
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrengthened d–p orbital hybridization and hydrogen diffusion in a hollow N-doped porous carbon/Ru cluster catalyst system for hydrogen evolution reactions†
Ruidong
Li‡
a,
Hongyu
Zhao‡
a,
Lin
Wang
b,
Qingqu
Zhou
a,
Xiong
Yang
a,
Linbo
Jiang
a,
Xu
Luo
a,
Jun
Yu
 *a,
Jingwen
Wei
a and
Shichun
Mu
*a
*a,
Jingwen
Wei
a and
Shichun
Mu
*a
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, China. E-mail: msc@whut.edu.cn
bNRC (Nanostructure Research Centre), Wuhan University of Technology, Wuhan 430070, China
First published on 24th January 2025
Abstract
Developing advanced catalysts with rapid hydrogen evolution reaction (HER) kinetics in alkaline media is vital for hydrogen production. Through the d–p orbital hybridization effect, the electronic structure and H* adsorption can be optimized on metal species. Herein, a N-doped hollow carbon (H-NPC)-supported Ru cluster (c-Ru@H-NPC) catalyst was constructed via carbonization of well-defined hollow metal–organic frameworks, followed by etching and anchoring of Ru clusters. The hollow structure could not alter the coordination number of Ru while exhibiting higher-level electron transfer, thereby strengthening the orbital hybridization. Additionally, finite element simulations indicated the acceleration of H2 diffusion for hollow structures. Furthermore, the N-doping strengthened the electron interaction of Ru–C by the d–p hybridization effect, which was confirmed by theoretical calculations and in situ Raman spectroscopy. Therefore, in alkaline/alkaline seawater media, c-Ru@H-NPC needed only 10/12 mV overpotentials and 1.52/1.55 V cell voltages to drive the HER and overall water splitting, respectively, at a current density of 10 mA cm−2, exhibiting outstanding catalytic activity. Meanwhile, the attenuation of current density was very small towards successive stability tests for >55 h at 10 mA cm−2. This work permits new insights into the design of high-performance metal cluster catalysts for the HER and other conversion reactions.
1 Introduction
The extensive consumption of fossil-fuel energy has led to worsening energy and environmental crises.1–3 Hydrogen (H2), a potential form of secondary energy, has attracted worldwide attention due to its “green” and renewable properties and high caloric value.4–7 However, the hydrogen evolution reaction (HER),8–11 as the critical half-reaction of water electrolysis, has sluggish kinetics in alkaline media. Hence, noble metal catalysts are required to obtain high hydrogen production performance. Even so, such catalysts suffer from obvious degradation during the HER.12–14 An appropriate support and electronic structure are crucial for catalysts to obtain the highest HER performance, with a low amount of noble metals,15–17 which facilitates the development of water electrolysis technology.Hollow carbon materials have been considered to be important hierarchical supports in chemical industry and energy and environmental applications.18–22 Hollow metal–organic frameworks (H-MOFs), as an emerging porous material constructed from metal ions/clusters and polyfunctional organic linkers, have attracted increasing interest in the construction of hollow carbon materials due to their ability to retain a specific morphology and structural characteristics during carbonization.23–29 In addition, the diversity of tunable organic linkers and metal nodes makes it possible to prepare hollow MOF-derived carbon materials with faster mass transport, multiple active components, more exposed active sites and higher specific surface area than solid carbon materials.
Ruthenium (Ru), with about 1/30 of the cost of Pt metal, displays high activity towards the HER owing to a unique electronic structure and the ability to adsorb intermediates.30–32 Through means of the strong metal–support interaction (SMSI), the interaction of a hierarchical MOF-derived carbon support and Ru can induce orbital hybridization, which modulates the electronic structure and enhances spin–orbital splitting, thereby boosting the intrinsic activity of Ru.33–39 Moreover, the introduction of heteroatoms (such as N atoms) into the Ru@MOF-derived carbon system would strongly tune the electron structure of Ru and C, and further strengthen the d–p hybridization of Ru–C, which is conducive to the HER.40–44
However, the precise synthesis of hollow carbon nanocages and modulation of the interaction between the metal and support to optimize HER performance remain challenging. Exploring the interaction through matrix regulation and mechanistic studies is an effective approach to addressing the aforementioned issues. Herein, the direct pyrolysis of a hierarchical hollow UiO-66-NH2 (H-UiO-66-NH2) produced a carbon-based composite consisting of nitrogen-doped carbon (NC) and ZrO2 with a wrinkled surface. Subsequently, it was treated with HF solution to obtain a well-defined hollow N-doped porous (H-NPC) structure, and then Ru clusters were anchored to obtain a c-Ru@H-NPC catalyst with maintained morphology and microporosity/mesoporosity. A finite element method (FEM) simulation indicated that the void within the hollow structure could be treated as the nanoreactor to produce more H2, which rapidly became saturated, thus promoting the outward diffusion of hydrogen. Additionally, the N doping strengthened the d–p hybridization effect, and then facilitated electron transfer and optimized the electronic structure, which was confirmed by density functional theory (DFT) calculations and in situ Raman spectroscopy. Due to the N-enhanced d–p effect, together with a hierarchically hollow structure of the carbon support, c-Ru@H-NPC exhibited high activity and durability towards the HER in both alkaline and alkaline seawater media.
