DOI:
10.1039/D4SC07803A
(Edge Article)
Chem. Sci., 2025,
16, 4342-4351
Regioselective late-stage functionalization of tetraphenylenes: rapid construction of double helical architectures and potential hole transport materials†‡
Received
18th November 2024
, Accepted 24th January 2025
First published on 25th January 2025
Abstract
Herein we report a novel approach for diversification of tetraphenylene via regioselective late-stage iodination followed by atom(s) insertion into the resulting cyclic iodonium salts. Thus, the steric hindrance effect of tert-butyl facilitates the regioselective synthesis of two cyclic iodonium salts of 2,7,10,15-tetra-tert-butyltetraphenylene. In addition, two more cyclic iodonium salts of 2,7,10,15-tetranitrotetraphenylene were also readily synthesized due to the meta-position induced effect of the electron-withdrawing NO2 group. Subsequent functionalization of these tetraphenylene-based cyclic iodonium salts via diverse atom(s) insertion processes led to several tetraphenylene-based [8 + n] and [n + 8 + n] fused rings including fascinating double helical architectures. This newly developed late-stage functionalization methodology was also successfully applied to rapid synthesis of potential hole transport materials, thereby demonstrating its robust synthetic value in both tetraphenylene chemistry and materials science.
Introduction
Tetraphenylene (1) is a nonplanar molecule featuring two opposite pairs of benzene rings that are oriented alternatively above and below the plane of the central cyclooctatetraene, featuring a distinct saddle-shaped structure with two types of C–C bonds in different lengths (Fig. 1).1,2 It is noteworthy that appropriate substitutions on the benzene rings of 1 impair the symmetry of 1, providing versatile chiral tetraphenylene derivatives.1d,3 With the unique three-dimensional geometry and chiral properties, studies of tetraphenylenes have led to extensive applications in a variety of research fields, such as supramolecular chemistry,4 asymmetric catalysis,5 materials science,6 covalent-organic frameworks,7etc.
 |
| | Fig. 1 Structure of tetraphenylene (1). | |
Since the first synthesis of 1 was reported in 1943,2a numerous approaches towards the bottom-up construction of the tetraphenylene core have been reported.1g–i Generally, there are mainly five methods, which are summarized in Scheme 1A: (1) Diels–Alder cycloaddition between 1,2,5,6-dibenzocycloocta-3,7-diynes and furans followed by deoxygenation,8 (2) metal-mediated pyrolysis of biphenylenes followed by annulative dimerization,9 (3) rhodium-catalyzed double [2 + 2 + 2] cycloaddition of triynes,10 (4) metal-catalyzed or metal-mediated (cross)coupling of 2,2′-disubstituted biphenyls or 2-halobiphenyls,3,4d,5b,11 and (5) cyclization of o-quateraryls by the Scholl reaction.12 Indeed, over several decades, these methods have their merits and were employed efficiently in the synthesis of various tetraphenylenes. However, the limitations and disadvantages of these methods are also obvious because the starting materials rely on pre-functionalized building blocks and are usually accompanied by synthetic difficulties. Therefore, an efficient synthesis of highly functionalized tetraphenylenes has always been a synthetic challenge.
 |
| | Scheme 1 (A) Typical methods for bottom-up construction of tetraphenylenes; (B) examples of previous work on late-stage functionalization of tetraphenylenes; (C) representative examples for preparation of cyclic iodonium salts; (D) proposed tetraphenylenes for regioselective synthesis of cyclic iodonium salts; (E) late-stage functionalization of tetraphenylenes via regioselective iodination followed by atom(s) insertion, and application in synthesis of hole transport materials. | |
Late-stage functionalization has emerged as an appealing and versatile strategy for expanding the diversity of complex scaffolds.13 Apart from the aforementioned bottom-up methods for the construction of the cyclooctatetraene core, direct late-stage functionalization of tetraphenylenes is a concise and valuable strategy to address the diversity of tetraphenylenes, particularly for unsymmetrically substituted tetraphenylenes.14 As shown in Scheme 1B, in 2016, Zhang and co-workers reported the late-stage transition-metal-catalyzed halogenation, acetoxylation and carbonylation of 1, providing a variety of 2-substituted tetraphenylenes.14h Later, they further developed carbonyl directed ruthenium-catalyzed ortho C–H functionalization of 2-acetyltetraphenylene, leading to a variety of 2,3-disubstituted tetraphenylene derivatives.14i Recently, our group has devised a reliable method for the realization of a library of 2,15-disubstituted 1,16-dihydroxytetraphenylenes, whose pathway involves a late-stage lithiation/boronation process to afford a key 2,15-bis-borylated intermediate, allowing for subsequent Suzuki coupling reactions.5f Although these late-stage functionalization methodologies have provided excellent results for the synthesis of various tetraphenylenes, there are still significant limitations such as (1) electrophilic aromatic substitutions are only applicable to the synthesis of monosubstituted tetraphenylenes, (2) the access to multi-substituted tetraphenylenes relies on the use of ortho-directing groups, (3) it is usually hard to precisely predict the substituted positions of tetraphenylenes due to the random reaction sites of four benzene rings, and so on. Thus, to date, reliable and practical strategies on late-stage functionalization of tetraphenylenes to afford their respective highly functionalized derivatives remain sparse. It is still desirable to develop new late-stage functionalization strategies for the precise and divergent synthesis of polyfunctional tetraphenylenes.
Over the past decade, the applications of cyclic diaryliodonium salts in the construction of complex molecules as an effective late-stage functionalization methodology have witnessed significant progress; therefore the preparation of cyclic diaryliodonium salts has been well established.15 One of the attractive and efficient methods for the synthesis of cyclic diaryliodonium salts is that 4,4′-disubstituted diaryls were directly used to react with iodosyl sulfate (Scheme 1C).16 Inspired by the synthetic simplicity of cyclic diaryliodoniums and their abundant and various transformations,15a we opine that meticulous manipulation of tetraphenylenes bearing suitable substituents (such as sterically bulky tBu groups and electron-withdrawing NO2 groups) could regioselectively afford similar cyclic iodonium salts for further functionalization, leading to structurally diverse tetraphenylene derivatives (Scheme 1D). Herein we report a novel approach for diversification of 1via regioselective late-stage iodination followed by insertion of atoms into the resulting cyclic iodonium salts. These approaches afforded various structurally well-defined tetraphenylene-based [8 + n] and [n + 8 + n] fused-ring compounds with aesthetic architectures such as fascinating double helical frameworks. Furthermore, this methodology was also successfully applied to a rapid synthesis of several potential hole transport materials (Scheme 1E).
Results and discussion
Synthesis of cyclic iodonium salts of tetraphenylenes
To date, a number of methods for preparing cyclic aryliodoniums have been established.15a For example, early in the 1950s, Hwang and co-workers built up a very simple and efficient method to prepare six-membered cyclic diaryliodonium salts via the reaction of 4,4′-disubstituted diaryls and iodosyl sulfate in concentrated sulfuric acid.16a,b Later, five-membered cyclic diaryliodonium salts of 4,4-dinitrobiphenyl were readily achieved under the same reaction conditions.16c On the other hand, a literature report revealed that the sterically bulky tBu group on the benzene ring could hinder iodination of the ortho-positions.17 Thus, these elegant synthetic studies featuring operational simplicity have inspired us to attempt to realize similar tetraphenylene-based cyclic iodonium salts.
As shown in Scheme 2, initially we adopted a cobalt-catalyzed homo-coupling reaction of in situ generated magnesium bromide of 1-bromo-3-tert-butylbenzene (2) to provide biaryl 3 with good yield (86%).18 Subsequently, biaryl 3 was treated with I2, m-CPBA and TfOH in CH2Cl2 at 0 °C, generating the cyclic iodonium salt 4 in 72% yield under mild conditions.19 Then, a palladium-catalyzed annulative dimerization of 4 in the presence of NaHCO3 offered 2,7,10,15-tetra-tert-butyltetraphenylene (5) in 36% yield.11e,20 Finally, treatment of 5 with I2, m-CPBA and TfOH successfully furnished cyclic iodonium salt A in 53% yield.19 Under similar conditions, bis-cyclic iodonium salt A′ was also obtained in a yield of 32% by increasing the amounts of I2, m-CPBA and TfOH.
 |
| | Scheme 2 Synthetic route of tetraphenylene 5 and its cyclic iodonium salts A and A′. | |
Likewise, 2,7,10,15-tetranitrotetraphenylene-based cyclic iodonium salts B and B′ were synthesized as described in Scheme 3. Initially, 1-iodo-4-nitrobenzene (6) went through a Cu-mediated Ullmann coupling to generate 4,4′-dinitrobiphenyl (7) with a yield of 80%. Subsequent treatment of 7 with NaIO3 in concentrated sulfuric acid at 110 °C directly provided cyclic iodonium salt 8 in 81% yield.21 However, a straightforward palladium-catalyzed annulative dimerization of 8 did not give expected 2,7,10,15-tetranitrotetraphenylene (9).11e,20 Alternatively, reduction of 8 with piperidine successfully delivered 2-iodo-4,4′-dinitrobiphenyl (10) with 87% yield.22 Then, biphenyl 10 underwent a palladium-catalyzed annulative dimerization in the presence of NaHCO3 at 130 °C for 48 h to furnish 9 with a yield of 26%.11e Of note, a microwave-assisted reaction under the same conditions could significantly shorten the reaction time from 48 h to 3.5 h, but did not improve the reaction yield (28%). Finally, tetranitrotetraphenylene 9 was subjected to 1.1 equivalents of NaIO3 in concentrated sulfuric acid at 110 °C, successfully giving the cyclic iodonium salt B in 62% yield.21 The bis-cyclic iodonium salt B′ was successfully obtained in 60% yield by using 3.0 equivalents of NaIO3 under the same conditions.
 |
| | Scheme 3 Synthetic route of tetraphenylene 9 and its cyclic iodonium salts B and B′. | |
Functionalization of tetraphenylenes enabled by atom(s) insertion into cyclic iodonium salts
With cyclic iodonium salt A in hand, we tried a series of reactions to form a variety of [8 + n] fused rings. Thus, cyclic iodonium salt A was first employed for the synthesis of a family of [8 + 5] fused rings by insertion of various atoms including nitrogen, oxygen, sulfur, and selenium (Scheme 4). 4-Methoxyaniline (11) readily reacted with A under the catalyzed system of Cu(OAc)2 with Na2CO3 refluxing in ethylene glycol/iPrOH, providing a nitrogen insertion compound 12 with a yield of 52%.23 Then, a copper-catalyzed oxygen insertion of A was also realized, yielding an oxygen-bridged tetraphenylene 13 (50%).24 Notably, H2O was vital, acting as the oxygen source in this reaction. Treatment of A with Na2S·9H2O using FeCl3 as a catalyst in DMSO at 100 °C generated a sulfur-containing compound 14 in 64% yield.25 Additionally, the transition-metal-free selenium insertion into A in DMSO with KOtBu at 100 °C delivered 15 with an acceptable yield of 33%.26
 |
| | Scheme 4 Syntheses of [8 + 5] fused rings. | |
After a family of [8 + 5] fused rings had been synthesized, the synthesis of [8 + 6] fused rings was also carried out (Scheme 5). The double Suzuki reaction between cyclic iodonium salt A and 1,2-diborylated 16 (ref. 27) at a lower temperature (40 °C) efficiently offered a [8 + 6] fused ring 17 in 68% yield.11c In addition, the [4 + 2] benzannulation between A and 1,2-diphenylethyne (18), utilizing a palladium-catalyzed protocol, delivered a [8 + 6] fused ring 19 in an acceptable yield of 54%.28
 |
| | Scheme 5 Syntheses of [8 + 6] fused rings. | |
To expand the reaction scope, it was worth noting that the synthesis of [8 + 7], [8 + 8], and [8 + 9] fused rings by employing palladium-catalyzed coupling annulative reactions of cyclic iodonium salt A and various coupling blocks was also investigated (Scheme 6). The palladium-catalyzed double Suzuki coupling reaction between cyclic iodonium salt A and naphthalene-1,8-diboronic acid anhydride (20) led to the formation of 21 in 58% yield.11c Treatment of A with 1,1′-biphenyl-2,2′-diboronic acid anhydride (22) under the same Suzuki coupling conditions delivered the desired [8 + 8] fused ring 23 in a yield of 39%. Notably, dibenzoxaborininol 24, which was obtained from diaryl ether following Fu's procedure,29 was allowed to react with A to give a [8 + 9] fused ring 25 in 41% yield.
 |
| | Scheme 6 Syntheses of [8 + 7], [8 + 8], and [8 + 9] fused rings. | |
Moreover, palladium-catalyzed Suzuki coupling reactions of bis-cyclic iodonium salt A′ and various coupling partners for the construction of [n + 8 + n] fused rings were also investigated (Scheme 7). First, a [7 + 8 + 7] fused ring 26 was obtained in 40% yield by a palladium-catalyzed coupling reaction between bis-cyclic iodonium salt A′ and naphthalene-1,8-diboronic acid anhydride (20).11c Then, the palladium-catalyzed coupling reaction between A′ and diboronic acid anhydride 22 afforded an octaphenylene 27 in 35% yield. It is interesting to note that a [9 + 8 + 9] fused ring 28 was easily obtained in an acceptable yield (44%) by a twofold atom insertion process. The fascinating double helical structure 28 (CCDC 2381518) was revealed by an X-ray diffraction analysis (Fig. 2).30 Unluckily, the palladium-catalyzed coupling reaction between A′ and naphthalene carboxamide 29 could not deliver the expected diimide-containing 30.31
 |
| | Scheme 7 Syntheses of [7 + 8 + 7], [8 + 8 + 8], and [9 + 8 + 9] fused rings. | |
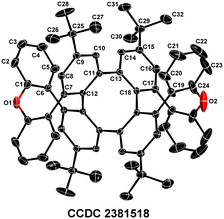 |
| | Fig. 2 X-ray diffraction analysis of 28. Displacement ellipsoids are drawn at the 50% probability level. H atoms are omitted for clarity. | |
Oligotetraphenylenes are a class of interesting molecules, featuring an artificial double helical architecture with fascinating molecular configurations and extensive application prospects.6j–l,32 As shown in Scheme 8, Rajca and coworkers first reported a convergent synthetic route to octaphenylene 33 based upon stepwise connection and annelation of two molecules of 1,16-dibromotetraphenylene 31 bearing tert-butyl substituents. In their work, double lithium–bromine exchange of 31 followed by the oxidative coupling of the resulting dilithiotetraphenylene 32 with CuBr2 gave the target 33 in a mere 4% yield, suffering from tedious and time-consuming purification steps.32a Notably, our new method shows robust synthetic advantage on the construction of double helical structures. In comparison, the microwave-assisted palladium-catalyzed annulative dimerization of A streamlines the synthetic steps of 33 and eventually delivered the double helical octaphenylene 33 in a yield of 21% with operational simplicity.
 |
| | Scheme 8 Application in synthesis of double helical structure 33. | |
Application in rapid synthesis of potential hole transport materials
Over the last two decades, there has been considerable progress in the development of perovskite solar cells (PSCs).33 Hole transport materials (HTMs) are a key component of PSCs due to their vital role in adjusting the charge dynamics.34 Generally, an ideal HTM candidate for PSCs should possess the following features: (1) appropriately aligned energy levels with perovskites: the highest occupied molecular orbital (HOMO) level must be less negative than the valence band of perovskites for efficient hole capture, while the high-lying lowest unoccupied molecular orbital (LUMO) level ensures low electron affinity to block electron recombination at interfaces, (2) high hole mobility and conductivity: HTMs should have excellent transporting ability of free charge carriers for reducing charge losses, (3) good solubility: HTMs should be well dissolved in nonpolar and aprotic solvents for facilitating hole transport layer formation, and (4) high thermal and photochemical stability and high hydrophobicity: the stability of HTMs and their protection of perovskites from water are an efficient strategy for retarding the degradation of PSCs in a long-term durable photovoltaic operation; a high glass transition temperature of the HTM enables structural changes induced by continuous sunlight exposure to be avoided. In addition, development of HTMs requires a simple synthetic process in order to reduce the cost.
It is well known that spiro-OMeTAD is the most widely used HTM in PSCs due to its rigid spiro structure, which suppresses molecular aggregation, thus resulting in amorphous film-forming performance.35 Similarly, the tetraphenylene core offers a nonpolar and rigid molecular backbone to avoid π–π stacking and forms a thermally stable amorphous phase. Therefore, the tetraphenylene framework in replacement of the spiro core would lead to a new family of HTMs. In this regard, Wang and co-workers developed two tetraphenylene-based HTMs (OTP-OMeDPA and OTPE-OMeDPA) with good power conversion efficiency (PCE) for PSCs.6g As shown in Fig. 3, by employing OTP-OMeDPA and OTPE-OMeDPA respectively with the same dopant in the hole transporting layer, PSC devices were fabricated with an excellent PCE of 20.6% and 21.5%, respectively, in comparison with that of 19.4% for spiro-OMeTAD. Notably, OTPE-OMeTPA presents an improved PCE in comparison with its counterpart OTP-OMeDPA probably due to its extended π-conjugation. These results opened a promising avenue for the modification of tetraphenylenes for the development of higher performance HTMs. Inspired by our methodology enabling rapid diversification of tetraphenylenes, we further investigated the modification of OTP-OMeDPA with the aim of improving the performance of potential HTMs.
 |
| | Fig. 3 Structure of representative hole transport materials. | |
Thus, a series of OTP-OMeDPA-based molecules including 2Me-OTP-OMeDPA, S-OTP-OMeDPA, and 2S-OTP-OMeDPA were synthesized via π-conjugation modification of the central eight-membered ring (Scheme 9).14e A palladium-catalyzed double Stille coupling of cyclic iodonium salt B and SnMe4 led to the formation of dimethyltetranitrotetraphenylene (34),36 which was allowed to undergo a FeCl3-catalyzed reduction by hydrazine hydrate11h to give tetraaminotetraphenylene 35 followed by Buchwald–Hartwig amination37 with bromide 36 to furnish 2Me-OTP-OMeDPA (Scheme 9a). Then, the sulfur atom was chosen as the bridging unit of the tetraphenylene core. Consequently, the reaction involving an Fe(III)-catalyzed sulfur insertion into B afforded a sulfur-bridged product 38 in a yield of 47%.25 Subsequently, 38 was allowed to undergo similar reduction and amination procedures to furnish the desired S-OTP-OMeDPA (Scheme 9b). In addition, by treatment of bis-cyclic iodonium salt B′ with Na2S·9H2O catalyzed by FeCl3 in DMSO at 180 °C for 8 h, the disulfur-bridged compound 41 was also obtained in 61% yield. Likewise, subsequent FeCl3-catalyzed reduction of 41 and Buchwald–Hartwig amination of 42 worked smoothly to deliver 2S-OTP-OMeDPA in an acceptable yield (Scheme 9c).
 |
| | Scheme 9 Rapid synthesis of potential hole transport materials (a) 2Me-OTP-OMeDPA, (b) S-OTP-OMeDPA, and (c) 2S-OTP-OMeDPA. | |
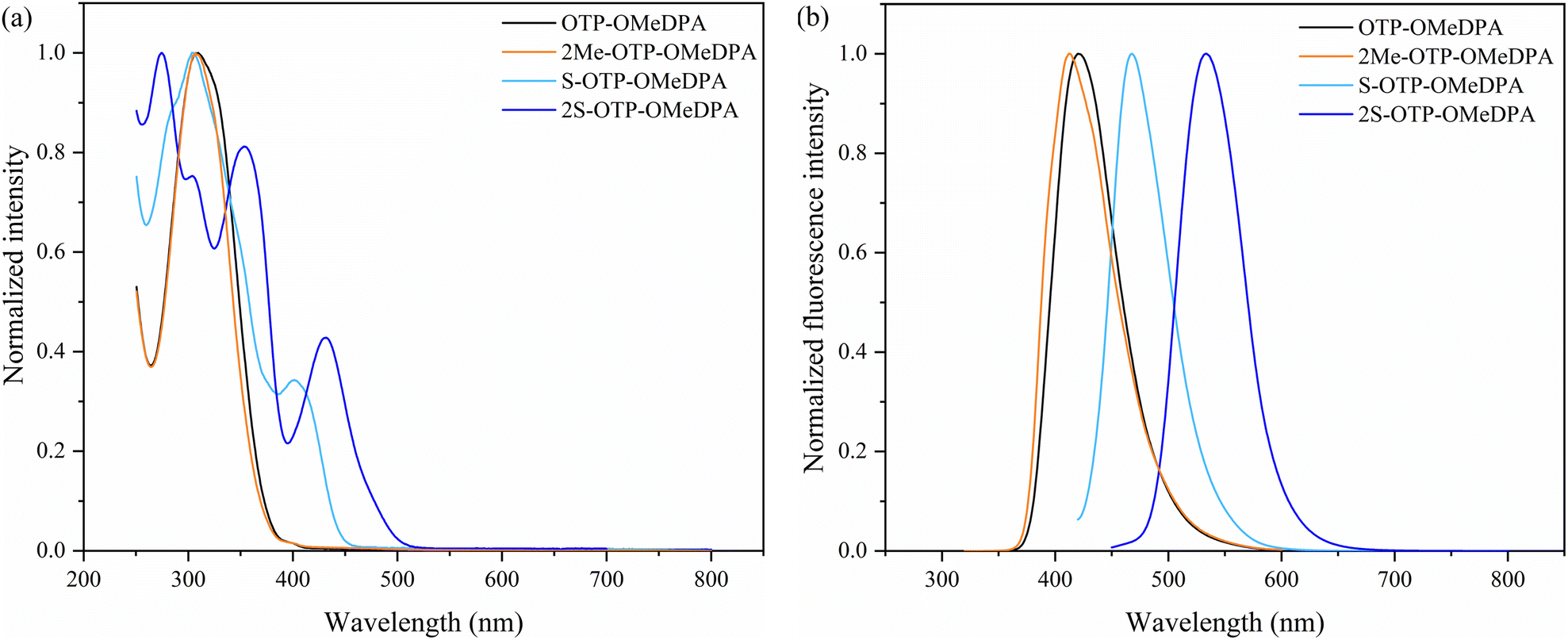
UV-vis absorption and fluorescence spectra of the potential hole transport materials 2Me-OTP-OMeDPA, S-OTP-OMeDPA, and 2S-OTP-OMeDPA as well as OTP-OMeDPA in CH2Cl2 were investigated (Fig. 4). The absorption maximum (λmax) of OTP-OMeDPA at 307 nm showed a conspicuous blue shift but a narrow absorption band in comparison with that of spiro-OMeTAD (λmax = 383 nm, see Fig. S1 in the ESI‡).38 The absorption curves of 2Me-OTP-OMeDPA (λmax = 307 nm) similar to that of OTP-OMeDPA indicated that the insertion of the two methyl groups did not affect the π-conjugation of the tetraphenylene skeleton much. Sulfur inserted derivatives S-OTP-OMeDPA (λmax = 420 nm) and 2S-OTP-OMeDPA (λmax = 432 nm) exhibited a significant red shift in comparison with OTP-OMeDPA, which is attributed to the formation of larger conjugated fragments after sulfur was inserted into the bay area of the tetraphenylene core.14e Fluorescence spectra showed that OTP-OMeDPA, 2Me-OTP-OMeDPA and 2S-OTP-OMeDPA had relatively large Stokes shifts of around 80–113 nm, while S-OTP-OMeDPA showed a small Stokes shift of 48 nm. All of them possessed higher Stokes shifts than spiro-OMeTAD, whose Stokes shift was 43 nm (see Fig. S2 in the ESI‡). These results suggested a more significant change in the geometric configuration of the tetraphenylene-based HTMs than that of spiro-OMeTAD and thus benefited in the infiltration and pore-filling for forming compact hole-transporting layers to protect perovskites.39
 |
| | Fig. 4 (a) UV-vis absorption spectra normalized at the peak value in solution (5 × 10−5 M in CH2Cl2); (b) fluorescence spectra normalized at the peak value in solution (5 × 10−5 M in CH2Cl2); the fluorescence spectra of OTP-OMeDPA, 2Me-OTP-OMeDPA, S-OTP-OMeDPA and 2S-OTP-OMeDPA were recorded with excitation at 307, 307, 420, and 432 nm, respectively. | |
To preliminarily evaluate their potential application in HTMs, parameters including half-wave potentials for the oxidation waves (Eox), HOMO energy levels (EHOMO), and LUMO energy levels (ELUMO) of tetraphenylene-based OTP-OMeDPA, 2Me-OTP-OMeDPA, S-OTP-OMeDPA, and 2S-OTP-OMeDPA as well as spiro-OMeTAD were determined and are summarized in Table 1.6g,34f Cyclic voltammograms (CVs) of the tetraphenylenes (Fig. 5) and spiro-OMeTAD (see details in Fig. S3 in the ESI‡) were measured in THF solutions with 0.1 M nBu4NPF6 employing FeCp2+/FeCp2 as the internal standard to obtain the Eox values. From the CV curves shown in Fig. 5, the Eox values for the modified tetraphenylenes (2Me-OTP-OMeDPA, S-OTP-OMeDPA, and 2S-OTP-OMeDPA) were found to be 0.27, 0.33 and 0.43 eV vs. FeCp2+/FeCp2, which are equal to or slightly more positive than that of OTP-OMeDPA (0.27 eV vs. FeCp2+/FeCp2) and therefore appropriate for energy alignment with the perovskite layer.34f The HOMO energy levels (EHOMO) could be calculated from the Eoxvs. FeCp2+/FeCp2 using the equation EHOMO = −(5.10 eV + eEox).40 In comparison, the HOMO energy levels of 2Me-OTP-OMeDPA (−5.37 eV), S-OTP-OMeDPA (−5.43 eV) and 2S-OTP-OMeDPA (−5.53 eV), which are equal to or slightly lower than that of OTP-OMeDPA (−5.37 eV) and spiro-OMeTAD (−5.45 eV), would enable efficient hole capture. Noteworthy is that the lower HOMO levels of S-OTP-OMeDPA and 2S-OTP-OMeDPA are apparently associated with their extended S-bridged connection, leading to the n–π conjugation. In addition, the optical energy bandgap (Egap) could be estimated from the longest absorption wavelength λedge, according to the equation Egap = 1240/λedge eV.39 As a result, the LUMO energy levels (ELUMO) could be estimated from the Egap and EHOMO according to the equation ELUMO = EHOMO + Egap. The LUMO energy levels of 2Me-OTP-OMeDPA (−2.06 eV), S-OTP-OMeDPA (−2.62 eV) and 2S-OTP-OMeDPA (−3.05 eV) were found to be close to or lower than that of OTP-OMeDPA (−2.03 eV), exhibiting suitable reduction potentials for hole extraction from the perovskite layer.
Table 1 Oxidation potentials and frontier molecular orbital energy levels of HTM candidates
| Compound |
λ
edge
(nm) |
E
gap
(eV) |
E
ox
(V) |
E
HOMO
(eV) |
E
LUMO
(eV) |
|
The wavelength of the absorption edge (λedge) recorded in a solution (5 × 10−5 M in CH2Cl2).
HOMO–LUMO energy bandgap estimated from λedge, Egap = 1240/λedge.
The half-wave potential versus FeCp2+/FeCp2 for the oxidation wave.
E
HOMO = −(5.10 eV + eEox).
Calculated from the HOMO–LUMO gap (Egap) and the HOMO energy level (EHOMO).
|
|
spiro-OMeTAD
|
414.6 |
2.99 |
0.35 |
−5.45 |
−2.46 |
|
OTP-OMeDPA
|
371.6 |
3.34 |
0.27 |
−5.37 |
−2.03 |
|
2Me-OTP-OMeDPA
|
380.6 |
3.31 |
0.27 |
−5.37 |
−2.06 |
|
S-OTP-OMeDPA
|
442.0 |
2.81 |
0.33 |
−5.43 |
−2.62 |
|
2S-OTP-OMeDPA
|
499.7 |
2.48 |
0.43 |
−5.53 |
−3.05 |
 |
| | Fig. 5 Cyclic voltammograms of (a) OTP-OMeDPA; (b) 2Me-OTP-OMeDPA; (c) S-OTP-OMeDPA; and (d) 2S-OTP-OMeDPA recorded in THF with FeCp2+/FeCp2 as the internal standard. | |
Conclusions
In summary, we reported a rapid and modular approach for late-stage atom(s) insertion into the tetraphenylene-based cyclic iodonium salts. The synthesis of tetraphenylene-based cyclic iodonium salts was successfully achieved through regioselective oxidative iodination oriented by both tert-butyl and nitro substituents. Atom(s) insertion reactions yielded a series of complex polycyclic hydrocarbons, featuring tetraphenylene-based [8 + n] and [n + 8 + n] fused-ring skeletons. It is worthy of note that functionalization of tetraphenylene cyclic iodonium salts could rapidly generate double helical architectures. Such appealing double helical frameworks with intrinsic chirality and potentially expandable π-conjugated systems should be of interest in the development of a new generation of advanced materials with interesting chiroptical properties. We believe that this work not only expands the diversity of tetraphenylene molecules, but also serves to inspire future widespread applications of tetraphenylene-based chiral systems in multiple fields of chiral optoelectronic materials. In addition, this late-stage atom(s) insertion strategy also enables a modular modification of the tetraphenylene-based hole transport materials. The three modified molecules show suitable properties for potential application as HTMs, indicating the robust synthetic potential of this strategy for the development of next generation HTMs.
Data availability
The data supporting this article have been included as part of the ESI.‡ Crystallographic data for 28 have been deposited at the CCDC under 2381518 and can be obtained from http://www.ccdc.cam.ac.uk/data_request/cif, or by emailing E-mail: data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author contributions
X. Xu and H.-R. Ma performed the experimental work and data analysis. J.-F. Cui, X.-S. Peng and H. N. C. Wong conceived the project, supervised the investigation and wrote the manuscript. All authors discussed the results and contributed to the preparation of the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (22271243, 21971219), the Research Program and Guangdong Leading Talent Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2021ZT090195), the Shenzhen Science, Technology and Innovation Committee for the Shenzhen Key Laboratory Scheme (ZDSYS20220507161600001), The Leading Talent Scheme of the “Pearl River Talent Recruitment Program” of Guangdong Province (2021CX020028), the Research Grants Council (Hong Kong) (GRF CUHK14304819), and the University Development Fund Grants from The Chinese University of Hong Kong, Shenzhen.
References
-
(a) X.-L. Hou, H. Huang and H. N. C. Wong, Synlett, 2005, 2005, 1073–1089 Search PubMed;
(b) A. Rajca, S. Rajca, M. Pink and M. Miyasaka, Synlett, 2007, 2007, 1799–1822 CrossRef;
(c) H. Huang, C.-K. Hau, C. C. M. Law and H. N. C. Wong, Org. Biomol. Chem., 2009, 7, 1249–1257 RSC;
(d) A. Rajca and S. Rajca, Angew. Chem., Int. Ed., 2010, 49, 672–674 CrossRef CAS PubMed;
(e) J.-W. Han, J.-X. Chen, X. Li, X.-S. Peng and H. N. C. Wong, Synlett, 2013, 24, 2188–2198 CrossRef CAS;
(f) J.-W. Han, X. Li and H. N. C. Wong, Chem. Rec., 2015, 15, 107–131 CrossRef CAS PubMed;
(g) J.-W. Han, X.-S. Peng and H. N. C. Wong, Natl. Sci. Rev., 2017, 4, 892–916 CrossRef CAS;
(h) G. González Miera, S. Matsubara, H. Kono, K. Murakami and K. Itami, Chem. Sci., 2022, 13, 1848–1868 RSC;
(i) H.-R. Ma, X.-S. Peng, J.-F. Cui and H. N. C. Wong, Tetrahedron Lett., 2023, 119, 154429 CrossRef CAS.
-
(a) W. S. Rapson, R. G. Shuttleworth and J. N. Van Niekerk, J. Chem. Soc., 1943, 326–327 RSC;
(b) I. L. Karle and L. O. Brockway, J. Am. Chem. Soc., 1944, 66, 1974–1979 CrossRef CAS;
(c) H. Irngartinger and W. R. K. Reibel, Acta Crystallogr., Sect. B, 1981, 37, 1724–1728 CrossRef.
-
(a) A. Rajca, A. Safronov, S. Rajca and J. Wongsriratanakul, J. Am. Chem. Soc., 2000, 122, 3351–3357 CrossRef CAS;
(b) A. Rajca, H. Wang, P. Bolshov and S. Rajca, Tetrahedron, 2001, 57, 3725–3735 CrossRef CAS.
-
(a) N. Z. Huang and T. C. W. Mak, J. Chem. Soc., Chem. Commun., 1982, 543–544 RSC;
(b) Y. M. Man, T. C. W. Mak and H. N. C. Wong, J. Org. Chem., 1990, 55, 3214–3221 CrossRef CAS;
(c) H.-Y. Peng, C.-K. Lam, T. C. W. Mak, Z. Cai, W.-T. Ma, Y.-X. Li and H. N. C. Wong, J. Am. Chem. Soc., 2005, 127, 9603–9611 CrossRef CAS PubMed;
(d) J.-F. Cui, C. Chen, X. Gao, Z.-W. Cai, J.-W. Han and H. N. C. Wong, Helv. Chim. Acta, 2012, 95, 2604–2620 CrossRef CAS;
(e) C. Cheng, Z. Cai, X.-S. Peng and H. N. C. Wong, J. Org. Chem., 2013, 78, 8562–8573 CrossRef CAS PubMed;
(f) C.-L. Deng, X.-D. Xiong, D. T. W. Chik, Z. Cai, X.-S. Peng and H. N. C. Wong, Chem.–Asian J., 2015, 10, 2342–2346 CrossRef CAS PubMed;
(g) A. Sygula, F. R. Fronczek, R. Sygula, P. W. Rabideau and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 3842–3843 CrossRef CAS PubMed.
-
(a) C.-K. Hau, H. He, A. W. M. Lee, D. T. W. Chik, Z. Cai and H. N. C. Wong, Tetrahedron, 2010, 66, 9860–9874 CrossRef CAS;
(b) G.-L. Chai, J.-W. Han and H. N. C. Wong, Synthesis, 2017, 49, 181–187 CAS;
(c) G.-L. Chai, B. Zhu and J. Chang, J. Org. Chem., 2019, 84, 120–127 CrossRef CAS PubMed;
(d) G.-L. Chai, A. Q. Sun, D. Zhai, J. Wang, W.-Q. Deng, H. N. C. Wong and J. Chang, Org. Lett., 2019, 21, 5040–5045 CrossRef CAS PubMed;
(e) G.-L. Chai, Y. Qiao, P. Zhang, R. Guo, J. Wang and J. Chang, Org. Lett., 2020, 22, 8023–8027 CrossRef CAS PubMed;
(f) J. Guo, W.-B. Xiong, H.-R. Ma, L. Fan, Y.-Y. Zhou, H. N. C. Wong and J.-F. Cui, Chem. Sci., 2022, 13, 4608–4615 RSC;
(g) J. Guo, H.-R. Ma, W.-B. Xiong, L. Fan, Y.-Y. Zhou, H. N. C. Wong and J.-F. Cui, Chem. Sci., 2022, 13, 13914–13921 RSC.
-
(a) B. Ejlli, P. Nußbaum, F. Rominger, J. Freudenberg, U. H. F. Bunz and K. Müllen, Angew. Chem., Int. Ed., 2021, 60, 20220–20224 CrossRef CAS PubMed;
(b) Y. Wang, Y. Zhang, S. Wang and D. Cao, Chem.–Eur. J., 2021, 27, 12012–12018 CrossRef CAS PubMed;
(c) C.-K. Hau, S. S.-Y. Chui, W. Lu, C.-M. Che, P.-S. Cheng, T. C. W. Mak, Q. Miao and H. N. C. Wong, Chem. Sci., 2011, 2, 1068–1075 RSC;
(d) C. Hägele, E. Wuckert, S. Laschat and F. Giesselmann, ChemPhysChem, 2009, 10, 1291–1298 CrossRef PubMed;
(e) E. Wuckert, M. Dix, S. Laschat, A. Baro, J. L. Schulte, C. Hägele and F. Giesselmann, Liq. Cryst., 2004, 31, 1305–1309 CrossRef CAS;
(f) Y. Byun, L. S. Xie, P. Fritz, T. Ashirov, M. Dincă and A. Coskun, Angew. Chem., Int. Ed., 2020, 59, 15166–15170 CrossRef CAS PubMed;
(g) J. Wang, X. Xie, Y. Cai, L. He, Y. Yuan, W. Fei, L. Wang and P. Wang, J. Mater. Chem. A, 2021, 9, 9927–9936 RSC;
(h) R. Rathore, P. Le Magueres, S. V. Lindeman and J. K. Kochi, Angew. Chem., Int. Ed., 2000, 39, 809–812 CrossRef CAS;
(i) C. B. Nielsen, T. Brock-Nannestad, T. K. Reenberg, P. Hammershøj, J. B. Christensen, J. W. Stouwdam and M. Pittelkow, Chem.–Eur. J., 2010, 16, 13030–13034 CrossRef CAS PubMed;
(j) Y. Liu, Z. Ma, Z. Wang and W. Jiang, J. Am. Chem. Soc., 2022, 144, 11397–11404 CrossRef CAS PubMed;
(k) K. Chen, Y. Liu, Z. Wang, S. Hu, Y. Zhao, W. Wang, G. Liu, Z. Wang and W. Jiang, J. Am. Chem. Soc., 2024, 146, 13499–13508 CrossRef CAS PubMed;
(l) Y. Liu, Z. Li, M.-W. Wang, J. Chan, G. Liu, Z. Wang and W. Jiang, J. Am. Chem. Soc., 2024, 146, 5295–5304 CrossRef CAS PubMed;
(m) F. Lin, H.-Y. Peng, J.-X. Chen, D. T. W. Chik, Z. Cai, K. M. C. Wong, V. W. W. Yam and H. N. C. Wong, J. Am. Chem. Soc., 2010, 132, 16383–16392 CrossRef CAS PubMed.
-
(a) Y. Zhang, Y. Zhu, D. Lan, S. H. Pun, Z. Zhou, Z. Wei, Y. Wang, H. K. Lee, C. Lin, J. Wang, M. A. Petrukhina, Q. Li and Q. Miao, J. Am. Chem. Soc., 2021, 143, 5231–5238 CrossRef CAS PubMed;
(b) S. N. Talapaneni, J. Kim, S. H. Je, O. Buyukcakir, J. Oh and A. Coskun, J. Mater. Chem. A, 2017, 5, 12080–12085 RSC;
(c) S. Wang, X.-X. Li, L. Da, Y. Wang, Z. Xiang, W. Wang, Y.-B. Zhang and D. Cao, J. Am. Chem. Soc., 2021, 143, 15562–15566 CrossRef CAS PubMed.
-
(a) J.-F. Wen, W. Hong, K. Yuan, T. C. W. Mak and H. N. C. Wong, J. Org. Chem., 2003, 68, 8918–8931 CrossRef CAS PubMed;
(b) Y. D. Xing and N. Z. Huang, J. Org. Chem., 1982, 47, 140–142 CrossRef CAS;
(c) H. N. C. Wong, Y.-M. Man and T. C. W. Mak, Tetrahedron Lett., 1987, 28, 6359–6362 CrossRef CAS;
(d) C. W. Hui, T. C. W. Mak and H. N. C. Wong, Tetrahedron, 2004, 60, 3523–3531 CrossRef CAS;
(e) S. H. Pun, C. K. Chan, Z. Liu and Q. Miao, Org. Mater., 2020, 02, 248–252 CrossRef CAS;
(f) H. Chen and Q. Miao, ChemPlusChem, 2019, 84, 627–629 CrossRef CAS PubMed.
-
(a) J. J. Eisch, A. M. Piotrowski, K. I. Han, C. Kruger and Y. H. Tsay, Organometallics, 1985, 4, 224–231 CrossRef CAS;
(b) H. Schwager, S. Spyroudis and K. P. C. Vollhardt, J. Organomet. Chem., 1990, 382, 191–200 CrossRef CAS;
(c) D. Masselot, J. P. H. Charmant and T. Gallagher, J. Am. Chem. Soc., 2006, 128, 694–695 CrossRef CAS PubMed.
- T. Shibata, T. Chiba, H. Hirashima, Y. Ueno and K. Endo, Angew. Chem., Int. Ed., 2009, 48, 8066–8069 CrossRef CAS PubMed.
-
(a) J.-F. Cui, H. Huang and H. N. C. Wong, Synlett, 2011, 2011, 1018–1022 CrossRef;
(b) K. Ouyang and Z. Xi, Chin. J. Catal., 2015, 36, 24–32 CrossRef CAS;
(c) X. Li, J.-W. Han and H. N. C. Wong, Asian J. Org. Chem., 2016, 5, 74–81 CrossRef CAS;
(d) H. Jiang, Y. Zhang, D. Chen, B. Zhou and Y. Zhang, Org. Lett., 2016, 18, 2032–2035 CrossRef CAS PubMed;
(e) C. Zhu, Y. Zhao, D. Wang, W.-Y. Sun and Z. Shi, Sci. Rep., 2016, 6, 33131 CrossRef PubMed;
(f) S. Matsubara, Y. Koga, Y. Segawa, K. Murakami and K. Itami, Nat. Catal., 2020, 3, 710–718 CrossRef CAS;
(g) Y. Zhang, J. Han and Z.-J. Liu, J. Org. Chem., 2016, 81, 1317–1323 CrossRef CAS PubMed;
(h) C. W. Lai, C. K. Lam, H. K. Lee, T. C. W. Mak and H. N. C. Wong, Org. Lett., 2003, 5, 823–826 CrossRef CAS PubMed.
-
(a) X. Yang, M. Gao, H. Chen, Q. Gong and Q. Miao, Org. Chem. Front., 2024, 11, 2624–2631 RSC;
(b) Y. Sakamoto and T. Suzuki, J. Am. Chem. Soc., 2013, 135, 14074–14077 CrossRef CAS PubMed.
-
(a) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS PubMed;
(b) J. Börgel and T. Ritter, Chem, 2020, 6, 1877–1887 CrossRef;
(c) N. J. Castellino, A. P. Montgomery, J. J. Danon and M. Kassiou, Chem. Rev., 2023, 123, 8127–8153 CrossRef CAS PubMed;
(d) E. King-Smith, F. A. Faber, U. Reilly, A. V. Sinitskiy, Q. Yang, B. Liu, D. Hyek and A. A. Lee, Nat. Commun., 2024, 15, 426 CrossRef CAS PubMed.
-
(a) D. Gust, G. H. Senkler and K. Mislow, J. Chem. Soc., Chem. Commun., 1972, 1345a RSC;
(b) H. P. Figeys and A. Dralants, Tetrahedron Lett., 1971, 12, 3901–3904 CrossRef;
(c) G. H. Senkler Jr, D. Gust, P. K. Riccobono and K. Mislow, J. Am. Chem. Soc., 1972, 94, 8626–8627 CrossRef;
(d) C.-N. Feng, M.-Y. Kuo and Y.-T. Wu, Angew. Chem., Int. Ed., 2013, 52, 7791–7794 CrossRef CAS;
(e) X.-D. Xiong, C.-L. Deng, X.-S. Peng, Q. Miao and H. N. C. Wong, Org. Lett., 2014, 16, 3252–3255 CrossRef CAS PubMed;
(f) X. Xiong, C.-L. Deng, B. F. Minaev, G. V. Baryshnikov, X.-S. Peng and H. N. C. Wong, Chem.–Asian J., 2015, 10, 969–975 CrossRef CAS PubMed;
(g) P. Rashidi-Ranjbar, Y. M. Man, J. Sandstroem and H. N. C. Wong, J. Org. Chem., 1989, 54, 4888–4892 CrossRef CAS;
(h) S. Pan, H. Jiang, Y. Zhang, Y. Zhang and D. Chen, Beilstein J. Org. Chem., 2016, 12, 1302–1308 CrossRef CAS PubMed;
(i) S. Pan, H. Jiang, Y. Zhang, D. Chen and Y. Zhang, Synlett, 2016, 27, 1997–2002 CrossRef CAS.
-
(a) X. Peng, A. Rahim, W. Peng, F. Jiang, Z. Gu and S. Wen, Chem. Rev., 2023, 123, 1364–1416 CrossRef CAS PubMed;
(b) X. Zhang, K. Zhao and Z. Gu, Acc. Chem. Res., 2022, 55, 1620–1633 CrossRef CAS PubMed;
(c) K. Zhao, L. Duan, S. Xu, J. Jiang, Y. Fu and Z. Gu, Chem, 2018, 4, 599–612 CrossRef CAS;
(d) J. Ke, B. Zu, Y. Guo, Y. Li and C. He, Org. Lett., 2021, 23, 329–333 CrossRef CAS PubMed;
(e) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed.
-
(a) W.-K. Hwang, Acta Chim. Sin., 1956, 22, 292–298 Search PubMed;
(b) W.-K. Hwang, Q.-H. Wang and S.-Y. Chen, Chin. Sci. Bull., 1957, 8, 49–50 CrossRef;
(c) W.-K. Hwang and T.-W. Tung, Lanzhou Daxue Xuebao, Ziran Kexueban, 1961, 18–24 Search PubMed.
- S. Stavber, P. Kralj and M. Zupan, Synthesis, 2002, 2002, 1513–1518 CrossRef.
- S.-Y. Chen, J. Zhang, Y.-H. Li, J. Wen, S.-Q. Bian and X.-Q. Yu, Tetrahedron Lett., 2009, 50, 6795–6797 CrossRef CAS.
-
M. Bielawski and B. Olofsson, in Organic Syntheses, pp. , pp. 308–314 Search PubMed.
- J. B. Lee, M. H. Jeon, J. K. Seo, G. von Helden, J.-U. Rohde, B. S. Zhao, J. Seo and S. Y. Hong, Org. Lett., 2019, 21, 7004–7008 CrossRef CAS PubMed.
- C.-C. Lee, M.-K. Leung, G.-H. Lee, Y.-H. Liu and S.-M. Peng, J. Org. Chem., 2006, 71, 8417–8423 CrossRef CAS PubMed.
- Z. Hou and J. Zhang, Lanzhou Daxue Xuebao, Ziran Kexueban, 1990, 142–144 CAS.
- D. Zhu, Q. Liu, B. Luo, M. Chen, R. Pi, P. Huang and S. Wen, Adv. Synth. Catal., 2013, 355, 2172–2178 CrossRef CAS.
-
(a) D. Zhu, Z. Wu, B. Luo, Y. Du, P. Liu, Y. Chen, Y. Hu, P. Huang and S. Wen, Org. Lett., 2018, 20, 4815–4818 CrossRef CAS PubMed;
(b) D. Zhu, M. Li, Z. Wu, Y. Du, B. Luo, P. Huang and S. Wen, Eur. J. Org Chem., 2019, 2019, 4566–4571 CrossRef CAS.
- L. Liu, J. Qiang, S. Bai, Y. Li and J. Li, Appl. Organomet. Chem., 2017, 31, e3810 CrossRef.
- Q. Tan, D. Zhou, T. Zhang, B. Liu and B. Xu, Chem. Commun., 2017, 53, 10279–10282 RSC.
- L. Biesen, J. Krenzer, N. Nirmalananthan-Budau, U. Resch-Genger and T. J. J. Müller, Chem. Sci., 2022, 13, 5374–5381 RSC.
- Y. Wu, F. Wu, D. Zhu, B. Luo, H. Wang, Y. Hu, S. Wen and P. Huang, Org. Biomol. Chem., 2015, 13, 10386–10391 RSC.
- L. Niu, H. Yang, Y. Jiang and H. Fu, Adv. Synth. Catal., 2013, 355, 3625–3632 CrossRef CAS.
- CCDC 2381518 contain the supplementary crystallographic data of compound 28 for this paper.
- K. Kantarod, T. Worakul, D. Soorukram, C. Kuhakarn, V. Reutrakul, P. Surawatanawong, W. Wattanathana and P. Leowanawat, Org. Chem. Front., 2021, 8, 522–530 RSC.
-
(a) A. Rajca, A. Safronov, S. Rajca and R. Shoemaker, Angew. Chem., Int. Ed., 1997, 36, 488–491 CrossRef CAS;
(b) J.-X. Chen, J.-W. Han and H. N. C. Wong, Org. Lett., 2015, 17, 4296–4299 CrossRef CAS PubMed.
-
(a) J. Y. Kim, J.-W. Lee, H. S. Jung, H. Shin and N.-G. Park, Chem. Rev., 2020, 120, 7867–7918 CrossRef CAS PubMed;
(b) P. Chen, Y. Xiao, S. Li, X. Jia, D. Luo, W. Zhang, H. J. Snaith, Q. Gong and R. Zhu, Chem. Rev., 2024, 124, 10623–10700 CrossRef CAS PubMed.
-
(a) K. Rakstys, C. Igci and M. K. Nazeeruddin, Chem. Sci., 2019, 10, 6748–6769 RSC;
(b) T. H. Schloemer, J. A. Christians, J. M. Luther and A. Sellinger, Chem. Sci., 2019, 10, 1904–1935 RSC;
(c) F. Wang, Y. Cao, C. Chen, Q. Chen, X. Wu, X. Li, T. Qin and W. Huang, Adv. Funct. Mater., 2018, 28, 1803753 CrossRef;
(d) L. Calió, S. Kazim, M. Grätzel and S. Ahmad, Angew. Chem., Int. Ed., 2016, 55, 14522–14545 CrossRef PubMed;
(e) H. D. Pham, T. C.-J. Yang, S. M. Jain, G. J. Wilson and P. Sonar, Adv. Energy Mater., 2020, 10, 1903326 CrossRef CAS;
(f) V. A. Chiykowski, Y. Cao, H. Tan, D. P. Tabor, E. H. Sargent, A. Aspuru-Guzik and C. P. Berlinguette, Angew. Chem., Int. Ed., 2018, 57, 15529–15533 CrossRef CAS PubMed.
-
(a) Z. Hawash, L. K. Ono and Y. Qi, Adv. Mater. Interfaces, 2018, 5, 1700623 CrossRef;
(b) U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissörtel, J. Salbeck, H. Spreitzer and M. Grätzel, Nature, 1998, 395, 583–585 CrossRef CAS.
- N. A. Bumagin, L. I. Sukhomlinova, S. O. Igushkina, A. N. Banchikov, T. P. Tolstaya and I. P. Beletskaya, Russ. Chem. Bull., 1992, 41, 2128–2129 CrossRef.
- L. Cai, X. Qian, W. Song, T. Liu, X. Tao, W. Li and X. Xie, Tetrahedron, 2014, 70, 4754–4759 CrossRef CAS.
-
(a) L. Nakka, Y. Cheng, A. G. Aberle and F. Lin, Adv. Energy Sustainability Res., 2022, 3, 2200045 CrossRef CAS;
(b) M. Jeong, I. W. Choi, E. M. Go, Y. Cho, M. Kim, B. Lee, S. Jeong, Y. Jo, H. W. Choi, J. Lee, J.-H. Bae, S. K. Kwak, D. S. Kim and C. Yang, Science, 2020, 369, 1615–1620 CrossRef CAS PubMed.
- H. Li, K. Fu, A. Hagfeldt, M. Grätzel, S. G. Mhaisalkar and A. C. Grimsdale, Angew. Chem., Int. Ed., 2014, 53, 4085–4088 CrossRef CAS PubMed.
- L. Leonat, S. Beatrice Gabriela and I. V. Branzoi, UPB Sci. Bull. B: Chem. Mater. Sci., 2013, 75, 111–118 CAS.
Footnotes |
| † This article is dedicated to the memory of Professor Chin-Kang Sha. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2381518. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07803a |
| § These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Xiao-Shui
Peng
*a,
Xiao-Shui
Peng