
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing organic cathodes of aqueous zinc-ion batteries via nitro group modification†
Donghong
Wang
 *a,
Mengxuan
Qin
a,
Changyou
Zhang
b,
Mengxue
Li
b,
Chao
Peng
c,
Chunyi
Zhi
*df,
Qing
Li
e and
Lei
Zhu
*b
*a,
Mengxuan
Qin
a,
Changyou
Zhang
b,
Mengxue
Li
b,
Chao
Peng
c,
Chunyi
Zhi
*df,
Qing
Li
e and
Lei
Zhu
*b
aSchool of Materials Science and Engineering, Anhui Province Key Laboratory of Efficient Conversion and Solid-State Storage of Hydrogen & Electricity, Anhui University of Technology, Ma'anshan 243032, Anhui, China. E-mail: dhwang@ahut.edu.cn
bSchool of Chemistry and Chemical Engineering, Anhui University of Technology, Ma'anshan 243032, Anhui, China. E-mail: leizhu2022@ahut.edu.cn
cMultiscale Crystal Materials Research Center, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China
dDepartment of Materials Science and Engineering, City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong 999077, China. E-mail: cy.zhi@cityu.edu.hk
eInstitute of Applied Physics and Materials Engineering, University of Macau, Macau 999078, China
fSongshan Lake Materials Laboratory, Dongguan, Guangdong, China
First published on 21st January 2025
Abstract
Organic compounds present promising options for sustainable zinc battery electrodes. Nevertheless, the electrochemical properties of current organic electrodes still lag behind those of their inorganic counterparts. In this study, nitro groups were incorporated into pyrene-4, 5, 9, 10-tetraone (PTO), resulting in an elevated discharge voltage due to their strong electron-withdrawing capabilities. Additionally, a novel electrochemical conversion of nitro to azo groups was observed in aqueous electrolytes. This transformation can be leveraged to enhance cycling stability, especially at low current densities. The electrochemical process of nitro-PTO during discharge comprises three distinct steps. Initially, two stages of H+/Zn2+ coordination to the carbonyl groups led to a high capacity of ∼284 mA h g−1 above 0.80 V—significantly higher than that of PTO. Further discharge irreversibly transformed –NO2 groups into N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds, resulting in exceptionally high specific capacities of approximately 695 mA h g−1 and 905 mA h g−1 for PTO decorated with single and double –NO2 groups, respectively. As –NO2 was continuously reduced to NN, the resultant azo-conjugated PTO (PTO-Azo) demonstrated reversible H+/Zn2+ co-storage and release during subsequent charge/discharge cycles, with improved capacity retention and higher kinetics. This work, therefore, elucidates the impact of nitro group chemistry on the electrochemical performance of carbonyl-rich organic electrodes.
N bonds, resulting in exceptionally high specific capacities of approximately 695 mA h g−1 and 905 mA h g−1 for PTO decorated with single and double –NO2 groups, respectively. As –NO2 was continuously reduced to NN, the resultant azo-conjugated PTO (PTO-Azo) demonstrated reversible H+/Zn2+ co-storage and release during subsequent charge/discharge cycles, with improved capacity retention and higher kinetics. This work, therefore, elucidates the impact of nitro group chemistry on the electrochemical performance of carbonyl-rich organic electrodes.
Introduction
Aqueous batteries are characterized by their inherently high safety and facile manufacturing requirements.1,2 Due to these merits, the development of aqueous batteries for large-scale energy storage applications has been revived in recent years. Especially, aqueous zinc batteries (AZBs) have garnered significant interest for grid-scale energy storage due to their numerous advantages, including low cost, low redox potential, and the high energy density of zinc metal.3 Extensive research has focused on addressing critical challenges such as zinc dendrite formation,4,5 hydrogen evolution,6,7 and the development of high-performance cathode materials.8–10 Among these, the cathode materials are particularly crucial, as they often limit the device's energy density and cycling stability.11–13 To date, a variety of inorganic cathode candidates, such as Prussian blue analogs14 and oxides based on vanadium15 and manganese,16–18 have been explored. However, the intercalation/conversion mechanisms in these materials frequently result in irreversible structural damage or dissolution during discharge.19 Moreover, enhancements in energy density are limited by the crystal lattice and structural stability of these cathodes, which necessitate counterions of specific sizes and valences to maintain compatibility with the crystal structures.20Recently, organic materials have garnered significant attention as they offer considerable promise as redox species, attributed to their chemical diversity, abundance of earth-sourced elements, and low crossover effects.3,21–23 To date, a variety of molecules have been explored as potential redox species for AZB applications, such as quinone,24–26 phenazine,27,28 thianthrene,29 and TEMPOs.30 Among these, quinones stand out owing to their potentially high specific capacity; nevertheless, they are challenged by rapid capacity fading at low current density or limited voltages, even with the aid of molecular engineering.24,31,32 Extending the conjugation plane and introducing high-electron withdrawing components have been identified as efficient strategies to enhance stability and increase discharge voltage.27,33–35 However, the incorporation of additional inactive components can reduce the specific capacity.36 Therefore, the development of small joint segments with electron withdrawing ability is highly desirable for constructing highly conjugated cathode materials for AZBs.
In this study, we found that pyrene-4, 5, 9, 10-tetraone (PTO), when used as cathode materials for AZBs, exhibited fast capacity decay at a current density of 0.2 A g−1. By introducing nitro groups, which serve as both active sites and high electron-withdrawing moieties, we observed a significant increase in discharge voltage as the number of nitro groups increased. This modification also led to a substantial enhancement in the first discharge capacity, rising from ∼330 mA h g−1 for PTO to ∼695 mA h g−1 for PTO with one nitro group (PTO-NO2), and reaching ∼905 mA h g−1 for PTO with two nitro groups (PTO-2NO2) when the voltage limit was set to 0.1 V, due to the four-electron transfer reactions on each nitro group. Furthermore, within the voltage range of 0.80 V to 1.5 V, the specific capacity was boosted from ∼115 mA h g−1 for PTO to over 284 mA h g−1 for PTO-2NO2. Upon deep electroreduction, the nitro groups were converted into azo with the neighboring nitro groups, thereby extending the PTO segments and forming PTO-Azo products. As a result, the specific capacity after 400 cycles was significantly raised from only 107 mA h g−1 (PTO) to 216 mA h g−1 (PTO-Azo), facilitated by the implanted nitro groups. Additionally, the conjugated PTO-Azo outperformed pristine PTO in both the rate capability and cycle reliability.
Results and discussion
In this work, the electrochemical activity of nitro groups and their effects on PTO in AZBs were investigated by implanting varying numbers of nitro groups: without nitro group (PTO), with a nitro group (PTO-NO2), and two nitro groups (PTO-2NO2) (Fig. 1a). The structure and the bonding in these compounds were characterized using X-ray diffraction (XRD) (Fig. S1†), Fourier transform infrared (FTIR) spectra (Fig. S2†) and nuclear magnetic resonance (NMR) (Fig. S3–S5†). The three samples show crystalline structure pattern in XRD. The signal of carbonyl groups located at ∼1690 cm−1. Compared to pure PTO, the presence of ∼1530 and ∼1350 cm−1 vibration peaks in the FTIR spectrum certified the successful decoration of nitro groups onto the skeleton of PTO. As displayed in Fig. S3,† two peaks are visible in the 1H NMR of PTO, with an integral area ratio of 2/1, which is consistent with its symmetric structure. The 1H NMR spectrum of PTO-NO2 shows three peaks, with two of them having nearly equal integral areas at chemical shifts of 2.04 and 1.00 ppm (Fig. S4†), and the PTO-2NO2 possesses one peak (Fig. S5†), which matches with the symmetric structure of PTO-NO2 and PTO-2NO2, respectively.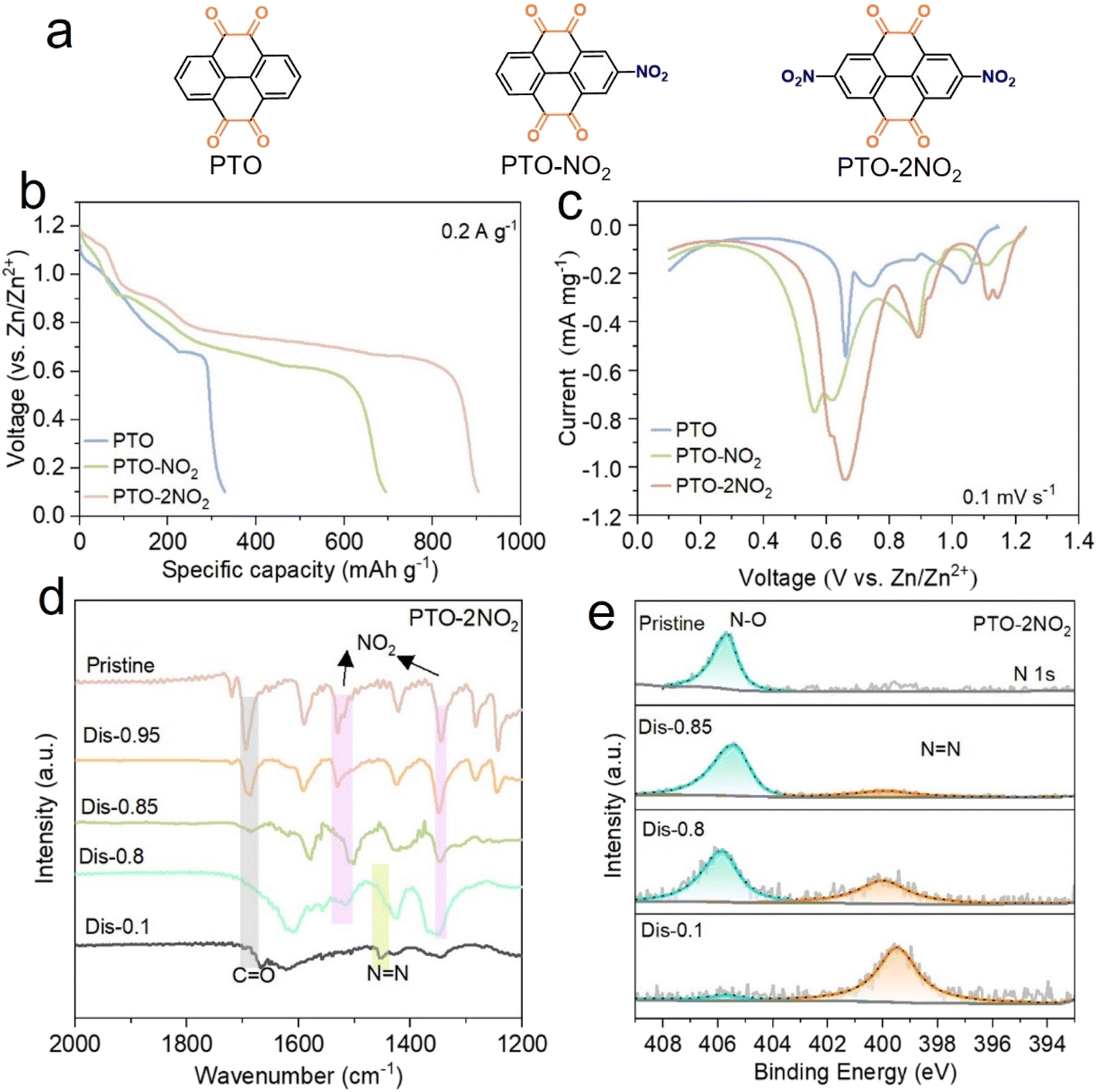 | ||
| Fig. 1 (a) Molecular structures for PTO, PTO-NO2, and PTO-2NO2; (b) the discharge profiles of different cathode materials upon the first discharge at 0.2 A g−1; (c) the cathodic spectra of different cathode materials; (d) FT-IR spectra of PTO-2NO2 electrode at different discharge states; (e) N 1s XPS spectra of PTO-2NO2 electrode at different discharge states. | ||
The electrochemical performance of PTO, PTO-NO2 and PTO-2NO2 was evaluated in coin cells using 2 M aqueous ZnSO4 electrolyte, with Zn metal as counter electrode. Fig. 1b displays the electrochemical behaviors of these electrode materials during the first discharge cycle. PTO delivered a long, sloping plateau centered at 0.87 V, followed by a short and flat plateau at 0.66 V (Fig. 1b). After introducing a nitro group, the sloping discharge profile was divided into three distinct stages, centered at 1.05 V, 0.82 V and 0.67 V, respectively. The flat plateau was also extended, resulting in a discharge capacity of 694.6 mA h g−1. Further incorporation of a second nitro group lifted the discharge plateaus and contributed to an ultrahigh discharge capacity of 904.8 mA h g−1. Similar to the galvanostatic charge–discharge (GCD) curves, the cyclic voltammetry (CV) results for PTO reveal two primary reduction peaks at ∼0.66 and ∼1.0 V, corresponding to the reduction reaction of carbonyl groups. In comparison, the PTO-NO2 electrode exhibits positively shifted peaks at 1.11/1.05 and 0.88 V, along with the emergence of new peaks at 0.62 and 0.56 V during the initial cathodic scan (Fig. 1c). Notably, these reduction peaks associated with carbonyl groups shift to even higher potentials (1.15/1.11 and 0.89 V) when the number of nitro groups is doubled, confirming the electron-withdrawing ability of nitro groups. The number of electron transfers was determined based on the high capacities observed during the first discharge, as exhibited in eqn (S1) and Table S1.† Each nitro group contributed approximately four additional electrons to the transfer capacity, likely due to the conversion of nitro groups to azo groups.
To monitor the evolution of the functional groups, ex situ FT-IR was conducted on the discharged products at different stages. Upon the first discharge, the intensity of carbonyl groups gradually reduced with the discharge voltage and disappeared after being discharged to 0.8 V (Fig. 1d), signifying the carbonyl groups in PTO-2NO2 nearly fully converted to C–O groups. Upon further discharge to 0.1 V, the nitro signals at approximately 1350 and 1530 cm−1 nearly disappeared, and a new signal around 1450 cm−1 emerged, corresponding to the stretching mode of azo groups.37 This indicates a transformation of nitro groups into azo groups. It demonstrated that the carbonyl groups were mainly responsible for energy storage above 0.80 V in PTO-2NO2. X-ray photoelectron spectroscopy (XPS) was subsequently performed to investigate the structural evolution of nitro groups during the first discharge process. The XPS spectra of N 1s revealed that NN started to emerge around 0.85 V, and a small portion of NN (399.8 eV) appeared upon discharging to 0.80 V, due to the high electron-withdrawing ability of nitro groups and the conjugated structure of PTO-NO2. Further discharge (0.1 V) converts most of the nitro groups into NN groups (Fig. 1e). The PTO-2NO2 electrode subjected to multiple cycles was also characterized using FT-IR, with results presented in Fig. S6.† The presence of NN groups and the absence of nitro groups clearly corroborated the electrochemical reduction of nitro to azo. The corresponding structures of discharged products are displayed in Fig. S7.† The electrochemical reaction of nitro to azo was also observed in various aqueous electrolytes when the solute was switched to different zinc salts (Zn(CF3SO3)2, Zn(ClO4)2, Zn(BF4)2), as well as MgSO4 and Na2SO4, due to their similarly high discharge capacities. Additional discharge profiles are presented in Fig. S8–S10.†
The high electron-withdrawing ability of nitro groups effectively increased the discharge voltage, as evidenced by the positively shifted reduction peaks from 1.0 V and 0.66 V to 1.15 V and 0.89 V (Fig. 1c), respectively. To further investigate this electron-inductive effect, PTO-2NO2 was selected as a representative sample, and the cut-off voltages were set above 0.80 V and 0.85 V, respectively. For comparison, PTO and 2,7-dibromopyrene-4,5,9,10-tetraone (PTO-2Br) were evaluated. As illustrated in the first and second discharge profiles (Fig. S11a and b†), PTO delivered an average discharge voltage of 0.96 V and a specific capacity of 114.6 mA h g−1 at 0.2 A g−1 within the voltage range of 0.80–1.5 V. The introduction of electron-withdrawing Br groups increased the discharge voltage to 1.07 V, achieving a discharge capacity of 136 mA h g−1 above 0.80 V. In sharp contrast, PTO-2NO2 demonstrated a significantly higher capacity, surpassing 284.2 mA h g−1, along with two distinct voltage plateaus at 1.15 and 1.05 V. However, the introduction of nitro groups also increased the solubility of the reduced product due to their hydrophilic nature, resulting in a gradual decline in cycling stability, as shown in Fig. S11c.† The slower degradation observed within the voltage range of 0.80–1.5 V should be attributed to the partially conjugated structure formed during the sequential conversion of nitro groups to azo groups. The structural evolution during discharge was also evaluated via O 1s XPS spectra at various stages. As illustrated in Fig. S12,† the CO groups evolved into C–O–H upon discharging to 0.95 V, while O–Zn groups emerged with further discharge to 0.85 V. Upon discharge to 0.80 V, all CO groups were converted to C–O–H and O–Zn, with a ratio of 1.7, indicating an initial coordination with H+ ions followed by Zn2+ coordination during the discharge process mediated by the carbonyl groups.
To evaluate the impact of the azo-bridged structure on electrochemical performance, the samples were cycled four times within the voltage range of 0.1–1.5 V and were denoted as 2PTO-Azo and PTO-Azo, originating from PTO-NO2 and PTO-2NO2, respectively. The CV curves of PTO, 2PTO-Azo, and PTO-Azo are shown in Fig. S13–S15.† Notably, the two pairs of redox peaks gradually converged as the number of nitro groups increased, ultimately merging into a broadly single peak for PTO-Azo. Correspondingly, the electrochemical behavior transitioned from being ion-dominated to being primarily controlled by a pseudo-capacitive process, as evidenced by the significant increase in b values from 0.5–0.59 to 0.80–0.98 (Fig. 2a). The effects of the azo-conjugated structure on cycling stability were also assessed (Fig. S16† and 3b). Although the PTO presents a higher discharge capacity in the initial 10 cycles than both 2PTO-Azo and PTO-Azo, it experienced rapid capacity fading and only retained a capacity of 107 mA h g−1 over 400 cycles. After introducing a single nitro group, the resulting 2PTO-Azo dimers enhanced the specific capacity to 186.5 mA h g−1 after 400 cycles. Impressively, PTO-Azo maintained a specific capacity of 216.3 mA h g−1 with a slow capacity decay rate of 0.034% (Fig. 2b). The self-discharge performance was also evaluated; the battery was first charged to 1.5 V, after rested for 48 h, the battery was then discharged to 0.1 V. As shown in Fig. S17,† a significant improvement in the coulombic efficiency can be observed after the formation of azo bridges, reflecting a much-enhanced durability. Similar cycling performance was observed when the electrodes were cycled at 0.5 A g−1 for 700 cycles (Fig. S18†), with PTO-Azo demonstrating superior capacity retention of 80.6%. Among the three samples, PTO-Azo exhibited the best cycling stability and fastest kinetics. Such excellent cycling durability of PTO-Azo at low current density is superior to previously reported materials incorporating quinone units (Fig. S19†). Consequently, PTO-Azo was further evaluated for its rate capability, with PTO serving as the reference. As shown in Fig. 2c and S20,† PTO-Azo retained a reversible capacity of 120 mA h g−1 even when the current density was increased to 10.0 A g−1, with nearly 100% capacity recovery after the current density returned to 0.1 A g−1. In contrast, PTO retained only 50 mA h g−1 at 10 A g−1, with a capacity retention of just 60% at 0.1 A g−1. Furthermore, the average discharge plateau of PTO-Azo at 0.1 A g−1 was approximately 0.8 V, higher than that of pure PTO (0.75 V). As the current density increased, both cathodes exhibited a decrease in average discharge voltage, with PTO-Azo showing a slower decline compared to PTO (Fig. 2d). The rate performance also surpasses that of most organic cathodes with quinone unit structure for AZBs, including DTT,25 C4Q,24 TCBQ,38 C8Q,39 HqTp,39 TAQ-BQ,40 and PBQS,41 as exhibited in Fig. S21.† These results indicate that PTO-Azo possesses faster charge–discharge behavior and a more stable structure. To further confirm these findings, Zn‖PTO-Azo cells were cycled at 2 and 5 A g−1 (Fig. 2e and f). An initial capacity of 218.5 mA h g−1 and a retention rate of 76% were obtained after 3000 cycles at a current density of 2 A g−1. At 5 A g−1, the cell achieved a capacity of 138.4 mA h g−1 after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, with a minimal decay rate of 0.00247% per cycle (Fig. 2f).
000 cycles, with a minimal decay rate of 0.00247% per cycle (Fig. 2f).
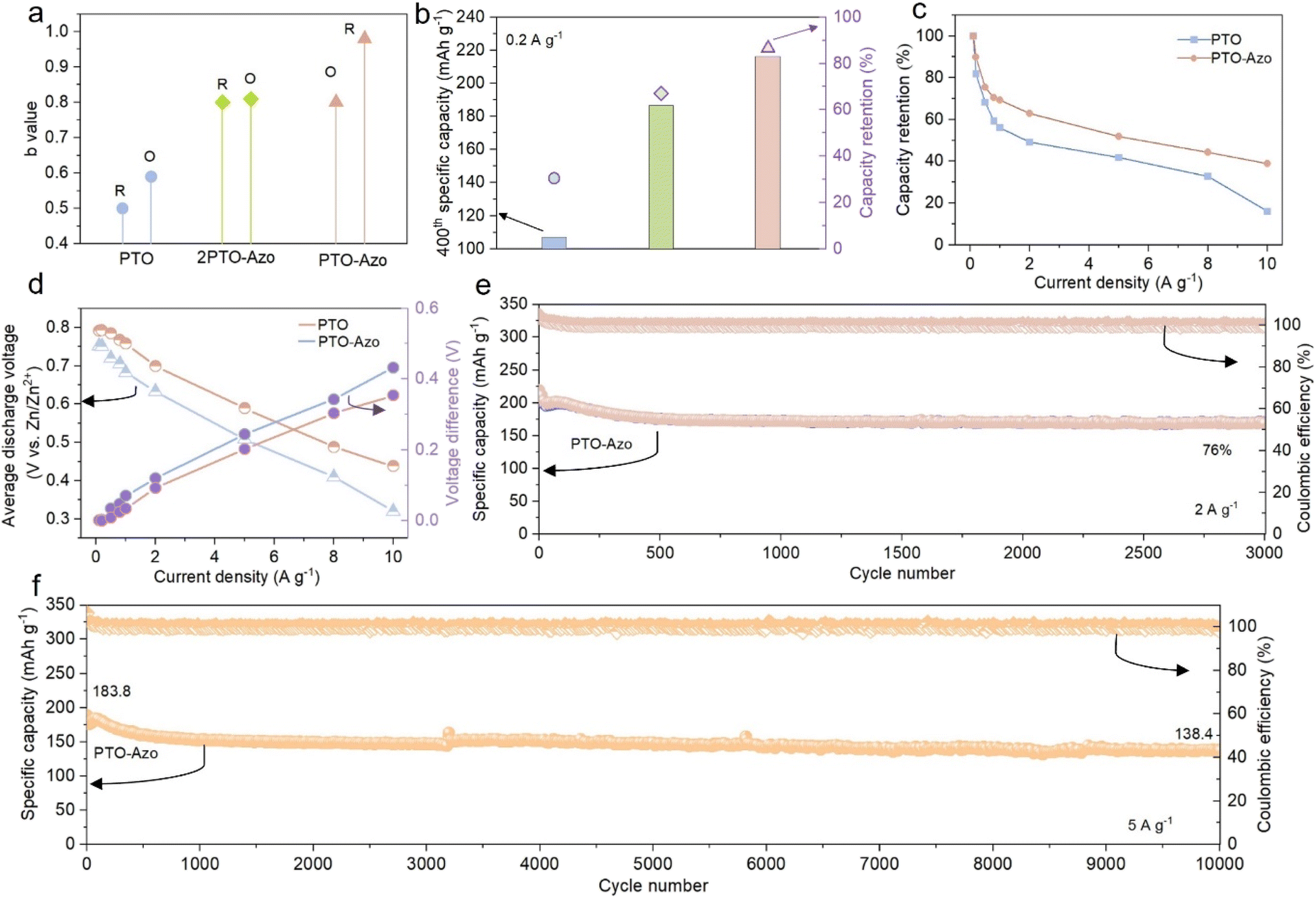 | ||
| Fig. 2 (a) The b values obtained from the CV curves for three different cathode materials; (b) the 400th cycle specific capacity of the three cathode materials and their corresponding capacity retention at the 400th cycle; (c) the rate performance of PTO-Azo cathode, with PTO as the comparison; (d) the average discharge voltages and the corresponding voltage difference between the average charge and discharge voltages for the Zn‖PTO and Zn‖PTO-Azo cells; (e and f) cycling performance of PTO-Azo at 2 A g−1 (e) and 5 A g−1 (f). | ||
To elucidate the enhanced rate capability of azo compounds, electrochemical impedance spectroscopy (EIS) was employed to measure the evolution of PTO and PTO-Azo electrodes during charge/discharge cycling. As shown in Fig. 3a and b, since the depressed semicircle represents the charge transfer resistance (Rct), the PTO-Azo electrode witnessed a notable decrease from approximately 647 Ω to 183 Ω after the first cycle, and further to 149 Ω after four cycles. This reduction is attributed to the lower impedance of the newly generated azo compound, which accounts for the enhanced charge/discharge capability observed in PTO-Azo. In contrast, the PTO electrode exhibited higher interphase resistance, as illustrated in Fig. 3c. To further investigate the interfacial reaction resistances at different stages of charge and discharge, the galvanostatic intermittent titration technique (GITT) was utilized. During testing, a current pulse of 0.2 A g−1 was applied for 2 min, as shown in Fig. 3d and S22,† followed by a relaxation period of 30 minutes. The abrupt potential changes observed during GITT correspond to charge transfer and ohm resistance, while the gradual potential changes are attributed to ion diffusion.42 As displayed in Fig. 3e, the derived reaction resistance in the PTO-Azo cathode exhibited only a slight increase during the initial discharge potential range (1.5–0.73 V) and a small rise towards the end of the discharging stage (0.6–0.1 V). In comparison, the PTO cathode demonstrated a significant increase in reaction resistance across the discharge voltage ranges of 1.0–0.8 V and 0.7–0.1 V. Moreover, the reaction resistance of the PTO-Azo cathode was approximately half that of the PTO cathode. The ionic diffusion coefficient (D), calculated and displayed in Fig. 3f, indicated much faster ion diffusion in PTO-Azo, further confirming the significantly accelerated reaction kinetics after the formation of azo-conjugated polymers. To understand the superior performance of PTO-Azo compared to PTO, first-principles calculations were conducted to access the electronic properties of these materials. The molecular electrostatic potential (ESP) of the PTO and azo-bridged PTO electrode material is illustrated in Fig. 3g. The oxygen atoms (red regions), which have a more negative ESP, exhibit higher electronegativity, making them more likely to attract electrophilic cations (Zn2+ or H+), while the nitrogen and carbon atoms display positive ESP (blue regions). This indicates that the carbonyl groups act as the primary redox-active sites. Furthermore, the energy levels, including the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) were computed. As shown in Fig. 3h, the LUMO energy level of the PTO-Azo molecule (−4.66 eV) is significantly lower than that of PTO (−4.10 eV), suggesting that PTO-Azo has a higher electron affinity and higher reduction potential, which correlates with the elevated average discharge plateaus observed in Fig. 2d. Additionally, the electrical conductivity of the materials is indicated by the energy gap (Eg) between HOMO and LUMO, with smaller gaps suggesting better conductivity. As anticipated, the gap in PTO-Azo (2.76 eV) is smaller than that of PTO (3.47 eV), implying that PTO-Azo exhibits superior electrical conductivity, facilitating rapid electron transmission and exceptional reaction kinetics. This conclusion aligns well with the observed results for reaction resistance and rate performance.
 | ||
| Fig. 3 (a) EIS curves of Zn‖PTO-Azo cells at different states; (b) the corresponding charge transfer resistance; (c) EIS curves of Zn‖PTO cells at different states; (d) GITT curves of Zn‖PTO-Azo battery; (e) the reaction resistance change and (f) ionic diffusion coefficient during the discharge process of Zn‖PTO-Azo and Zn‖PTO coin cells at 0.2 A g−1; (g) the ESP distributions for the PTO and PTO-Azo electrode materials; (h) illustrations of the energy levels along with the corresponding optimized molecular structures. | ||
To figure out the energy storage mechanism of the PTO-Azo in aqueous ZnSO4 solution, electrolytes containing only H+ and Zn2+ ions were prepared. PTO-Azo electrodes, disassembled from the Zn‖PTO-2NO2 cell after four cycles, with the sample characterized using XPS, recoverable CO groups and firmly stable NN groups (Fig. S23†) certified the stable molecule structure of PTO-Azo. The PTO-Azo electrodes were subsequently evaluated in an organic electrolyte (0.5 M Zn(CF3SO3)2 in acetonitrile, Zn(OTF)2/ACN) and in a 0.5 M H2SO4 electrolyte. The CV curves in the organic zinc-ion electrolyte revealed a pair of redox peaks that partially overlapped with those observed in aqueous ZnSO4 (Fig. 4a). In the strong acid electrolyte, the CV curves displayed one pair of small peaks and one pair of sharp peaks (Fig. 4b). After adjusting the CV curves obtained in H2SO4 to the same pH level as the ZnSO4 solution (pH 4.45) using the Nernst equation (eqn (S2) and (S3)†), it was observed that the positions of the oxidation/reduction peaks partially overlapped with those measured in 2 M ZnSO4 (Fig. 4b). Moreover, the GCD curves showed comparable specific capacities across the three different electrolytes (Fig. S24 and S25†). FTIR and XPS analyses were conducted to monitor the behavior of carbonyl groups. The points indicated on the GCD curve in Fig. 4c represent the states chosen for the ex situ tests. As shown in Fig. 4d, the vibrational signals of carbonyl groups at ∼1665 cm−1 gradually weakened upon discharging to 0.1 V, indicating coordination between CO motifs and Zn2+/H+ ions. The O 1s spectra (Fig. 4e) reveal that, during the discharge process, the intensity of the CO signal gradually diminishes, while signals corresponding to C–O–H and C–O⋯Zn emerge at higher and lower binding energy, respectively.43–45 Note that, the signal of C–O–H appears upon discharging to 1.0 V (B), and becomes quite obvious after discharge to 0.9 V (C) and more pronounced upon a further discharge, while the C–O⋯Zn signal emerges after discharge to 0.9 V and becomes apparent after further discharge. Quantitative analysis of the XPS data shows that, at the initial discharge stages, H+ contributes twice as much as Zn2+, highlighting a H+-dominated electrochemical process at the early stage due to the faster kinetics of H+. Even after full discharge, H+ remains the predominant contributor at 53%, compared to Zn2+ at 47% (Fig. 4f). Additionally, the azo groups exhibit a gradual shift of their peak to lower binding energies (Fig. 4g) during the electron injection throughout the discharge process. This shift is attributed to the enhanced electronic delocalization resulting from the extended conjugation of the azo bridging group.
 | ||
| Fig. 4 (a and b) CV curves of PTO-Azo in different electrolytes; (c) discharge profile of PTO-Azo cathode with different discharge depths at the 5th cycle; (d) ex situ FT-IR spectra; (e) ex situ XPS spectra of O element; (f) statistical chart of capacity contribution; (g) ex situ XPS spectra of N element. | ||
Conclusions
In summary, the functionalization of PTO skeletons with nitro groups, serving as both strong electron-withdrawing and active moieties, led to significant improvements in specific capacity, voltage plateaus, and cycling stability. Specifically, the two-nitro-functionalized PTO exhibited a high specific capacity of 284.2 mA h g−1 within a relatively high voltage range of 0.80–1.5 V, a marked increase compared to the 114.6 mA h g−1 achieved by pure PTO. Additionally, the discharge voltage plateaus were elevated to 1.15 and 1.05 V. When cycled within a broader voltage range of 0.1–1.5 V, the specific capacity increased from 330 mA h g−1 to over ∼695 mA h g−1 and even reached ∼905 mA h g−1 with a higher number of nitro groups per PTO skeleton. The nitro groups were found to undergo irreversible reduction to azo groups during deep discharge cycles, forming bridges that extend the conjugation of PTO segments. This transformation not only prolonged the cycle life but also enhanced electrochemical kinetics. Furthermore, the charge-storage mechanism was revealed to involve the co-storage of H+ and Zn2+ ions, with H+ as the predominant contributor.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
M. X. Qin carried out the experiment, materials characterization, D. H. Wang contributed to the data analysis, writing – original draft, writing – review & editing and supervision, C. Y. Zhi and L. Zhu directed and supervised the project. C. Y. Zhang, M. X. Li, C. Peng and Q. Li contributed to the data analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by Scientific Research Foundation of Education Department of Anhui Province of China (2023AH051109), Anhui Provincial Natural Science Foundation (no. 2308085QB46), the open research fund of Songshan Lake Materials Laboratory (no. 2023SLABFN18), National Natural Science Foundation of China (22409002).References
- M. Xia, J. Zhou and B. Lu, Adv. Energy Mater., 2024, 2404032 CrossRef
.
- C. Han, J. Zhu, C. Zhi and H. Li, J. Mater. Chem. A, 2020, 8, 15479–15512 RSC
- Z. Li, J. Tan, Y. Wang, C. Gao, Y. Wang, M. Ye and J. Shen, Energy Environ. Sci., 2023, 16, 2398–2431 RSC
- X. Zheng, Z. Liu, J. Sun, R. Luo, K. Xu, M. Si, J. Kang, Y. Yuan, S. Liu, T. Ahmad, T. Jiang, N. Chen, M. Wang, Y. Xu, M. Chuai, Z. Zhu, Q. Peng, Y. Meng, K. Zhang, W. Wang and W. Chen, Nat. Commun., 2023, 14, 76 CAS
- X. Zhang, J. Li, Y. Liu, B. Lu, S. Liang and J. Zhou, Nat. Commun., 2024, 15, 2735 CrossRef CAS PubMed
- C.-C. Kao, C. Ye, J. Hao, J. Shan, H. Li and S.-Z. Qiao, ACS Nano, 2023, 17, 3948–3957 CAS
- C. Guo, J. Zhou, Y. Chen, H. Zhuang, Q. Li, J. Li, X. Tian, Y. Zhang, X. Yao, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202210871 CAS
- T. Sun, W. Zhang, Q. Nian and Z. Tao, Nanomicro Lett., 2023, 15, 36 CAS
- F. Wan, L. Zhang, X. Wang, S. Bi, Z. Niu and J. Chen, Adv. Funct. Mater., 2018, 28, 1804975 CrossRef
- Y. Liu, S. Guo, W. Ling, M. Cui, H. Lei, J. Wang, W. Li, Q. Liu, L. Cheng and Y. Huang, J. Energy Chem., 2023, 76, 11–18 CrossRef CAS
- P. Ruan, S. Liang, B. Lu, H. J. Fan and J. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202200598 CAS
- X. Wang, Z. Zhang, B. Xi, W. Chen, Y. Jia, J. Feng and S. Xiong, ACS Nano, 2021, 15, 9244–9272 CAS
- C. Liu, Z. G. Neale and G. Cao, Mater. Today, 2016, 19, 109–123 CAS
- L. Ma, S. Chen, C. Long, X. Li, Y. Zhao, Z. Liu, Z. Huang, B. Dong, J. A. Zapien and C. Zhi, Adv. Energy Mater., 2019, 9, 1902446 CAS
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS
- Y. Liu, K. Wang, X. Yang, J. Liu, X.-X. Liu and X. Sun, ACS Nano, 2023, 17, 14792–14799 CrossRef CAS PubMed
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS
- M. Chuai, J. Yang, M. Wang, Y. Yuan, Z. Liu, Y. Xu, Y. Yin, J. Sun, X. Zheng, N. Chen and W. Chen, eScience, 2021, 1, 178–185 CrossRef
- Y. Mei, Y. Liu, W. Xu, M. Zhang, Y. Dong and J. Qiu, Chem. Eng. J., 2023, 452, 139574 CrossRef CAS
- T. Sun, J. Xie, W. Guo, D.-S. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS
- H. Cui, L. Ma, Z. Huang, Z. Chen and C. Zhi, SmartMat, 2022, 3, 565–581 CrossRef CAS
- J. Chen, Y. Wang, Y. Yu, J. Wang, J. Liu, H. Ihara and H. Qiu, Exploration, 2023, 3, 20220144 CrossRef PubMed
- R. Shi, S. Jiao, Q. Yue, G. Gu, K. Zhang and Y. Zhao, Exploration, 2022, 2, 20220066 CrossRef CAS PubMed
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 CrossRef PubMed
- Y. Wang, C. Wang, Z. Ni, Y. Gu, B. Wang, F. Wan, J. Ma and Y. Wang, Adv. Mater., 2020, 32, e2000338 CrossRef PubMed
- Q. Wang, Y. Liu and P. Chen, J. Power Sources, 2020, 468, 228401 Search PubMed
- J. Fu, J. Y. Liu, G. H. Zhang, Q. H. Zhu, S. L. Wang, S. Qin, L. He and G. H. Tao, Small, 2023, 19, 2302570 CrossRef CAS PubMed
- Z. Tie, L. Liu, S. Deng, D. Zhao and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 4920–4924 CAS
- H. Cui, T. Wang, Z. Huang, G. Liang, Z. Chen, A. Chen, D. Wang, Q. Yang, H. Hong, J. Fan and C. Zhi, Angew. Chem., Int. Ed., 2022, 61, e202203453 CrossRef CAS PubMed
- Y. Luo, F. Zheng, L. Liu, K. Lei, X. Hou, G. Xu, H. Meng, J. Shi and F. Li, ChemSusChem, 2020, 13, 2239–2244 CrossRef CAS PubMed
- J. Kumankuma-Sarpong, S. Tang, W. Guo and Y. Fu, ACS Appl. Mater. Interfaces, 2021, 13, 4084–4092 CrossRef CAS PubMed
- B. Yang, Y. Ma, D. Bin, H. Lu and Y. Xia, ACS Appl. Mater. Interfaces, 2021, 13, 58818–58826 CrossRef CAS PubMed
- J. Liu, J. Jiang, Q. Zhou, Z. Chen, R. Zhang, X. Xu, X. Han, S. Yang, Z. Zhou, P. Cheng and W. Shi, eScience, 2023, 3, 100094 CrossRef
- Z. Ye, S. Xie, Z. Cao, L. Wang, D. Xu, H. Zhang, J. Matz, P. Dong, H. Fang, J. Shen and M. Ye, Energy Storage Mater., 2021, 37, 378–386 CrossRef
- T. Sun, W. Zhang, Z. Zha, M. Cheng, D. Li and Z. Tao, Energy Storage Mater., 2023, 59, 102778 CrossRef
- H. Peng, J. Xiao, Z. Wu, L. Zhang, Y. Geng, W. Xin, J. Li, Z. Yan, K. Zhang and Z. Zhu, CCS Chem., 2023, 5, 1789–1801 CrossRef CAS
- Q. Li, H. Wang, H. G. Wang, Z. Si, C. Li and J. Bai, ChemSusChem, 2020, 13, 2449–2456 Search PubMed
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 Search PubMed
- C. Ding, Y. Wang, C. Li, J. Wang, Q. Zhang and W. Huang, Chem. Sci., 2024, 15, 4952–4959 RSC
- Z. Lin, L. Lin, J. Zhu, W. Wu, X. Yang and X. Sun, ACS Appl. Mater. Interfaces, 2022, 14, 38689–38695 CrossRef CAS PubMed
- G. Dawut, Y. Lu, L. Miao and J. Chen, Inorg. Chem. Front., 2018, 5, 1391–1396 RSC
- X. Fan, Y. Zhu, C. Luo, L. Suo, Y. Lin, T. Gao, K. Xu and C. Wang, ACS Nano, 2016, 10, 5567–5577 CrossRef CAS PubMed
- H. Cui, J. Zhu, R. Zhang, S. Yang, C. Li, Y. Wang, Y. Hou, Q. Li, G. Liang and C. Zhi, J. Am. Chem. Soc., 2024, 146, 15393–15402 CrossRef CAS PubMed
- L. Lin, Z. Lin, J. Zhu, K. Wang, W. Wu, T. Qiu and X. Sun, Energy Environ. Sci., 2023, 16, 89–96 RSC
- M. Gu, H. Fu, A. M. Rao, J. Zhou, Y. lin, S. Wen, L. Fan and B. Lu, Adv. Funct. Mater., 2024, 34, 2407867 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08514k |
| This journal is © The Royal Society of Chemistry 2025 |