2 Results and discussion
2.1 Synthesis of catalysts
As illustrated in Scheme 1, the hierarchical c-Ru@H-NPC was constructed through direct carbonization of hollow UiO-66-NH2 (H-UiO-66-NH2), followed by the as-synthesis of the hollow N-doped porous carbon (H-NPC) to support Ru clusters. Scanning electron microscopy (SEM) images showed an octahedral shape and wrinkled surface, which demonstrated no dynamic morphological transformation from solid UiO-66-NH2 (Fig. 1a), ZrO2/NC (Fig. S1a†), NPC (Fig. S1b†) to c-Ru@NPC (Fig. 1b). Similarly, after propionic acid (PA) etching, the hollow morphology of H-UiO-66-NH2 (Fig. S1c†), H-ZrO2/NC (Fig. S1d†) and H-NPC (Fig. S1e†) and the c-Ru@H-NPC catalyst (Fig. 1c) did not change obviously. The transmission electron microscopy (TEM) image, as displayed in Fig. 1d, suggested the solid structure of c-Ru@NPC, and the lattice spacing of 0.239 nm was indexed to the (111) crystal face of Ru (Fig. 1e) according to high-resolution transmission electron microscopy (HRTEM). Furthermore, aberration-corrected scanning transmission electron microscopy (ac-STEM) was used to identify the morphological presence of Ru. Ru existed in the form of clusters of size <1.5 nm in c-Ru@NPC (Fig. 1f). Fig. 1g verified the preparation of c-Ru@H-NPC with a shell of thickness ∼25 nm. From the HRTEM results of c-Ru@H-NPC, the crystal plane spacing of 2.14 Å could be attributed to Ru (002) lattice plane (Fig. 1h), demonstrating the reduction of Ru precursors in c-Ru@H-NPC. Based on Fig. 1i, the size of a Ru cluster was ∼1.0 nm. These results suggested that the hollow structure did not affect the state of Ru clusters on a N-doped carbon matrix. Ru clusters of diminished size exhibited higher specific surface area and strengthened capacity for water dissociation, making c-Ru@H-NPC more advantageous towards catalytic reactions. Additionally, HAADF-STEM and EDS mapping images both displayed a solid nanostructure and hollow nanostructure with homogeneous distribution of C, O, N and Ru elements. Zr element could not be found in c-Ru@NPC and c-Ru@H-NPC (Fig. 1j and k) because of the HF etching of MOF-derived ZrO2, which corresponded to the results of the atomic fraction (Fig. S2†). | ||
| Scheme 1 Synthesis of a hollow c-Ru@H-NPC catalyst. | ||
 | ||
| Fig. 1 SEM image of UiO-66-NH2 (a), c-Ru@NPC (b) and c-Ru@H-NPC (c). (d) (g) TEM image of c-Ru@NPC and c-Ru@H-NPC. (e) (h) High-resolution TEM image of c-Ru@NPC and c-Ru@H-NPC. (f) (i) ac-STEM image of c-Ru@NPC and c-Ru@H-NPC. (j) (k) HAADF-STEM image and corresponding EDS mapping images of N, C, Ru and O of c-Ru@NPC and c-Ru@H-NPC. | ||
As shown in Fig. 2a, compared with pristine UiO-66-NH2, the typical X-ray diffraction (XRD) peaks at 7.43° and 8.56° became obviously weak and broadened, indicating the decline of crystallinity for H-UiO-66-NH2 due to PA etching. The XRD pattern of ZrO2/NC and H-/ZrO2/NC, obtained through the pyrolysis of UiO-66-NH2 and H-UiO-66-NH2 correspondingly, matched well with ZrO2 (JCPDS no. 49-1642). After treatment with HF solution, the obtained solid N-doped porous carbon (NPC) and hollow N-doped porous carbon (H-NPC) had no characteristic peak of ZrO2, consistent with EDS results. In addition, a diffraction peak corresponding to crystalline Ru could not be found in c-Ru@NPC or c-Ru@H-NPC, which could be ascribed to highly dispersed Ru clusters, consistent with TEM results.
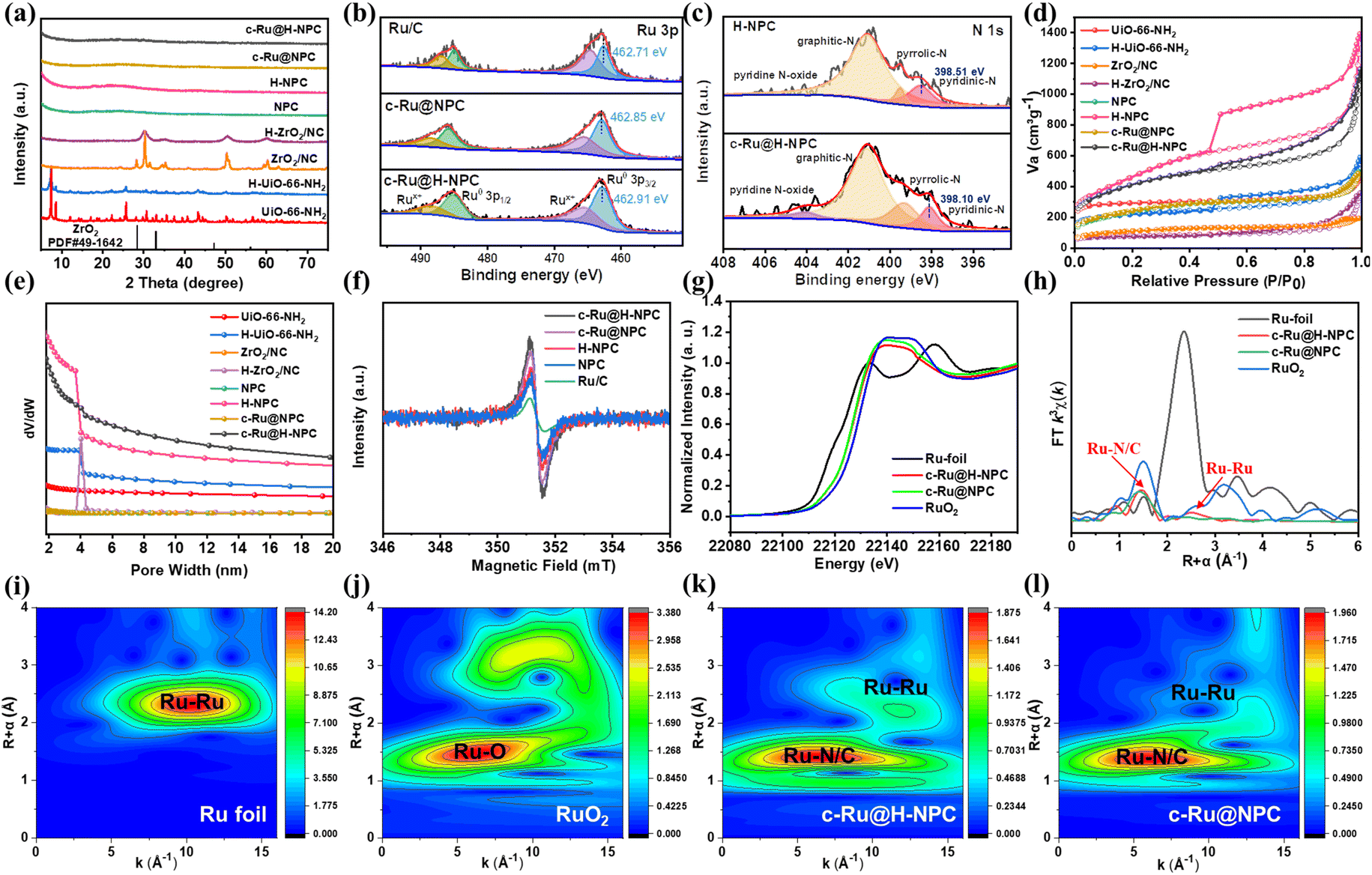 | ||
| Fig. 2 (a) XRD patterns of various samples. (b) High-resolution Ru 3p XPS spectra, (c) N 1s XPS spectra, (d) N2 adsorption–desorption isotherms and (e) corresponding pore size distribution. (f) EPR spectra of c-Ru@H-NPC, H-NPC, NPC and commercial Ru/C. (g) XANES, (h) EXAFS spectra and (i–l) wavelet transform (WT) analyses of Ru foil, RuO2, c-Ru@H-NPC and c-Ru@NPC. | ||
As shown in Ru 3p X-ray photoelectron spectroscopy (XPS), the peak position of Ru 3p3/2 at 462.85 eV for both c-Ru@NPC and 462.91 eV for c-Ru@H-NPC suggested a positive shift of 0.14 eV and 0.20 eV, respectively, compared with that of commercial Ru/C (Fig. 2b). This could have been caused by the formation of a d–p orbital hybridization effect, possibly because Ru element (2.2) has a lower electronegativity compared with that of N (3.0) and C (2.5).33,45,46 The more-shifted Ru peak of c-Ru@H-NPC indicated that the optimized hollow structure could expose more active sites and, thus, intrinsically enhance d–p hybridization.47–49 In particular, the N 1s spectra in Fig. 2c could be deconvoluted into four peaks, corresponding to pyridinic N (398.10 eV), pyrrolic N (399.30 eV), graphitic N (401.10 eV) and pyridine N oxide (404.20 eV). The binding energies of pyridinic N for c-Ru@H-NPC exhibited a negative shift of 0.40 eV compared with H-NPC, while the binding energy of pyridine N of c-Ru@NPC was negatively shifted by ∼0.19 eV (Fig. S3†), which further indicated electron transfer from Ru to the defective NPC and H-NPC matrix. Additionally, the peak of pyridine N oxide within NPC and H-NPC was too weak to be observed, possibly due to the removal of the oxide by HF processing.
The hierarchical pore of hollow c-Ru@H-NPC was also confirmed by N2 sorption isotherms and corresponding pore size distribution curves (Fig. 2d and e). Compared with initial UiO-66-NH2, the content of mesoporous pores increased largely after propionic acid treatment. After the removal of ZrO2, the surface area (Table S1†) of both solid NPC and hierarchical H-NPC increased largely. However, slight surface area loss occurred, and their previous pore structure was maintained, respectively, after the addition of highly dispersed Ru clusters. Moreover, the total incorporation of Ru in c-Ru@H-NPC, as detected by inductively coupled plasma-optical emission spectrometry (ICP-OES), was ∼4.39 wt% (Table S2†), which was higher than that in c-Ru@NPC and consistent with the results of BET calculations.
Moreover, electron paramagnetic resonance (EPR) spectroscopy was conducted to further confirm the effect of the hollow structure and the existence of d–p hybridization within c-Ru@H-NPC. As illustrated in Fig. 2f, the intensity of the EPR signal for solid NPC was lower than that of H-NPC, while c-Ru@NPC was lower than that of c-Ru@H-NPC, suggesting the high content of unpair electrons as result of the exposed sites within the hollow structure.50,51 Notably, compared with that of H-NPC, stronger single Lorentzian line signals could be observed for the spectra of c-Ru@H-NPC, demonstrating that the target catalyst possessed a higher content of unpaired electron contents which originated from enhanced d–p hybridization. Meanwhile, a similar phenomenon could also be observed between c-Ru@H-NPC, c-Ru@NPC and Ru/C, illustrating that N doping increased the defects of catalysts.
Meanwhile, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements were conducted to further analyze the local coordination structure and valence states of Ru clusters. Fig. 2g revealed that the dominant valence states of Ru in both c-Ru@H-NPC and c-Ru@NPC were 2.1+ and 2.0+, accordingly located between those of standard Ru foil and RuO2, because of the very close absorption edges of the Ru K-edge XANES spectrum. These data suggested that Ru clusters transferred their electrons to the carbon support and that the hollow structure could not modify the coordination structure of Ru species. The EXAFS spectra for c-Ru@H-NPC and c-Ru@NPC showed pronounced peaks at around 1.5 Å and 2.6 Å, which could be attributed to the Ru–N/C bond of the atomic Ru coordinated with N and C within the carbon support and Ru–Ru coordination with an average coordination number of ∼1.4, respectively. It could be further concluded that the structural features of Ru clusters were in agreement with the results of ac-STEM (Fig. 2h). The wavelet-transformed (WT) patterns of the samples further confirmed the coordination information (Fig. 2i–l). The good fitting results toward R-space and k-space suggested the reliability of the data (Fig. S4 and Table S3†). Thus, all of these results suggested that the hollow structure could not alter the coordination number of Ru clusters and that Ru tended to transfer electrons to N-doped carbon materials, thus enhancing orbital hybridization. This is crucial for fundamental improvement of catalytic activity.
2.2 Hydrogen evolution performance of c-Ru@H-NPC catalysts
The HER performance of c-Ru@H-NPC was first evaluated in 1 M KOH by a typical three-electrode system. Fig. 3a showed that the polarization curves of c-Ru@H-NPC, c-Ru@NPC, H-NPC, NPC, and commercial Pt/C and Ru/C. NPC and H-NPC could barely perform well during the HER. After introducing the Ru atom, c-Ru@NPC showed a lower HER overpotential (22 mV) than Ru/C (48 mV) at a current density of 10 mA cm−2. This indicated that the strengthened d–p hybridization effect due to N doping optimize the electronic structure of Ru. Notably, among the samples, c-Ru@H-NPC (10 mV) exhibited the lowest overpotential at 10 mA cm−2. Compared with c-Ru@NPC, the hierarchical hollow structure of c-Ru@H-NPC could expose more active sites and promote electron transfer, leading to higher HER activity. Additionally, Fig. 3b showed that the overpotential advantage of c-Ru@H-NPC over commercial Ru/C became larger with increasing current density (Δη = 38@10, 88@50 mV@mA cm−2). In addition, c-Ru@H-NPC showed higher mass activity (3.232 A mgRu−1) than c-Ru@NPC (2.066 A mgRu−1) and Pt/C (0.1707 A mgPt−1) at 50 mV. | ||
| Fig. 3 HER performance of c-Ru@H-NPC, c-Ru@NPC, H-NPC, NPC, commercial Pt/C and Ru/C in alkaline media. (a) LSV polarization curves with iR correction, (b) relevant overpotentials at different current densities (@10 and 50 mA cm−2) and (c) Tafel plots of the HER. (d) Double-layer capacitances (Cdl) versus various scan rates and (e) equivalent circuit and fitted EIS curves of various samples. (f) Stability tests of c-Ru@H-NPC. (g) Comparisons of η10 with different catalysts in 1 M KOH. | ||
As presented in Fig. 3c, c-Ru@H-NPC exhibited a minimal Tafel slope of 10.7 mV dec−1, which indicated an optimal dynamic mechanism compared with H-NPC (24.6 mV dec−1), Pt/C (31.2 mV dec−1) and Ru/C (68.5 mV dec−1). Besides, the electrochemical active surface area (ECSA) was measured via double-layer capacitance (Cdl) from cyclic voltammetry (CV) curves within the non-faradaic range (Fig. 3d and S5†). Compared with c-Ru@NPC (85.4 mF cm−2), H-NPC and NPC (60.1 mF cm−2), c-Ru@H-NPC had the largest Cdl (103.8 mF cm−2) and the largest ECSA (0.173 m2) accordingly. Electrochemical impedance spectroscopy (EIS) was performed to explore the improved electrochemical kinetics (Fig. 3e). The supporting of Ru clusters lowered the charge transfer resistance (Rct) of H-NPC and NPC. Moreover, c-Ru@NPC (9.7 Ω) exhibited better charge transfer capability than commercial Ru/C (18.7 Ω) because N-doping further enhanced the d–p effect. By contrast, c-Ru@H-NPC (5.4 Ω) had the smallest Rct among all the mentioned catalysts, implying the fastest charge transfer process. Furthermore, c-Ru@H-NPC was superior to other reported Ru-based or other electrocatalysts in alkaline media, as shown in Fig. 3g and Table S4.†
Stability is also one of the important features for catalysts. Fig. 3f shows a good long-term stability for c-Ru@H-NPC with steady current output at 10 mA cm−2 for 100 h. The inset indicated that c-Ru@H-NPC maintained activity with negligible decay after 3000 CV cycles. Moreover, as shown in Fig. S6a–c,† after successive long-term tests, no obvious change in c-Ru@H-NPC could be observed in the representative HRTEM images (Fig. S6d†), implying that c-Ru@H-NPC remained well-maintained, with preservation of good dispersion of Ru clusters (Fig. S6e and f†), and small amounts of Ru leaching according to ICP data (Table S2†). The Ru XPS profile indicated that the Ru 3p3/2 peak at 463.6 eV in c-Ru@H-NPC shifted and positively and the valence state of Ru was not significantly elevated after the HER test (Fig. S7†). As shown in Fig. S8,† the XRD patterns of the used catalyst remained the same as that of before the catalysis. Thus, it could be concluded that c-Ru@H-NPC had excellent stability.
Encouraged by the excellent activity of c-Ru@H-NPC under alkaline media, the alkaline seawater electrolysis of the catalyst was applied. We noted that c-Ru@H-NPC maintained the lowest overpotential of 12 and 33 mV, at current densities of 10 and 50 mA cm−2, respectively (Fig. 4a and b), far lower than that of commercial Pt/C, Ru/C and other samples, which also illustrated the higher HER activity in seawater media than most of the other reported catalysts (Fig. 4g and Table S5†). c-Ru@H-NPC presented higher mass activity (3.119 A mgRu−1) than c-Ru@NPC (1.872 A mgRu−1) or Pt/C (0.1620 A mgPt−1) at 50 mV. Similarly, c-Ru@H-NPC displayed a lower Tafel slope (13.3 mV dec−1) than commercial Ru/C (48.6 mV dec−1), Pt/C (23.3 mV dec−1) or c-Ru@NPC (22.1 mV dec−1) (Fig. 4c). Additionally, the Cdl (Fig. 4d) and ECSA of c-Ru@H-NPC, calculated from a series of CV curves outside the HER voltage range (Fig. S9†), was the largest. Meanwhile, the EIS results are also shown in Fig. 4e, which maintained the same trend as in the alkaline condition. After 3000 continuous CV cycles, c-Ru@H-NPC merely increased by 5 mV in the overpotential at 10 mV cm−2, and the i–t chronoamperometric response test showed a steady current output for 55 h, still maintaining excellent activity in seawater (Fig. 4f). Taken together, these data indicated that c-Ru@H-NPC owns high HER activity and stability in alkaline seawater.
 | ||
| Fig. 4 HER performance of c-Ru@H-NPC, c-Ru@NPC, H-NPC, NPC, and commercial Pt/C and Ru/C in alkaline seawater. (a) LSV polarization curves with iR correction, (b) relevant overpotentials at different current densities and (c) Tafel plots of the HER. (d) Current density variation versus various scan rates and (e) equivalent circuit and fitted EIS curves of various samples. (f) Stability tests of c-Ru@H-NPC. (g) Comparisons of η10 with different catalysts in alkaline seawater medium. | ||
Considering the excellent HER catalytic performance of c-Ru@H-NPC, we further combined the catalyst with commercial RuO2 to form a two-electrode system, c-Ru@H-NPC‖RuO2, for overall water/seawater splitting, in which c-Ru@H-NPC and RuO2 was the cathode and anode, respectively. As shown in Fig. S10a and S11a,† it needed only low voltages of 1.52 and 1.55 V to realize a current density of 10 mA cm−2 in 1 M KOH and alkaline seawater, respectively. The faradaic efficiency of c-Ru@H-NPC in 1 M KOH (Fig. S10b and c†) and alkaline seawater (Fig. S11b and c†) was also measured to investigate charge transfer efficiency during the HER. The good consistency between the measured and theoretical H2 amounts revealed a Faraday efficiency of nearly 100% within such alkaline conditions. Meanwhile, c-Ru@H-NPC‖RuO2 coupling shows good long-term durability of at least 40 h for overall water splitting in both 1 M KOH (Fig. S10d†) and alkaline seawater (Fig. S11d†). The above results illustrated that the d–p effect and hollow structure could expose more active sites but also optimize the electronic structure, thus resulting in its excellent HER performance.
2.3 Mechanism discussion
A finite element simulation (FEM) was conducted to investigate the advantages toward mass transfer of hollow structures. Compared with the solid material (Fig. 5b), the inside of the hollow structure provided extra active sites and, thus, the generated H2 in the cavity could diffuse more readily to the outside of the catalyst (Fig. 5a). The hydrogen concentration at the center within hollow structures was significantly superior to that of solid structures (Fig. 5c). During the initial stages of the HER, hydrogen within the hollow structure will diffuse into the void but also throughout the electrolyzer, in contrast with a solid structure. Subsequently, the hollow structure facilitates H2 diffusion as the internal concentration increases to saturation, and finally has a higher mole flow rate (Fig. 5d). | ||
| Fig. 5 Finite element method (FEM) simulation results of the H2 concentration in c-Ru@H-NPC (a) and c-Ru@NPC (b). (c) Temporal evolution of the H2 concentration at the center point and (d) the surface H2 diffusion rates of samples. (e) Model of c-Ru@NPC with charge density difference and (f) corresponding two-dimensional slice. (g) Partial density of states (PDOS) of ruthenium active sites. (h) Comparison of the outermost electron number of bonded Ru and C atom in c-Ru@H-NPC and Ru/C. (i) Gibbs free energy diagram for c-Ru@H-NPC, Ru/C and Ru. (j) ΔEH2O values on surfaces of various structure models. In situ Raman spectra of interfacial water on (k) c-Ru@H-NPC and (l) c-Ru@NPC (E versus RHE). | ||
Furthermore, density functional theory (DFT) calculations were carried out for c-Ru@H-NPC to unveil the effect of d–p hybridization on an alkaline HER. Due to the similar Ru and N-doped carbon support coordination between c-Ru@NPC and c-Ru@H-NPC, an approximation treatment was applied. The optimized models, such as undoped graphite supported Ru, Ru/C and c-Ru@H-NPC (c-Ru@NPC), are shown in Fig. S12a–c.† The differential charge density calculated with DFT suggested electron transfer within c-Ru@H-NPC, with a reduced charge density on Ru atoms, while the charge density on the N and C atoms increased. These data implied that some of the electrons were transferred from Ru to N and C atoms (Fig. 5e and f). Such a strong Ru and H-NPC interaction led to electronic rearrangement near the interface affected by d–p hybridization, in agreement with the results of XPS.
As can be seen from the partial density of states (PDOS) of various samples, the d-band center (−1.660 eV) of c-Ru@H-NPC negatively drifted compared with that of Ru/C (−1.434 eV) and Ru (−1.409 eV), which indicated weakened hydrogen adsorption behavior (Fig. 5g). This was because the N doping further enhanced the hybridization level between the d orbit of Ru and the p orbit of C, thereby increasing the HER chemical activity and improving HER reaction kinetics. According to the outermost electron structures of Ru 4d75s1 and C 2s22p2 (Fig. 5h), compared with the theoretical electrons of the outermost shell of Ru (14) and C (4) atoms, the outermost electron numbers of Ru atoms in c-Ru@H-NPC and Ru/C were 13.51 and 13.67, respectively, while they were 3.74 and 4.11 for C atoms in c-Ru@H-NPC and Ru/C, respectively. This also agreed with the result from Fig. S13† that more partial electrons of Ru (0.49–) in c-Ru@H-NPC were lost than that of Ru/C (0.33–), which was also consistent with electronegativity theory.
Meanwhile, due to the optimized electronic structure, c-Ru@H-NPC had the lower Gibbs free energy (−0.13 eV) of hydrogen intermediate (ΔGH*) than that of Ru/C (−0.24 eV) and Ru (−0.46 eV), indicating the optimal H* adsorption and desorption strength (Fig. 5i). In addition, the adsorption energy of H2O molecules (ΔEH2O) on the surface of c-Ru@H-NPC, Ru/C and Ru was also calculated. c-Ru@H-NPC presented a lower ΔEH2O of −0.62 eV than those of Ru/C (−0.59 eV) and Ru (−0.51 eV), demonstrating that N doping played an important part in the adsorption energy of water, and that initially H2O was easily captured on the surface of c-Ru@H-NPC to facilitate the Volmer reaction (Fig. 5j).13 Thus, the strong d–p hybridization by the modulation of the electronic structure of c-Ru@H-NPC could synergistically boost the HER process.
Furthermore, in situ Raman tests were conducted to confirm the configuration of interfacial water during the HER in 1 M KOH. The Raman diffraction peaks corresponding to 2970–3800 cm−1 were attributed to the O–H stretching band of c-Ru@H-NPC (Fig. 5k) and Ru/C (Fig. 5l). The characteristic peak deconvoluted into three peaks of 3220, 3430 and 3600 cm−1 could be defined as four hydrogen-bonded water (4-HBW), two hydrogen-bonded water (2-HBW) and K+ ion hydrated water (KW), respectively.4,52,53 Compared with Ru/C, c-Ru@H-NPC obviously had the larger KW proportion in any according voltage value, and acted as the co-catalyst to accelerate the electron transfer of the HER Volmer process.54 It could be concluded that the N doping of the catalyst could optimize the d–p effect and improve the intrinsic activity of the active site, promoting the reaction efficiency and catalytic activity.
3 Conclusions
We presented hollow N-doped porous carbon through direct carbonization of hollow metal–organic frameworks and subsequent HF etching to confine the fine Ru clusters for the HER. The enhanced d–p hybridization of Ru–C, along with a well-defined hollow N-doped porous carbon structure, had a key role in substantially boosting the alkaline hydrogen electrocatalysis performance. Experimental and finite element method simulation results demonstrated that the hollow structure endowed it with a large surface area, exposing a greater number of active sites, which effectively assisted in the enhancement of hydrogen diffusion. DFT calculations further indicates that Ru clusters transferred more electrons to the N-doped carbon support regulated by the d–p hybridization effect, significantly optimizing the electronic structure of Ru clusters, and improving the H* adsorption energy. Additionally, the hybridization effect enhanced the adsorption energy of water on Ru according to in situ Raman spectroscopy, thereby accelerating the Volmer reaction, which greatly improved the HER activity. This work presents new dimensions of the design of hollow structure catalysts with d–p hybridization toward high-performance HER or other practical applications.Data availability
The data supporting the conclusions reached in this study have been included as part of the ESI.†Author contributions
Ruidong Li: conceptualization, methodology, writing (original draft), data curation, and visualization. Jingwen Wei: methodology and data analysis. Hongyu Zhao: software. Xu Luo: investigation. Jun Yu: supervision. Lin Wang and Xiong Yang: project administration. Linbo Jiang and Qingqu Zhou: validation. Shichun Mu: writing (review and editing), funding acquisition, and resources.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially sponsored by the National Natural Science Foundation of China (22179104, 22379117), and the State Key Laboratory of Advanced Technology for Materials Synthesis and Processing (Wuhan University of Technology) (2023-ZT-1).Notes and references
- P. Achakulwisut, P. Erickson, C. Guivarch, R. Schaeffer, E. Brutschin and S. Pye, Nat. Commun., 2023, 14, 5425 CrossRef CAS PubMed.
- H. Q. Fu, M. Zhou, P. F. Liu, P. Liu, H. Yin, K. Z. Sun, H. G. Yang, M. Al-Mamun, P. Hu, H.-F. Wang and H. Zhao, J. Am. Chem. Soc., 2022, 144, 6028–6039 CrossRef CAS PubMed.
- J. Zhang, D. Chen, J. X. Jiao, W. H. Zeng and S. Mu, Green Energy Environ., 2024 DOI:10.1016/j.gee.2024.10.006.
- Y.-H. Wang, S. Zheng, W.-M. Yang, R.-Y. Zhou, Q.-F. He, P. Radjenovic, J.-C. Dong, S. Li, J. Zheng, Z.-L. Yang, G. Attard, F. Pan, Z.-Q. Tian and J.-F. Li, Nature, 2021, 600, 81–85 CrossRef CAS PubMed.
- Y. Zhao, P. V. Kumar, X. Tan, X. Lu, X. Zhu, J. Jiang, J. Pan, S. Xi, H. Y. Yang, Z. Ma, T. Wan, D. Chu, W. Jiang, S. C. Smith, R. Amal, Z. Han and X. Lu, Nat. Commun., 2022, 13, 2430 CrossRef CAS PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- Z. Cai, Z. Wang, Y. Xia, H. Lim, W. Zhou, A. Taniguchi, M. Ohtani, K. Kobiro, T. Fujita and Y. Yamauchi, Angew. Chem., Int. Ed., 2021, 133, 4797–4805 CrossRef.
- D. Chen, R. Yu, D. Wu, H. Zhao, P. Wang, J. Zhu, P. Ji, Z. Pu, L. Chen, J. Yu and S. Mu, Nano Energy, 2022, 100, 107445 CrossRef CAS.
- T. Chao, W. Xie, Y. Hu, G. Yu, T. Zhao, C. Chen, Z. Zhang, X. Hong, H. Jin, D. Wang, W. Chen, X. Li, P. Hu and Y. Li, Energy Environ. Sci., 2024, 17, 1397–1406 RSC.
- F. Li, M. Du, X. Xiao and Q. Xu, ACS Nano, 2022, 16, 19913–19939 CrossRef CAS PubMed.
- L. Zong, F. Lu, P. Li, K. Fan, T. Zhan, P. Liu, L. Jiang, D. Chen, R. Zhang and L. Wang, Adv. Mater., 2024, 2403525 CrossRef CAS PubMed.
- Z. Tao, H. Zhao, N. Lv, X. Luo, J. Yu, X. Tan and S. Mu, Adv. Funct. Mater., 2024, 34, 2312987 CrossRef CAS.
- G. Li, H. Jang, S. Liu, Z. Li, M. G. Kim, Q. Qin, X. Liu and J. Cho, Nat. Commun., 2022, 13, 1270 CrossRef CAS PubMed.
- S. He, Y. Tu, J. Zhang, L. Zhang, J. Ke, L. Wang, L. Du, Z. Cui and H. Song, Small, 2024, 20, 2308053 CrossRef CAS PubMed.
- J. Zhang, G. Chen, Q. Liu, C. Fan, D. Sun, Y. Tang, H. Sun and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202209486 CrossRef CAS PubMed.
- L. Gong, F. Xia, J. Zhu, X. Mu, D. Chen, H. Zhao, L. Chen and S. Mu, Angew. Chem., Int. Ed., 2024, 63, e202411125 CrossRef CAS PubMed.
- M. Kim, S. Kim, J. Park, S. Lee, I. Jang, S. Kim, C. Y. Lee, O. J. Kwon, H. C. Ham, J. T. Hupp, N. Jung, S. J. Yoo and D. Whang, Adv. Funct. Mater., 2023, 33, 2300673 CrossRef CAS.
- Y. Hu, C. Wang, Y. Liu, H. Lin and K. Zhang, J. Mater. Chem. A, 2024, 12, 6438–6445 RSC.
- Z. Zhang, Z. Zhang, X. Chen, H. Wang, H. Lu, Z. Shi and S. Feng, CCS Chem., 2024, 6, 1324–1337 CrossRef CAS.
- S. Feng, K. Li, P. Hu, C. Cai, J. Liu, X. Li, L. Zhou, L. Mai, B.-L. Su and Y. Liu, ACS Nano, 2023, 17, 23152–23159 CrossRef CAS PubMed.
- R. Li, L. Wu, G. Chang, S. Ke, Y. Wang, Y. Yao, Y. Zhang, J. Li, X. Yang and B. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 5887–5896 CrossRef CAS PubMed.
- H.-H. He, J.-P. Yuan, P.-Y. Cai, K.-Y. Wang, L. Feng, A. Kirchon, J. Li, L.-L. Zhang, H.-C. Zhou and Y. Fang, J. Am. Chem. Soc., 2023, 145, 17164–17175 CrossRef CAS PubMed.
- A. Radwan, H. Jin, D. He and S. Mu, Nano-Micro Lett., 2021, 13, 132 CrossRef CAS PubMed.
- Z. Cai, Z. Wang, J. Kim and Y. Yamauchi, Adv. Mater., 2019, 31, 1804903 CrossRef PubMed.
- H.-F. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC.
- M. Kim, R. Xin, J. Earnshaw, J. Tang, J. P. Hill, A. Ashok, A. K. Nanjundan, J. Kim, C. Young, Y. Sugahara, J. Na and Y. Yamauchi, Nat. Protoc., 2022, 17, 2990–3027 CrossRef CAS.
- T. Qiu, S. Gao, Z. Liang, D. Wang, H. Tabassum, R. Zhong and R. Zou, Angew. Chem., Int. Ed., 2021, 60, 17314–17336 CrossRef CAS PubMed.
- A. Salah, H.-D. Ren, N. Al-Ansi, F.-Y. Yu, Z.-L. Lang, H. Tan and Y.-G. Li, J. Mater. Chem. A, 2021, 9, 20518–20529 RSC.
- Q. Wang and D. Astruc, Chem. Rev., 2020, 120, 1438–1511 CrossRef CAS PubMed.
- B. Zhang, J. Wang, G. Liu, C. M. Weiss, D. Liu, Y. Chen, L. Xia, P. Zhou, M. Gao, Y. Liu, J. Chen, Y. Yan, M. Shao, H. Pan and W. Sun, Nat. Catal., 2024, 7, 441–451 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Y. Hu, C. Wang, Y. Liu, H. Lin and K. Zhang, J. Mater. Chem. A, 2024, 12, 6438–6445 RSC.
- Y.-X. Xiao, J. Ying, J.-B. Chen, Y. Dong, X. Yang, G. Tian, J. Wu, C. Janiak, K. I. Ozoemena and X.-Y. Yang, Chem. Mater., 2022, 34, 3705–3714 CrossRef CAS.
- W. Li, H. Zhang, K. Zhang, W. Hu, Z. Cheng, H. Chen, X. Feng, T. Peng and Z. Kou, Appl. Catal., B, 2022, 306, 121095 CrossRef CAS.
- X. Wang, X. Yang, G. Pei, J. Yang, J. Liu, F. Zhao, F. Jin, W. Jiang, H. Ben and L. Zhang, Carbon Energy, 2024, 6, e391 CrossRef CAS.
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi, Q. Huo, H. Yang, J. Liu and C. He, Nat. Commun., 2022, 13, 3958 CrossRef CAS PubMed.
- Y. Zhu, K. Fan, C. Hsu, G. Chen, C. Chen, T. Liu, Z. Lin, S. She, L. Li, H. Zhou, Y. Zhu, H. M. Chen and H. Huang, Adv. Mater., 2023, 35, 2301133 CrossRef CAS PubMed.
- X. Cao, Y. Tian, J. Ma, W. Guo, W. Cai and J. Zhang, Adv. Mater., 2024, 36, 2309648 CrossRef CAS PubMed.
- J. Zhao, Y. Zhang, Z. Zhuang, Y. Deng, G. Gao, J. Li, A. Meng, G. Li, L. Wang, Z. Li and D. Wang, Angew. Chem., Int. Ed., 2024, 63, e202404968 CrossRef CAS PubMed.
- L. Wu, L. Su, Q. Liang, W. Zhang, Y. Men and W. Luo, ACS Catal., 2023, 13, 4127–4133 CrossRef CAS.
- W. Zhu, F. Yao, Q. Wu, Q. Jiang, J. Wang, Z. Wang and H. Liang, Energy Environ. Sci., 2023, 16, 2483–2493 RSC.
- J. Ying, G. Jiang, Z. Paul Cano, L. Han, X.-Y. Yang and Z. Chen, Nano Energy, 2017, 40, 88–94 CrossRef CAS.
- D. Gao, S. Li, X. Wang, L. Xi, K. M. Lange, X. Ma, Y. Lv, S. Yang, K. Zhao, H. M. Loussala, A. Duan, X. Zhang and G. Chen, J. Catal., 2019, 370, 385–403 CrossRef CAS.
- C. Tian, R. Liu, Y. Zhang, W. Yang and B. Wang, Nano Res., 2024, 17, 982–1002 CrossRef CAS.
- K. Gao, B. Wang, L. Tao, B. V. Cunning, Z. Zhang, S. Wang, R. S. Ruoff and L. Qu, Adv. Mater., 2019, 31, 1805121 CrossRef PubMed.
- Q. Hu, G. Li, Z. Han, Z. Wang, X. Huang, X. Chai, Q. Zhang, J. Liu and C. He, Adv. Energy Mater., 2019, 9, 1901130 CrossRef.
- X. Han, T. Zhang, X. Wang, Z. Zhang, Y. Li, Y. Qin, B. Wang, A. Han and J. Liu, Nat. Commun., 2022, 13, 2900 CrossRef CAS PubMed.
- Y. Hou, Y.-L. Liang, P.-C. Shi, Y.-B. Huang and R. Cao, Appl. Catal., B, 2020, 271, 118929 CrossRef CAS.
- L. Yu, H. Hu, H. B. Wu and X. W. (David) Lou, Adv. Mater., 2017, 29, 1604563 CrossRef PubMed.
- D. Xu, Y. Jin, B. He, X. Fang, G. Chen, W. Qu, C. Xu, J. Chen, Z. Ma, L. Chen, X. Tang, X. Liu, G. Wei and Y. Chen, Nat. Commun., 2024, 15, 8614 CrossRef CAS PubMed.
- Y. Guo, Y. Xue, B. Shen, Y. Dong, H. Zhang, J. Yuan, Z. Liu, L. Li and K. Ren, ACS Appl. Mater. Interfaces, 2024, 21, 27511–27522 CrossRef PubMed.
- S. R. Lowry and K. A. Mauritz, J. Am. Chem. Soc., 1980, 102, 4665–4667 CrossRef CAS.
- C. Cai, K. Liu, L. Zhang, F. Li, Y. Tan, P. Li, Y. Wang, M. Wang, Z. Feng, D. Motta Meira, W. Qu, A. Stefancu, W. Li, H. Li, J. Fu, H. Wang, D. Zhang, E. Cortés and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202300873 CrossRef CAS PubMed.
- M. Xing, S. Zhu, X. Zeng, S. Wang, Z. Liu and D. Cao, Adv. Energy Mater., 2023, 13, 2302376 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08498e |
| ‡ Ruidong Li and Hongyu Zhao contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |