
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-objective drug design with a scaffold-aware variational autoencoder†
Tiejun
Dong
 a,
Linlin
You
*a and
Calvin Yu-Chian
Chen
*bcd
a,
Linlin
You
*a and
Calvin Yu-Chian
Chen
*bcd
aIntelligent Medical Research Center, School of Intelligent Systems Engineering, Sun Yat-sen University, Shenzhen 518107, China. E-mail: youllin@mail.sysu.edu.cn; dongtj@mail2.sysu.edu.cn
bSchool of AI for Science, Peking University Shenzhen Graduate School, Shenzhen, Guangdong 518055, China. E-mail: cy@pku.edu.cn
cState Key Laboratory of Chemical Oncogenomics, Key Laboratory of Chemical Genomics, Peking University Shenzhen Graduate School, Shenzhen, Guangdong 518055, China
dDepartment of Medical Research, China Medical University Hospital, Taichung, 40447, Taiwan
First published on 25th June 2025
Abstract
Designing molecules with multiple properties of interest is a fundamental challenge in drug development. To tackle this, we have developed ScafVAE, an innovative scaffold-aware variational autoencoder designed for the in silico graph-based generation of multi-objective drug candidates. By integrating our proposed bond scaffold-based generation with perplexity-inspired fragmentation, we expand the accessible chemical space of the conventional fragment-based approach while preserving its high chemical validity. ScafVAE was pre-trained on a large dataset of molecules and further augmented through contrastive learning and molecular fingerprint reconstruction, resulting in high accuracy in predicting various computationally and experimentally measured molecular properties. Only a few of its parameters are task-specific, facilitating easy adaptation to new desired properties. ScafVAE was employed to generate dual-target drug candidates against drug resistance in cancer therapy, considering four distinct resistance mechanisms, with or without additional properties such as drug-likeness or toxicity. The generated molecules exhibited strong binding strength to target proteins using molecular docking or experimentally measured affinity while maintaining optimized extra properties. Further molecular dynamics simulations confirmed the stable binding interactions between the generated molecules and target proteins. These findings position ScafVAE as a promising alternative to conventional generation approaches.
1 Introduction
Small-molecule drugs play a crucial role in various aspects of public health care,1 from the treatment of COVID-19 to cancer therapies.2,3 A typical drug development pathway involves evaluating a vast space of pharmacologically relevant molecules, which was estimated to be up to 1060.4 Furthermore, in real-world scenarios, the designed molecule often needs to satisfy two or more properties of interest,5e.g., binding to two different proteins (dual-target drugs), achieving a high drug-likeness score and low toxicity. These multi-objective drugs can demonstrate advantageous pharmacological effects, e.g., dual-target drugs can leverage synthetic lethality to treat cancer with high specificity.6 However, for decades, many multi-objective drugs were discovered serendipitously rather than through rational design,7 making multi-objective drug design an open challenge in the field of drug development.Computer-aided drug design (CADD) has been extensively utilized across various stages of drug discovery to accelerate development and reduce costs,8 which typically encompasses molecular modeling and computational chemistry.8 Recently, artificial intelligence (AI), particularly deep learning (DL), has achieved remarkable advancements in several CADD domains, such as structural modeling9 and drug screening,10–12 predominantly through string-based and graph-based methodologies. In the field of drug design (i.e., molecular generation), the string-based approach, which employs sequence-based molecular representations (e.g., simplified molecular input line entry specification, SMILES) coupled with advanced language models, has gained widespread adoption alongside deep language models,13,14 such as MolGPT15 and Chemformer.16 Conversely, graph-based molecular generation, which represents a molecule as a collection of nodes and edges, is regarded as a more natural and interpretable approach,17–21 such as JT-VAE,20 GraphAF22 and GEGL.23 However, the graph-based approach has not been fully developed, especially within the context of multi-objective drug design.24,25
Three significant barriers exist in graph-based multi-objective molecular generation. First, three conventional molecular graph generation approaches (atom-based, fragment-based, and reaction-based) exhibit conflicting trade-offs regarding the accessible chemical space and the control over chemical validity or synthetic accessibility. Fragment-based and reaction-based approaches generate molecules fragment by fragment, ensuring high validity and synthetic accessibility, but their novelty and diversity are constrained by a predefined fragment set.26,27 In contrast, the atom-based approach generates molecules atom by atom with high chemical space accessibility while often requiring much more effort to control chemical validity.26,27 Second, the accuracy of property predictions is often hampered by data scarcity, particularly concerning experimentally measured properties.28,29 This limitation adversely affects the quality of the generated molecules. Third, models are required to be easily adaptable to new tasks14 (i.e., new properties), which presents a challenge for models where all parameters are task-specific.
To address the aforementioned challenges, we propose ScafVAE, a graph-based variational autoencoder (VAE) framework designed for the de novo design of multi-objective molecules with a scaffold-aware generating process. It learns to encode each molecule into a Gaussian-distributed latent vector, which can be decoded back into the original molecule (Fig. 1a). Surrogate models can be trained on the latent space for downstream predictions of molecular properties, enabling the sampling of molecules in latent space that exhibit desired properties. ScafVAE is built upon three key novel designs: bond scaffold-based molecular generation (Fig. 1e), perplexity-inspired fragmentation (Fig. 1c), and surrogate model augmentation (Fig. 1d). Unlike atom- or fragment-based generation approaches, bond scaffold-based generation first assembles fragments without specifying atom types (referred to as bond scaffolds) before decorating them with atom types to produce valid molecules. This approach offers a novel compromise that blurs the boundaries between atom- and fragment-based approaches, preserving the advantages of fragment-based generation while expanding its accessible chemical space. In particular, bond scaffolds are derived in a data-driven manner, utilizing bond perplexity as an indicator for fragmentation. Additionally, only a small machine learning (ML) module in the surrogate model is task-specific, and the remainder of the model is augmented through contrastive learning and fingerprint reconstruction, which improves its capacity to predict molecular properties.
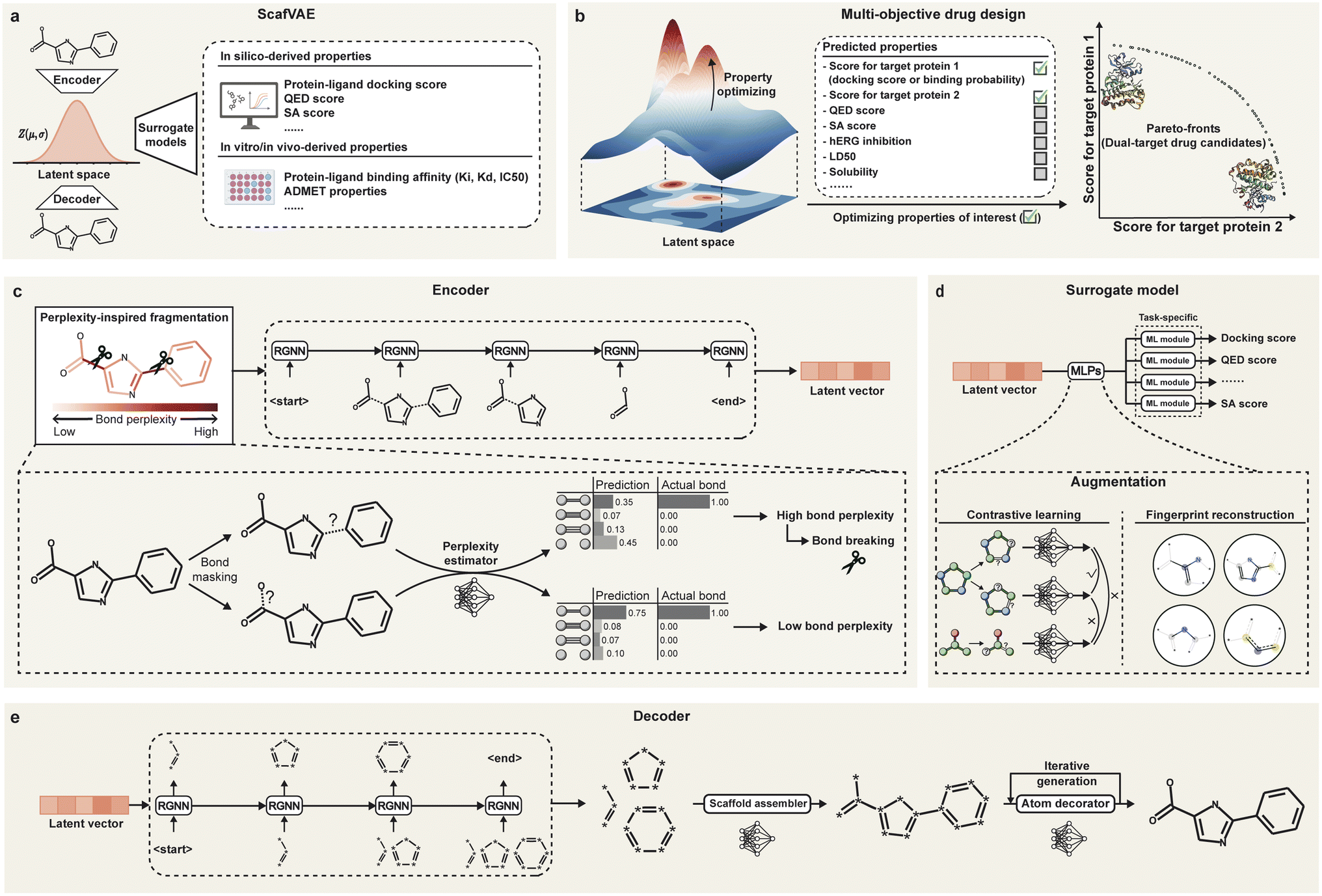 | ||
| Fig. 1 Overview of the ScafVAE framework. (a) The conception of ScafVAE, a graph-based VAE-style framework, consists of an encoder, a decoder, and a surrogate model. The encoder converts the molecule into a latent vector with an isotropic Gaussian distribution, followed by a decoder, which is capable of reconstructing the molecule from the latent vector. Various in silico or in vitro/in vivo biomedical datasets can be applied to the surrogate models to predict properties based on the latent space. (b) Molecules with desired properties can be generated through multi-objective optimization in the latent space, utilizing the predicted properties. For example, dual-target drug candidates can be obtained using the predicted docking score or probability of binding to two target proteins. (c) The encoder learns the molecule in a sequential manner using perplexity-inspired fragmentation, where perplexity serves as an indicator for bond breaking. The perplexity of each bond is estimated with a pre-trained masked graph model (i.e., the perplexity estimator), which reflects the uncertainty associated with each bond. (d) The surrogate model is composed of MLPs and task-specific ML modules. Contrastive learning and fingerprint reconstruction are employed for augmentation. (e) The decoder generates bond scaffolds sequentially, followed by a scaffold assembler and an atom decorator. Bond scaffolds are assembled by specifying connected bonds, and atoms are generated iteratively. | ||
ScafVAE was evaluated for predicting molecular properties and generating multi-objective molecules using various computational or experimentally measured datasets (Fig. 1a), including the molecular docking score, protein–ligand binding affinity, absorption, distribution, metabolism, excretion, toxicity (ADMET), drug-likeness (QED) score, and synthetic accessibility (SA) score. Specifically, we applied ScafVAE to generate dual-target molecules against drug resistance in cancer therapy based on four different resistance mechanisms, using either molecular docking or experimentally measured binding affinity. We further extended it to multi-objective molecular generation by incorporating more properties of interest. The results demonstrated that our model maintained a Gaussian-distributed latent space and outperformed the tested graph models on the GuacaMol benchmark,30 while being comparable to advanced string-based models. Benefiting from the surrogate model augmentation, our model achieved high accuracy in predicting 20 ADMET properties. The generated dual-target molecules exhibited a strong docking score or high predicted probability of binding. Further experiments revealed that it is feasible to generate dual-target molecules while optimizing their QED score, SA score, and ADMET properties. Molecular dynamics (MD) simulations confirmed strong and stable binding between the target proteins and the generated molecules. These results demonstrate a novel graph-based AI-driven drug design (AIDD) approach for multi-objective molecular design.
2 Results
2.1 Overview of ScafVAE
ScafVAE is a graph-based VAE-style framework for de novo multi-objective drug design (Fig. 1a). It mainly comprises three components: the encoder, the decoder, and the surrogate model. The encoder converts each molecule into a 64-D vector (known as the latent vector) with an isotropic Gaussian distribution in a sequential manner (Fig. 1c), followed by applying a decoder to reconstruct the molecule from the latent vector (Fig. 1e). Surrogate models can be trained using the latent vector for various downstream tasks (Fig. 1d). Once trained, single or multiple objective molecules can be generated by sampling vectors from the latent space (Fig. 1b).The encoder and decoder are stacks of graph neural network (GNN) and recurrent graph neural network (RGNN) blocks, where each molecule is represented as a graph. The graph is a set of nodes connected by edges, with the features (i.e., vectors) of nodes and edges initialized using the one-hot encoding of atom elements and bond types, respectively. The GNN block performs message passing to update features of a graph, where each node aggregates features from its adjacent nodes and corresponding edges.31 The updated features are selectively memorized by the RGNN block, with its gated recurrent unit (GRU).31 In contrast to the encoder and decoder, the surrogate model is much more light weight, predicting molecular properties by applying two shallow multilayer perceptrons (MLPs) followed by a task-specific conventional ML module. Except for the task-specific ML module, the entire model was pre-trained on a large molecular dataset (see Section 4.4). Only the task-specific ML module was trained with various downstream tasks, allowing our model to be rapidly adapted to new tasks.
Our framework contains three key designs that enhance its accuracy and generalizability: bond scaffold-based molecular generation (Fig. 1e), perplexity-inspired fragmentation (Fig. 1c), and surrogate model augmentation (Fig. 1d).
2.2 ScafVAE embeds molecules into Gaussian-distributed representations based on the bond scaffold
We first demonstrate the advantages of the bond scaffold-based strategy in terms of molecular chemical validity using a graph perturbation experiment. Atoms and bonds of the molecule were randomly edited (e.g., replacing atom elements or bond type, see Section 4.6 for details). As shown in Fig. 2a, perturbations involving bonds significantly decrease the validity of the molecules, particularly when bonds are randomly added or removed. In contrast, mutations to the atoms have a minimal impact on the overall validity. This reveals a major challenge faced by the atom-based generation process: a single incorrect bond assignment can lead to the generation of invalid molecules. By employing the bond scaffold-based generation process, this issue can be mitigated. | ||
| Fig. 2 ScafVAE learns molecules based on bond scaffolds while maintaining a Gaussian-distributed latent space. (a) Molecular chemical validity in different perturbation steps. (b) Perplexity-inspired fragmentation of three molecules. The perplexity of each bond is colored red, with a deeper red indicating higher perplexity. Molecules are fragmented by breaking bonds with high perplexity, and the bond scaffolds are obtained by subsequently removing atoms. (c) The most frequently occurring bond scaffolds in the training set. (d) Distribution of bond perplexity across different bond types in the training set. (e) The number of unique fragments (red, i.e., fragments without removing atoms) and unique bond scaffolds (black, i.e., fragments with removing atoms) in the training set obtained by perplexity-inspired fragmentation. (f) Visualization of the two dimensions of the latent space, with 3000 randomly selected molecules for each group. | ||
The bond scaffold set was collected with perplexity-inspired fragmentation, enabling the model to focus on learning bonds associated with high uncertainty. As shown in Fig. 1c and 2b, most bonds exhibit low perplexity (Fig. A1†), and the triple bonds show the highest perplexity (Fig. 2d). The fragments were converted to bond scaffolds through the removal of atoms, with the most frequent bond scaffolds shown in Fig. 2c. By grouping fragments into identical bond scaffolds, ScafVAE largely reduces the vocabulary size (i.e. the number of predicted categories in the network) in the generating process by one to two orders of magnitude (Fig. 2e). Furthermore, we compared the perplexity-inspired fragmentation with BRICS,33 a conventional rule-based fragmentation method (Table A5†). BRICS failed to cleave 0.96% of the molecules, while our method successfully cleaves all molecules and obtains more fragments with more than two connection points, allowing for more branching possibilities. Moreover, after removing the attachment points, our method resulted in only 6010 fragments, which is significantly less than that obtained with BRICS (23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 116). This indicates that combining perplexity-inspired fragmentation with the bond scaffold strategy is effective in achieving a smaller vocabulary size.
116). This indicates that combining perplexity-inspired fragmentation with the bond scaffold strategy is effective in achieving a smaller vocabulary size.
The pre-trained ScafVAE was first evaluated for its performance of reconstruction with the distribution of its latent space. As listed in Table A2,† the reconstruction accuracy for bond scaffold and molecule reach 0.987 and 0.683, respectively. This indicates that nearly all bond scaffolds can be reversibly mapped to the latent space, while the model is able to perfectly reconstruct the majority of the molecules. Moreover, the model maintains a Gaussian-distributed latent space (Fig. 2f), and the further t-SNE analysis indicates that the model can learn a smoother latent space (Fig. A18a†) compared with that of the JT-VAE20 (Fig. A18b†). Additionally, the latent space exhibits a gradient in relation to molecular weight, even without direct supervision regarding this aspect (Fig. A18a†). Furthermore, we constructed a grid visualization of the local neighborhood of a molecule by decoding its two random orthogonal unit vectors (Fig. A19†). The neighboring molecules display a smooth transition related to the atoms (Fig. A19b†) and bond scaffold (Fig. A19c†). These results are advantageous for the subsequent molecular generation processes performed within this latent space (see Section 2.4 and 2.5).
We further evaluated ScafVAE on the unconditional molecular generation using the distribution-learning benchmark from GuacaMol30 (Tables A3 and A4†). It includes five metrics for assessing the quality of generated molecules: the validity, uniqueness, novelty, KL-divergence41 (KLD) and Fréchet ChemNet distance42 (FCD) scores. The KLD and FCD scores are measures of the similarity of distributions between generated molecules and those used for training. ScafVAE showed high validity (0.987/0.997), uniqueness (1.000/1.000), and novelty (1.000/1.000) on the ChEMBL and ZINC datasets, respectively, which indicates that our model can generate valid molecules that were never present in the training set. ScafVAE achieves superior KLD (0.959) and FCD scores (0.338) on the ChEMBL dataset compared to the best-performing graph-based model VGAE-MCTS (KLD: 0.659 and FCD: 0.016). Although its FCD score is lower than that of the best SMILES-based model (0.913), its KLD score is comparable to that of the best SMILES-based model (0.991), and its validity, uniqueness, and novelty surpass those of all tested SMILES-based models. On the ZINC dataset, ScafVAE achieves the best FCD score (0.622), an increase of 0.304 compared to the second-best model, which scores 0.318. Additionally, our model has the second-best KLD score (0.857), which is comparable to the best model's score of 0.882. These results indicate the superiority of ScafVAE as a deep generative model for graph-based molecular generation.
2.3 Accurate prediction of molecular properties with the augmented surrogate model
The task-specific ML module in the surrogate model was trained to predict various molecular properties based on the latent space (Fig. 1a), and their accuracy is crucial for the follow-up molecular design. We comprehensively tested it on various datasets, including ADMET, QED score, SA score, protein–ligand binding, and protein–ligand docking.ScafVAE was first evaluated on 20 different ADMET tasks and demonstrated high accuracy compared to the JT-VAE,20 benefiting from the surrogate model augmentation, as shown in Fig. 3. For each ADMET task, five popular ML algorithms were employed, both with and without our augmentation, including Adaboost, support vector machine (SVM), K-nearest neighbor (KNN), random forest (RF), and MLP. For comparison, the performances of ML algorithms trained based on the latent space of JT-VAE20 are also shown. Full ScafVAE (i.e., with augmentation, orange in Fig. 3) outperforms the model without augmentation (green) and JT-VAE (blue) across most tasks. For the average performance of all ADMET experiments of regression tasks, ScafVAE achieves Spearman's ρ values of 0.73, while the values for the model without augmentation and JT-VAE are 0.70 and 0.69, respectively. For the average performance of all classification tasks, the values of the receiver operating characteristic curve (ROC-AUC) are 0.56, 0.48, and 0.46 for ScafVAE, ScafVAE without augmentation, and JT-VAE, respectively. These results indicate that ScafVAE achieves higher accuracy across various ADMET tasks through surrogate model augmentation. Further testing reveals that the time cost for augmentation (i.e., passing through two shallow pre-trained MLPs) was 1.82 × 10−4 s for single samples using one CPU core on average, which is near-negligible.
 | ||
| Fig. 3 ScafVAE accurately predicts molecular ADMET properties through surrogate model augmentation. (a–e) The performance of ScafVAE on absorption (a), distribution (b), excretion (c), metabolism (d), and toxicity (e) tasks, respectively. Each task was evaluated five times with random scaffold splitting, and the size of each dataset is listed in Table A6.† Five popular ML algorithms are used for evaluation: Adaboost, SVM, KNN, RF, and MLP. The performance of ML algorithms trained based on the latent space of JT-VAE20 is also shown for comparison. | ||
ScafVAE was further applied to predict properties related to protein–ligand interactions, as small molecules usually exert their bioactivity by binding to disease-associated proteins. Two types of datasets were adopted: computational docking datasets and experimentally measured binding datasets. The docking datasets used the molecular docking tool to generate potential binding strength (i.e. predicted binding free energy) for protein–ligand interactions (see Section 4.7), and the binding datasets were collected from the BindingDB database43 as described in Section 4.1, where inhibitors were identified using the IC50, Ki, and Kd values. Eight proteins, which play a vital role in drug resistance in cancer therapy,44–46 were selected for evaluation: epidermal growth factor receptor (EGFR), receptor tyrosine-protein kinase erbB-2 (HER2), P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), poly(ADP-ribose) polymerase 1 (PARP1), phosphatidylinositol 3-kinase (PI3K), histone deacetylases (HDAC), and bromodomain containing protein 4 (BRD4). As shown in Fig. A2,† ScafVAE achieves Spearman's ρ values ranging from 0.73 to 0.92 and mean absolute error (MAE) values ranging from 0.37 to 0.96 kcal mol−1 on the test set of the docking dataset, indicating strong correlations between the predicted docking score and actual docking score. For binding tasks (Fig. A3†), ScafVAE reaches ROC-AUC values ranging from 0.88 to 0.97, indicating that ScafVAE can accurately classify most active binders. Furthermore, ScafVAE achieves Spearman's ρ values of 0.93 and 0.92 in predicting QED and SA scores (Fig. A4†). Such high accuracy in predicting various properties supports the follow-up accurate and efficient molecular generation.5
2.4 Generating dual-target drug candidates against drug resistance in cancer
We applied ScafVAE in dual-target drug design against drug resistance in cancer, which has demonstrated potential in clinical therapy through synergistic or additive mechanisms.44,45,47 Eight proteins related to four mechanisms underlying drug resistance were selected: epigenetic alteration (EGFR/HER2) (Fig. 4a), drug efflux (P-gp/BCRP) (Fig. 4e), DNA damage repair (PARP1/PI3K) (Fig. 4i), and deregulated cell death (HDAC/BRD4) (Fig. 4m) for evaluation. Two types of generation routes were considered: the first is based on molecular docking datasets, aiming to generate molecules with improved docking scores (docking score-based generation). The second relies on binding datasets, with the goal of generating molecules with high probabilities of binding (binding probability-based generation). | ||
| Fig. 4 Design of dual-target drug candidates against drug resistance in cancer based on four different resistance mechanisms. Four resistance mechanisms are included: epigenetic alteration (a), drug efflux (e), DNA damage repair (i), and deregulated cell death (m). For two proteins related to each resistance mechanism, their sequence, structure, and binding pocket similarity are presented for comparison. (b, f, j and n) Visualization of actual docking scores of ten generated molecules in docking score-based generation for the four resistance mechanisms, respectively. The generated molecules were re-docked into proteins to obtain their actual docking scores. The actual docking scores of generated molecules are shown in orange, and the distribution of the training set is shown in black. (c, d, g, h, k, l, o and p) Visualization of interactions of re-docked structures for the four resistance mechanisms, respectively. The hydrophobic contact, π–π interaction, and hydrogen bond are colored green, cyan and blue, respectively. | ||
By performing Bayesian optimization (BO) in the latent space, molecules can be sampled with desired properties (i.e., the docking score or binding probability). However, this bi-objective optimization task often results in multiple optimal molecules due to potential internal conflicts between objectives, resulting in a solution set known as the Pareto front.48,49 In brief, for all molecules observed during the optimization, the Pareto front comprises molecules in which one of the objectives (e.g., the EGFR docking score) cannot be improved without worsening another objective (e.g., the HER2 docking score) (Fig. 1b). We addressed this bi-objective optimization problem using the non-dominated sorting genetic algorithm-II50,51 (NGSA-II) and selected the final molecule for evaluation from the Pareto front utilizing the pseudo-weight vector approach,52 which scores each molecule based on its weighted normalized distance to the worst molecule regarding each objective.
Our experiments demonstrate that ScafVAE can efficiently generate dual-target drug candidates for proteins with varying sequence, structure, or binding pocket similarity, with docking or binding datasets, respectively. We conducted ten independent runs for each task. The results indicate that the model has effectively converged to the Pareto front on docking score-based generation (Fig. A5†). We selected molecules (colored red) from the Pareto front by the pseudo-weight vector approach, assigning equal importance to both target proteins with weights of 0.5:0.5 (selected molecules are shown in Fig. A7–A10†). Since we used the predicted docking score for BO, the selected molecules were re-docked into the protein to obtain their actual docking scores. Most of the generated molecules exhibit strong actual docking scores (Fig. 4b, f, j and n). For instance, the generated molecules against drug efflux (Fig. 4f) achieve actual docking scores ranging from −11.5 kcal mol−1 to −15.8 kcal mol−1 for two proteins, which are strong and much better than the majority of those in the training set (black in Fig. 4f). The molecules with superior docking scores closely match the shape of the pocket surface and demonstrated strong hydrophobic interactions (Fig. 4c, d, g, h, k, l, o and p). Furthermore, we also performed binding probability-based generation by maximizing their probability of binding to the two target proteins (Fig. A6, generated molecules are shown in Fig. A11–A14†), similar to the docking score task. For all four resistance mechanisms, the generated molecules achieve a binding probability of greater than 0.95 with respect to two target proteins. These results indicate that ScafVAE exhibits outstanding capability in the generation of dual-target molecules with strong docking scores or high binding probability against two target proteins.
2.5 Multi-objective molecular design is possible using ScafVAE
We further attempted to govern the dual-target molecular generation by incorporating various extra properties (e.g., the QED score and ADMET properties) through multi-objective optimization. The previously generated molecules (Fig. A7–A14†) reveal that, in the absence of additional constraints, most of the generated molecules did not comply with the empirical rules of drug design proposed in previous studies.53–55 For instance, docking score-based generation (Fig. 4 and A7–A10†) often featured large rings, multiple rotatable bonds, and high molecular weight, which violates Lipinski's rule of five.53 This trend can be attributed to that docking tools often tend to assign better scores to molecules with more non-covalent interactions with the proteins (e.g., hydrogen bonding and hydrophobic interaction),56 consequently leading to an increase in molecule volume. Therefore, it is essential to incorporate more constraints in the design of dual-target molecules, resulting in a multi-objective optimization.ScafVAE was evaluated for generating dual-target drug candidates for EGFR/HER2 with docking score and QED score optimization, which constitutes a tri-objective optimization task. The QED score takes into account molecular weight and surface area,57 which helps implicitly control the size of the generated molecules. We employed the NGSA-II50,51 algorithm for this task. The Pareto front is shown in Fig. 5a. Molecules on the Pareto front with better docking scores exhibit lower QED scores, indicating an internal conflict between docking scores and molecular drug-likeness. We selected the molecules using the pseudo-weight vector approach with weights of 0.33:0.33: 0.33 (Fig. A15†). Compared with the molecules generated without QED score optimization (Fig. A7†), the generated tri-objective molecules resembled pre-drugs more closely, featuring fewer rotatable bonds and reduced molecular weight while still achieving a favorable actual docking score (Fig. 5b). Two representative generated molecules are shown in Fig. 5c, with actual QED scores of 0.70 and 0.76 and EGFR docking scores of −11.0 kcal mol−1 and −13.3 kcal mol−1, indicating promising potential for binding.
 | ||
| Fig. 5 Generation of multi-objective molecules. (a) Visualization of the Pareto front of the tri-objective optimization task (EGFR/HER2 docking score and QED score). (b) Visualization of actual docking scores of ten generated molecules for the tri-objective optimization task. The actual docking scores of generated molecules are shown in orange, and the distribution of the training set is shown in black. (c) Visualization of the re-docked structure of two generated molecules (compounds 1 and 2), along with their actual docking score and QED score. (d) Comparison of the predicted properties of the generated multi-objective molecules with those of the training set, including the HDAC/BRD4 docking score, the QED score, absorption, metabolism, and toxicity properties. (e–p) MD simulation of two generated multi-objective molecules, compound 3 (e) and compound 4 (k), along with their ligand pairwise RMSD (f–h and l–n) and protein RMSF with contact frequency between the protein and ligand (i, j, o and p). | ||
Furthermore, we applied ScafVAE to design multi-objective molecules targeting HDAC and BRD4 by optimizing various extra 12 properties, including the QED score, absorption, metabolism, and toxicity. The NGSA-III51,58,59 algorithm was employed for optimizing these 14 objectives, followed by selecting molecules using the pseudo-weight vector approach, with weights listed in Table A9.† We aimed to minimize the toxicity (hERG inhibition, AMES mutagenicity, drug-induced liver injury, and skin reaction), inhibition of metabolism systems (CYP2C19, CYP2D6, CYP3A4, CYP1A2, and CYP2C9), and docking scores, while maximizing the QED score, LD50, and solubility. Compared to the training set, the generated multi-objective molecules outperform in most predicted properties (Fig. 5d). On average, the properties of generated molecules achieve the following: inhibition of hERG (0.60/0.45), CYP2C19 (0.52/0.23), CYP2D6 (0.39/0.35), CYP3A4 (0.50/0.26), CYP1A2 (0.42/0.20), CYP2C9 (0.47/0.26), AMES mutagenicity (0.40/0.37), drug-induced liver injury (0.66/0.35), skin reaction (0.34/0.38), HDAC2 docking score (−6.66/– 7.03 kcal mol−1), and BRD4 docking score (−8.01/– 8.05 kcal mol−1) for the training set and the generated molecules, respectively, where the properties of generated molecules are lower (i.e. better) than those of the training set, except for the skin reaction. For the properties that need to be maximized, molecules achieve the following: QED score (0.58/0.52), LD50 (2.74/3.28), and solubility (−3.99/– 3.00) for the training set and the generated molecules, respectively, where the properties of generated molecules are higher than those of the training set, except for the QED score. These results indicate that ScafVAE makes it possible to design multi-objective molecules by combining different surrogate models, demonstrating promising potential for customized drug design.
We conducted MD simulations and retrosynthesis planning for the generated multi-objective drug candidates targeting HDAC and BRD4. Starting from the re-docked structure, we performed MD simulations for 20 ns for two molecules (compound 3 in Fig. 5e and compound 4 in Fig. 5k). The binding free energy over the simulation trajectories was calculated using the MM/PBSA60 approach. Compound 3 and compound 4 achieved average binding free energies of −20.22 kcal mol−1 and −22.45 kcal mol−1 for HDAC and −11.47 kcal mol−1 and −19.60 kcal mol−1 for BRD4, respectively. The ligand pairwise (root-mean-square deviation) RMSD calculated with the same trajectories shows average values of 0.61/0.87 Å, 0.92/0.87 Å for HDAC and BRD4 (Fig. 5f, g, l and m), respectively. In contrast, the ligand RMSD between different trajectories (i.e., binding to different proteins) achieves higher average RMSD values of 1.99/1.45 Å. This indicates that the generated molecules maintained stability when binding to a protein while demonstrating the ability to adopt different binding conformations when simulating with different proteins (Fig. A17†). Additionally, root-mean-square fluctuation (RMSF) analysis reveals low structural fluctuations in the proteins when bound to the generated molecules (Fig. 5i, j, o and p). Most residues with high contact frequency with the ligand exhibit low RMSF, indicating stable protein–ligand interactions. These findings suggest that the generated multi-objective molecules have the potential to stably bind to both target proteins while possessing multiple optimized molecular properties. Furthermore, we performed retrosynthesis planning for the two molecules (Fig. A24a and b†) with ASKCOS.61 Both molecules successfully identified potential reaction routes with a depth of less than 3. The average plausibility scores are 0.81 and 0.96, respectively, indicating good retrosynthetic feasibility.
3 Discussion
Our research introduces a novel deep graph-based generative model named ScafVAE, which is capable of generating multi-objective molecules from an innovative perspective. ScafVAE achieved high scores in validity, uniqueness, novelty, and KLD for molecular generation on the GuacaMol benchmark. Benefiting from the bond scaffold-based molecular generation and perplexity-inspired fragmentation, ScafVAE, our model, can cover a broader chemical space compared to conventional fragment-based methods. Moreover, the augmentation strategy enhances the accuracy of predictions across various molecular property tasks, thereby facilitating subsequent molecule generation. We applied ScafVAE to generate multi-objective molecules, focusing on the design of dual-target molecules by leveraging molecular docking scores or binding probabilities. Additionally, we considered extra molecular properties of interest, such as the QED score, the SA score, and ADMET. Subsequent MD simulations indicated that the generated dual-target molecules stably bind to the two targets through different binding modes while maintaining optimized extra properties. These results demonstrate that ScafVAE presents a promising and efficient alternative for de novo multi-objective drug design, surpassing conventional CADD methodologies.Our proposed bond scaffold-based generation approach bridges the gap between traditional atom- and fragment-based methods, demonstrating advanced performance on a dataset featuring larger molecules. Many popular graph-based models were used and evaluated only with small molecules.17,20,57 For example, the widely used molecular datasets ZINK250k62 and QM963 have a maximum number of heavy atoms of 38 and 9, respectively. In contrast, the dataset adopted in our study contains larger molecules (with a maximum of 128 heavy atoms). We found that ScafVAE is nearly perfect in reconstructing molecular scaffolds from the latent space, although it shows lower accuracy in reconstructing entire molecules. This indicates that the atom decoration process has a higher error rate, likely due to accumulated errors arising from the iterative atom generation process. This issue is expected to be addressed in future work, potentially through a one-shot generation process.
ScafVAE employs multiple designs to facilitate its application to new downstream tasks and to improve the quality of the generated multi-objective molecules. Recent studies have shown that VAE models are capable of learning representative latent distributions related to property values through joint training (i.e., full end-to-end) for property prediction.64–66 However, it is impractical to apply this approach to many molecular properties, especially those that are experimentally measured (e.g., ADMET), as the sizes of the available labeled datasets are much smaller compared to the vast chemical space of drug-like molecules, which is estimated at 1060.4 Besides, joint training could make it difficult to tune for new tasks since all parameters are task-specific. By contrast, only a small ML module in ScafVAE is task-specific, allowing for easier application to new tasks. The time required to train a machine learning module on a new molecular property is nearly negligible compared to the time required for tuning the entire model; for example, a KNN module can be trained within one second for 10000 data samples using a single CPU core. Furthermore, the augmentation of the surrogate model enhances the accuracy of property predictions, thereby increasing the model's generalizability. Subsequent experiments demonstrate that generating dual-target molecules with multiple additional properties of interest is feasible.
However, experiments with ScafVAE revealed several limitations in multi-objective drug design. First, objectives often conflict with one another,67–69 and this conflict intensifies with the increase of the objectives. For example, our results show a significant internal conflict between the docking score and the QED score. This conflict may arise because optimizing for better non-covalent interactions could lead to a substantial increase in molecular volume, which is unfavorable for its drug-likeness. Second, the quality of the generated molecules heavily relies on the accuracy of property predictions. Although several properties (e.g., the QED score and SA score) can be easily calculated with their actual values, desired properties are often either measured experimentally (e.g., ADMET) or require computationally intensive processes (e.g., molecular docking). This indicates that using predicted properties remains a more practical approach, and improving the accuracy of property prediction is still a major concern that needs to be addressed. Third, molecular geometry is not considered in this study. ScafVAE employs bond scaffold-based generation, which can incorporate geometries naturally as they are a set of fragments without specifying atom types. Future work is expected to investigate molecular geometry with bond scaffolds to learn the complex distribution of molecular geometry properties such as structure design14 and MD simulation.70
4 Materials and methods
4.1 Dataset collection
737964 molecules from the ChEMBL database and 218651 from ZINC250K were finally used in our study, respectively.
000 randomly selected molecules from the ChEMBL dataset were docked for each protein by following the docking process described in Section 4.7. The top-1 docking score was used for each molecule.
4.2 Data preprocessing
Each molecule was converted from a SMILES string into a complete graph (each node is connected to every other node), where each node represents a heavy atom and the bonds between them are represented as edges. The nodes and edges were initialized using one-hot encoding for the atom elements and the bond types, respectively. The available elements in the encoding include C, O, N, S, F, Cl, Br, B, I, and P. Atoms that do not belong to any of these element types are encoded as ‘UNK’. Similarly, the available bond types consist of single, double, triple, and aromatic bonds. If two atoms are not bonded, a ‘dummy bond’ is assigned to represent this absence of a connection. Chirality is not included in this study. To control the complexity, molecules with more than 128 heavy atoms or having a ring of size exceeding 16 are excluded. We used RDKit90 to convert molecules between SMILES strings and graphs.4.3 ScafVAE architecture
ScafVAE follows a VAE-style design network with a bond scaffold-based coding strategy and perplexity-inspired fragmentation, which are outlined in Section 2.1. Its neural networks mainly consist of GNN and RGNN blocks, which leverage the memory mechanism to encode and decode a molecular graph in a sequential manner. The detailed architecture of the encoder, decoder, GNN block, RGNN block, and surrogate model is described in Section A.2.2–A.2.6,† respectively. Additionally, the masked graph model used in perplexity-inspired fragmentation with its detailed settings is described in Section A.2.1.†4.4 Pre-training
The entire model, except for the task-specific ML module in the surrogate model, was first pre-trained with three losses: molecular reconstruction loss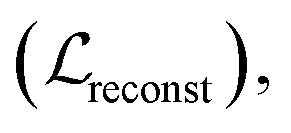 Kullback–Leibler loss
Kullback–Leibler loss  and surrogate model augmentation loss
and surrogate model augmentation loss  . The final loss can be written as:
. The final loss can be written as: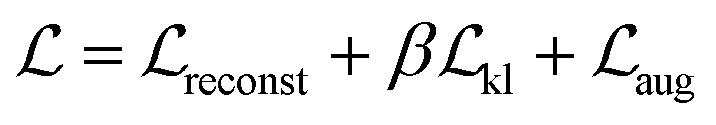 | (1) |
 is the sum of the reconstruction error calculated based on the input graph and the output graph, which is calculated as follows:
is the sum of the reconstruction error calculated based on the input graph and the output graph, which is calculated as follows: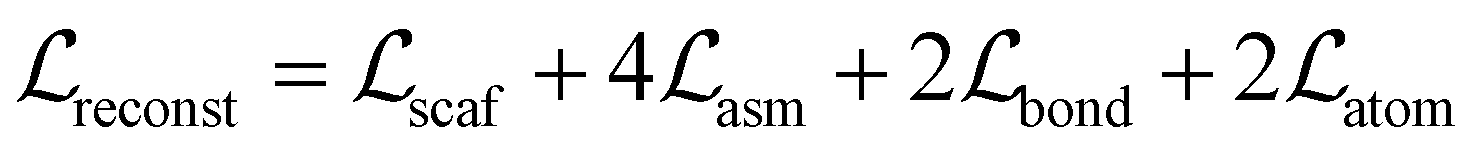 | (2) |
• Bond scaffold reconstruction loss  is used to train the decoder to reconstruct input bond scaffolds from the latent vector, which is a cross-entropy loss between the predicted distribution and the ground truth.
is used to train the decoder to reconstruct input bond scaffolds from the latent vector, which is a cross-entropy loss between the predicted distribution and the ground truth.
• Assembling loss 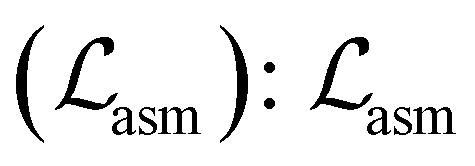 is used to train the assembler to reconstruct the connection (bond formation) between two bond scaffolds. We used the reverse order of the node removal to train the decoder. For each bond scaffold, the decoder predicts the probability of it being connected to each of the previously generated bond scaffolds. Benefiting from the settings of our model (see perplexity-inspired fragmentation in Section A.2.1 and BFS ordering in Section A.2.2†), each bond scaffold can only form a bond to one of the previously generated bond scaffolds. Therefore,
is used to train the assembler to reconstruct the connection (bond formation) between two bond scaffolds. We used the reverse order of the node removal to train the decoder. For each bond scaffold, the decoder predicts the probability of it being connected to each of the previously generated bond scaffolds. Benefiting from the settings of our model (see perplexity-inspired fragmentation in Section A.2.1 and BFS ordering in Section A.2.2†), each bond scaffold can only form a bond to one of the previously generated bond scaffolds. Therefore,  is calculated as a classification task with a cross-entropy loss.
is calculated as a classification task with a cross-entropy loss.
• Bond reconstruction loss  is used to train the assembler to reconstruct bond formation between two bond scaffolds. The decoder predicts the probability of bond formation for every atom pair between two bond scaffolds. Owing to our procedure (see Section A.2.1†), only one bond can be formed between two bond scaffolds, and therefore,
is used to train the assembler to reconstruct bond formation between two bond scaffolds. The decoder predicts the probability of bond formation for every atom pair between two bond scaffolds. Owing to our procedure (see Section A.2.1†), only one bond can be formed between two bond scaffolds, and therefore,  is also calculated as a classification task with a cross-entropy loss.
is also calculated as a classification task with a cross-entropy loss.
• Atom reconstruction loss  is used to train the atom decorator to reconstruct the atom element types based on the bond scaffolds, which is a cross-entropy loss between the predicted distribution and the input atom element types. It is achieved by using atom masking, i.e. the atoms requiring prediction are masked. We used a mask rate that was uniformly sampled from 0 to 1 during training.
is used to train the atom decorator to reconstruct the atom element types based on the bond scaffolds, which is a cross-entropy loss between the predicted distribution and the input atom element types. It is achieved by using atom masking, i.e. the atoms requiring prediction are masked. We used a mask rate that was uniformly sampled from 0 to 1 during training.
 is a regularization term between the normal distribution and the distribution predicted by the encoder, which is calculated using the Kullback–Leibler divergence. The β is a hyperparameter for balancing (β = 0.003 in this study).
is a regularization term between the normal distribution and the distribution predicted by the encoder, which is calculated using the Kullback–Leibler divergence. The β is a hyperparameter for balancing (β = 0.003 in this study).
 includes two losses for surrogate model augmentation:
includes two losses for surrogate model augmentation: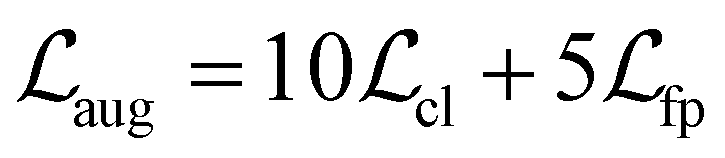 | (3) |
• Contrastive loss  contrastive learning38 aims at learning meaningful representation by contrasting similar and dissimilar pairs of molecules. Given a molecular graph, we randomly masked its nodes and edges with a probability of 15%. The InfoNCE loss91 was adopted for loss calculation. Masked graphs derived from the same molecules are considered positive pairs, while those derived from different molecules are treated as negative pairs (Fig. 1d).
contrastive learning38 aims at learning meaningful representation by contrasting similar and dissimilar pairs of molecules. Given a molecular graph, we randomly masked its nodes and edges with a probability of 15%. The InfoNCE loss91 was adopted for loss calculation. Masked graphs derived from the same molecules are considered positive pairs, while those derived from different molecules are treated as negative pairs (Fig. 1d).
• Fingerprint reconstruction loss  is used to encourage the model to decode the molecular fingerprint, which is calculated with MACCS and Morgan fingerprints (radius of 4, 512 bits) with a binary cross-entropy loss.
is used to encourage the model to decode the molecular fingerprint, which is calculated with MACCS and Morgan fingerprints (radius of 4, 512 bits) with a binary cross-entropy loss.
The model was pre-trained with the Adam optimizer.92 For the ChEMBL dataset, it underwent an initial training of 93800 iterations, with a maximum number of nodes set at 64 (learning rate of 5 × 10−4 and a batch size of 200). Subsequently, it underwent additional training of 10600 iterations, with a maximum number of nodes increased to 128 (learning rate of 1 × 10−4 and a batch size of 128). 50000 randomly sampled molecules are used as the test set, and the rest are used for training. For the ZINC dataset, the model was trained for 200000 iterations, employing a cosine annealing learning rate schedule (learning rate of 5 × 10−4 and a batch size of 24). 21785 randomly sampled molecules, which are approximately 10% of the ZINC dataset, are used as the test set. All bond scaffolds were ensured to appear at least once in the training set.
4.5 Task-specific training and evaluation
After pre-training, we trained the ML module with various conventional algorithms. To optimize the hyperparameters of ML modules used for ADMET tasks, we conducted a grid search using fivefold cross-validation on the training set. The search space was detailed in Table A8.† For other tasks, we employed the SVM algorithm with an RBF kernel and C = 1.0. Standard normalization was applied on the training set for all regression tasks, and the SMOTE93 algorithm was applied for balancing the number of different categories in the training set of all classification tasks. Scaffold splitting was applied for ADMET tasks, with each task being repeated five times, while single random splitting was applied for other tasks. For the docking, QED, and SA datasets, 45000 molecules were used for training and 5000 molecules were used for testing. For binding datasets, the training and test datasets were split with a ratio of 0.8:0.2. For experiments without surrogate model augmentation shown in Fig. 3, we used the latent vector as the input for the task-specific ML module (i.e., removed the pre-trained MLPs).
4.6 Molecular perturbation
Four basic perturbation processes were employed: atom mutation, bond mutation, bond removal and bond addition. Atom mutation involves randomly changing the element type of an atom, while bond mutation randomly alters the bond type of a bond, similar to atom mutation. Bond removal randomly eliminates an existing bond, and bond addition randomly adds a bond between two atoms with randomly assigned bond types. In the “bond removal/addition” group, there is a 50% chance of performing either removal or addition. In the “bond mutation + removal/addition” group, there is a one-third chance of performing mutation, removal, or addition, respectively. We randomly sampled 100000 molecules from the ChEMBL dataset used for this experiment. Each molecule was applied to 1 to 10 steps of perturbation.
4.7 Docking program settings
Molecular docking was performed with Smina94 with 50000 randomly sampled molecules from the ChEMBL dataset. It was allowed to generate conformations up to 20 with default parameters. Pockets were selected based on native ligands in crystal structures.
4.8 Molecular dynamics simulation
MD simulation was performed with the OpenMM95 package with the AMBER ff99sb force field.96 Protein and ligand topology files were generated using AmberTools97 and ACPYPE.98,99 pK values of ionizable groups were calculated using the H++100 at pH = 7.0. Each complex was dissolved with TIP3P water models in a cubic periodic box and placed at least 15 Å away from the periodic wall. The electroneutral system was guaranteed by adding the Na+ or Cl−. After energy minimization, the system was equilibrated at the NPT ensemble for 5000 ps at 298.15 K and 1.0 atm. The initial velocities were sampled from a Boltzmann distribution at 298.15 K. The production run was performed at the NPT ensemble for 20 ns at 298.15 K and 1.0 atm. The binding free energy was calculated with MM-GB/PBSA using gmx_MMPBSA.101 Analysis of trajectory was performed with MDTraj,102 and the contacts between proteins and compounds were identified with a distance cutoff of 4.5 Å. Protein–ligand interactions in frame shots were identified and visualized using PoseEdit.1034.9 Protein similarity evaluation
The similarity of the two proteins was shown for evaluating tasks of dual-target compound design in Fig. 4. The sequence, structure, and pocket similarity were calculated as the identity or score usinh NW-align,104,105 TM-align,106 and PocketMatch,107 respectively. The binding pocket was selected with a distance cutoff (4 Å) to the native ligand that was provided in the crystal structures.Data availability
All datasets used in this study are publicly available as described in Section 4.1. The source code of ScafVAE is available under an open-source license at https://github.com/tiejundong/ScafVAE.Author contributions
Tiejun Dong and Calvin Yu-Chian Chen led the research. Tiejun Dong contributed the idea, developed the method and performed the data processing and analytics. Tiejun Dong, Linlin You and Calvin Yu-Chian Chen wrote the manuscript together.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 62176272), Research and Development Program of Guangzhou Science and Technology Bureau (No. 2023B01J1016), and Key-Area Research and Development Program of Guangdong Province (No. 2020B1111100001).References
- F. D. Makurvet, Biologics vs. small molecules: drug costs and patient access, Med. Drug Discovery, 2021, 9, 100075 CrossRef CAS.
- S. Hoelder, P. A. Clarke and P. Workman, Discovery of small molecule cancer drugs: successes, challenges and opportunities, Mol. Oncol., 2012, 6(2), 155–176 CrossRef CAS PubMed.
- S. Lei, X. Chen, J. Wu, X. Duan and K. Men, Small molecules in the treatment of COVID-19, Signal Transduction Targeted Ther., 2022, 7(1), 387 CrossRef CAS PubMed.
- R. S. Bohacek, C. McMartin and W. C. Guida, The art and practice of structure-based drug design: a molecular modeling perspective, Med. Res. Rev., 1996, 16(1), 3–50 CrossRef CAS PubMed.
- P. Schneider, W. P. Walters, A. T. Plowright, N. Sieroka, J. Listgarten and Jr R. A. Goodnow, et al., Rethinking drug design in the artificial intelligence era, Nat. Rev. Drug Discovery, 2020, 19(5), 353–364 CrossRef CAS PubMed.
- N. J. O'Neil, M. L. Bailey and P. Hieter, Synthetic lethality and cancer, Nat. Rev. Genet., 2017, 18(10), 613–623 CrossRef PubMed.
- K. Maddeboina, B. Yada, S. Kumari, C. McHale, D. Pal and D. L. Durden, Recent advances in multitarget-directed ligands via in silico drug discovery, Drug Discovery Today, 2024, 103904 CrossRef CAS PubMed.
- L. Zhao, H. L. Ciallella, L. M. Aleksunes and H. Zhu, Advancing computer-aided drug discovery (CADD) by big data and data-driven machine learning modeling, Drug discovery today, 2020, 25(9), 1624–1638 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green and A. Pritzel, et al., Accurate structure prediction of biomolecular interactions with AlphaFold 3, Nature, 2024, 1–3 Search PubMed.
- G. Zhou, D. V. Rusnac, H. Park, D. Canzani, H. M. Nguyen and L. Stewart, et al., An artificial intelligence accelerated virtual screening platform for drug discovery, Nat. Commun., 2024, 15(1), 7761 CrossRef CAS PubMed.
- T. Dong, Z. Yang, J. Zhou and C. Y. C. Chen, Equivariant flexible modeling of the protein–ligand binding pose with geometric deep learning, J. Chem. Theory Comput., 2023, 19(22), 8446–8459 CrossRef CAS PubMed.
- M. W. Mullowney, K. R. Duncan, S. S. Elsayed, N. Garg, J. J. van der Hooft and N. I. Martin, et al., Artificial intelligence for natural product drug discovery, Nat. Rev. Drug Discovery, 2023, 22(11), 895–916 CrossRef CAS PubMed.
- Y. Du, A. R. Jamasb, J. Guo, T. Fu, C. Harris and Y. Wang, et al., Machine learning-aided generative molecular design, Nat. Mach. Intell., 2024, 1–16 Search PubMed.
- D. B. Catacutan, J. Alexander, A. Arnold and J. M. Stokes, Machine learning in preclinical drug discovery, Nat. Chem. Biol., 2024, 20(8), 960–973 CrossRef CAS PubMed.
- V. Bagal, R. Aggarwal, P. Vinod and U. D. Priyakumar, MolGPT: molecular generation using a transformer-decoder model, J. Chem. Inf. Model., 2021, 62(9), 2064–2076 CrossRef PubMed.
- R. Irwin, S. Dimitriadis, J. He and E. J. Bjerrum, Chemformer: a pre-trained transformer for computational chemistry, Mach. Learn.: Sci. Technol., 2022, 3(1), 015022 Search PubMed.
- D. Flam-Shepherd, K. Zhu and A. Aspuru-Guzik, Language models can learn complex molecular distributions, Nat. Commun., 2022, 13(1), 3293 CrossRef CAS PubMed.
- J. Lim, S. Y. Hwang, S. Moon, S. Kim and W. Y. Kim, Scaffold-based molecular design with a graph generative model, Chem. Sci., 2020, 11(4), 1153–1164 RSC.
- W. J. Godinez, E. J. Ma, A. T. Chao, L. Pei, P. Skewes-Cox and S. M. Canham, et al., Design of potent antimalarials with generative chemistry, Nat. Mach. Intell., 2022, 4(2), 180–186 CrossRef.
- W. Jin, R. Barzilay and T. Jaakkola, Junction tree variational autoencoder for molecular graph generation, in International conference on machine learning, PMLR, 2018, pp. 2323–2332 Search PubMed.
- X. Kong, W. Huang, Z. Tan and Y. Liu, Molecule generation by principal subgraph mining and assembling, Adv. Neural Inf. Process. Syst., 2022, 35, 2550–2563 Search PubMed.
- C. Shi, M. Xu, Z. Zhu, W. Zhang, M. Zhang and J. Tang, Graphaf: a flow-based autoregressive model for molecular graph generation, arXiv, 2020, preprint, arXiv:200109382, DOI:10.48550/arXiv.2001.09382.
- S. Ahn, J. Kim, H. Lee and J. Shin, Guiding deep molecular optimization with genetic exploration, Adv. Neural Inf. Process. Syst., 2020, 33, 12008–12021 Search PubMed.
- A. N. Abeer, N. M. Urban, M. R. Weil, F. J. Alexander and B. J. Yoon, Multi-objective latent space optimization of generative molecular design models, Patterns, 2024, 5(10), 101042 CrossRef PubMed.
- S. Luukkonen, H. W. van den Maagdenberg, M. T. Emmerich and G. J. van Westen, Artificial intelligence in multi-objective drug design, Curr. Opin. Struct. Biol., 2023, 79, 102537 CrossRef CAS PubMed.
- P. Polishchuk, CReM: chemically reasonable mutations framework for structure generation, J. Cheminf., 2020, 12(1), 28 Search PubMed.
- J. Meyers, B. Fabian and N. Brown, De novo molecular design and generative models, Drug discovery today, 2021, 26(11), 2707–2715 CrossRef CAS PubMed.
- B. Dou, Z. Zhu, E. Merkurjev, L. Ke, L. Chen and J. Jiang, et al., Machine learning methods for small data challenges in molecular science, Chem. Rev., 2023, 123(13), 8736–8780 CrossRef CAS PubMed.
- A. Nandy, C. Duan and H. J. Kulik, Audacity of huge: overcoming challenges of data scarcity and data quality for machine learning in computational materials discovery, Curr. Opin. Chem. Eng., 2022, 36, 100778 CrossRef.
- N. Brown, M. Fiscato, M. H. Segler and A. C. Vaucher, GuacaMol: benchmarking models for de novo molecular design, J. Chem. Inf. Model., 2019, 59(3), 1096–1108 CrossRef CAS PubMed.
- K. Cho, Learning Phrase Representations using RNN Encoder–Decoder for Statistical Machine Translation, arXiv, 2014, preprint, arXiv:14061078, DOI:10.48550/arXiv.1406.1078.
- Q. Liu, M. Allamanis, M. Brockschmidt and A. Gaunt, Constrained graph variational autoencoders for molecule design, Adv. Neural Inf. Process. Syst., 2018, 31, 7795–7804 Search PubMed.
- J. Degen, C. Wegscheid-Gerlach, A. Zaliani and M. Rarey, On the art of compiling and using ‘drug-like’ chemical fragment spaces, ChemMedChem, 2008, 3(10), 1503 CrossRef CAS PubMed.
- S. R. Gadre, R. N. Shirsat and A. C. Limaye, Molecular tailoring approach for simulation of electrostatic properties, J. Phys. Chem., 1994, 98(37), 9165–9169 CrossRef CAS.
- S. Jinsong, J. Qifeng, C. Xing, Y. Hao and L. Wang, Molecular fragmentation as a crucial step in the AI-based drug development pathway, Commun. Chem., 2024, 7(1), 20 CrossRef CAS PubMed.
- M. A. Collins, Systematic fragmentation of large molecules by annihilation, Phys. Chem. Chem. Phys., 2012, 14(21), 7744–7751 RSC.
- M. A. Collins, Molecular forces, geometries, and frequencies by systematic molecular fragmentation including embedded charges, J. Chem. Phys., 2014, 141(9), 094108 CrossRef PubMed.
- K. He, H. Fan, Y. Wu, S. Xie and R. Girshick, Momentum contrast for unsupervised visual representation learning, in Proceedings of the IEEE/CVF conference on computer vision and pattern recognition, 2020, pp. 9729–9738 Search PubMed.
- J. Deng, Z. Yang, H. Wang, I. Ojima, D. Samaras and F. Wang, A systematic study of key elements underlying molecular property prediction, Nat. Commun., 2023, 14(1), 6395 CrossRef CAS PubMed.
- B. Zagidullin, Z. Wang, Y. Guan, E. Pitkänen and J. Tang, Comparative analysis of molecular fingerprints in prediction of drug combination effects, Briefings Bioinf., 2021, 22(6), bbab291 CrossRef PubMed.
- S. Kullback and R. A. Leibler, On information and sufficiency, Ann. Math. Stat., 1951, 22(1), 79–86 CrossRef.
- K. Preuer, P. Renz, T. Unterthiner, S. Hochreiter and G. Klambauer, Fréchet ChemNet distance: a metric for generative models for molecules in drug discovery, J. Chem. Inf. Model., 2018, 58(9), 1736–1741 CrossRef CAS PubMed.
- BindingDB Search PubMed.
- J. Ye, J. Wu and B. Liu, Therapeutic strategies of dual-target small molecules to overcome drug resistance in cancer therapy, Biochim. Biophys. Acta, Rev. Cancer, 2023, 1878(3), 188866 CrossRef CAS PubMed.
- L. Tan, J. Zhang, Y. Wang, X. Wang, Y. Wang and Z. Zhang, et al., Development of dual inhibitors targeting epidermal growth factor receptor in cancer therapy, J. Med. Chem., 2022, 65(7), 5149–5183 CrossRef CAS PubMed.
- N. M. Raghavendra, D. Pingili, S. Kadasi, A. Mettu and S. Prasad, Dual or multi-targeting inhibitors: the next generation anticancer agents, Eur. J. Med. Chem., 2018, 143, 1277–1300 CrossRef CAS PubMed.
- L. Hu, M. Fan, S. Shi, X. Song, F. Wang and H. He, et al., Dual target inhibitors based on EGFR: promising anticancer agents for the treatment of cancers (2017-), Eur. J. Med. Chem., 2022, 227, 113963 CrossRef CAS PubMed.
- D. A. Van Veldhuizen and G. B. Lamont, et al., Evolutionary computation and convergence to a Pareto front, in Late breaking papers at the genetic programming 1998 conference, Citeseer, 1998, pp. 221–228 Search PubMed.
- H. Yao, Z. Xu, Y. Hou, Q. Dong, P. Liu and Z. Ye, et al., Advanced industrial informatics towards smart, safe and sustainable roads: a state of the art, J. Traffic Transp. Eng., Engl. Ed., 2023, 10(2), 143–158 Search PubMed.
- K. Deb, A. Pratap, S. Agarwal and T. Meyarivan, A fast and elitist multiobjective genetic algorithm: NSGA-II, IEEE Trans. Evol. Comput., 2002, 6(2), 182–197 CrossRef.
- J. Jazzbin, et al., Geatpy: the genetic and evolutionary algorithm toolbox with high performance in python, https://www/geatpy/com/, accessed 2020, 2020 Search PubMed.
- C. Coello, G. Lamont and D. van Veldhuizen, Multi-objective optimization using evolutionary algorithms, John Wiley & Sons, Inc., NY, USA, 2nd edn, 2007 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23(1–3), 3–25 CrossRef CAS.
- C. A. Lipinski, Lead-and drug-like compounds: the rule-of-five revolution, Drug Discovery Today: Technol., 2004, 1(4), 337–341 CrossRef CAS PubMed.
- I. Muegge, Selection criteria for drug-like compounds, Med. Res. Rev., 2003, 23(3), 302–321 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31(2), 455–461 CrossRef CAS PubMed.
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Quantifying the chemical beauty of drugs, Nat. Chem., 2012, 4(2), 90–98 CrossRef CAS PubMed.
- K. Deb and H. Jain, An evolutionary many-objective optimization algorithm using reference-point-based nondominated sorting approach, part I: solving problems with box constraints, IEEE Trans. Evol. Comput., 2013, 18(4), 577–601 Search PubMed.
- H. Jain and K. Deb, An evolutionary many-objective optimization algorithm using reference-point based nondominated sorting approach, part II: Handling constraints and extending to an adaptive approach, IEEE Trans. Evol. Comput., 2013, 18(4), 602–622 Search PubMed.
- S. Genheden and U. Ryde, The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities, Expert Opin. Drug Discovery, 2015, 10(5), 449–461 CrossRef CAS PubMed.
- Z. Tu, S. J. Choure, M. H. Fong, J. Roh, I. Levin and K. Yu, et al., ASKCOS: an open source software suite for synthesis planning, arXiv, 2025, preprint, arXiv:250101835, DOI:10.48550/arXiv.2501.01835.
- T. Sterling and J. J. Irwin, ZINC 15–ligand discovery for everyone, J. Chem. Inf. Model., 2015, 55(11), 2324–2337 CrossRef CAS PubMed.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. Von Lilienfeld, Quantum chemistry structures and properties of 134 kilo molecules, Sci. Data, 2014, 1(1), 1–7 Search PubMed.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling and D. Sheberla, et al., Automatic chemical design using a data-driven continuous representation of molecules, ACS Cent. Sci., 2018, 4(2), 268–276 CrossRef PubMed.
- A. F. Oliveira, J. L. Da Silva and M. G. Quiles, Molecular property prediction and molecular design using a supervised grammar variational autoencoder, J. Chem. Inf. Model., 2022, 62(4), 817–828 CrossRef CAS PubMed.
- J. Boitreaud, V. Mallet, C. Oliver and J. Waldispuhl, OptiMol: optimization of binding affinities in chemical space for drug discovery, J. Chem. Inf. Model., 2020, 60(12), 5658–5666 CrossRef CAS PubMed.
- C. A. Nicolaou and N. Brown, Multi-objective optimization methods in drug design, Drug Discovery Today: Technol., 2013, 10(3), e427–e435 CrossRef PubMed.
- G. Lambrinidis and A. Tsantili-Kakoulidou, Challenges with multi-objective QSAR in drug discovery, Expert Opin. Drug Discovery, 2018, 13(9), 851–859 CrossRef PubMed.
- C. Von Lücken, B. Barán and C. Brizuela, A survey on multi-objective evolutionary algorithms for many-objective problems, Comput. Optim. Appl., 2014, 58, 707–756 Search PubMed.
- N. Yao, X. Chen, Z. H. Fu and Q. Zhang, Applying classical, ab initio, and machine-learning molecular dynamics simulations to the liquid electrolyte for rechargeable batteries, Chem. Rev., 2022, 122(12), 10970–11021 CrossRef CAS PubMed.
- B. Zdrazil, E. Felix, F. Hunter, E. J. Manners, J. Blackshaw and S. Corbett, et al., The ChEMBL database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods, Nucleic Acids Res., 2024, 52(D1), D1180–D1192 CrossRef CAS PubMed.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, ZINC: a free tool to discover chemistry for biology, J. Chem. Inf. Model., 2012, 52(7), 1757–1768 CrossRef CAS PubMed.
- K. Huang, T. Fu, W. Gao, Y. Zhao, Y. Roohani and J. Leskovec, et al., Therapeutics data commons: machine learning datasets and tasks for drug discovery and development, arXiv, 2021, preprint, arXiv:210209548, DOI:10.48550/arXiv.2102.09548.
- T. Hou, J. Wang, W. Zhang and X. Xu, ADME evaluation in drug discovery. 7. Prediction of oral absorption by correlation and classification, J. Chem. Inf. Model., 2007, 47(1), 208–218 CrossRef CAS PubMed.
- N. N. Wang, J. Dong, Y. H. Deng, M. F. Zhu, M. Wen and Z. J. Yao, et al., ADME properties evaluation in drug discovery: prediction of Caco-2 cell permeability using a combination of NSGA-II and boosting, J. Chem. Inf. Model., 2016, 56(4), 763–773 CrossRef CAS PubMed.
- M. Wenlock and N. Tomkinson, Experimental in vitro DMPK and physicochemical data on a set of publicly disclosed compounds, EMBL-EBI, 2015 Search PubMed.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse and A. S. Pappu, et al., MoleculeNet: a benchmark for molecular machine learning, Chem. Sci., 2018, 9(2), 513–530 RSC.
- M. C. Sorkun, A. Khetan and S. Er, AqSolDB, a curated reference set of aqueous solubility and 2D descriptors for a diverse set of compounds, Sci. Data, 2019, 6(1), 143 CrossRef PubMed.
- D. L. Mobley and J. P. Guthrie, FreeSolv: a database of experimental and calculated hydration free energies, with input files, J. Comput.-Aided Mol. Des., 2014, 28, 711–720 CrossRef CAS PubMed.
- I. F. Martins, A. L. Teixeira, L. Pinheiro and A. O. Falcao, A Bayesian approach to in silico blood–brain barrier penetration modeling, J. Chem. Inf. Model., 2012, 52(6), 1686–1697 CrossRef CAS PubMed.
- F. Lombardo and Y. Jing, In silico prediction of volume of distribution in humans. Extensive data set and the exploration of linear and nonlinear methods coupled with molecular interaction fields descriptors, J. Chem. Inf. Model., 2016, 56(10), 2042–2052 CrossRef CAS PubMed.
- R. S. Obach, F. Lombardo and N. J. Waters, Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 670 drug compounds, Drug Metab. Dispos., 2008, 36(7), 1385–1405 CrossRef CAS PubMed.
- H. Veith, N. Southall, R. Huang, T. James, D. Fayne and N. Artemenko, et al., Comprehensive characterization of cytochrome P450 isozyme selectivity across chemical libraries, Nat. Biotechnol., 2009, 27(11), 1050–1055 CrossRef CAS PubMed.
- S. Wang, H. Sun, H. Liu, D. Li, Y. Li and T. Hou, ADMET evaluation in drug discovery. 16. Predicting hERG blockers by combining multiple pharmacophores and machine learning approaches, Mol. Pharmaceutics, 2016, 13(8), 2855–2866 CrossRef CAS PubMed.
- C. Xu, F. Cheng, L. Chen, Z. Du, W. Li and G. Liu, et al., In silico prediction of chemical Ames mutagenicity, J. Chem. Inf. Model., 2012, 52(11), 2840–2847 CrossRef CAS PubMed.
- Y. Xu, Z. Dai, F. Chen, S. Gao, J. Pei and L. Lai, Deep learning for drug-induced liver injury, J. Chem. Inf. Model., 2015, 55(10), 2085–2093 CrossRef CAS PubMed.
- V. M. Alves, E. Muratov, D. Fourches, J. Strickland, N. Kleinstreuer and C. H. Andrade, et al., Predicting chemically-induced skin reactions. Part I: QSAR models of skin sensitization and their application to identify potentially hazardous compounds, Toxicol. Appl. Pharmacol., 2015, 284(2), 262–272 CrossRef CAS PubMed.
- H. Zhu, T. M. Martin, L. Ye, A. Sedykh, D. M. Young and A. Tropsha, Quantitative structure-activity relationship modeling of rat acute toxicity by oral exposure, Chem. Res. Toxicol., 2009, 22(12), 1913–1921 Search PubMed.
- S. K. Burley, C. Bhikadiya, C. Bi, S. Bittrich, H. Chao and L. Chen, et al., RCSB Protein Data Bank (RCSB. org): delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning, Nucleic Acids Res., 2023, 51(D1), D488–D508 CrossRef CAS PubMed.
- RDKit: open-source cheminformatics, https://www.rdkit.org Search PubMed.
- O. Avd, Y. Li and O. Vinyals, Representation learning with contrastive predictive coding, arXiv, 2018, preprint, arXiv:180703748, DOI:10.48550/arXiv.1807.03748.
- D. P. Kingma, Adam: a method for stochastic optimization, arXiv, 2014, preprint, arXiv:14126980, DOI:10.48550/arXiv.1412.6980.
- N. V. Chawla, K. W. Bowyer, L. O. Hall and W. P. Kegelmeyer, SMOTE: synthetic minority over-sampling technique, J. Artif. Intell. Res., 2002, 16, 321–357 CrossRef.
- D. R. Koes, M. P. Baumgartner and C. J. Camacho, Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise, J. Chem. Inf. Model., 2013, 53(8), 1893–1904 CrossRef CAS PubMed.
- P. Eastman, R. Galvelis, R. P. Peláez, C. R. Abreu, S. E. Farr and E. Gallicchio, et al., OpenMM 8: molecular dynamics simulation with machine learning potentials, J. Phys. Chem. B, 2023, 128(1), 109–116 CrossRef PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Comparison of multiple Amber force fields and development of improved protein backbone parameters, Proteins: Struct., Funct., Bioinf., 2006, 65(3), 712–725 CrossRef CAS PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, D. S. Cerutti, G. A. Cisneros and V. W. D. Cruzeiro, et al., AmberTools, J. Chem. Inf. Model., 2023, 63(20), 6183–6191 CrossRef CAS PubMed.
- A. W. Sousa da Silva and W. F. Vranken, ACPYPE-Antechamber python parser interface, BMC Res. Notes, 2012, 5, 1–8 CrossRef PubMed.
- L. Kagami, A. Wilter, A. Diaz and W. Vranken, The ACPYPE web server for small-molecule MD topology generation, Bioinformatics, 2023, 39(6), btad350 CrossRef CAS PubMed.
- R. Anandakrishnan, B. Aguilar and A. V. Onufriev, H++ 3.0: automating p K prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations, Nucleic Acids Res., 2012, 40(W1), W537–W541 CrossRef CAS PubMed.
- M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente and E. Moreno, gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS, J. Chem. Theory Comput., 2021, 17(10), 6281–6291 CrossRef PubMed.
- R. T. McGibbon, K. A. Beauchamp, M. P. Harrigan, C. Klein, J. M. Swails and C. X. Hernández, et al., MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories, Biophys. J., 2015, 109(8), 1528–1532 CrossRef CAS PubMed.
- K. Diedrich, B. Krause, O. Berg and M. Rarey, PoseEdit: Enhanced ligand binding mode communication by interactive 2D diagrams, J. Comput.-Aided Mol. Des., 2023, 37(10), 491–503 CrossRef CAS PubMed.
- S. B. Needleman and C. D. Wunsch, A general method applicable to the search for similarities in the amino acid sequence of two proteins, J. Mol. Biol., 1970, 48(3), 443–453 CrossRef CAS PubMed.
- Y. Zhang, NW-align, https://zhanglab.dcmb.med.umich.edu/NW-align Search PubMed.
- Y. Zhang and J. Skolnick, TM-align: a protein structure alignment algorithm based on the TM-score, Nucleic Acids Res., 2005, 33(7), 2302–2309 CrossRef CAS PubMed.
- D. Nagarajan and N. Chandra, PocketMatch (version 2.0): a parallel algorithm for the detection of structural similarities between protein ligand binding-sites, in 2013 National Conference on Parallel Computing Technologies (PARCOMPTECH), IEEE, 2013, pp. 1–6 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08736d |
| This journal is © The Royal Society of Chemistry 2025 |