
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnravelling the pH-dependent mechanism of ferroelectric polarization on different dynamic pathways of photoelectrochemical water oxidation†
Xing
Ji‡
 a,
Zhouhao
Zhu‡
b,
Ming
Zhou
a,
Ying
Zhang
c,
Liyong
Gan
*b,
Yunhuai
Zhang
*a and
Peng
Xiao
*b
a,
Zhouhao
Zhu‡
b,
Ming
Zhou
a,
Ying
Zhang
c,
Liyong
Gan
*b,
Yunhuai
Zhang
*a and
Peng
Xiao
*b
aCollege of Chemistry and Chemical Engineering, Chongqing University, Chongqing 401331, China. E-mail: zyh2031@cqu.edu.cn
bCollege of Physics, Chongqing University, Chongqing 401331, China. E-mail: ganly@cqu.edu.cn; xiaopeng@cqu.edu.cn
cCollege of Chemical and Environmental Engineering, Xinjiang Institute of Engineering, Urumqi 830023, China
First published on 14th January 2025
Abstract
Ferroelectric polarization is considered to be an effective strategy to improve the oxygen evolution reaction (OER) of photoelectrocatalysis. The primary challenge is to clarify how the polarization field controls the OER dynamic pathway at a molecular level. Here, electrochemical fingerprint tests were used, together with theoretical calculations, to systematically investigate the free energy change in oxo and hydroxyl intermediates on TiO2–BaTiO3 core–shell nanowires (BTO@TiO2) upon polarization in different pH environments. We demonstrate that the adsorbate evolution mechanism (AEM) dominated in acidic environments, and both positive and negative polarization resulted in a reduction in the oxo-free energy, which inhibited the reaction kinetics. In the oxide path mechanism (OPM) that occurs in alkaline conditions, the ferroelectric polarization exhibits repulsive adsorbate–adsorbate interaction for OH− coverage and free energy shift of the OH− groups. We elucidate that a weakly alkaline electrolyte is the optimal environment for ferroelectric polarization because the positive polarization promotes OH− coverage and facilitates reaction pathway transfer from AEM to OPM; therefore, BTO@TiO2 exhibited a record polarization enhancement to 0.52 mA cm−2 at 1.23 VRHE in pH = 11. This work provides a more accurate insight into the pH-dependent effect of ferroelectric polarization on the OER dynamic pathway than conventional models that are based solely on the regulation of band bending.
Introduction
The oxygen evolution reaction (OER) from water transforms renewable energy, such as solar radiation, into chemical energy, such as H2, through sustainable electro- or photo-electrochemical approaches. At present, the predominant hurdle for the OER is the sluggish kinetics of the four electron/four proton transfers. This necessitates the application of large overpotentials at meaningful rates.1,2 Although the “Sabatier Principle” has been proposed to compare catalysts,3 understanding the intrinsic rules governing the reaction steps and intermediates evolution is central to clarifying the basis of the kinetics of the catalysts in detail.According to the widely accepted adsorbate evolution mechanism (AEM),4,5 the OER process involves the transformation of multiple oxygen intermediates (i.e., OH*, O*, and OOH*) on a single active site, as shown in Scheme 1a. The rate-determining step (RDS) for this path is the reaction of adsorbed O* with water to form an OOH*.6 Strategies, such as lattice doping and interface engineering, have been widely applied to modulate the spin/charge density or proton acceptor sites, thus optimizing the binding energy of intermediates and kinetics in the RDS.7–10 However, a high overpotential (η) is required to drive the OER in this pathway due to a theoretical limit of approximately 370 ± 100 mV.11,12 Recently, a novel oxide path mechanism (OPM) was put forward to address this limitation.13 This mechanism allows the direct coupling of O–O species on two active sites without the generation of extra OOH* intermediates (Scheme 1b), thus breaking the scaling relationship and accelerating the OER kinetics further as compared to AEM.14 Symmetric dual-metal configurations with appropriate atomic distances are expected to be advantageous in promoting O–O coupling with a low energy barrier.15 In an exciting recent breakthrough, a “two-dimensional volcano” plot was reported to elucidate the intrinsic OER kinetics that can be further enhanced by optimizing both the binding energy and the interaction strength between the catalytically active states.16 This finding provides a more accurate description of the OER dynamic pathway than traditional models, which are based solely on the binding energetics.
 | ||
| Scheme 1 Comparison of reported OER mechanisms. Schematic illustration of (a) AEM and (b) OPM. (c) The reduction process to realize the intermediates, (d) the light–dark electrochemical scan test, and (e) the reduction potential of hydroxyl and oxo intermediates. | ||
Ferroelectric (FE) materials exhibit spontaneous electric polarization due to changes in bonding (such as BiFeO3 (ref. 17)) or the displacement of ions from their equilibrium position (such as BaTiO3 (ref. 18)). Over the past few decades, FE polarization has shown great promise in the electro- or photoelectron- OER process. One respectable view is that spontaneous polarization offers an internal electric field or band-engineering effects to propel the charge transport behaviors.19,20 In addition, the FE materials may attract electrically charged species from the solvent environment on their poled surfaces to screen the built-in electric field for the sake of charge neutrality.21,22 Therefore, an ideal catalytic system to break the Sabatier principle can be realized on FE materials by executing reversible polarization, which achieves efficient adsorption of reaction substrates and rapid desorption of products.23 First-principles computations have been used to identify the adsorption-free energy of the OER intermediates on FE-based heterojunctions. For example, Jun et al. found that positive polarization effectively reduces the energy required for the formation of OH* and O*.24 Iyer et al. indicated that polarization in the opposite direction causes an asymmetric response to the OER Gibbs free energy profile due to the modulation of interface chemistry and surface reconstructions.25 The challenge is that experimentally probing the intermediate's evolution is extremely difficult because these reactive species form within a hydrogen-bonded network and at a buried metal–oxide interface that obscures them.
Spectroscopy has been employed to directly probe the configurations of OER intermediates, such as OH* and O*. For example, X-ray absorption spectroscopy (XAS) can monitor the change in the local electronic state near the metal atoms and oxygen K-edge, and oxygen species (O− and O2−) have been reported to form during the OER.26 Coupled with optical regimes (i.e., ultraviolet-visible, infrared, and Raman), the vibrational energy of the oxygen intermediates, such as OH*, O* and OOH*, can be determined after efficient photoexcitation.27 However, these complex spectroscopic techniques cannot characterise the change in free energy of the OER intermediates across the full pH range, which is a critical factor for AEM and OPM kinetics. Additional analytical tools, such as isotope labelling and mass spectrometry, should be combined to obtain further insight into the nature of the intermediates.28
In a photoelectrocatalytic OER process, H2O or OH− adsorbed on a photoanode surface undergo the sequential transformation of intermediates under the action of photo-induced holes to eventually produce oxygen. These intermediates do not form on the photoanode surface before the OER cycle; instead, they only appear dynamically on active sites under photoexcitation.29 This process exhibits non-equilibrium and the isolation of the intermediate configurations often requires photoexcitation. Once the light source is ceased, some of the intermediates cannot proceed with complete conversion to oxygen and remain on the photoanode surface. Accordingly, negative electrochemical scanning in the dark facilitates the gain of electrons by the residual intermediates, resulting in a reduction peak on the scanning curve,30,31 as shown in Scheme 1c and d. The peak potentials can be employed to quantify the degree of oxidation of the intermediates, which is related to their free energy. We have verified that the reduction peak (Ep) at 0.40 VRHE on TiO2 at pH = 0 is related to the reduction of O* during AEM, and the Ep for OH* appeared at 0.29 VRHE on TiO2 at pH = 14, which corresponded to the OPM (Scheme 1e).32 It has been reported that the surface structure of FE materials, BaTiO3 (BTO) or SrTiO3, is terminated by a TiO2 double layer.33 The configuration of the OER intermediates on these ferroelectrics should be similar to TiO2. Therefore, if we can probe the intermediates on the FE, the polarization regulates the energy of the intermediate and its evolution can be clarified, which is extremely important to explore the inherent origins of enhanced kinetics.
Hence, TiO2 nanowire (NWs) arrays covered with a thin layer of BaTiO3 (denoted as BTO@TiO2) were constructed using a two-step hydrothermal method. The oxo and hydroxyl intermediates on BTO@TiO2 and their reduction peak potentials were probed using the light–dark electrochemical scan test under different pH conditions. The reduction potential shift of the oxo and hydroxyl intermediates that was related to the free energy shift of the intermediates was studied in detail under polarization experiments, and a possible mechanism was proposed by DFT calculations. Based on the AEM and OPM dynamic pathways, we gained a clear understanding of the mechanism behind the improved OER kinetics when they are affected by ferroelectric polarization. We elucidate that ferroelectric polarization not only controls the free energy of the oxo and hydroxyl intermediates but also transforms the catalytic pathway between AEM and OPM by altering the OH− coverage density on the photoanode, resulting in asymmetric changes of the OER performance. We verify that the optimal application environment for ferroelectric polarization is in a weakly alkaline electrolyte, where the OH− coverage on the photoanode does not reach saturation, and positive polarization can promote OH− coverage and facilitate the O–O coupling during OPM. Under this condition, the photocurrent of BTO@TiO2_P exhibits a remarkable enhancement of 0.52 mA cm−2 at 1.23 VRHE in the OER performance under positive polarization. Taken together, these effects reveal the mechanistic origins of the ferroelectric polarization in different OER dynamic pathways and provide rational strategies for the application of ferroelectric catalysts.
Results and discussion
In this work, BaTiO3 was chosen as the epitaxial layer for the TiO2 NWs mainly due to its small lattice mismatch and strong ferroelectric polarization intensity at room temperature (15–30 μC cm−2).34 The synthesis of BTO@TiO2 involved seed-layer-assisted hydrothermal growth of TiO2 NWs on an FTO substrate, following a previously reported hydrothermal-driven cation exchange strategy,35 as shown in Fig. 1a. A pristine TiO2 NWs reference sample has been prepared by the same method without the cation exchange. X-ray diffraction patterns (XRD, Fig. S1†) appearing at 35.8° confirmed the rutile phase of the pristine TiO2 with (011) face. Upon a second hydrothermal reaction at 150 °C, a novel diffraction peak at 31.1° was observed on the XRD pattern, corresponding to the (001) face of the tetragonal-phase BaTiO3.36 The diffraction peak exhibited an increasing intensity with an extension of the reaction time, suggesting that the transformation of TiO2 to BaTiO3 was effectively achieved. Linear sweep voltammetry (LSV) demonstrated that BTO@TiO2 prepared with 2 hours of hydrothermal cation exchange exhibited the highest OER photocurrent density (Fig. S2†). Subsequent characterization and measurements were conducted using this sample. The introduction of Ba2+ was further confirmed by XPS (Fig. S3†), indicating the formation of BTO@TiO2.37 In contrast to the pure TiO2 NWs (Fig. S4†), BTO@TiO2 exhibited an identical morphology with no discernible surface roughness (Fig. 1b and c), implying that Ba2+ diffused into the TiO2 surface layer in a smooth and orderly manner to form BaTiO3 at the surface. Furthermore, the specific capacitances of TiO2 and BTO@TiO2 were found to be similar to each other based on the electrochemically active specific surface area test (Fig. S5†). This finding further indicates that the morphological structures of the two are not significantly different. Consequently, the BTO@TiO2 can be regarded as a heterojunction structure, where TiO2 serves as the inner core and BaTiO3 as the outer shell. Furthermore, the atomic-scale structure of the individual BTO@TiO2 NW's outer layer was corroborated by HAADF-STEM characterization as shown in Fig. 1h. The boundaries between TiO2 and BaTiO3 can be observed clearly, where the thickness of the outer BaTiO3 was approximately 2 nm after 2 h of hydrothermal reaction, and crystallinity was inadequate due to hydrothermal transformation. The atomic structure of the outer layer of BaTiO3 after undergoing annealing is shown in Fig. 1i. Obviously, the BaTiO3 surface was encapsulated by two distinct layers of TiO2, which is consistent with prior observations.33 This finding provided further evidence that, for the BTO@TiO2 heterojunction, the intermediate states of the OER process will be predominantly affected by the TiO2-like surface, rather than the BTO surface. | ||
| Fig. 1 Characterization of the BTO@TiO2 photoanode. (a) Schematic of the synthesis of BTO@TiO2. (b) SEM images of BTO@TiO2 with the top (c) and side view, (d–g) EDX elemental distribution of a single BTO@TiO2 NW, (h) the HAADF-STEM image of BTO@TiO2, and (i) an annealed surface layer of BTO. | ||
Fig. 2a–c shows the photoelectrocatalytic OER performance of TiO2 and BTO@TiO2 in different pH electrolytes under AM 1.5 G solar irradiation with a power density of 100 mW cm−2. The geometric photocurrent densities of TiO2 were 1.06, 0.87, and 0.98 mA cm−2 at 1.23 VRHE in electrolytes with pH = 0, pH = 7, and pH = 14 respectively. The photocurrent of BTO@TiO2 increased to 1.76, 1.53, and 1.71 mA cm−2 for the corresponding pH electrolytes with growth rates of 39.5, 41.0, and 38.9%, respectively. The photoelectrocatalytic current density J on a photoanode is determined by:
| J = Jabs × ηsep × ηoxi |
 | ||
| Fig. 2 The recognition of intermediates on TiO2 and BTO@TiO2 photoanodes. The photocurrent curves of TiO2 and BTO@TiO2 in (a) pH = 0, (b) pH = 7, and (c) pH = 14. The Jtotal and Jrec of TiO2 and BTO@TiO2 at different bias voltages in (d) pH = 0, (e) pH = 7, and (f) pH = 14. The negative scan curves of TiO2 and BTO@TiO2 photoanodes in the dark at (g) pH = 0, (h) pH = 7, and (i) pH = 14. | ||
J abs, ηsep, and ηoxi are described in the ESI.†Jabs is the photocurrent density when completely converting the absorbed photons into current. ηsep is related to the hole transport dynamics in the photoanode bulk, and ηoxi is connected with the surface reaction kinetics. We first compared the UV-visible absorption of BTO@TiO2 relative to TiO2, and the results (Fig. S6†) showed that the light absorption ability for both samples is almost no different, meaning that Jabs are the same. Then, ηsep and ηoxi were determined based on sulfite oxidation (Fig. S7†). As shown in Fig. S6f,† the improvement in ηsep for BTO@TiO2 was only about 10%, which is significantly lower than that in ηoxi (40–90%). This suggests that the increased performance of BTO@TiO2 was attributable to the enhancement of catalytic kinetics at the photoanode surface.
A transient photocurrent test was also conducted to investigate hole recombination at the surface. As illustrated in Fig. S8,† the bias voltage drives the photogenerated hole to the photoanode surface, where it binds to the reaction substrate and generates a total photocurrent (Jtotal). However, the intermediates can be reduced by photogenerated electrons in an inverse reaction, thus causing current decay. The area of the corresponding decay peak represents the surface recombination current (Jrec) or the hole recombination degree.38,39 Following the measurement of transient photocurrent spectra in three different pH electrolytes (Fig. S9–11†), the values of Jtotal and Jrec were calculated at corresponding bias voltages, and the difference in their variation with a bias voltage from 1.03 to 1.23 V is plotted in Fig. 2d–f. It can be observed that there was no notable disparity between the Jrec of TiO2 and BTO@TiO2 under the three different electrolytes tested. Thus, the increase in Jtotal, which represents the number of holes involved in the reaction is closely related to the acceleration of the OER kinetics itself. The OER is a sluggish reaction involving four-hole/proton transfer steps and multiple intermediates. Regulating the energy barrier of intermediates can effectively enhance OER kinetics.
Subsequently, light–dark electrochemical scanning was conducted on TiO2 and BTO@TiO2, respectively, to obtain the intermediate states formed during OER. It should be noted that both AEM and OPM mechanisms coexist when OER occurs in the whole pH range,32,40 while AEM is the dominant reaction in acidic electrolytes and OPM reaction ascends in the basic electrolyte. In a neutral environment, the competition between AEM and OPM results in a decline in the photocurrent performance. The different RDS in AEM and OPM allow the detection of residual intermediates, oxo and hydroxyl species, under acidic and basic conditions, respectively. As illustrated in Fig. 2g–i, as compared with the reduction peaks from pristine TiO2 in different electrolytes, the reduction peaks at 0.33 and 0.31 VSHE for BTO@TiO2 correspond to the oxo groups when pH = 0 and 7. The reduction peak at −0.45 VSHE is related to the hydroxyl group on BTO@TiO2 in pH = 14. The reduction peak potential, Ep, of both the oxo and hydroxyl groups are positively shifted for BTO@TiO2 in both acidic and alkaline conditions in comparison to pristine TiO2; this shift is accompanied by the significantly enhanced photocurrent of BTO@TiO2. Even in the neutral case, where dual-mechanism coexistence is seen, the photocurrent of BTO@TiO2 is significantly enhanced with a positive shift in the Ep of oxo species, as shown in Fig. 2h. As the potential of the reduction peak is related to the free energy of the intermediates, in the acidic condition, the positive shift in the reduction peak indicates that the free energy of the oxo is elevated, which effectively shrinks the RDS energy barrier between the oxo and hydroperoxo in the AEM pathway, thus improving the OER kinetics and enhancing the catalytic reaction rate, as shown in Fig. 3a. Similarly, in the alkaline environment, the elevated hydroxyl energy reduces the difference in free energy between the hydroxyl and oxo groups in the OPM pathway, as shown in Fig. 3b. It seems that the positive shift in the Ep of the oxo and hydroxyl groups is associated with enhanced OER performance. The question is whether it can be deduced that the positive shift Ep of the two intermediates is a descriptor of the acceleration of OER kinetics, regardless of the oxygen evolution mechanism being AEM or OPM?
 | ||
| Fig. 3 Influence of intermediate states on the catalytic mechanism. (a) Effect of the oxo group energy change on the AEM. (b) The effect of hydroxyl energy change on OPM. (The grey arrows represent the mechanism that occurred on TiO2, and the yellow arrows represent the mechanism that occurred on BTO@TiO2) (c) mechanism transformation between AEM and OPM induced by excessive amorphization. The variation in the photocurrent at 1.23 VRHE and Ep of intermediates with reduction potential at (d) pH = 0 and (e) pH = 14. The TEM image of (f) pristine TiO2 and (g) TiO2 after electrochemical reduction treatment. | ||
To further verify this deduction, we reconstructed an amorphous TiO2 thin layer on the TiO2 NWs surface by the electrochemical reduction method (Fig. S12†) without changing its UV-visible absorption ability (Fig. S13†). In addition, we obtained electrochemical fingerprints of the two intermediates in different pH electrolytes (Fig. S14†). The comparison between the photocurrent J and the intermediate potential Epvs. electrochemical reduction voltage through light–dark electrochemical scanning tests is shown in Fig. 3d and e. Fig. 3d shows that under different reduction voltage treatments, the trend in OER performance change is consistent with the oxo Ep change in an acidic environment. This further confirms that increased oxo energy reduces the RDS potential during AEM; the Ep of oxo species can be used as a descriptor of the kinetic rate in AEM.16,41 The larger the value of Ep, the faster the catalytic kinetics of the AEM. However, in an alkaline environment, the tendency for performance change and the hydroxyl Ep change are not entirely consistent, as shown in Fig. 3e. In particular, the photocurrent of TiO2 NWs that had been treated by a reduction voltage larger than −0.5 VRHE decreases, but the energy of the hydroxyl group continues to increase. For the OPM pathway, the O–O coupling that occurs at two active sites needs more stringent requirements for the geometric configuration of metal active sites.15 It has been reported that both the O* interaction strength and O* binding energetics control the kinetics of water oxidation.16 Therefore, the kinetics of the OPM reaction depend not only on the hydroxyl energy but also on the interatomic resistance of the oxo groups. Excessive amorphization (Fig. 3f and g) will enlarge the distance between the active sites and increase the interatomic resistance for O–O coupling, leading to the conversion of OPM into AEM as shown in Fig. 3c.28,42 Although the free energy of hydroxyl enhanced, the performance of TiO2 reduced at more negative voltage and decreased in pH = 14. Briefly, the oxo energy can be regarded as a descriptor for the modulation of the OER kinetics in AEM as the reaction proceeds on a single active site. However, in OPM, the modulation of the OER kinetics must be considered for the hydroxyl energy, as well as the interatomic resistance effect.
According to the above analysis, the intrinsic correlation between the energy change of the intermediates and the reaction kinetics, with the aid of ferroelectric polarization, can be further studied. Fig. 4a and b demonstrate that the tetragonal BaTiO3 layer on TiO2 is a ferroelectric phase with reversible polarization; in addition, the amplitude-biased curves in butterfly shape were obtained by a piezoelectric force microscopy, and 180° phase transitions were observed. Then, BTO@TiO2 was polarized by applying an external bias voltage in a two-electrode system as shown in Fig. S16.† In this system, the positive electrode of the power source was connected to the photoanode to perform positive polarization (marked as BTO@TiO2_P). Conversely, the negative pole is connected to the photoanode for negative polarization (labelled as BTO@TiO2_N). Following the completion of the polarization process, a series of light–dark electrochemical scans were conducted. The reduction peaks of oxo exhibited a negative shift in both BTO@TiO2_P and BTO@TiO2_N in pH = 0 as shown in Fig. 4e. The negative shift in the reduction peak indicates a decrease in the oxo energy, which undoubtedly results in an increased in the energy difference between the oxo and hydroperoxo intermediates that consequently leads to an elevated AEM barrier and the inhibition of the OER kinetics. The slower reaction kinetics results in a more challenging hole capture on the photoanode surface, which in turn, leads to a notable reduction in photocurrent, as illustrated in Fig. 4c. Furthermore, in alkaline environments, the reduction peak of the hydroxyl group is almost unchanged on the BTO@TiO2_P, while the photocurrent curves essentially overlap before and after positive polarization, as shown in Fig. 4d and f. Meanwhile, the increase in the hydroxyl Ep on BTO@TiO2_N resulted in a further decrease in photocurrent. It is shown that negative polarization triggers a decrease in the performance at pH = 14.
 | ||
| Fig. 4 The effect of ferroelectric polarization on photocurrent, as well as intermediate states. The piezoelectric force response of BTO@TiO2 with (a) butterfly loop of the amplitude and (b) hysteresis loop of the phase. The photocurrent and electrochemical fingerprints scan of BTO@TiO2 in (c and e) pH = 0 and (d and f) pH = 14 after positive and negative polarization. | ||
In acidic or neutral electrolytes, the polarization field merely affects the adsorption of H+, Na+, or SO42− groups, which do not take part in the OER process (Fig. 5a, b, d and e). In an alkaline electrolyte (pH = 14), the dominant groups are OH− anions and Na+ cations, therefore, the OER mechanism mainly follows the OPM pathway. After negative polarization, the negatively charged BTO@TiO2_N attracts Na+ cations and reduces the coverage of OH− on the surface, as shown in Fig. 5c.43
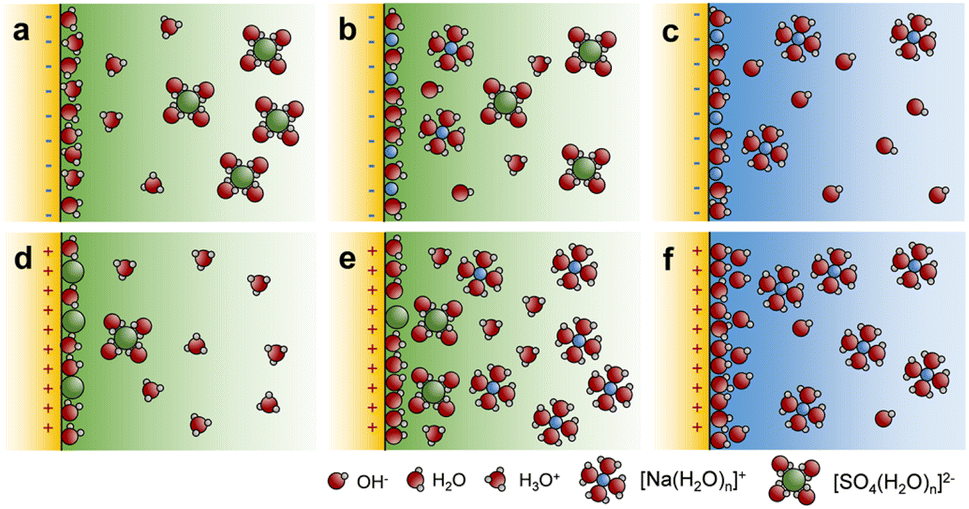 | ||
| Fig. 5 The effect of the polarization on interfacial ion adsorption. Diagrams of ion adsorption at the BTO@TiO2-electrolyte interface in (a and d) acidic, (b and e) neutral, and (c and f) basic electrolytes after negative and positive polarization. | ||
The reduced OH− coverage results in lower surface hydroxyl density, which aggravates the interatomic resistance of the O–O coupling, failing to sustain the OPM, and thus, the OER mechanism shifts to the AEM and the photocurrent declines, as shown in Fig. 4d. Recently, it has been reported that oxygen species formed at the OER exhibit repulsive adsorbate–adsorbate interactions, and increasing their coverage weakens their binding, thereby promoting O–O bond formation.16 Here we demonstrated that the reduced OH− coverage on BTO@TiO2_N, when combined with the increase in the energy of the hydroxyl group, as shown in Fig. 4f, which is in accordance with the above literature.
Therefore, we verified that polarization not only alters the energy of the intermediates of AEM and OPM but also controls the mechanism transformation between AEM and OPM when pH > 7. Negative polarization results in the conversion of OPM to AEM, and positive polarization promotes the conversion of AEM to OPM as shown in Fig. 6a. Thus, we can speculate that in a weakly alkaline environment, where the OH− coverage on the photoanode does not reach saturation, the positive polarization may promote OH− coverage and facilitate the OPM kinetics. As shown in Fig. 6b, the photocurrent of TiO2/BTO_P increased by 0.52 mA cm−2 at 1.23 VRHE in the electrolyte with pH = 11, which is a notable enhancement compared to the reported works (Fig. 6d). The OER was able to progress more expeditiously through the OPM in this case, thereby enhancing catalytic kinetics and performance, while the energy of the hydroxyl group decreased. Overall, ferroelectric polarization alters the catalytic kinetics by transforming the catalytic mechanism via controlled OH− coverage and the free energy of the intermediate state, which leads to asymmetric changes in performance.
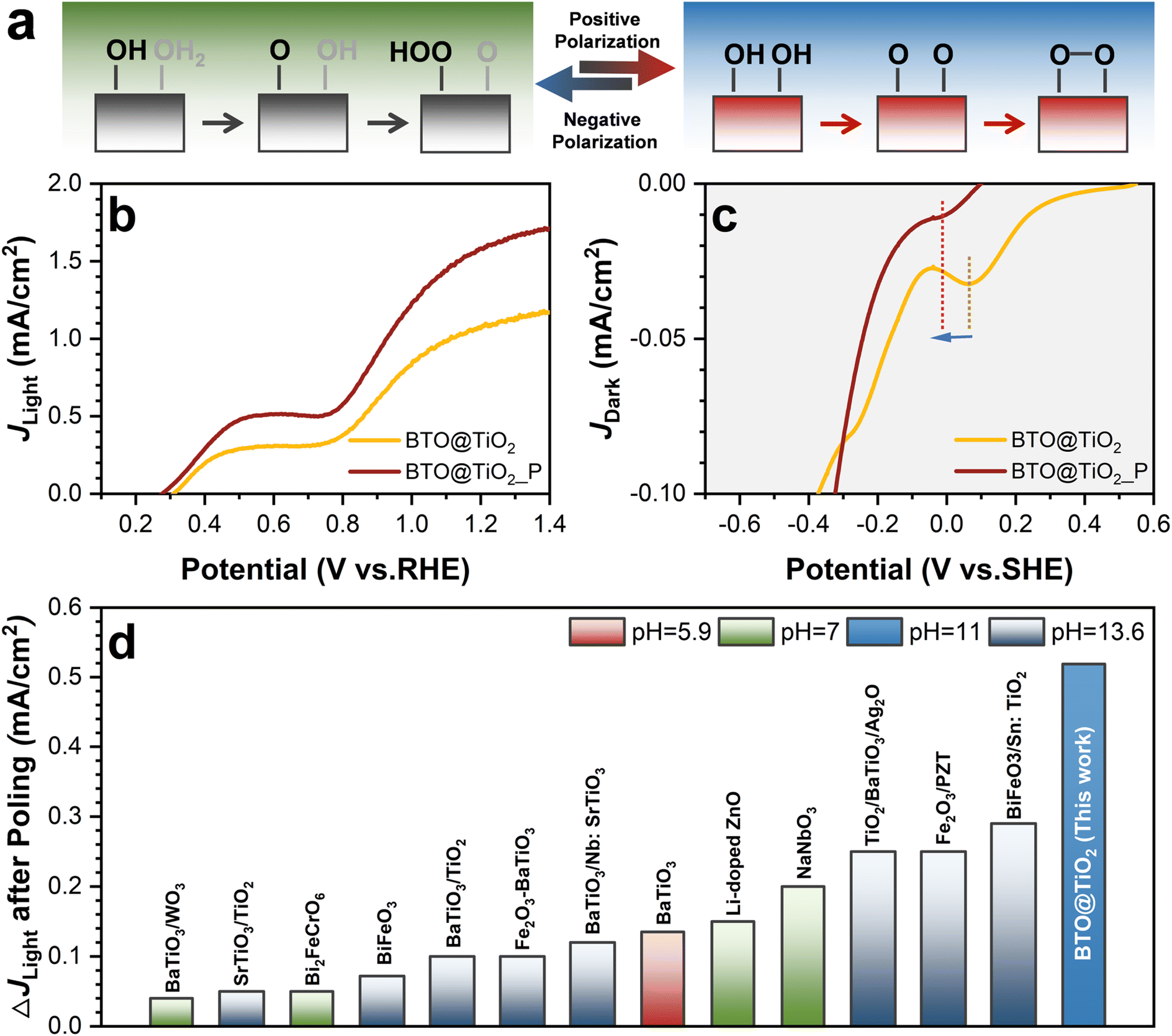 | ||
| Fig. 6 The validation of the catalytic mechanisms transformation induced by polarization when pH > 7. (a) Mechanism transformation induced by positive polarization. (b) The photocurrent and (c) electrochemical fingerprints curve of BTO@TiO2 at pH = 11 after positive polarization. (d) Comparison of the enhancement of J at 1.23 VRHE for BTO@TiO2 after polarization in this work with other ferroelectric-based photoanodes (full data are listed in Table S1†). | ||
First-principles calculations were performed to explore how polarization affects the energetics of the OER intermediates. Based on the AC-TEM results (Fig. 1i), we constructed a heterojunction structure that consisted of one TiO2 (001) and two BTO layers (Fig. S17†) to model the intrinsic (w/o) and polarized (±Pz) BTO@TiO2 layer. Two layers of the Pt (001) plane were constructed under the BTO to screen the dipole moment, according to the previous study. The free energy changes from OH* to O*, which can be detected with light–dark electrochemical scanning, were calculated for non-polarized, positively (+Pz) and negatively polarized (−Pz) BTO@TiO2, as shown in Fig. 7a. The free energies of O* decreased under both +Pz and −Pz, while the free energy of OH* increased under −Pz and decreased under +Pz. This trend is consistent with the results based on the experimental tests (Fig. 4e and f). As a result, the decrease in free energy of OH* in the case of +Pz, relative to that without polarization, can be partially attributed to the concordant dipole direction of the OH* group and BTO, resulting in a Coulomb attraction, and thus, stronger adsorption. In contrast, for −Pz, the dipole direction of OH* and BTO was opposite, leading to Coulomb repulsion, and thus, an increase in free energy. Although both positive and negative polarization leads to a decrease in the energy of O*, the causes of the decrease are different. As illustrated in Fig. 7b, the electron localization function reveals that the O* is bonded to the surface titanium atoms exclusively, establishing a distinct covalent bond. +Pz only results in a reduction in the bond length from 1.73 to 1.65 Å, which is corroborated by Fig. 7c. However, −Pz significantly alters the O* adsorption configuration. Fig. 7b and c show that the O* forms a bond with not only the Ti atom but also with the O atoms on the surface. Furthermore, to understand the different adsorption behaviour of O* on the −Pz polarized TiO2-BTO, the configuration, where O* was adsorbed on only one Ti site was also investigated, as shown in Fig. S19.† It turns out that this adsorption configuration is energetically less favourable by 1.65 eV. The Crystal Orbital Hamilton Populations (COHP) analysis (Fig. S20†) reveals a much stronger bonding in the O–O* than that in the Ti–O* bond, indicating that O–O* bonding plays a dominant role in the adsorption of O* on the −Pz polarized BTO@TiO2.
 | ||
| Fig. 7 Theoretical analyses of the influence of polarization on the intermediates. (a) Free energy change from OH* to O* on BTO@TiO2 without (w/o) and with positive (+Pz) and negative polarization (−Pz). (b) Electron localization function (ELF) and (c) charge density difference of O* among the three heterojunctions. (d) Local density of states of the TiO2 surface layer at the three heterojunctions. The Fermi level (vertical dashed lines) is set to zero. (e) Schematic diagram of polarization-modulated electron transfer direction (denoted by arrows) between TiO2 and BTO. | ||
Furthermore, the local density of states of the surface TiO2 layer was analysed to explore the origin of the polarization effect on the adsorption behaviour of O* (Fig. 7d). Clearly, +Pz leads to the electron occupation in the conduction band minimum (CBM) of TiO2, while −Pz results in unoccupied states in the valence band maximum (VBM) of TiO2. These features should originate from the polarization-modulated electron transfer direction between TiO2 and BTO. According to the planar average charge differences (Fig. S21†), the electron transfer directions in the two polarized heterojunctions are schematically illustrated in Fig. 7e. Clearly, +Pz results in electron transfer from BTO to TiO2, while the transfer direction is opposite in the −Pz BTO@TiO2. It is well-known that CBM and VBM of TiO2 are dominated by Ti and O, respectively.44 Thus, electron accumulation is around Ti sites (mainly in the dxy orbital) in the +Pz BTO@TiO2, as presented in Fig. S22b,† which renders the Ti sites partially reduced. In the case of −Pz, electrons are depleted around the O sites, leading to several unoccupied states, mainly in the O py orbital (Fig. S22f†). As a result, more electrons were donated from Ti to O* on the +Pz BTO@TiO2, which is the origin of the stronger bonding than that on the non-polarized system. In comparison, the unoccupied states can activate surface O sites in the −Pz-polarized BTO@TiO2, which enables the formation of a strong covalent bond to O*. Therefore, it becomes responsible for the dual-site adsorption configuration of O*. Consequently, these calculated results demonstrate that opposite polarizations in BTO result in opposite electron transfer directions between TiO2 and BTO, which subsequently affect the chemical states of the surface Ti and O sites and ultimately give rise to sharply contrasted energetics in the OER intermediates.
Conclusions
In conclusion, our work combines experimental and theoretical techniques to re-evaluate the pH-dependent mechanism of ferroelectric polarization during the AEM and OPM pathways of the OER by unravelling the energy shift in the hydroxyl and oxo intermediates. In detail, based on the core–shell structure of the BTO@TiO2 photoanode, we identified the shift in the reduction peak of the hydroxyl and oxo intermediates, which is correlated with the free energy shift, on BTO@TiO2 under polarization through light–dark state electrochemical scanning. We note that the AEM pathway predominantly occurs in acidic environments, and the energy shift of the oxo intermediates is a descriptor for the acceleration of the OER kinetics. The OPM occurred mainly in the basic electrolyte, and thus, the modulation of the OER kinetics must consider the hydroxyl energy shift, as well as the spatial site resistance effect of the O–O coupling. We clarified that both +Pz and −Pz polarization reduces the free energy of the oxo group, thereby inhibiting the reaction kinetics of the AEM in acidic conditions. By contrast, in the OPM, ferroelectric polarization demonstrates a repulsive adsorbate–adsorbate interaction for OH− coverage and energy shift of OH*, increasing OH− coverage under +Pz weakens the free energy of OH* and improves O–O coupling, which is a key factor affecting the OPM kinetics. Thus, the optimal operation environment for ferroelectric polarization is a weakly alkaline electrolyte, where +Pz promotes OH− coverage and facilitates the transformation of the OER pathway from the AEM to OPM. We obtained a 47.7% improvement in the OER performance for BTO@TiO2 at 1.23 VRHE under this condition. We also used DFT to provide a molecular-level interpretation of the free energy shift of the two intermediates. Our work highlights the pH-dependent effects of ferroelectric polarization on the free energy of intermediates and the transformation of the catalytic mechanism. Our work leads to a more accurate description of the ferroelectric polarization mechanism during the OER than conventional models that are based solely on regulating band bending.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Peng Xiao and Yunhuai Zhang designed and guided the project. Liyong Gan carried out the DFT calculations and data analysis. Xing Ji and Zhouhao Zhu performed most experiments. Ming Zhou and Ying Zhang reviewed the manuscript and provided advice on the revised manuscript. Peng Xiao and Yunhuai Zhang wrote the manuscript with contributions from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the financial support from the Fundamental Research Funds for the Central Universities (Project No. 2024CDJXY010 and Grant No. 2024CDJXY022) and the National Natural Science Foundation of China (52471196).References
- S. Z. Oener, M. J. Foster and S. W. Boettcher, Science, 2020, 369, 1099–1103 CrossRef PubMed.
- A. Li, S. Kong, C. Guo, H. Ooka, K. Adachi, D. Hashizume, Q. Jiang, H. Han, J. Xiao and R. Nakamura, Nat. Catal., 2022, 5, 109–118 CrossRef.
- M. Busch, N. B. Halck, U. I. Kramm, S. Siahrostami, P. Krtil and J. Rossmeisl, Nano Energy, 2016, 29, 126–135 CrossRef.
- J. O. Bockris, J. Chem. Phys., 1956, 24, 817–827 CrossRef.
- J. O. Bockris and T. Otagawa, J. Electrochem. Soc., 1984, 131, 290–302 CrossRef.
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spöri, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Pérez-Ramírez, B. Roldan Cuenya, R. Schlögl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408 CrossRef PubMed.
- Y. Yao, S. Hu, W. Chen, Z.-Q. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang, G. Wu, W. Yuan, T. Yuan, B. Zhu, W. Liu, Z. Li, D. He, Z. Xue, Y. Wang, X. Zheng, J. Dong, C.-R. Chang, Y. Chen, X. Hong, J. Luo, S. Wei, W.-X. Li, P. Strasser, Y. Wu and Y. Li, Nat. Catal., 2019, 2, 304–313 CrossRef.
- J. Chen, C. Chen, M. Qin, B. Li, B. Lin, Q. Mao, H. Yang, B. Liu and Y. Wang, Nat. Commun., 2022, 13, 5382 CrossRef PubMed.
- D. Zhang, M. Li, X. Yong, H. Song, G. I. N. Waterhouse, Y. Yi, B. Xue, D. Zhang, B. Liu and S. Lu, Nat. Commun., 2023, 14, 2517 CrossRef PubMed.
- Y. Lin, Z. Tian, L. Zhang, J. Ma, Z. Jiang, B. J. Deibert, R. Ge and L. Chen, Nat. Commun., 2019, 10, 162 CrossRef PubMed.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Norskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef.
- Z.-F. Huang, J. Song, S. Dou, X. Li, J. Wang and X. Wang, Matter, 2019, 1, 1494–1518 CrossRef.
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465–468 CrossRef CAS PubMed.
- W. Zhou, H. Su, W. Cheng, Y. Li, J. Jiang, M. Liu, F. Yu, W. Wang, S. Wei and Q. Liu, Nat. Commun., 2022, 13, 6414 CrossRef CAS.
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, Nat. Catal., 2021, 4, 1012–1023 CrossRef CAS.
- C. W. Liang, R. R. Rao, K. L. Svane, J. H. L. Hadden, B. Moss, S. B. Scott, M. Sachs, J. Murawski, A. M. Frandsen, D. J. Riley, M. P. Ryan, J. Rossmeisl, J. R. Durrant and I. E. L. Stephens, Nat. Catal., 2024, 7, 763–775 CrossRef CAS.
- J. B. Neaton, C. Ederer, U. V. Waghmare, N. A. Spaldin and K. M. Rabe, Phys. Rev. B, 2005, 71, 014113 CrossRef.
- W. J. Merz, Phys. Rev., 1954, 95, 690–698 CrossRef CAS.
- L. Pan, S. Sun, Y. Chen, P. Wang, J. Wang, X. Zhang, J.-J. Zou and Z. L. Wang, Adv. Energy Mater., 2020, 10, 2000214 CrossRef CAS.
- C. Hu, S. Tu, N. Tian, T. Ma, Y. Zhang and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 16309–16328 CrossRef CAS.
- X. B. Li, W. W. Wang, F. Dong, Z. Q. Zhang, L. Han, X. D. Luo, J. T. Huang, Z. J. Feng, Z. Chen, G. H. Jia and T. R. Zhang, ACS Catal., 2021, 11, 4739–4769 CrossRef CAS.
- F. Chen, H. W. Huang, L. Guo, Y. H. Zhang and T. Y. Ma, Angew. Chem., Int. Ed., 2019, 58, 10061–10073 CrossRef CAS.
- T. L. Wan, L. Ge, Y. Pan, Q. Yuan, L. Liu, S. Sarina and L. Kou, Nanoscale, 2021, 13, 7096–7107 RSC.
- J. H. Lee and A. Selloni, Phys. Rev. Lett., 2014, 112, 196102 CrossRef PubMed.
- A. A. Iyer and E. Ertekin, Phys. Chem. Chem. Phys., 2017, 19, 5870–5879 RSC.
- V. Pfeifer, T. E. Jones, S. Wrabetz, C. Massue, J. J. V. Velez, R. Arrigo, M. Scherzer, S. Piccinin, M. Haevecker, A. Knop-Gericke and R. Schloegl, Chem. Sci., 2016, 7, 6791–6795 RSC.
- E. Pastor, Z. Lian, L. Xia, D. Ecija, J. R. Galán-Mascarós, S. Barja, S. Giménez, J. Arbiol, N. López and F. P. García de Arquer, Nat. Rev. Chem., 2024, 8, 159–178 CrossRef.
- H. Yu, Y. Ji, C. Li, W. Zhu, Y. Wang, Z. Hu, J. Zhou, C.-W. Pao, W.-H. Huang, Y. Li, X. Huang and Q. Shao, J. Am. Chem. Soc., 2024, 146, 20251–20262 CrossRef CAS PubMed.
- P. You, D. Chen, X. Liu, C. Zhang, A. Selloni and S. Meng, Nat. Mater., 2024, 23, 1100–1106 CrossRef CAS.
- O. Zandi and T. W. Hamann, J. Phys. Chem. Lett., 2014, 5, 1522–1526 CrossRef CAS PubMed.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, Energy Environ. Sci., 2012, 5, 7626–7636 RSC.
- X. Ji, Y. Ou, L. Huang, L. Gan, Y. Zhang and P. Xiao, ACS Catal., 2023, 13, 5841–5849 CrossRef CAS.
- L. A. Crosby, B.-R. Chen, R. M. Kennedy, J. Wen, K. R. Poeppelmeier, M. J. Bedzyk and L. D. Marks, Chem. Mater., 2018, 30, 841–846 CrossRef CAS.
- Y. Wang, L. Zhang, J. Wang, Q. Li, H. Wang, L. Gu, J. Chen, J. Deng, K. Lin, L. Huang and X. Xing, J. Am. Chem. Soc., 2021, 143, 6491–6497 CrossRef CAS.
- W. Yang, Y. Yu, M. B. Starr, X. Yin, Z. Li, A. Kvit, S. Wang, P. Zhao and X. Wang, Nano Lett., 2015, 15, 7574–7580 CrossRef CAS.
- F. Wu, Y. Yu, H. Yang, L. N. German, Z. Li, J. Chen, W. Yang, L. Huang, W. Shi, L. Wang and X. Wang, Adv. Mater., 2017, 29, 1701432 CrossRef.
- Z. Liu, L. Wang, X. Yu, J. Zhang, R. Yang, X. Zhang, Y. Ji, M. Wu, L. Deng, L. Li and Z. L. Wang, Adv. Funct. Mater., 2019, 29, 1807279 CrossRef CAS.
- F. Le Formal, K. Sivula and M. Grätzel, J. Phys. Chem. C, 2012, 116, 26707–26720 CrossRef.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, J. Am. Chem. Soc., 2012, 134, 16693–16700 CrossRef.
- L. Su, J. Chen, F. Yang, P. Li, Y. Jin, W. Luo and S. Chen, J. Am. Chem. Soc., 2023, 145, 12051–12058 CrossRef.
- C. Liang, Y. Katayama, Y. Tao, A. Morinaga, B. Moss, V. Celorrio, M. Ryan, I. E. L. Stephens, J. R. Durrant and R. R. Rao, J. Am. Chem. Soc., 2024, 146, 8928–8938 CrossRef PubMed.
- N. Wang, P. Ou, R. K. Miao, Y. Chang, Z. Wang, S.-F. Hung, J. Abed, A. Ozden, H.-Y. Chen, H.-L. Wu, J. E. Huang, D. Zhou, W. Ni, L. Fan, Y. Yan, T. Peng, D. Sinton, Y. Liu, H. Liang and E. H. Sargent, J. Am. Chem. Soc., 2023, 145, 7829–7836 CrossRef PubMed.
- G. Gonella, E. H. G. Backus, Y. Nagata, D. J. Bonthuis, P. Loche, A. Schlaich, R. R. Netz, A. Kühnle, I. T. McCrum, M. T. M. Koper, M. Wolf, B. Winter, G. Meijer, R. K. Campen and M. Bonn, Nat. Rev. Chem., 2021, 5, 466–485 CrossRef PubMed.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08291e |
| ‡ Xing Ji and Zhouhao Zhu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |