecular fingerprints to accurately predict excitation and emission wavelengths. Our model demonstrated high accuracy with mean relative errors (MRE) of 0.1% for excitation and 0.4% for emission wavelengths. Advancing this, we implemented a closed-loop strategy that integrates experimental feedback to iteratively enhance the design algorithm's accuracy, thereby improving the probes' performance and reliability. This method not only accelerates the probe development cycle but also facilitates the creation of spectrally customized fluorescent probes, offering a significant advancement in the field of bioanalytical chemistry.
Introduction
Fluorescent probes are engineered to absorb light at specific wavelengths and subsequently emit light at typically longer wavelengths.1 These probes are meticulously designed for selective binding to target molecules or cellular structures within biological samples, rendering them indispensable in the realms of biosensing and bioimaging.2,3 Upon interaction with analytes, these probes undergo structural, physical, or chemical transformations, which manifest as alterations in their fluorescent properties, such as spectral shifts or changes in the extinction coefficient and quantum yield.4–9 A comprehensive understanding of the spectral property changes during the response process is essential for the development of high-performance fluorescent probes.Fluorescent probe design conventionally necessitates an in-depth understanding of chemistry, biochemistry, and the principles of fluorescence, which is heavily contingent on the expertise of scientists.10,11 The synthesis and spectral characterization of these probes typically demand considerable time and labor. Consequently, there is a pressing need for predictive tools that can forecast the spectral alterations of probes in response to various analytes and under diverse environmental conditions, thereby facilitating a more expeditious and efficient design process. However, empirical rules and theoretical frameworks, such as the Woodward–Fieser rules, first-principles methods and time-dependent density functional theory (TD-DFT), possess certain limitations. They are often applicable to specific molecular systems, and there is a lack of comprehensive theories for calculating certain properties.12–15
Artificial intelligence (AI) is revolutionizing the field of chemistry by leveraging “big data” techniques and machine learning (ML) algorithms, enabling programs to autonomously discern complex patterns from extensive datasets that elude manual analysis. This paradigm shift from traditional, expert-rule-based methods to data-driven insights is significantly enhancing the efficiency and accuracy of chemical research.16,17 The integration of AI in the simulation and optimization of molecular structures has accelerated the screening process for molecules with potential therapeutic effects or unique properties, addressing the historical inefficiencies and high costs associated with conventional molecular design.18,19 Transforming chemical data into formats compatible with AI models is crucial for progressively refining algorithms and enhancing predictive accuracy.20–22
In a pioneering study, Par et al. introduced a deep learning strategy for the expeditious and dependable prediction of optical and photophysical attributes of organic compounds. This was accomplished by training the model on an extensive experimental database encompassing over 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 permutations of chromophores and solvents.23 Subsequently, in 2023, Tsuda et al. harnessed a massively parallelized variant of ChemTS to engineer fluorescent molecules. They adeptly employed DFT to assess potential energy surfaces, thereby achieving a judicious balance between reliability and computational economy.24 Despite these advancements, the realm of fluorescent probe design remains uncharted territory for AI models. Incorporating a feedback mechanism into the design algorithm facilitates iterative refinement and optimization, bolstering the system's efficacy and dependability through empirical validation.25–27 In light of these considerations, the development of AI-driven tools designed to demystify the complexities of predicting excitation and emission spectra in response to probe interactions is of paramount importance.
000 permutations of chromophores and solvents.23 Subsequently, in 2023, Tsuda et al. harnessed a massively parallelized variant of ChemTS to engineer fluorescent molecules. They adeptly employed DFT to assess potential energy surfaces, thereby achieving a judicious balance between reliability and computational economy.24 Despite these advancements, the realm of fluorescent probe design remains uncharted territory for AI models. Incorporating a feedback mechanism into the design algorithm facilitates iterative refinement and optimization, bolstering the system's efficacy and dependability through empirical validation.25–27 In light of these considerations, the development of AI-driven tools designed to demystify the complexities of predicting excitation and emission spectra in response to probe interactions is of paramount importance.
In this study, we have meticulously curated a novel dataset, designated as RFP1, comprising over 614 rhodamine-based fluorescent probes that span the UV–Vis–NIR spectra in terms of excitation and emission wavelengths. Utilizing three distinct molecular fingerprints, we have engaged five state-of-the-art AI models to predict the maximum excitation and emission wavelengths with remarkable accuracy (Scheme 1). Our evaluation involving ∼100 unseen samples has demonstrated exceptional predictive performance, with Morgan fingerprints and the Multilayer Interaction Convolutional Network (MICNet) achieving mean relative errors (MRE) of 0.1% and 0.4%, respectively. Notably, the predictive accuracy of our AI model has been significantly enhanced through iterative updates with new experimental data, underscoring the profound advantages of the iterative approach in AI-based rhodamine probe design.
 | ||
| Scheme 1 Schematic overview of the AI-enhanced rhodamine probe design strategy. We have meticulously compiled a novel dataset, designated RFP1, encompassing a diverse array of over 614 rhodamine-based fluorescent probes, spanning the UV–Vis–NIR spectrum in terms of excitation and emission wavelengths. Drawing from literature and patents, we collated data including molecular structures, excitation/emission spectra, solvent effects, pH dependencies, and more, converting this wealth of information into a computer-readable format utilizing the simplified molecular-input line-entry system (SMILES) and molecular fingerprints. This comprehensive database was subsequently employed to train five sophisticated deep learning models, which were continuously refined with incoming experimental data to enhance predictive accuracy. | ||
Results and discussion
Data processing and database construction
A comprehensive dataset of over 614 rhodamine-based fluorescent probes has been curated from literature and patent sources, reflecting the spectra of molecular architectures, excitation/emission profiles, pH responsiveness, and solvent interactions.28,29 Compounds were prioritized for the compilation of excitation and emission data if they exhibited multiple spectral peaks with the most red-shifted wavelength or the greatest intensity. pH data were coded as ‘0’ for neutral, ‘1’ for basic, and ‘−1’ for acidic conditions. The dataset was further refined by addressing missing values and outliers, and by applying normalization techniques, preparing it for training the neural network model. Following a rigorous filtering process, a refined dataset, RFP1, was constructed, encompassing rhodamine probes with excitation and emission spectra ranging from the UV to the NIR (Fig. 1A). The absorption wavelength range from 300 to 1100 nm and the corresponding emission wavelength range from 300 to 700 nm are illustrated in Fig. 1B and C. The majority of absorption bands peak between 300 and 700 nm, with approximately 70% surpassing 600 nm. In contrast, emission bands are primarily confined to the 500 to 700 nm range (Fig. 1B–D). The molecular structures of the collected probes were converted into computational descriptors, including RDKit descriptors, Morgan fingerprints, and MACCSKeys fingerprints. After excluding non-recognizable and invalid molecules, the curated database comprised a total of 614 entries. | ||
| Fig. 1 Database assembly of rhodamine probes. (A) Core structures of rhodamine fluorophores, highlighting the diversity of fluorescent scaffolds. (B) Distribution of the excitation band dataset, detailing the count of rhodamine probes within defined spectral intervals. (C) Corresponding distribution of the emission band dataset, showcasing the spectral diversity of the probes. (D) Quantitative representation of rhodamine probes allocated for detection and biological imaging, indicating their applicational prevalence. | ||
By utilizing existing databases containing excitation and emission spectral data of rhodamine derivatives and applying computational learning algorithms to analyze the influence of various functional groups on molecular fragments, a robust database of larger structural moieties can be established. This comprehensive resource could be a valuable asset for the design of optical probes. The database enables predictive modeling of excitation and emission spectra for rhodamine molecules when provided with the machine-encoded structural formula of an unidentified rhodamine species.
Assessment of molecular probes' fluorescence prediction models and fingerprints
Predicting the intricate fluorescence properties of molecular probes, which are defined by their complex spatial configurations, necessitates a sophisticated analysis of the interactions among multiple molecular targets. We then introduce MICNet to discern the subtle correlations between molecular architecture and spectral characteristics across various temporal dimensions (Fig. 2). The core of MICNet is the MIC-block, a modular unit that dissects the input molecular probe data into distinct subsequences. Each subsequence is subjected to dedicated convolutional processing, thereby extracting pivotal temporal features. Arranging multiple MIC-blocks in a binary tree architecture facilitates an exhaustive and profound dissection of the molecular probes' multifaceted attributes. By aligning MICNet's architectural design with the intrinsic features of molecular probe data, our approach enhances the precision of fluorescence property predictions for molecular probes (Fig. 2). The stacking of K layers of MICNets can significantly enhance the accuracy of fluorescence property predictions for molecular probes, which possess complex spatial structures. The stacking of multiple MICNet layers is particularly effective in teasing out the intricate relationships between molecular architecture and spectral behavior. | ||
| Fig. 2 Schematic representation of the MICNet architecture. This model is carefully designed to elucidate the complex relationships between molecular structures and their corresponding fluorescence properties. The figure illustrates the data processing sequence within MICNet, beginning with the division of molecular probe data into distinct subsequences in the MIC-block, followed by a series of convolutional operations specifically tailored to extract temporal features. The arrangement of MIC-blocks in a binary tree structure ensures comprehensive analysis, aligning with the detailed characteristics of molecular probe data and culminating in the accurate prediction of fluorescence properties as visualized in the diagram. | ||
We evaluated five artificial intelligence models, comprising CNN-LSTM, RESNet, VGG, VMD-Attention-LSTM (VATTL), and MICNet, for their ability to predict the fluorescence properties of molecular probes based on their spatial structures, utilizing the RFP1 database.30–32 Each model was trained on molecular fingerprint data encoded through RDKit descriptors, Morgan fingerprints, and MACCSKeys fingerprints, targeting the maximum excitation (Fig. 3A and S1†) and emission (Fig. 3B and S2†) values. To achieve the optimal balance between model performance and computational cost, 20-fold cross-validation allows for a more granular evaluation on the training data, thereby reducing the variability introduced by different training/validation dataset splits and enhancing the model's robustness. Predictive performance across all models improved with increasing training iterations, reaching a plateau at 50 iterations. Although RESNet exhibits the quickest convergence, MICNet has been chosen for further research due to the less time-sensitive nature of the task described in this article. MICNet demonstrates higher predictive accuracy after convergence, which is more relevant in the context of designing rhodamine probes where time sensitivity is low. The Mean Absolute Error (MAE) for each model stabilized after a certain number of training steps. Detailed results for mean relative error (MRE), MAE, and mean square error (MSE) are depicted in Fig. 3C, S3 and S4,† respectively. MICNet, in conjunction with Morgan fingerprint encoding, achieved the lowest MRE (0.001 for excitation, 0.004 for emission), MAE (0.9 for excitation, 1.6 for emission), and MSE (133.3 for excitation, 130.3 for emission). These values suggest that MICNet combined with Morgan fingerprints provides predictions that closely approximate the actual fluorescence properties of rhodamine probes. This optimal performance is ascribed to MICNet's recursive architecture and interactive learning mechanism, which efficiently captures the complex spatial features of molecular probes, thus enhancing the model's predictive accuracy for their fluorescence characteristics.33,34
 | ||
| Fig. 3 AI model construction and molecular fingerprint matching for fluorescence prediction. (A) maximum excitation and (B) emission values predicted by AI models are plotted against observed data across 20-fold cross-validation. The analysis incorporates three molecular fingerprinting methods: RDKit descriptors, Morgan fingerprints, and MACCSKeys fingerprints, subsequently processed by MICNet, CNN-LSTM, RESNet, VGG, and VATTL algorithms. (C) Comprehensive predictive performance is detailed for maximum excitation and emission, showcasing the impact of various fingerprinting methods on the accuracy of different AI algorithms. | ||
The impact of learning environmental related datasets on prediction accuracy
To enhance the prediction accuracy of the MICNet model, we have extended our analysis to encompass not only the molecular structural modeling but also the solvation effects on the absorption and fluorescence properties of rhodamine probes.35,36 The dataset, comprising a series of rhodamine probes designated as ‘test probe’, was meticulously assembled to validate the optical properties predicted by the MICNet model against empirical data. The predictive capabilities of the model were scrutinized under varying conditions, including the influence of pH, solvents, and their combined effects during the training phase. These findings are delineated in Fig. 4, which presents a comparative analysis of the model's predictive performance across three distinct environmental scenarios. In scenarios where pH or solvents were considered individually as training variables, the MICNet model exhibited MAE values of 1.919 for maximum excitation and 0.886 for maximum emission predictions. Conversely, when both pH and solvent properties were concurrently integrated into the training framework, the model's predictive accuracy significantly improved, with MAE values reduced to 0.474 for maximum excitation and 0.567 for maximum emission predictions, as depicted in Fig. 4B and C. Experimental evidence suggests that the model's predictive prowess is optimized when it concurrently accounts for both pH and solvent properties, as illustrated in Fig. S5–S7.†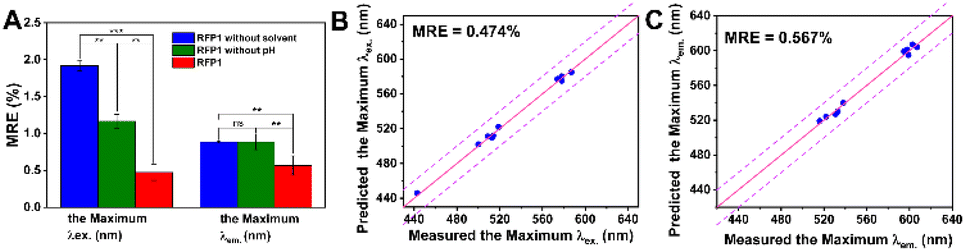 | ||
| Fig. 4 Enhanced predictive accuracy of spectral properties in rhodamine probes by MICNet. (A) Comparative analysis of the mean relative errors (MRE) for MICNet predictions with and without consideration of solvent and pH effects. (B) Maximum excitation and (C) emission values of the test probe, depicted as point-line plots, showcase the model's predictive accuracy under varying environmental conditions. The MRE is confined within a range of 0 to 3% (*p < 0.05, **p < 0.005, and ***p < 0.0005). | ||
AI model assisting the design of pH-response rhodamine probes
In pursuit of enhancing the AI system's role in the design of novel probes, we have developed a series of previously uncharacterized, pH-responsive rhodamine fluorescent probes. Fig. 6 illustrates the synthesis of four rhodamine-based structures, each appended with a diethylenetriamine spacer and linked to fluorescein, resulting in the creation of innovative pH-responsive probes: Rh640-fluorescein, Rh100-fluorescein, Rh19-fluorescein, and RhB-fluorescein. These probes are designed to exhibit an increase in rhodamine fluorescence and a concomitant decrease in fluorescein fluorescence in response to acidic conditions, with an inverse response in basic environments. The synthesis of these four pH-responsive rhodamine probes was successfully achieved, and their spectral characteristics in both acidic and basic media were meticulously measured and compared against predicted values. Notably, Rh640-fluorescein demonstrated the most significant spectral shifts, with excitation and emission peaks at 514/521 nm and 582/607 nm in acidic and alkaline solutions, respectively, highlighting its potential for pH sensing applications. The AI system's predictions were in close accord with experimental findings, with discrepancies consistently beneath 0.4%, thereby underscoring the system's reliability in forecasting spectral alterations associated with probe-analyte interactions, and its utility in the strategic design of fluorescent probes (Fig. 5). | ||
| Fig. 5 De novo design of pH-responsive rhodamine probes using MICNet AI modeling. The accuracy of MICNet is validated by comparing predicted and observed excitation/emission maxima. A schematic depicts the structural transitions of rhodamine probes under varying pH. Mean absolute errors (MAE) for excitation and emission predictions further highlight the system’s reliability in probe design. | ||
Assessment of pH-responsive properties of the Rh640-fluorescein
Four pH-responsive rhodamine probes were synthesized by integrating a diethylenetriamine segment with both tetrahydrochloride and fluorescein frameworks. The structural elucidation of these probes was thoroughly conducted in Fig. S8–S15.† The photophysical characteristics of the Rh640-fluorescein probe were examined in aqueous media at 37 °C. Fig. 6A illustrates the reversible pH-dependent fluorescence switching of Rh640-fluorescein between acidic and basic conditions, facilitating its utility as a pH indicator. Specifically, under acidic conditions, Rh640-fluorescein presented a distinct absorption peak at 582 nm and a pronounced emission peak at 607 nm. Under basic conditions, these values shifted to 514 nm and 521 nm, respectively (Fig. 6B and C). As pH increased, the fluorescence intensity at 521 nm (FL521) rose, while that at 607 nm (FL607) diminished, effecting a visible transition from red to green fluorescence. Within the pH range of 2.0–12.0, the ratio of fluorescence intensities (FL521/FL607) exhibited a 103-fold increase, from 0.08 to 8.25 (Fig. 1D). Notably, a linear correlation between this fluorescence intensity ratio and pH was established across the pH range of 4–8, which encompasses the majority of biologically relevant pH environments. Moreover, the reversible fluorescence response of Rh640-fluorescein to pH fluctuations between 4 and 10, as shown in Fig. 6E, underscores its potential for monitoring dynamic pH changes in biological systems. | ||
| Fig. 6 Comprehensive evaluation of Rh640-fluorescein for pH detection. (A) Schematic representation of the pH-induced fluorescence response mechanism of Rh640-fluorescein. (B) Absorbance spectra comparison at pH 3 and pH 10 for Rh640-fluorescein. (C) Fluorescence emission spectra of Rh640-fluorescein (5.0 μM) across a pH gradient (arrows indicate the spectral shift with increasing pH from 3 to 10). (D) Calibration curve depicting the ratio of fluorescence intensities at 521 nm (FL521) to 607 nm (FL607) as a function of pH. (E) Demonstration of the reversible fluorescence intensity ratio (FL521/FL607) upon alternating pH conditions between 4 and 10. (F) Confocal microscopic images of HepG2 cells treated with Rh640-fluorescein, showcasing fluorescence changes in a range of pH 4 to 10 solutions. | ||
Subsequently, fluorescence imaging of cells in different PBS buffers (pH 4–10) was determined using the Rh640-fluorescein probe through confocal microscopy (Fig. 6F). Spectral analysis of HepG2 cells labeled with Rh640-fluorescein revealed two distinguishable emission spectra corresponding to fluorescein at 510–530 nm (green) and rhodamine 640 at 600–620 nm (red). As shown in the overlay images, as the pH increased, the red fluorescence in HepG2 cells gradually decreased while the green fluorescence increased, which is consistent with the conclusions drawn from the in vitro solution, further confirming that Rh640-fluorescein is suitable for pH detection. The pH-responsive probe Rh640-fluorescein, developed through an integrated approach of MICNet model performance prediction, screening, synthesis, and verification, is of significant importance.
Enhancing the predictive precision of MICNet through iterative database refinement
The AI system integrates an evolutionary algorithm, enabling the model to incrementally refine its predictive capabilities with each database update. The enhancement in predictive performance following the incorporation of empirical data from pH-responsive rhodamine probes resulted into the RFP1 database (Fig. 7A). By rigorously controlling the experimental process and ensuring consistency with the training protocol of RFP1, we ensured that the RFP2 data exhibited the same feature space distribution, thereby avoiding potential conflicts between the two datasets. This upgrade facilitates the AI model's continuous learning during operational deployment, allowing for the accumulation of data and the refinement of molecular probe property features, thereby augmenting the model's predictive accuracy. The comparative analysis of relative errors and P values for the excitation prediction of the test probe, both pre- and post-database update indicates a reduction in the mean absolute error (MAE) of predicted excitation values by 0.9 upon model updating (Fig. 7B and S16A†). A corresponding improvement is observed in the prediction of emission values, with a decrease in relative errors and P values by 1.5 (Fig. 7C and S16B†). These findings substantiate that database updates significantly bolster the predictive accuracy of the MICNet model, providing robust support for the design of molecular probes. | ||
| Fig. 7 Enhancement of predictive performance in the RFP2 dataset upon incorporation of Rh640-fluorescein measurements. (A) The dataset updating protocol facilitates the refinement of the iterative AI model, thereby augmenting its predictive accuracy. Comparative analysis of the relative prediction errors for the excitation (B) and emission (C) wavelengths of the test probe, both pre- and post-dataset enhancement. | ||
Experimental section
All experimental methods are provided in the ESI.† Statistical analyses were performed using GraphPad Prism 8 software. Statistical significance was determined by performing either Student's t-test (two-tailed) or one-way ANOVA to calculate p-values between groups. A p-value less than 0.05 was considered statistically significant (*, p < 0.05; **, p < 0.01; ***, p < 0.001).Conclusions
Our study introduces a groundbreaking methodology employing iterative deep learning neural models for the de novo synthesis of fluorescent probes, marking a significant advancement in accelerating the development of probes with tailored spectral characteristics. The MICNet model demonstrates exceptional precision in predicting excitation and emission wavelengths, achieving average relative errors as low as 0.1% and 0.4%, respectively. This highlights the potential to drastically expedite the design process of such probes. Additionally, the integration of a closed-loop strategy that continuously refines the design algorithms based on experimental feedback enhances the predictive accuracy and reliability of the system.However, it is crucial to acknowledge the inherent limitations of our study. A primary concern is the scale and quality of the dataset, which serves as the foundation for training our AI models. Despite our efforts to assemble an extensive and representative dataset of rhodamine fluorescent probes, potential biases or omissions within the data may influence the predictive accuracy of our models. Moreover, while promising outcomes have been achieved in rhodamine probe design, further validation is necessary to establish the broader applicability and generalizability of our approach across different classes of fluorescent probes. To enhance the practical utility of our findings, several strategic initiatives can be pursued. First, expanding and refining the dataset for AI model training is essential, ensuring a more comprehensive representation of various fluorescent probe classes and their spectral characteristics. Additionally, fostering interdisciplinary collaboration between computational and experimental scientists is key to validating AI-generated predictions and further improving model accuracy. Developing accessible software tools based on our methodology would also facilitate the broader adoption and integration of AI-driven probe design processes, further strengthening the impact of our research.
Furthermore, AI techniques such as generative models and reinforcement learning show significant potential in addressing key challenges in fluorescent probe design, particularly photostability and cell permeability. For photostability, generative models, especially deep learning-based approaches, can predict novel structures with enhanced resistance to photobleaching by learning the relationship between molecular features and photostability. Reinforcement learning can further refine these designs by adjusting molecular structures based on simulated or experimental feedback, thereby improving their stability under different lighting conditions. Regarding cell permeability, AI can optimize the physicochemical properties that govern a probe's ability to cross cell membranes. Machine learning algorithms can identify patterns in molecular descriptors related to permeability, while generative models can propose new compounds with improved membrane penetration. By integrating molecular dynamics simulations with AI tools, further insights into probe-membrane interactions can be gained, allowing for the design of probes with enhanced permeability.
By leveraging these AI-driven approaches, we can accelerate the design of fluorescent probes that exhibit both high photostability and improved cell permeability, making them more effective for biological applications. These methods represent a promising frontier for the development of next-generation fluorescent probes. We will expand upon these AI solutions in the revised manuscript to provide a more comprehensive discussion on how AI can address the unique challenges of fluorescent probe design, further advancing their applicability in real-world scenarios.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
G. Ma, Q. Ding, Y. Zhang, and X. Zeng equally contributed to this work. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was partially supported by National Key R&D Program of China (2023YFC3605502), This work was supported by the National Natural Science Foundation of China (52403206, 82273796, 22477129, U22A20184, 52250077, and 52272080). The Program for The Doctoral Scientific Research Foundation of Xinxiang Medical University (300/505541). Henan Province Key Science and Technology under Grant (242102211076). The Jilin Province Natural Science Foundation of China (20220201093GX), and the Fundamental Research Funds for the Central Universities. The Shenzhen Science and Technology Program (KQTD20210811090115019), Shenzhen Basic Research (key project) (China) (JCYJ20210324120011030), the Major Instrumentation Development Program of the Chinese Academy of Sciences (Project Number: ZDKYYQ20220008). Shandong Laboratory Program (SYS202205), Natural Science Foundation of Shandong Province (ZR2023MB085, ZR2024QH253). We also acknowledge Prof. Pengfei Zhang from Shenzhen Institutes of Advanced Technology (SIAT), Chinese Academy of Sciences for his help on the prediction the optical properties of fluorescent materials based on AI models.References
- M. S. T. Gonçalves, Chem. Rev., 2009, 109, 190–212 CrossRef PubMed.
- L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC.
- Q. Ding, X. Wang, Y. Luo, X. Leng, X. Li, M. Gu and J. S. Kim, Coord. Chem. Rev., 2024, 508, 215772 CrossRef CAS.
- S. Tyagi, S. A. Marras and F. R. Kramer, Nat. Biotechnol., 2000, 18, 1191–1196 CrossRef CAS PubMed.
- L. Tu, C. Li, Q. Ding, A. Sharma, M. Li, J. Li, J. S. Kim and Y. Sun, J. Am. Chem. Soc., 2024, 146, 8991–9003 CrossRef CAS PubMed.
- J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906–918 CrossRef CAS PubMed.
- G. Ma, Q. Ding, Y. Zhang, Y. Wang, J. Xiang, M. Li, Q. Zhao, S. Huang, P. Gong and J. S. Kim, Chin. Chem. Lett., 2024, 35, 109293 CrossRef CAS.
- Y. Zhang, G. Zhang, Z. Zeng and K. Pu, Chem. Soc. Rev., 2022, 51, 566–593 RSC.
- S.-H. Park, N. Kwon, J.-H. Lee, J. Yoon and I. Shin, Chem. Soc. Rev., 2020, 49, 143–179 RSC.
- C. Liu, Q. Ding, Y. Liu, Z. Wang, Y. Xu, Q. Lu, X. Chen, J. Liu, Y. Sun and R. Li, Inorg. Chem., 2024, 63, 13059–13067 CrossRef CAS PubMed.
- L. Zeng, G. Ma, J. Lin and P. Huang, Small, 2018, 14, 1800782 CrossRef PubMed.
- R. B. Woodward, Structure and the absorption spectra of α, β-unsaturated ketones, J. Am. Chem. Soc., 1941, 63, 1123–1126 CrossRef CAS.
- C. C. J. Roothaan, Rev. Mod. Phys., 1951, 23, 69 CrossRef CAS.
- A. Charaf-Eddin, B. L. Guennic and D. Jacquemin, RSC Adv., 2014, 4, 49449–49456 RSC.
- R. O. Jones, Rev. Mod. Phys., 2015, 87, 897–923 CrossRef.
- A. F. De Almeida, R. Moreira and T. Rodrigues, Nat. Rev. Chem, 2019, 3, 589–604 CrossRef.
- Z. J. Baum, X. Yu, P. Y. Ayala, Y. Zhao, S. P. Watkins and Q. Zhou, J. Chem. Inf. Model., 2021, 61, 3197–3212 CrossRef CAS PubMed.
- A. Sahu, J. Mishra and N. Kushwaha, Comb. Chem. High Throughput Screening, 2022, 25, 1818–1837 CrossRef CAS PubMed.
- G. Schneider, Automating drug discovery, Nat. Rev. Drug Discovery, 2018, 17, 97–113 CrossRef CAS PubMed.
- W. Sha, Y. Guo, Q. Yuan, S. Tang, X. Zhang, S. Lu, X. Guo, Y.-C. Cao and S. Cheng, Intell. Syst., 2020, 2, 1900143 CrossRef.
- W. Li, Y. Wen, K. Wang, Z. Ding, L. Wang, Q. Chen, L. Xie, H. Xu and H. Zhao, Nat. Commun., 2024, 15, 2603 CrossRef CAS PubMed.
- Y. Yang, S. Zhang, K. Ranasinghe, O. Isayev and A. Roitberg, Annu. Rev. Phys. Chem., 2024, 75, 39371–39575 CrossRef.
- J. F. Joung, M. Han, J. Hwang, M. Jeong, D. H. Choi and S. Park, JACS Au, 2021, 1, 427–438 CrossRef CAS PubMed.
- M. Sumita, K. Terayama, N. Suzuki, S. Ishihara, R. Tamura, M. K. Chahal and D. T. Payne, Sci. Adv., 2022, 8, eabj3906 CrossRef CAS PubMed.
- R. P. Joshi and N. Kumar, Molecules, 2021, 26, 6761 CrossRef CAS PubMed.
- N. S. Hamzehkolaei and M. M. Miri Rashki, Eng. Comput., 2016, 32, 477–495 CrossRef.
- J. Dickerhoff, K. R. Warnecke, K. Wang, N. Deng and D. Yang, Int. J. Mol. Sci., 2021, 22, 10801 CrossRef CAS PubMed.
- L. Wang, W. Du, Z. Hu, K. Uvdal, L. Li and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 14026–14140 CrossRef CAS PubMed.
- J. Dai, C. Ma, P. Zhang, Y. Fu and B. Shen, Dyes Pigm., 2020, 177, 108321 CrossRef CAS.
- L. Boongasame, J. Boonpluk, S. Soponmanee, J. Muangprathub and K. Thammarak, Appl. Comput. Intell., 2024, 1, 8729440 Search PubMed.
- H. Chen, W. Xie and A. Vedaldi, IEEE, 2020, 721–725 Search PubMed.
- S. R. G. Reddy, G. P. S. Varma and R. L. Davuluri, Comput. Electr. Eng., 2023, 105, 108492 CrossRef.
- R. S. G. Sealfon, A. K. Wong and O. G. Troyanskaya, Nat. Rev. Mater., 2021, 6, 717–729 CrossRef.
- M. J. Volk, I. Lourentzou, S. Mishra, L. T. Vo, C. Zhai and H. Zhao, ACS Synth. Biol., 2020, 9, 1514–1533 CrossRef CAS PubMed.
- D. E. Jacquemin, A. Perpète and I. C. Ciofini Adamo, Acc. Chem. Res., 2009, 42, 326–334 CrossRef CAS PubMed.
- S. Chibani, D. Jacquemin and A. D. Laurent, Comput. Theor. Chem., 2014, 1040, 321–327 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08695c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |