
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulating the spin–flip rates and emission energies through ligand design in chromium(III) molecular rubies†
Yating
Ye
 a,
Maxime
Poncet
b,
Polina
Yaltseva
c,
Pablo
Salcedo-Abraira
a,
Antonio
Rodríguez-Diéguez
a,
Javier Heredia
Martín
a,
Laura
Cuevas-Contreras
a,
Carlos M.
Cruz
d,
Benjamin
Doistau
e,
Claude
Piguet
b,
Oliver S.
Wenger
c,
Juan Manuel
Herrera
a and
Juan-Ramón
Jiménez
*a
a,
Maxime
Poncet
b,
Polina
Yaltseva
c,
Pablo
Salcedo-Abraira
a,
Antonio
Rodríguez-Diéguez
a,
Javier Heredia
Martín
a,
Laura
Cuevas-Contreras
a,
Carlos M.
Cruz
d,
Benjamin
Doistau
e,
Claude
Piguet
b,
Oliver S.
Wenger
c,
Juan Manuel
Herrera
a and
Juan-Ramón
Jiménez
*a
aDepartment of Inorganic Chemistry, University of Granada and “Unidad de Excelencia enQuímica (UEQ)”, Avda Fuente Nueva s/n, Granada, 18071, Spain. E-mail: jrjimenez@ugr.es
bDepartment of Analytical and Inorganic Chemistry, University of Geneva, 30 Quai Ernest Ansermet, CH-1211 Geneva, Switzerland
cDepartment of Chemistry, University of Basel, St. Johanns-Ring 19, Basel, 4056, Switzerland
dDepartment of Organic Chemistry, University of Granada and “Unidad de Excelencia enQuímica (UEQ)”, Avda Fuente Nueva s/n, Granada, 18071, Spain
eLaboratoire de Chimie et de Biochimie Pharmacologiques et Toxicologiques, Université Paris Cité, CNRS, 45 Rue des Saint-Pères, F-75006 Paris, France
First published on 20th February 2025
Abstract
Three homoleptic spin–flip (SF) emitters, namely [Cr(Mebipzp)2]3+ (1), [Cr(IMebipzp)2]3+ (2) and [Cr(bip*)2]3+ (3), have been successfully synthesized and characterized. The weak distortion compared to a perfect octahedron imparts favourable structural properties to the three complexes, which display spin–flip (SF) luminescence at approximately 740 nm with quantum yields in the range of 9–11% for 1 and 2 in deaerated acetonitrile solutions at 25 °C. Time-resolved luminescence and transient UV-vis absorption experiments unveiled lifetimes for the lowest-lying 2MC (metal-centered) of 1.5 ms for 1 and 350 μs for 2. The incorporation of iodine atoms onto the ligand scaffold in 2 accelerates the 2MC → 4A2 relaxation process through simultaneous enhancements in the radiative and non-radiative rate constants. In agreement, the experimentally calculated absorption oscillator strength for the 2MC ← 4A2 transition amounts to 9.8 × 10−7 and 2.5 × 10−6 for 1 and 2, respectively. The 2.5 factor enhancement observed in the iodine derivative indicates a higher spin–flip transition probability, translating into higher values of radiative rate constant (krad). Interestingly, in compound 3, the substitution of the distal methyl-pyrazole with indazole rings causes an important bathochromic shift of the SF emission energy to 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 (830 nm). Likely, the extended π-system and the more covalent bond character induced by the indazole decrease the interelectronic repulsion further stabilizing the SF excited states. The recorded excited state lifetime of 111 μs in 3 remains among the longest for a molecular ruby emitting beyond 800 nm. These discoveries signify an underexplored avenue for modifying deactivation pathways and emission energy while retaining high quantum yields and long-lived excited states in molecular rubies.
000 cm−1 (830 nm). Likely, the extended π-system and the more covalent bond character induced by the indazole decrease the interelectronic repulsion further stabilizing the SF excited states. The recorded excited state lifetime of 111 μs in 3 remains among the longest for a molecular ruby emitting beyond 800 nm. These discoveries signify an underexplored avenue for modifying deactivation pathways and emission energy while retaining high quantum yields and long-lived excited states in molecular rubies.
Introduction
The increasing interest in and need for a more sustainable photochemistry has challenged scientists to develop photoactive metal complexes based on earth-abundant metal ions such as CoIII, VIII, FeII, FeIII, MnI, Cr0 or CrIII.1–17 Among these, CrIII has garnered significant attention due to its appealing physicochemical properties associated with (i) its relative kinetic inertness, enabling the manipulation of CrIII chromophores under different experimental conditions and (ii) the small excited-state distortions associated with the relevant spin–flip (SF) transition favouring radiative relaxation over non-radiative relaxation.18–23 Moreover, when surrounded by a strong near-octahedral ligand field, CrIII exhibits long-lived Cr(2T1, 2E) excited states showing lifetimes in the millisecond range with overall quantum yields (ϕ) attaining 17% for non-deuterated samples and up to 30% for deuterated ones.10,11,24–26 These appealing properties have been exploited in molecular upconversion and downshifting,27–32 molecular thermometry,33 circularly polarized luminescence (CPL)10,30,34–37 and photocatalysis.38,39 Most reported polypyridyl CrIII complexes show radiative emission within the range of 700–800 nm (14300–12500 cm−1)18,25 but recent examples demonstrated that increasing metal–ligand covalency favours the nephelauxetic effect (i.e. decreases the electron–electron repulsion) stabilizing the SF states and thus shifting the CrIII centered emission up to 1000–1067 nm (NIR-II region, ∼9400 cm−1).40–43 Conversely, Heinze and coworkers harnessed weak π-accepting ligands in the [Cr(bpmp)2]3+ complex (bpmp = 2,6-bis(2-pyridylmethyl)pyridine) which displays strong red emission (λem = 709 nm) (Fig. 1a).44 Since CrIII coordination chemistry has been mainly limited to polypyridyl organic ligands, Scattergood and co-workers introduced a novel class of ligands in which a central pyridine is functionalized at the 2,6 positions with 1,2,3-triazole donors.45 However, the photophysical properties of [Cr(btmp)2]3+ (btmp = 2,6-bis(4-phenyl-1,2,3-triazol-1-yl-methyl)-pyridine) remain comparatively modest (ϕ = 0.27%; τ = 37.9 μs) (Fig. 1b).
 | ||
| Fig. 1 Structure, luminescence quantum yield, emission lifetime (deaerated CH3CN/HClO4 solution at 293 K), wavelength of the emission band maximum, full width at half maximum of the emission band and radiative rate constant of some closely related emissive chromium(III) complexes: (a) [Cr(bpmp)2]3+, (b) [Cr(btmp)2]3+, (c) [Cr(Mebipzp)2]3+, (d) [Cr(IMebipzp)2]3+ and (e) [Cr(bip*)2]3+ (this work). | ||
Boosting the overall luminescence quantum yield in this type of emitters has traditionally been achieved by reducing high-energy oscillators (e.g., C–H, O–H, or N–H) near the metal center or encapsulating the compounds within a rigid silica matrix, thereby lowering non-radiative rates (knrad).26,35 However, beyond this common approach, other strategies such as enhancing the radiative rate constant (krad)—which describes the intrinsic rate of spontaneous emission from an excited state in the absence of any other non-radiative deactivating processes, remain comparatively underexplored.46,47 The radiative rate lifetime, τrad (krad−1), is a key parameter that governs the luminescence quantum yield, as shown in eqn (1), where ηsens is the sensitization efficiency, τobs is the measured (observed) luminescence lifetime, and knrad represents non-radiative rates. This equation reveals that higher values of krad relative to knrad could, in principle, promote greater emission efficiencies. The luminescence quantum yields of CrIII spin–flip emitters are currently constrained by the low radiative rates, a consequence of the intrinsically doubly forbidden nature of this transition. Therefore, enhancing krad presents an especially appealing strategy for improving the performance of this class of compounds.
 | (1) |
In this work, we present the modulation of spin–flip transition rates and the adjustment of emission energy through precise tuning of the organic ligands. We demonstrate that substituting the distal pyridines at the 2 and 6 positions of the ligand bpmp (Fig. 1a) with functionalized pyrazolyl or indazole groups (Fig. 1c–e) significantly broadens the spin–flip emission bandwidth, tunes the radiative rates and modifies the emission energy while maintaining long excited-state lifetimes and relatively high luminescence quantum yields compared to the related [Cr(btmp)2]3+. The syntheses, crystal structures, theoretical calculations and photophysical properties of these three novel molecular rubies: [Cr(Mebipzp)2]3+ (1) (Mebipzp = 2,6-bis((3,5-dimethyl-1H-pyrazol-1-yl)methyl)pyridine; Fig. 1c), [Cr(IMebipzp)2]3+ (2) (IMebipzp = 2,6-bis((4-iodo-3,5-dimethyl-1H-pyrazol-1-yl)methyl)pyridine; Fig. 1d) and [Cr(bip*)2]3+ (3) (bip* = 2,6-bis(indazol-2-ylmethyl)pyridine; Fig. 1e) are discussed hereunder.
Results and discussion
Synthesis and structural properties
Reacting CrII(SO3CF3)2·2H2O48 with two equivalents of Mebipzp, IMebipzp or bip* ligands, followed by oxidation with silver triflate (AgSO3CF3) in acetonitrile led to the desired compounds in a 90–95% yield (Fig. S1, further synthetic details in the ESI†). The alternative air oxidation resulted in undesirable redox reactions. Single crystals suitable for X-ray diffraction were obtained from slow diffusion of diethyl ether into concentrated acetonitrile solutions containing the CrIII complexes (Table S1†). For the three compounds, the two tridentate ligands coordinate in a twisted and meridional fashion (Fig. 2a and S2–S4†) around the central CrIII cation with dihedral angles of approximately 57° and 50° between the central pyridine ring and the flanking pyrazole or indazole units. Due to the distorted conformation adopted by the ligands, a decrease in the symmetry point group (D2d → D2) generates a pair of enantiomers (λλ and δδ; notation for the ligand disposition)49 that crystallize as racemic mixtures (PP and MM enantiomers). The four iodine atoms in 2 are in the equatorial plane of the molecule and the average Cr⋯I distance amounts to 6.22 Å. In all three compounds, the Cr–N(pyridine) bond lengths range from 2.0813(14) to 2.104(5) Å, consistent with those found in related polypyridyl-based molecular rubies and in the particular case of [Cr(bpmp)2]3+ with six coordinated pyridines (average Cr–N distance = 2.0731(13) Å). In contrast, the Cr–N(pyrazole) and Cr–N(indazole) bond lengths are shorter, spanning from 2.0470(15) to 2.069(5) Å in compounds 1 and 2, and reaching 1.999(7) Å in compound 3 (Fig. 2a and Tables S2–S4†). This reduction in bond length suggests a slightly greater covalent character than that of the Cr–N(pyridine) bonds, with this effect being more pronounced for the indazole-based ligand. Similar findings have been observed in [Cr(btmp)2]3+, where the Cr–N(triazole) bonds are as short as 1.995(2) Å.45 This structural feature induces compression in the equatorial plane of the complexes, resulting in a non-negligible structural distortion of the ground state.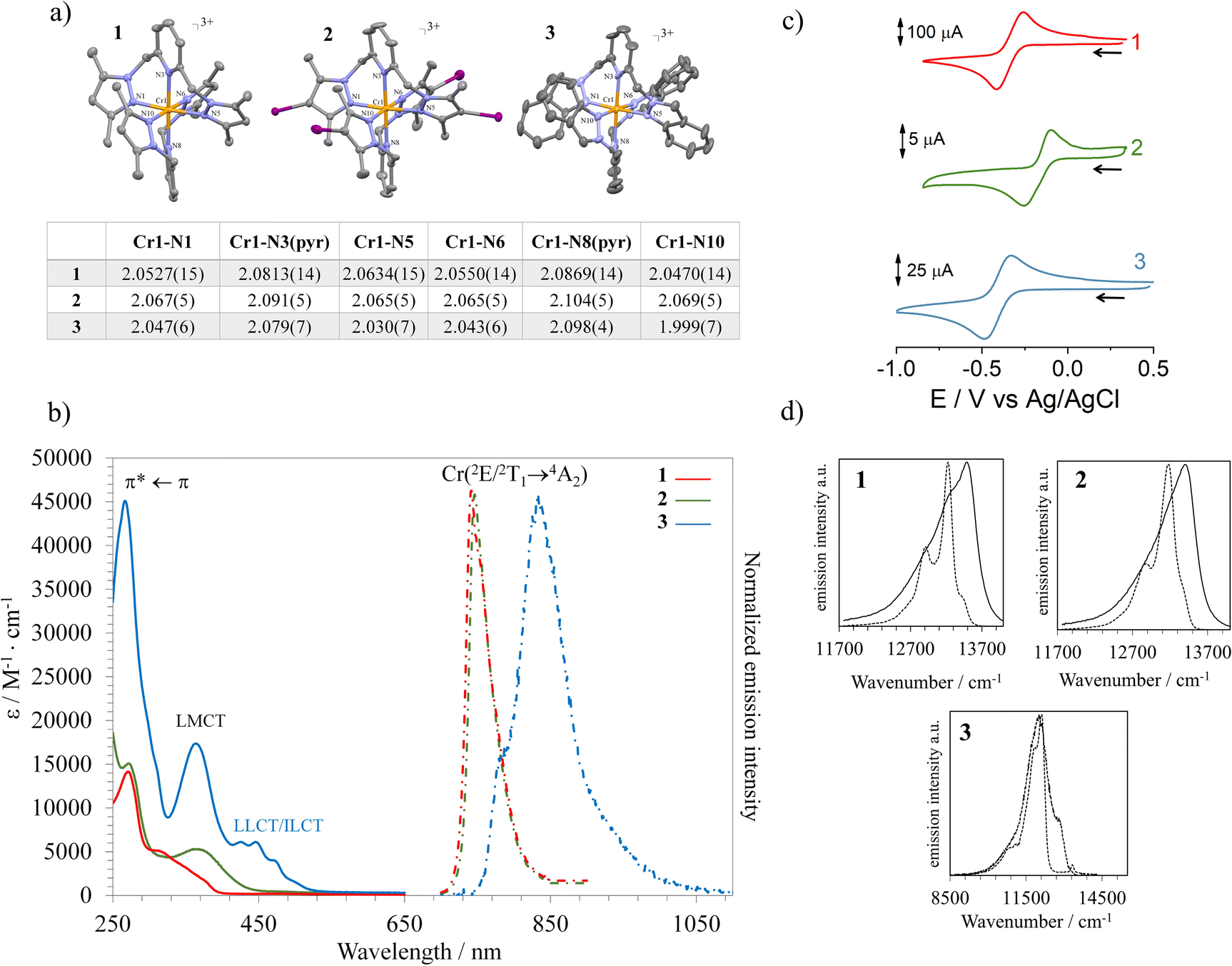 | ||
| Fig. 2 (a) Crystal structures of complexes 1, 2 and 3 including their Cr–N bond distances. Triflate anions, solvent molecules and hydrogen atoms were omitted for clarity. Colour codes: Cr (orange), N (blue), C (grey), I (purple). ORTEP ellipsoids are drawn at 50% probability. (b) Absorption (solid line) and emission spectra (dashed line) at room temperature of complexes 1 (red), 2 (green) and 3 (blue) in acetonitrile. (c) Cyclic voltammograms of compounds 1, 2 and 3 in acetonitrile and in the presence of nBu4NPF6 (0.1 M); (d) comparison of the emission spectra at 298 K (solid line) and 77 K (dashed line) for compounds 1–3. | ||
The N(terminal)–Cr–N(terminal) bite angles of 175–177° are close to the 180° expected for a perfect octahedron (Tables S2–S4†). A detailed geometrical analysis using  where ϕi stands for the cisoid N–Cr–N bond angles (Tables S2–S4†), gives ∑ = 31°, 33° and 33° for 1, 2 and 3, and 27° and 25° for [Cr(btmp)2]3+ and [Cr(bpmp)2]3+ respectively. Slightly more distorted geometries are observed for compounds 1–3, which may be attributed to steric hindrance caused by the methyl groups and fused heterocycles. In any case, this represents a small geometric distortion with respect to the ideal CrN6 octahedron and ensures an efficient orbital overlap between the metal and the ligands, thus inducing strong ligand field splitting destabilizing the 4T2 state and preventing, in principle, back-intersystem crossing with the SF excited states. Additionally, the coordination geometry adopted by the CrIII ion in the three compounds has been evaluated through the SHAPE software, which is based on the continuous shape measurements method (CShMs).50 The coordination sphere of compounds 1–3 closely resembles an ideal octahedral geometry (OC-6), typical of CrIII complexes with six ligands symmetrically arranged around the metal centre. However, compound 3 exhibits slight deviations (Table S5†), likely due to a more compressed octahedral structure resulting from the shortening of certain Cr–N bonds. In the solid state, CH⋯O hydrogen bonds are observed between the methylene bridges and the triflate anions (d(C⋯O) = 2.9–3.2 Å), which highlights the acidity of the CH2 bridge units in the cationic complexes (Fig. S5–S7†).44
where ϕi stands for the cisoid N–Cr–N bond angles (Tables S2–S4†), gives ∑ = 31°, 33° and 33° for 1, 2 and 3, and 27° and 25° for [Cr(btmp)2]3+ and [Cr(bpmp)2]3+ respectively. Slightly more distorted geometries are observed for compounds 1–3, which may be attributed to steric hindrance caused by the methyl groups and fused heterocycles. In any case, this represents a small geometric distortion with respect to the ideal CrN6 octahedron and ensures an efficient orbital overlap between the metal and the ligands, thus inducing strong ligand field splitting destabilizing the 4T2 state and preventing, in principle, back-intersystem crossing with the SF excited states. Additionally, the coordination geometry adopted by the CrIII ion in the three compounds has been evaluated through the SHAPE software, which is based on the continuous shape measurements method (CShMs).50 The coordination sphere of compounds 1–3 closely resembles an ideal octahedral geometry (OC-6), typical of CrIII complexes with six ligands symmetrically arranged around the metal centre. However, compound 3 exhibits slight deviations (Table S5†), likely due to a more compressed octahedral structure resulting from the shortening of certain Cr–N bonds. In the solid state, CH⋯O hydrogen bonds are observed between the methylene bridges and the triflate anions (d(C⋯O) = 2.9–3.2 Å), which highlights the acidity of the CH2 bridge units in the cationic complexes (Fig. S5–S7†).44
Electronic absorption properties
The UV-vis absorption spectra in acetonitrile (10−5 M) at room temperature show maxima between 250 and 300 nm (ε > 8000 M−1 cm−1), which can be attributed to ligand-centered π* ← π transitions, along with lower intensity bands between 300 and 550 nm (ε < 7000 M−1 cm−1) assigned to mixed metal-centered and ligand to metal charge transfer (MC/LMCT) transitions with a considerable red-shift for the iodine analog 2 (Fig. 2b). For compound 3 at this wavelength range, ε > 10000 M−1 cm−1, because of the admixture with intense ligand-to-ligand and intraligand charge transfer transitions (LLCT and ILCT) (Fig. 2b and S30–S32†). The band located at the lowest energy (∼450–480 nm) can be attributed to the 4T2 ← 4A2 transition according to TD-DFT calculations (Fig. 2b, S11 and S24–S32†). From this electronic transition, the value of the ligand field splitting (Δoct) is estimated at approximately 21500 cm−1 for the three compounds. This significant ligand-field splitting could potentially limit the thermally assisted back-intersystem crossing between the emissive excited states 2E/2T1 and 4T2.25
At much lower energies (12300–14500 cm−1), in solution and in solid state, the absorption spectra displayed two main bands with maxima at ∼740 nm, with a molar extinction coefficient (ε) of 0.58 M−1 cm−1 and 1.27 M−1 cm−1 for 1 and 2 respectively (Fig. 3 and S8†). For compound 3 the band maximum is red-shifted up to 773 with ε = 0.6 M−1 cm−1 (Fig. 3 and S8†). Weaker absorption bands at 690 nm with ε = 0.1–0.2 M−1 cm−1 for 1 and 2, and 717 nm for 3 with ε = 0.21 M−1 cm−1 have also been detected. These bands correspond to the spin-forbidden direct transitions from the quartet ground state (4A2) to the manifold low-lying doublet metal-centered excited states (2E and 2T1).
 | ||
| Fig. 3 Spin–flip absorption spectra of 1, 2 and 3 complexes in acetonitrile (c = 7.7 mM). Band assignment according to (CASSCF(7,12)/FIC-NEVPT2) calculations (see text and Fig. S23†). The oscillator strength (f) has been calculated according to eqn (2). | ||
Concerning these spin-forbidden transitions, it is worth noting that the decadic absorption coefficients ε for the lowest energy band for the three compounds are on average one order of magnitude greater (and at least 3 fold greater) than those previously reported for related complexes (0.02–0.3 M−1 cm−1).10,22,51,52 The even larger value found for complex 2 (1.27 M−1 cm−1) is attributed to the heavy-atom effect caused by the iodine atoms in the ligand scaffold, boosting the spin–orbit coupling and so partially relaxing the spin selection rule. The associated oscillator strength (fexp) for the lowest energy transition has been calculated using the Strickler–Berg relationship (eqn (2)), where m is the electron mass, c is the velocity of the light in a vacuum, Na is Avogadro's number, e is the electron charge, X = (n2 + 2)2/9n is the local field correction where n is the refractive index of the medium, ṽ is the wavenumber of the relevant electronic transition and ε is the molar absorption coefficient.
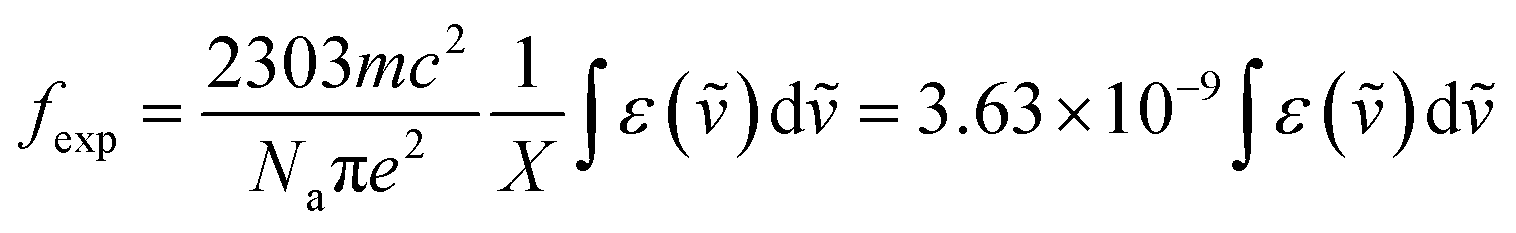 | (2) |
The obtained values of 9.8 × 10−7, 2.5 × 10−6 and 9.0 × 10−7 for 1, 2 and 3 respectively, are larger than those of previously reported related Cr(III) compounds.10,53 Compound 2 containing four iodine atoms reflects an increased value by a factor 2.5 in agreement with the abovementioned difference in molar extinction coefficient at the absorption maxima. The fexp in 2 lies in the order of magnitude found in lanthanide ions such as Yb(III), which is well known to act as a sensitizer in energy transfer upconversion processes (ETU).54 Although weak, the enhancement of the spin–flip transition cross-section (associated with fexp) would be the keystone to harness inert Cr(III) as an efficient NIR sensitizer for boosting ETU efficiency in (supra)molecular Cr(III)–Er(III) assemblies,27,32,55 and potentially also for enhancing the efficiency of red light-driven photoredox catalysis with Cr(III) complexes.56,57
Electrochemical experiments show reversible waves at E1/2 = −0.35 V, −0.16 V and −0.41 V vs. Ag/AgCl for compounds 1, 2 and 3 respectively (Fig. 2c). These reversible waves were likely associated with metal-centered processes as observed for compounds [Cr(bpmp)2]3+ and [Cr(btmp)2]3+. At lower potentials irreversible processes are observed probably due to complex dissociation (Fig. S21†). Additionally, no oxidation phenomena were observed within the electrochemical solvent window.
Theoretical calculations
The geometries of 1, 2 and 3 in their 4A2 ground states were optimized using unrestricted density functional theory (DFT) calculations at the UB3LYP/def2TZVPP level (see the ESI for technical details and Fig. S22†). The obtained geometries matched those obtained from the single-crystal diffraction, with minimal variations in their bite angles (Table S6†). The excited state landscape was evaluated using the complete active space self-consistent field method (CASSCF(7,12)/FIC-NEVPT2) for the three complexes. The calculations revealed a similar distribution of the excited states in the three compounds, where the microstates 2T1(1) and 2T1(2) are the lowest energy excited states (Fig. S23†). Similar results have been found in [Cr(bpmp)2]3+, [Cr(ddpd)2]3+, [Cr(btmp)2]3+ and [Cr(dqp)2]3+.10,44,45 Despite the admixture and small energy difference between the six microstates belonging to the 2T1 and 2E states, two distinguishable bands were experimentally observed in the absorption spectrum and were tentatively assigned to two different sets of doublet states (Fig. 3). Thus, according to the quantum chemical calculations and the experimental results, the band located at lower energy can be assigned to the [2E(1), 2T1(2), 2T1(1) ← 4A2] transitions and the higher one to [2E(2), 2T1(3) ← 4A2] which results in the Jablonski diagram depicted in Fig. S23.† Hundred spin-allowed vertical electronic transitions were computed using time dependent DFT (TD-DFT, Tables S7–S9†) at the UB3LYP/def2TZVP level of theory. The decomposition of these transitions suggested that the lowest energetic electronic transitions have a mainly metal centered (MC) character (Fig. S24, S27 and S30†), as expected. In 1, those transitions can be divided into two groups, three transitions falling under the observed band between 450 and 550 nm and three transitions between 370 and 450 nm (Fig. 2b). In 2 and 3, red-shifted ligand–metal charge transfer (LMCT) transitions intersperse with MC transitions; however, the set of three lowest energetic MC transitions is easily identified. Electron density difference maps (EDDM) assigned the orbitals involved in each transition, confirming their 4T2 ← 4A2 character (Fig. S25, S28 and S31†). In all cases, the calculated oscillator strengths are lower than 0.001, expected for parity-forbidden MC electric dipole transitions in chromium(III) complexes. According to the TD-DFT results, the observed band between 300 and 400 nm in 1 is composed of transitions with a strong ligand–metal charge transfer (LMCT) character, while the higher energetic transitions show an intraligand and ligand–ligand metal charge transfer (ILCT and LLCT, respectively) character. In 2 and 3, the LMCT transitions overlap with the higher energetic set of MC transitions. Remarkably, these transitions are mainly LMCT in 2, while in 3 they show a marked LLCT character. This fact could be due to the π-extension on the ligand, which could favour the LLCT between the indazole moieties of 3.Emission properties
At ambient temperature and after light excitation at 480 nm (4T2 ← 4A2 transition), the three complexes exhibit a broad SF emission band with FWHM ranging from 590 cm−1 (for 1 and 2) to 1070 cm−1 (for 3), and maxima located at 741 nm (13495 cm−1), 748 nm (13368 cm−1) and 830 nm (12000 cm−1) for 1, 2 and 3 respectively (Fig. 2b). The excitation spectra recorded upon monitoring the emission band coincide with the absorption spectra, demonstrating the participation of the 4T2 and 4LMCT excited states in the population of the emissive excited states of 1, 2 and 3 (Fig. S11†). Interestingly, the FWHM value is four times larger than those of the archetypal [Cr(ddpd)2]3+ and [Cr(dqp)2]3+ complexes (Fig. S9a†), and comparable to that of the analog [Cr(btmp)2]3+.45 At 77 K, the emission bands are more structured, as expected, but remain broad (dashed line in Fig. 2d and S9b†), which is in stark contrast with the sharper bands found at low temperature in the related [Cr(ddpd)2]3+, [Cr(dqp)2]3+ and [Cr(bpmp)2]3+ complexes (Fig. S9b†).44 The broad emission bands observed at both room temperature and low temperature are consistent with the important vibronic structure as a consequence of misalignments of the ground and excited potential wells (Fig. 4). The origin of this misalignment is in part due to the inhomogeneity of Cr–N bonds due to the presence of two different binding sites (pyrazole/indazole and pyridine), as similarly observed in fac-Cr(ppy)3 (ppy = 2-phenylpyridine) and [Cr(btmp)2]3+.45,58 Indeed, the compression of the octahedral geometry, resulting from the shorter Cr–N(pyrazole) and Cr–N(indazole) bond distances in the equatorial plane of the complexes, leads to a noticeable Jahn–Teller distortion. This compressed distortion causes a splitting of the t2 orbitals, which in turn contributes to the broadening of the emission. The associated increase of covalence of the t2 orbitals as stated before (in the O point group) can also be incriminated in the unusual Stokes shift of the SF transition reaching 340 cm−1 and 410 cm−1 respectively for 1 and 2 and an even larger value of 820 cm−1 for compound 3 (Fig. 4 and S10†). The latter Stokes shift reflected by the low temperature shifting of the emission band maxima, as also observed in [Cr(btmp)2]3+,45 is particularly uncommon for CrIII spin–flip transitions and mirrors misalignment between the potential wells of the excited 2MC and ground 4A2 states (Fig. 2d and 4). In addition, compared to the analogue [Cr(bpmp)2]3+, which features six pyridine ligands in the first coordination sphere around the CrIII with a SF emission at 709 nm, the incorporation of pyrazole and indazole results in a shift of the emission bands to ∼740 nm (for 1 and 2) and 830 nm (for 3). This bathochromic shift aligns with an increase in covalency and might be due to enhanced electronic delocalization of the metal-centered t2 electrons in the five membered aromatic heterocycle pyrazole and indazole in comparison to the six membered pyridine (nephelauxetic effect).
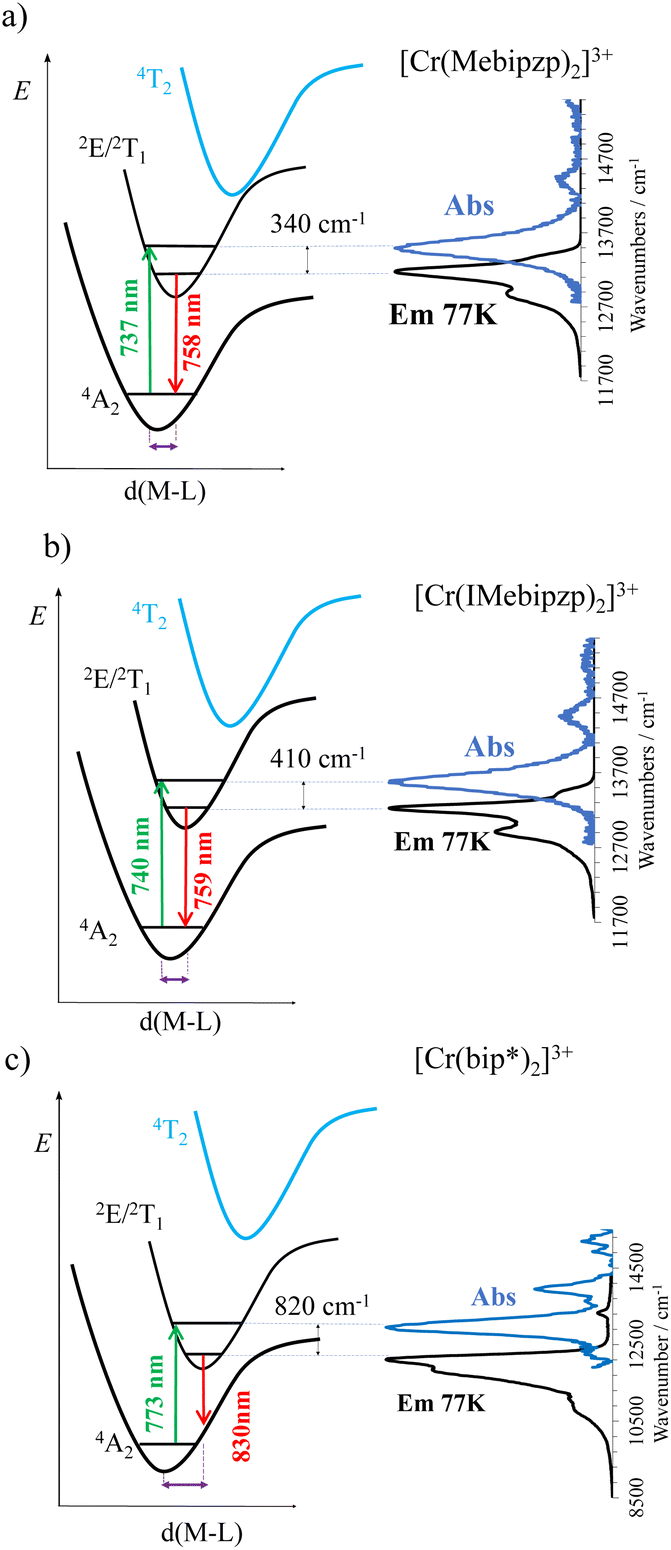 | ||
| Fig. 4 Schematic representation of the SF absorption and emission at 77 K (right-hand side) and potential energy curves for the quartet ground and excited state, 4A and 4T2, and the doublet excited state 2E and 2T1. | ||
The overall luminescence quantum yield (ϕ) at room temperature in non-acidified acetonitrile after excitation at 480 nm reaches (i) 9.2% (under Ar) and 0.7% (under air) for 1, (ii) 9.0% (under Ar) and 1.2% (under air) for 2 and 1.5% (under Ar) and 0.7% (under air) for 3. The obtained values for 1 and 2 represent substantial improvements compared to that of [Cr(bpmp)2]3+ (ϕ = 0.8%) which experiences quenching due to ground state deprotonation of the –CH2– moieties in pure acetonitrile.44 In acidic medium, complexes show a slight increase in luminescence quantum yields, in contrast to [Cr(bpmp)2]3+, where a luminescence quantum yield of 11.4% is measured (Table 1). This highlights the weaker acidic nature of the CH2 bridges upon replacing the lateral pyridine substituents in bpmp, with pyrazole or indazole rings. For 1 and 2, the luminescence quantum yield is one order of magnitude higher than that of the structurally most closely related [Cr(btmp)2]3+ complex (Fig. 1b) in both non-acidic and acidic media (Table 1). The freely rotating peripheral phenyl ring in the latter compound can likely activate additional nonradiative decay channels and could be responsible for its luminescence quenching.59
| Complex | λ abs/E(2MC) (nm cm−1) | λ em/E(2MC) (nm cm−1) | ϕ (%) | τ obs (μs) | τ obs (μs) 77 K | k rad /knrad (s−1) |
|---|---|---|---|---|---|---|
| a Emission quantum yield measured by using an integration sphere upon excitation at 480 nm in deaerated acetonitrile at room temperature. b Emission lifetime measured in deaerated acetonitrile solution at r.t. and upon excitation at 480 nm. c Excited state lifetime obtained by the time-correlated single photon counting (TCSPC) technique. d Excited state lifetime obtained through the transient absorption experiments. e k rad = Φ/τobs assuming ηISC = 1. | ||||||
| 1 – [Cr(Mebipzp)2]3+ | 738/13550 |
741/13500 |
9.2a | 1231b,c/1264d | 1500 | 74/738 |
| Acidic conditions | 11.0a | 1500 | ||||
| 2 – [Cr(IMebipzp)2]3+ | 742/13477 |
746/13404 |
9.0a | 313b,c/380d | 1063 | 287/2900 |
| Acidic conditions | 9.5a | 350 | ||||
| 3 – [Cr(bip*)2]3+ | 773/12940 |
830/12048 |
1.5a | 111b,c/110d | 145 | 135/8873 |
| Acidic conditions | 1.5a | 110 | ||||
| [Cr(bpmp)2]3+ | 706/14164 |
709/14100 |
0.8 | 840 | — | 88/686 |
| Acidic conditions | 11.4 | 1290 | ||||
| [Cr(btmp)2]3+ | — | 760/13157 |
0.27 | 37.9 | 782 | 131/24000 |
| Acidic conditions | 0.54 | 41.5 | ||||
The excited state lifetimes under argon in neat acetonitrile at room temperature are 1231 μs for 1, 313 μs for 2 and 111 μs for 3 (Fig. S12, S15 and S18†), becoming slightly longer in acidic medium (Table 1). Under an ambient (aerated) atmosphere, the excited state lifetimes for the three complexes are considerably shortened (Fig. S13, S16 and S19†), due to the quenching by atmospheric 3O2, which generates the reactive 1O2 species. kq values for energy transfer from doublet excited states to 3O2 of 3.8 × 107 M−1 s−1, 5.6 × 107 M−1 s−1 and 6.4 × 107 M−1 s−1 have been estimated for 1, 2 and 3 respectively (see the ESI† for details). These values are in line with those reported for related complexes.11,38,44,45 The efficient 1O2 generation has been demonstrated to be very useful in catalysis and for photodynamic therapy.25,38,39,60 In a rigid medium at 77 K (frozen acetonitrile solution), the excited-state lifetimes of 1500 μs for compound 1 and 145 μs for compound 3 are approximately 20% longer than the lifetimes observed at room temperature under deaerated conditions (Table 1, Fig. S14 and S20†). For compound 2, the excited state lifetime of 1064 μs at 77 K represents a threefold increase relative to 293 K (Fig. S17†), suggesting a significant and distinct thermally activated non-radiative relaxation mechanism compared to that of the structurally similar compound 1. This may be attributed to notable room temperature back-intersystem crossing, 2MC → 4T2, facilitated by the presence of heavy iodine atoms. This has been already observed in organic dyes and recently reported in chromium(III) complexes.61,62 The similar luminescence quantum yields of the compounds 1 and 2 (∼9.0% in non-acidic media) contrast with the shorter excited-state lifetime measured, in going from 1 to 2, highlighting a concomitant increase of the deactivation rate constants (radiative and non-radiative) for the spin–flip emission in complex 2. The radiative rate for the low energy 2MC excited state, krad, has been estimated assuming that the intersystem crossing between the 4MC and 2MC states is close to unity (i.e. η = 1 in eqn (1)).44,45,63 The computed values of 74 s−1 and 135 s−1 for 1 and 3 are larger than those found in [Cr(tpe)2]3+ (kCr,rad = 23–18 s−1), [Cr(CN)6]3− (kCr,rad = 25 s−1) and [Cr(phen)3]3+ (kCr,rad = 32–43 s−1), and similar to those of the related complexes such as [Cr(dqp)2]3+ (kCr,rad = 29–98 s−1) and [Cr(ddpd)2]3+ (kCr,rad = 135 s−1). For 2, a value of 271 s−1 has been calculated, which is significantly larger than those of the [Cr(dqp)2]3+ and [Cr(ddpd)2]3+ and the closet structurally related [Cr(bpmp)2]3+ (131 s−1) and [Cr(btmp)2]3+ (88 s−1) complexes as well as compounds 1 and 2. This observable increase of the radiative excited-state decay rate constant is consistent with the increased oscillator strength of the spin–flip transition in 2 in solution and solid state (Fig. 3 and S8†). The non-radiative de-excitation pathways of 2MC are independently evaluated by the non-radiative rate constants (knonrad) amounting in deaerated solutions to 738(50) s−1 and 2900(250) s−1 for 1 and 2 respectively.
Thus, the positive effect of heavy atoms on the radiative excited-state decay rate constant is balanced by a concomitant increase of the non-radiative de-excitation pathways, such as the thermally assisted back intersystem crossing. Moreover, the enhanced spin–orbit coupling will also favour vibrationally-induced nonradiative decay between strongly coupled potential energy surfaces, in other words, between the excited state and ground state.64
Nanosecond UV-vis transient absorption (TA) spectroscopy was further used to probe the photophysical properties of the doublet spin–flip excited states. Spectra for 1 and 2 were recorded in acetonitrile at room temperature following excitation at 355 nm (30 mJ pulses, ca. 10 ns duration) and for 3 at 450 nm (13 mJ per pulse). The TA spectrum of complex 1 features broad excited state absorption (ESA) bands with maxima at 410 nm and 515 nm and a ground state bleach (GSB) of the charge transfer band at 300 nm. Similarly, for complex 2, excited state absorption bands at 450 nm and 650 nm, as well as a ground state bleach at 350 nm, are observed. In compound 3, the ESA bands are at 510 and 660 nm, and the ground state bleach at 380 nm. In all cases, the ESA bands are tentatively attributed to electronic transitions from the 2E/2T1 states to higher-lying doublet excited states, most likely of charge transfer character. ESA lifetimes are consistent with the observed luminescence lifetimes (Table 1 and Fig. 5). Complex 2, with iodine-substituents, exhibits a three-fold decrease in the lifetime relative to 1, consistent with the luminescence lifetime experiments performed under identical conditions. Collectively, the photoluminescence decay and transient UV-vis absorption data support the hypothesis of a heavy atom effect, which accelerates radiative 2MC excited state decay.
 | ||
| Fig. 5 (a) and (b) Transient absorption spectra of compounds 1, 2 and 3 upon excitation at 355 nm (30 mJ per pulses, ca. 10 ns duration) for 1 and 2, and at 450 nm (13 mJ per pulse) for 3 with nanosecond pulses in deaerated acetonitrile solution at room temperature, time-integrated over 1 μs. (c) 410 nm, (d) 450 nm and (e) 520 nm decays of the normalized ESA bands for the three compounds. | ||
Conclusions
In conclusion, the straightforward synthesis of the [Cr(Mebipzp)2]3+ (1), [Cr(IMebipzp)2]3+ (2), and [Cr(bip*)2]3+ (3) complexes provides a simple route to substitute widely used saturated pyridine ligands with more versatile pyrazole and indazole heterocycles. The distinct bonding interactions introduced by the pyrazole/indazole and pyridine moieties induce significant energetic splitting of the lowest-lying doublet excited states, leading to a more pronounced distortion of the 2MC states relative to the electronic ground state. This results in broader emission profiles and unusually large Stokes shifts for the spin–flip transitions. Despite the broader emission spectrum, the luminescence quantum yields of compounds 1 and 2 (9–11% in deaerated solution) are among the largest reported for CrIII molecular rubies. Notably, the incorporation of iodine substituents in complex 2 improves the spin–orbit coupling via the heavy atom effect. This significantly increases (i) the oscillator strength (f), (ii) both the radiative and non-radiative rate constants for the 2MC ↔ 4A2 transition and (iii) facilitates the room temperature back-intersystem crossing. Finally, the substitution of distal methyl-pyrazole rings with indazole in compound 3 induces a significant bathochromic shift in the spin–flip emission to 830 nm (12000 cm−1), highlighting the impact of the extended π-system and increased covalent bond character of the indazole ligand. These findings reveal an underexplored approach for tuning deactivation pathways and emission energies while maintaining high quantum yields and long-lived excited states, offering exciting opportunities for the development of advanced molecular rubies with tailored photophysical properties.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: J. R. J.; data curation: Y. Y., M. P., P. Y., P. S. A., A. R. D., J. H. M., L. C. C., J. R. J.; formal analysis: C. P., O. W., B. D., J. M. H., J. R. J.; resources: M. P., C. P., J. R. J.; theoretical calculations: C. M. C.; funding acquisition: J. M. H., C. P., J. R. J.; investigation: Y. Y., P. Y., L. C. C., J. H. M.; methodology: Y. Y., J. R. J.; writing – original draft: J. R. J.; writing – review & editing: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the funding received from the grant TED2021.129598A.I00 funded by MCIN/AEI/10.13039/501100011033 and by European Union NextGenerationEU/PRTR and Junta de Andalucía. J. R. J. thanks Ministerio de Ciencia Innovación y Universidades for a Ramón y Cajal contract (grant RYC2022-037255-I) funded by MCIN/AEI/10.13039/501100011033 and FSE+. Financial support was received from the Ministerio de Ciencia e Innovación, Grant PID2022-138090NB-C21 funded by MICIU/AEI/10.13039/501100011033 and by ERDF/EU. This work was also financially supported by Junta de Andalucía (FQM-195, projects I + D + i A-FQM-172-UGR18 and B.FQM.328.UGR20) and the University of Granada. P. S.-A. acknowledges Juan de la Cierva Grant JDC2022-048964-I funded by MICIU/AEI/10.13039/501100011033 and by “European Union NextGenerationEU/PRTR”. We thank Centro de Instrumentación científica (CIC) and the Centro de Servicios de Informática y Redes de Comunicaciones (CSIRC), Universidad de Granada, for providing the technical facilities and the computing time. This work made use of the infrastructure services provided by S3IT (https://www.s3it.uzh.ch), the Service and Support for Science IT team at the University of Zurich. The authors would like to thank the S3IT team for their support. M. P., B. D., J. R. J. and C. P. were supported through grants from the Swiss National Science Foundation (grant 200020_207313).References
- C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057–1070 RSC.
- O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS PubMed.
- C. Wegeberg and O. S. Wenger, JACS Au, 2021, 1, 1860–1876 CrossRef CAS PubMed.
- S. Kaufhold, N. W. Rosemann, P. Chábera, L. Lindh, I. B. Losada, J. Uhlig, T. Pascher, D. Strand, K. Wärnmark, A. Yartsev and P. Persson, J. Am. Chem. Soc., 2021, 143, 1307–1312 CrossRef CAS PubMed.
- A. K. Pal, C. Li, G. S. Hanan and E. Zysman-Colman, Angew. Chem., 2018, 130, 8159–8163 CrossRef.
- M. Dorn, J. Kalmbach, P. Boden, A. Päpcke, S. Gómez, C. Förster, F. Kuczelinis, L. M. Carrella, L. A. Büldt, N. H. Bings, E. Rentschler, S. Lochbrunner, L. González, M. Gerhards, M. Seitz and K. Heinze, J. Am. Chem. Soc., 2020, 142, 7947–7955 CrossRef CAS PubMed.
- P. Herr, C. Kerzig, C. B. Larsen, D. Häussinger and O. S. Wenger, Nat. Chem., 2021, 13, 956–962 CrossRef CAS PubMed.
- L. A. Büldt, X. Guo, R. Vogel, A. Prescimone and O. S. Wenger, J. Am. Chem. Soc., 2017, 139, 985–992 CrossRef PubMed.
- B. C. Paulus, K. C. Nielsen, C. R. Tichnell, M. C. Carey and J. K. McCusker, J. Am. Chem. Soc., 2021, 143, 8086–8098 CrossRef CAS PubMed.
- J. R. Jiménez, B. Doistau, C. M. Cruz, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, J. Am. Chem. Soc., 2019, 141, 13244–13252 CrossRef PubMed.
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, Angew. Chem., Int. Ed., 2015, 54, 11572–11576 CrossRef CAS PubMed.
- J. Steube, A. Kruse, O. S. Bokareva, T. Reuter, S. Demeshko, R. Schoch, M. A. A. Cordero, A. Krishna, S. Hohloch, F. Meyer, K. Heinze, O. Kühn, S. Lochbrunner and M. Bauer, Nat. Chem., 2023, 15, 468–474 CrossRef CAS PubMed.
- K. S. Kjær, N. Kaul, O. Prakash, P. Chábera, N. W. Rosemann, A. Honarfar, O. Gordivska, L. A. Fredin, K. E. Bergquist, L. Häggström, T. Ericsson, L. Lindh, A. Yartsev, S. Styring, P. Huang, J. Uhlig, J. Bendix, D. Strand, V. Sundström, P. Persson, R. Lomoth and K. Wärnmark, Science, 2019, 363, 249–253 CrossRef PubMed.
- P. Chábera, Y. Liu, O. Prakash, E. Thyrhaug, A. El Nahhas, A. Honarfar, S. Essén, L. A. Fredin, T. C. B. Harlang, K. S. Kjær, K. Handrup, F. Ericson, H. Tatsuno, K. Morgan, J. Schnadt, L. Häggström, T. Ericsson, A. Sobkowiak, S. Lidin, P. Huang, S. Styring, J. Uhlig, J. Bendix, R. Lomoth, V. Sundström, P. Persson and K. Wärnmark, Nature, 2017, 543, 695–699 CrossRef PubMed.
- Y. Ye, P. Garrido-Barros, J. Wellauer, C. M. Cruz, R. Lescouëzec, O. S. Wenger, J. M. Herrera and J.-R. Jiménez, J. Am. Chem. Soc., 2024, 146, 954–960 CrossRef CAS PubMed.
- N. Sinha, C. Wegeberg, D. Häussinger, A. Prescimone and O. S. Wenger, Nat. Chem., 2023, 15, 1730–1736 CrossRef CAS PubMed.
- N. Sinha and O. S. Wenger, J. Am. Chem. Soc., 2023, 145, 4903–4920 CrossRef CAS PubMed.
- J. R. Jiménez, B. Doistau, M. Poncet and C. Piguet, Coord. Chem. Rev., 2021, 434, 213750 CrossRef.
- E. C. Constable, C. E. Housecroft, M. Neuburger, J. Schönle and J. A. Zampese, Dalton Trans., 2014, 43, 7227–7235 RSC.
- T. H. Maiman, Nature, 1960, 187, 493–494 CrossRef.
- W. R. Kitzmann, J. Moll and K. Heinze, Photochem. Photobiol. Sci., 2022, 21, 1309–1331 CrossRef CAS PubMed.
- B. Doistau, J. R. Jiménez, S. Guerra, C. Besnard and C. Piguet, Inorg. Chem., 2020, 59, 1424–1435 CrossRef CAS PubMed.
- J. Chong, C. Besnard, C. M. Cruz, C. Piguet and J. R. Jiménez, Dalton Trans., 2022, 51, 4297–4309 RSC.
- J.-R. Jiménez, M. Poncet, S. Miguez-Lago, S. Grass, J. Lacour, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, Angew. Chem., Int. Ed., 2021, 60, 10095–10102 CrossRef PubMed.
- S. Otto, M. Dorn, C. Förster, M. Bauer, M. Seitz and K. Heinze, Coord. Chem. Rev., 2018, 359, 102–111 CrossRef CAS.
- C. Wang, S. Otto, M. Dorn, E. Kreidt, J. Lebon, L. Sršan, P. Di Martino-Fumo, M. Gerhards, U. Resch-Genger, M. Seitz and K. Heinze, Angew. Chem., Int. Ed., 2018, 57, 1112–1116 CrossRef CAS PubMed.
- L. Aboshyan-Sorgho, C. Besnard, P. Pattison, K. R. Kittilstved, A. Aebischer, J. C. G. Bünzli, A. Hauser and C. Piguet, Angew. Chem., Int. Ed., 2011, 50, 4108–4112 CrossRef CAS PubMed.
- Y. Suffren, D. Zare, S. V. Eliseeva, L. Guénée, H. Nozary, T. Lathion, L. Aboshyan-Sorgho, S. Petoud, A. Hauser and C. Piguet, J. Phys. Chem. C, 2013, 117, 26957–26963 CrossRef CAS.
- B. Golesorkhi, H. Bolvin and C. Piguet, Dalton Trans., 2021, 50, 7955–7968 RSC.
- C. Dee, F. Zinna, W. R. Kitzmann, G. Pescitelli, K. Heinze, L. Di Bari and M. Seitz, Chem. Commun., 2019, 55, 13078–13081 RSC.
- J. Kalmbach, C. Wang, Y. You, C. Förster, H. Schubert, K. Heinze, U. Resch-Genger and M. Seitz, Angew. Chem., Int. Ed., 2020, 59, 18804–18808 CrossRef CAS PubMed.
- M. Poncet, C. Besnard, J. R. Jiménez and C. Piguet, Inorg. Chem., 2024, 63, 18345–18354 CrossRef CAS PubMed.
- S. Otto, N. Scholz, T. Behnke, U. Resch-Genger and K. Heinze, Chem.–Eur. J., 2017, 23, 12131–12135 CrossRef CAS PubMed.
- B. Doistau, J. R. Jiménez and C. Piguet, Front. Chem., 2020, 8, 1–27 CrossRef PubMed.
- J. R. Jiménez, S. Míguez-Lago, M. Poncet, Y. Ye, C. Lopez, C. M. Cruz, A. G. Campaña, E. Colacio, C. Piguet and J. M. Herrera, J. Mater. Chem. C, 2023, 2582–2590 RSC.
- Y. Cheng, J. He, W. Zou, X. Chang, Q. Yang and W. Lu, Chem. Commun., 2023, 59, 1781–1784 RSC.
- A. Gabbani, M. Poncet, G. Pescitelli, L. Carbonaro, J. Krzystek, E. Colacio, C. Piguet, F. Pineider, L. Di Bari, J.-R. Jiménez and F. Zinna, Chem. Sci., 2024, 15, 17217–17223 RSC.
- T. H. Bürgin, F. Glaser and O. S. Wenger, J. Am. Chem. Soc., 2022, 144, 14181–14194 CrossRef PubMed.
- S. Sittel, R. Naumann and K. Heinze, Front. Chem., 2022, 10, 340 Search PubMed.
- N. Sinha, J. R. Jiménez, B. Pfund, A. Prescimone, C. Piguet and O. S. Wenger, Angew. Chem., Int. Ed., 2021, 60, 23722–23728 CrossRef CAS PubMed.
- N. Sawicka, C. J. Craze, P. N. Horton, S. J. Coles, E. Richards and S. J. A. Pope, Chem. Commun., 2022, 58, 5733 RSC.
- Y. Cheng, Q. Yang, J. He, W. Zou, K. Liao, X. Chang, C. Zou and W. Lu, Dalton Trans., 2023, 52, 2561–2565 RSC.
- N. Sinha, P. Yaltseva and O. S. Wenger, Angew. Chem., Int. Ed., 2023, 202303864 Search PubMed.
- F. Reichenauer, C. Wang, F. Christoph, P. Boden, N. Ugur, B. Ricardo, J. Kalmbach, L. M. Carrella, E. Rentschler, C. Ramanan, G. Niedner-Schatteburg, M. Gerhards, M. Seitz, U. Resch-Genger and K. Heinze, J. Am. Chem. Soc., 2021, 143, 11843–11855 CrossRef CAS PubMed.
- R. W. Jones, A. J. Auty, G. Wu, P. Persson, M. V. Appleby, D. Chekulaev, C. R. Rice, J. A. Weinstein, P. I. P. Elliott and P. A. Scattergood, J. Am. Chem. Soc., 2023, 145, 12081–12092 CrossRef CAS PubMed.
- E. A. Juban and J. K. McCusker, J. Am. Chem. Soc., 2005, 127, 6857–6865 CrossRef CAS PubMed.
- J. N. Schrauben, K. L. Dillman, W. F. Beck and J. K. Mc Cusker, Chem. Sci., 2010, 1, 405–410 RSC.
- M. Cantuel, G. Bernardinelli, D. Imbert, J. C. G. Bünzli, G. Hopfgartnerd and C. Piguet, J. Chem. Soc., Dalton Trans., 2002, 1929–1940 RSC.
- M. Poncet, A. Benchohra, J. R. Jiménez and C. Piguet, ChemPhotoChem, 2021, 5, 880–892 CrossRef CAS.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE, version 2.1 Search PubMed.
- B. Golesorkhi, I. Taarit, H. Bolvin, H. Nozary, J. R. Jiménez, C. Besnard, L. Guénée, A. Fürstenberg and C. Piguet, Dalton Trans., 2021, 50, 7955–7968 RSC.
- S. Treiling, C. Wang, C. Förster, F. Reichenauer, J. Kalmbach, P. Boden, J. P. Harris, L. M. Carrella, E. Rentschler, U. Resch-Genger, C. Reber, M. Seitz, M. Gerhards and K. Heinze, Angew. Chem., Int. Ed., 2019, 58, 18075–18085 CrossRef CAS PubMed.
- K. DeArmond and L. S. Forster, Spectrochim. Acta, 1963, 19, 1687–1693 CrossRef CAS.
- W. T. Carnall, P. R. Fields and B. G. Wybourne, J. Chem. Phys., 1965, 42, 3797–3806 CrossRef CAS.
- I. Taarit, F. Alves, A. Benchohra, L. Guénée, B. Golesorkhi, A. Rosspeintner, A. Fürstenberg and C. Piguet, J. Am. Chem. Soc., 2023, 145, 8621–8633 CrossRef CAS PubMed.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS PubMed.
- F. Glaser and O. S. Wenger, JACS Au, 2022, 2, 1488–1503 CrossRef CAS PubMed.
- L. Stein, P. Boden, R. Naumann, C. Förster, G. Niedner-Schatteburg and K. Heinze, Chem. Commun., 2022, 58, 3701–3704 RSC.
- Z. Lou, Y. Hou, K. Chen, J. Zhao, S. Ji, F. Zhong, Y. Dede and B. Dick, J. Phys. Chem. C, 2018, 122, 185–193 CrossRef CAS.
- L. Gourdon, K. Cariou and G. Gasser, Chem. Soc. Rev., 2022, 51, 1167–1195 RSC.
- B. Pfund, V. Hutskalova, C. Sparr and O. S. Wenger, Chem. Sci., 2023, 14, 11180–11191 RSC.
- F. Reichenauer, R. Naumann, C. Förster, W. R. Kitzmann, A.-P. M. Reponen, S. Feldmann and K. Heinze, Chem. Sci., 2024, 15, 20251–20262 RSC.
- J. P. Zobel, H. Radatz and L. González, Molecules, 2023, 28, 1668 CrossRef CAS PubMed.
- C.-H. Ting, Photochem. Photobiol., 1969, 9, 17–31 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2266702, 2266703 and 2389781. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08021a |
| This journal is © The Royal Society of Chemistry 2025 |