
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMapping photoisomerization dynamics on a three-state model potential energy surface in bacteriorhodopsin using femtosecond stimulated Raman spectroscopy†
Ziyu
Wang‡
a,
Yu
Chen‡
a,
Jiaming
Jiang
a,
Xin
Zhao
b and
Weimin
Liu
 *a
*a
aSchool of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China. E-mail: liuwm@shanghaitech.edu.cn
bDepartment of Physics, East China Normal University, Shanghai, 200062, P. R. China
First published on 29th January 2025
Abstract
The process of proton translocation in Halobacterium salinarum, triggered by light, is powered by the photoisomerization of all-trans-retinal in bacteriorhodopsin (bR). The primary events in bR involving rapid structural changes upon light absorption occur within subpicoseconds to picoseconds. While the three-state model has received extensive support in describing the primary events between the H and K states, precise characterization of each excited state in the three-state model during photoisomerization remains elusive. In this study, we investigate the ultrafast structural dynamics of all-trans-retinal in bR using femtosecond stimulated Raman spectroscopy. We report Raman modes at 1820 cm−1 which arise from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch vibronic coupling and provide direct experimental evidence for the involvement of the I and J states with 2A−g symmetric character in the three-state model. The detection of the CC vibronic coupling mode, CN stretching mode (1700 cm−1), and hydrogen out-of-plane (HOOP) mode (954 cm−1) further supports the three-state model that elucidates the initial charge translocation along the conjugated chain accompanied by trans-to-cis photoisomerization dynamics through H(1B+u) → I(2A−g) → J(2A−g) → K(13-cis ground state) transitions in all-trans-retinal in bR.
C stretch vibronic coupling and provide direct experimental evidence for the involvement of the I and J states with 2A−g symmetric character in the three-state model. The detection of the CC vibronic coupling mode, CN stretching mode (1700 cm−1), and hydrogen out-of-plane (HOOP) mode (954 cm−1) further supports the three-state model that elucidates the initial charge translocation along the conjugated chain accompanied by trans-to-cis photoisomerization dynamics through H(1B+u) → I(2A−g) → J(2A−g) → K(13-cis ground state) transitions in all-trans-retinal in bR.
Introduction
The photophysical and photochemical properties of retinal proteins have been extensively investigated to comprehend the initial capture of visible light and subsequent structural modifications in the retinal chromophore.1–6 Bacteriorhodopsin (bR) is a photoreceptor protein discovered in the purple membrane of Halobacterium salinarum, which functions as a light-activated proton pump. Upon photon excitation, a complex photocycle process is initiated in the all-trans-retinal in bR. This intricate process involves multiple intermediates, including H → I → J → K → L → M → N → O states.7The ultrafast primary events involving temporal and structural changes upon light absorption in bR occur within subpicoseconds to a few picoseconds.8–10 Previous experimental findings and calculations strongly support the three-state model (see Fig. 1a) instead of the two-state model for describing the ultrafast primary events in bR.11–16 The all-trans-retinal molecule is formally classified under the C2h point group; the three-state model for all trans-retinal comprises one ground state and two excited states with Bu and Ag symmetries.17 It has been widely acknowledged that the absorption band observed in all trans-retinal in bR within the green spectral range is attributed to one photo-allowed transition from the S0 (1A−g) ground state to the S1 excited state with the 1B+u symmetry; while the S2 excited state, which is about 3500 cm−1 higher than the S1 (1B+u) state, shares the same symmetry (2A−g) as the ground state S0, resulting in a forbidden one-photon transition between them.17 The calculations suggest that after light absorption by the first excited state S1 (1B+u), retinal carries out a rapid torsion around its C13C14 bond, subsequently undergoing two nonadiabatic transitions between S1 (1B+u) and S2 (2A−g) as well as S2 (2A−g) and the cis ground state (see Fig. 1a).14 Despite conducting numerous time-resolved spectroscopic investigations with a high temporal resolution, there has been no direct experimental evidence to support the involvement of the S2 state with 2A−g symmetric character in the excited-state dynamics of bR.
 | ||
| Fig. 1 (a) Schematic potential energy curves of the three-state model along the torsional angle of the C13C14 bond of all-trans-retinal in bR. (b) The absorption spectrum of bR under light-adapted conditions with the actinic pump (AP) wavelength in FSRS depicted by a color spike. (c) The SADS from global analysis of the TA spectra of bR with the Raman pump (RP) wavelength in FSRS depicted by a color spike. (d) The chemical structures of the retinal molecule in bR in different intermediate states obtained by TR-SFX; θ represents the dihedral angle of C12–C13C14–C15. The PDB IDs for various intermediate states are as follows: H state (6G7H), I state (6G7I), J state (6G7J), and K state (6G7K).10 | ||
For short polyenes with a conjugation length of N ≤ 5, previous studies have identified a CC stretching Raman mode at 1750–1800 cm−1, attributed to an upshift in the frequency of the 2A−g state CC stretching Raman mode, resulting from the adiabatic vibronic coupling between the 2A−g and 1A−g states.18–21 Governed by the symmetry rules of the C2h point group in all-trans polyenes, this coupling results in a well-known frequency upshift of 100–150 cm−1 for the CC stretching mode in the 2A−g state, while simultaneously lowering the frequency of the CC stretching mode in the 1A−g state.19,22,23 More recently, femtosecond stimulated Raman spectroscopy (FSRS) has revealed a high-frequency Raman mode at 1712 cm−1 in UV-absorbing rhodopsin HKR1 (histidine kinase rhodopsin 1), attributed to the ethylenic CC stretch vibronic coupling mode of the 13-cis retinal molecule.5 These findings have motivated us to reexamine the ultrafast structural dynamics of all-trans-retinal in bR using FSRS, with a primary focus on high-frequency Raman signals (>1600 cm−1) associated with CC stretch vibronic coupling between the two A−g states.
In this study, the utilization of a Raman pump passing through a grating filter system in FSRS significantly enhances both spectral resolution and sensitivity, thereby facilitating the acquisition of more intricate insights into the excited state vibrational dynamics of all-trans-retinal in bR (see Fig. S1b and c†).24–28 The observation of the highest frequency Raman modes at 1820 cm−1, exhibiting biexponential decay dynamics, can be attributed to CC stretch vibronic coupling between I (2A−g)/J (2A−g) states and the 1A−g ground state. Furthermore, the identification of the vibronic coupling CC stretching mode, CN stretching mode (1700 cm−1), and the HOOP mode (954 cm−1) provides support for the three-state model that describes the initial charge transfer accompanied by transition of H (1B+u) → I (2A−g) → J (2A−g) → K (13-cis ground state) → L states in all-trans-retinal in bR.
Results and discussion
The absorption spectrum of all-trans retinal in bR under light-adapted conditions is illustrated in Fig. 1b. As shown in Fig. S3b and S4b,† the two-dimensional transient absorption (TA) spectra with an excitation energy of 200 nJ per pulse exhibit intricate excited state features between 400 nm and 750 nm. Global analysis with a sequential model was employed to obtain the species-associated difference spectra (SADS) (see Fig. 1c), yielding four characteristic lifetimes with time constants of τ1 = 200 fs (H → I state), τ2 = 510 fs (I → J state), τ3 = 8.5 ps (J → K state), and τ4 = an infinitely long lifetime (K → L state).9,29–31 Notably, the lifetime of the J state (τ3 = 8.5 ps) observed in our measurement exceeds the commonly recognized value of ∼3–4 ps.9,10,13,15 To explore this discrepancy, we collected TA spectra using a lower excitation energy of 50 nJ per pulse (see Fig. S3a and S4a†), where the SADS for the J-state lifetime (τ3 = 4.5 ps) were consistent with previous studies. It has been demonstrated that at sufficiently high light intensity, two-photon absorption occurs in bR, resulting in the excitation of the tryptophan residue (Trp86) close to the retinal chromophore.31,32 This excitation induces strong excitonic coupling between Trp86 and retinal, which significantly influences the structural dynamics of the retinal chromophore. To further validate our findings, we left the bR sample in the dark overnight following the TA measurement at 200 nJ per pulse. TA data were then recollected at low excitation energy (50 nJ per pulse), and the results were consistent with those from a fresh bR sample under identical excitation conditions (see Fig. S3c†).To gain insights into the structural dynamics beyond electronic features, we used FSRS to track the excited-state vibrational marker bands of bR. Fig. S5† shows the time-resolved FSRS spectra of bR across a frequency range of 900–2000 cm−1 at an excitation energy of 50 nJ per pulse. We strategically selected a 620 nm Raman pump to pre-resonantly enhance the signal on the Stokes side (Fig. 1b) by matching the transition of the ESA band of I,12 J, and K states (see Fig. S4†). The decay lifetimes for the three states are consistent with those observed in TA spectra (see Fig. S6†). However, due to the limited signal-to-noise ratio at this low excitation power, capturing more detailed Raman signal dynamics was challenging. In contrast, Fig. 2 presents time-resolved FSRS spectra of bR at a higher excitation energy of 200 nJ per pulse (with the baseline and raw data shown in Fig. S7†), revealing more pronounced excited-state Raman signals with intricate dynamics. The ground state spectrum (bottom of Fig. 2) reveals six major Raman peaks: the methyl rocking mode at 1006 cm−1, the C–C stretching doublet mode at 1168 and 1200 cm−1, and the ethylenic CC stretching mode at 1529 cm−1.9,33–37 Additionally, the HOOP mode at 958 cm−1 and the CN stretch mode at 1648 cm−1 are also discernible.33,37,38
 | ||
| Fig. 2 Excited-state FSRS spectra of all-trans-retinal in bR from 900–1850 cm−1 at an excitation energy of 200 nJ per pulse. The ground state FSRS spectrum is shown at the bottom for comparison. | ||
As depicted in Fig. 2, upon photoexcitation at 570 nm, the aforementioned five major Raman peaks in the excited state exhibit a significant redshift in frequency to 1162 and 1195 cm−1 (C–C stretch), 997 cm−1 (C–CH3 rocking), 954 cm−1 (HOOP), and to 1517 cm−1 (CC stretch) compared to their counterparts in the ground state.9 In contrast, the CN stretching mode associated with the Schiff base experiences a significant frequency blueshift to ∼1700 cm−1 in the excited state.39 The presence of a prominent Raman mode at approximately 1820 cm−1 has not been previously reported for all-trans-retinal in bR using FSRS,9,40,41 and its unusually high frequency suggests a specific origin. Notably, such a Raman mode was commonly observed in carotenoid molecules possessing linear conjugated polyene chains with conjugation lengths (N) ranging from 9 to 15.42–45 A similar excited state Raman mode at 1712 cm−1 has been reported for unprotonated 13-cis retinal in UV-absorbing rhodopsin,5 which is attributed to the CC stretching mode of the optically forbidden 2A−g state resulting from strong adiabatic vibronic coupling between 2A−g and 1A−g states. The Raman mode at 1820 cm−1 in our case is thereby assigned to a significant blueshift of the 2A−g state CC ethylenic stretch, arising from the vibronic coupling between the two A−g states in the all-trans retinal in bR.18 The discrepancy in frequencies between the vibronic-coupled CC stretching mode at 1820 cm−1 and the all-trans CC stretching mode (S0) at 1529 cm−1, i.e., 1820–1529 = 291 cm−1, exhibits a significantly higher magnitude compared to that observed for UV-absorbing rhodopsin (∼200 cm−1). This observation suggests a very strong vibronic coupling within the all-trans retinal of bR.46
We obtained SADS extracted from global analysis in a sequential mode using a kinetic scheme Hinvisible state → I state → J state → K state → of FSRS spectra,47,48 as shown in Fig. 3. Importantly, the H state exhibiting negligible FSRS signal intensity was designated as an invisible component in SADS analysis (i.e., a spectrally silent species48,49), primarily due to the experimental resonance Raman conditions where the H species is off-resonant with a 620 Raman pump. The FSRS spectra of bR are satisfactorily fitted in SADS using a model that includes three species of τ2 = 500 fs (I state), τ3 = 9.1 ps (J state), and τ4 = an infinitely long lifetime (K state), which reveals significant dynamics of formation and relaxation in the fingerprint Raman modes within the intermediate states I, J, and K.
 | ||
| Fig. 3 The SADS from global analysis of the excited state FSRS spectra of bR. Due to the experimental resonance Raman conditions, the H state with a negligible FSRS signal was set as an invisible component (not contributing to the spectra) in the global analysis, denoted by the dashed-line box. | ||
To quantitatively examine the temporal variation of stimulated Raman signals, we analyzed the transient amplitude observed in each Raman band. Fig. 4a, b, and S8† present the transient kinetic trace of the C–C and CC stretching bands at 1162, 1195, and 1517 cm−1.9 The area of these peaks was determined by fitting to a Gaussian line shape function. The time evolution of the peak area was fitted using least-squares fits with four-time components retrieved from SADS global fitting in FSRS spectra. All three Raman peaks exhibit a double exponential rise with time constants of τ1 = 200 fs and τ2 = 500 fs followed by two decay components of τ3 = 9.1 ps and τ4 = an infinitely long lifetime, indicating observation of the I state (green dashed line), J state (blue dashed line), and K state (purple dashed line) via the relaxation scheme 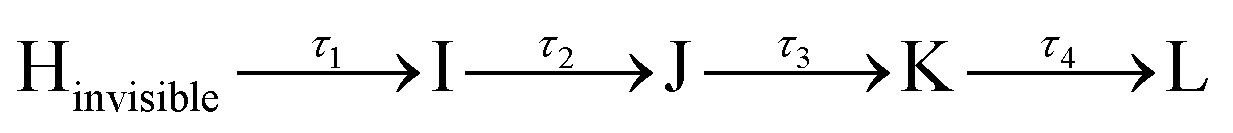 states.
states.
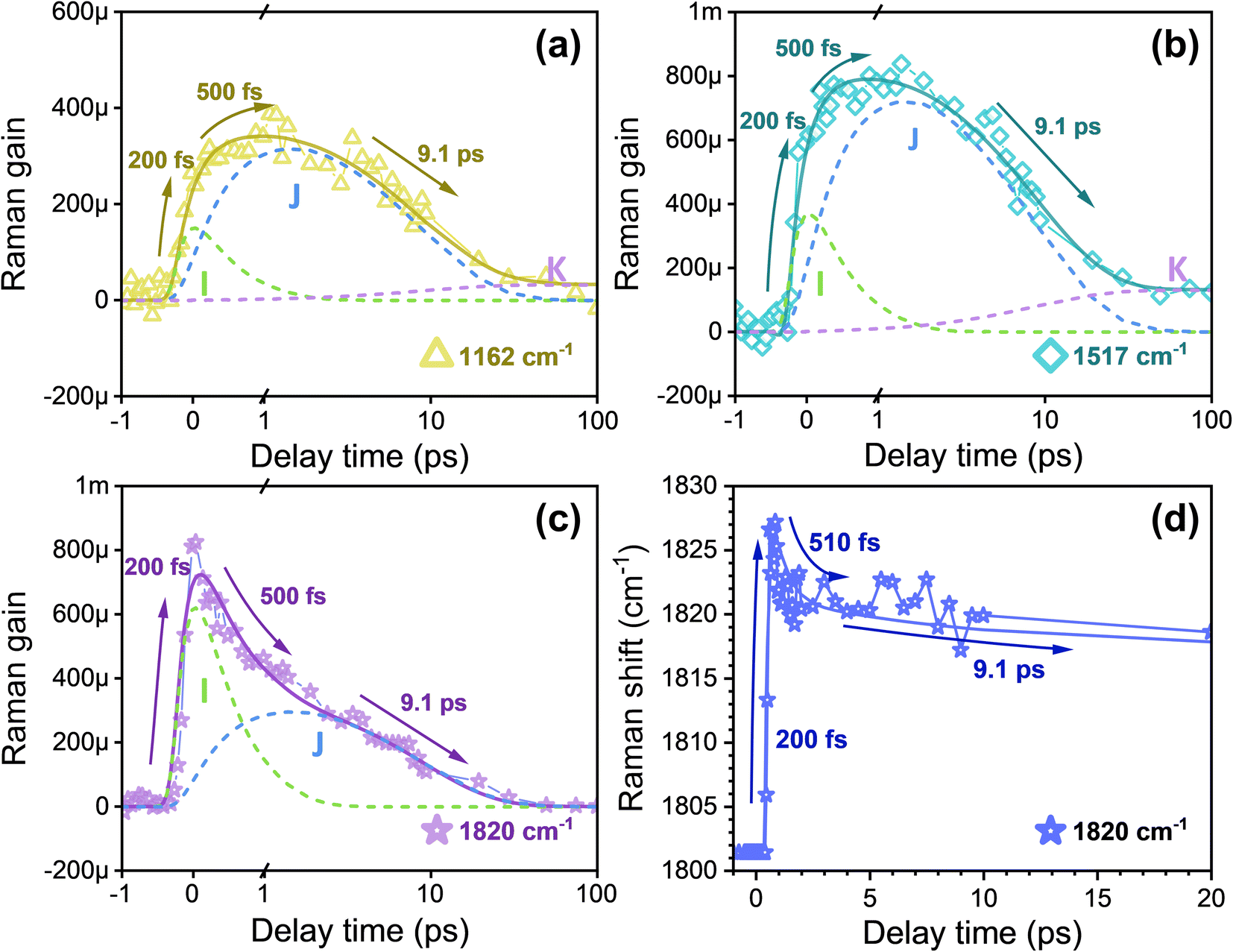 | ||
| Fig. 4 The transient Raman intensities of the (a) C–C stretching mode at 1162 cm−1 and (b) CC stretching mode at 1520 cm−1. The transient Raman intensities (c) and frequencies (d) of the CC vibronic coupling mode at 1820 cm−1. The dashed lines represent different intermediate states: the I state (green), J state (blue), and K state (purple). | ||
Notably, different from the C–C and CC stretching modes, the transient amplitude of the CC vibronic coupling Raman mode at 1820 cm−1 exhibits a rise time of 200 fs accompanied by two exponential decay components of 500 fs and 9.1 ps, as depicted in Fig. 4c. Based on previous studies, this behavior suggests that the excited states I and J can be assigned to the optically forbidden 2A−g state, which undergoes strong vibronic coupling with the trans-S0 ground state (1A−g). This coupling with the 2Ag−–1A−g states, shifts the CC stretching in the 2A−g (I and J) state upward to 1820 cm−1. Simultaneously, the vibronic interaction lowers the CC stretching of the 1A−g state to 1529 cm−1.19,22,23 The previous experimental and computational findings provide support for the three-state model,13–15 in which the optically allowed state, termed the single excited state π–π* 1Bu state, is positioned lower than an optically forbidden state (the 2Ag state with a double π–π* excitation) in the Frank–Condon geometry, based on the C2h symmetry. The primary determinant of the reaction coordinate is considered to be the torsional motion revolving around the C13C14 double bond involved in the reaction process.1,50 The vertical photoexcitation of the all-trans-retinal in bR initially leads to the formation of the intermediate excited state H (1B+u), which corresponds to a charge transfer state involving hole-pair (ionic) excitation.51–53 Consequently, the interaction between the H (1B+u) state and the upper 2A−g state surface results in a slight energy barrier on the H (1B+u) state surface caused by a small torsion process,14 after which an inversion of energy levels occurs between 1B+u and 2A−g at the first crossing point to form the intermediate state I (2A−g) after 200 fs. Importantly, the observed CC vibronic coupling in the I (2A−g) state suggests that the minor torsion following photoexcitation is unlikely to break the C2h symmetry of retinal in bR. This observation is consistent with the findings from time-resolved serial femtosecond crystallography (TR-SFX), which determined a change in torsion angle of about 30° via the C12–C13C14–C15 bonds (shifting from 15° in the H state to 45° in the I state), as shown in Fig. 1d.10 Having established the I (2A−g) state in 200 fs, further structure distortion is induced by twisting the C12–C13C14–C15 dihedral angle, and the observed τ2 = 500 fs ultrafast decay dynamics in Fig. 4c can be attributed to the torsion-induced weakening of the C2h symmetry in the 2A−g state along the I → J transition. However, this interpretation is challenged by TR-SFX data, which reveal a dihedral angle of approximately 97° in the J (2A−g) state (Fig. 1d), suggesting a potential loss of C2h symmetry.19 A possible explanation for this discrepancy may lie in the difference in experimental conditions. The TR-SFX experiments were conducted on bR in a crystalline state, where confinement and lower hydration levels may affect the structural dynamics differently than in physiological environments (in solution), potentially altering the torsional behavior.54 Our FSRS results for bR suggest that the dihedral angle in the J state is likely less than 90° under physiological conditions. Following the formation of the J (2A−g) state, the potential energy surfaces intersect with the K (cis-ground) state surface along the reaction coordinate at a second weakly crossing point.14 This reveals a J → K state transition by further twisting the C12–C13C14–C15 dihedral angle (to approximately 143°, see Fig. 1d), resulting in decreased vibronic coupling strength to zero due to the complete breaking of the C2h symmetry. Eventually, this leads to the formation of cis-retinal. As depicted in Fig. 4d, the transient frequency of the vibronic coupling mode reveals an ultrafast blueshift with a time constant of τ1 = 200 fs followed by two components of redshift with lifetimes of τ2 = 510 fs and τ3 = 9.1 ps. The frequency blueshift indicates an enhancement in the strength of vibronic coupling during the formation of the I (2A−g) state, while the biexponential redshift can be attributed to a weakening effect for vibronic coupling caused by torsion.55
The vibrational frequency at 954 cm−1 corresponds to the HOOP vibrational mode, which is highly sensitive to local torsion in the C13C14 region during retinal isomerization.3,9,25,37,38,56,57 As shown in Fig. 5a, akin to the transient dynamics observed in C–C and CC bonds, the C14–H HOOP mode exhibits two rising components at τ1 = 200 fs and τ2 = 500 fs followed by two decay components of τ3 = 9.1 ps and τ4 = an infinitely long lifetime, suggesting a direct probe of the isomerization in retinal during the I → J → K → L transition. Our findings are consistent with the previous reports using TR-SFX, as structural changes were already observed at the I state.10 Due to the sensitivity of the HOOP modes towards the tilting angle around the CC bond,15 the observed weak signal intensity in the I state indicates a twisting of the polyene backbone via C12–C13C14–C15 torsion at a small angle (see Fig. 1d).10,15 The increase in FSRS signal intensity observed during the development of the J state indicates a subsequent enhancement in the torsion angle of C12–C13C14–C15.10 During this period, there is an alteration in the torsion angles of the C13C14 and adjacent C11C12 and C15N bonds in opposing orientations, suggesting that the reaction follows a space-saving bicycle-pedal model of retinal isomerization.58 Subsequently, the decrease in transient intensity of the HOOP signal with a time constant of 9.1 ps arises due to a further twist in the C12–C13C14–C15 bond (to approximately 143°), indicating complete isomerization into its 13-cis configuration.58 The infinitely long decay lifetime is assigned to the decay from the K to the L state; observing this decay dynamics in the transient HOOP mode provides evidence for further conformation change during the K → L transition.59,60 This observation aligns with the TR-SFX diffraction data, which reveal that, during the K → L transition, the 13-cis retinal progressively adopts a planar configuration, ultimately achieving the essentially cis-planar configuration within approximately 290 ns.7
 | ||
| Fig. 5 The transient Raman intensities of the (a) HOOP vibrational mode at 954 cm−1 and (b) CN stretching vibrational mode at ∼1700 cm−1. | ||
As depicted in Fig. 5b, a vibration mode observed in the high-frequency region at ∼1700 cm−1 by FSRS corresponds to the CN stretching mode.39 The previous TA results have demonstrated excitonic coupling between the dipole moment of retinal and nearby tryptophan residues (Trp86); the experimental findings reflect a translocation of the positive charge from the protonated Schiff base toward the β-ionone end of retinal within 200 fs after photoexcitation.51,52 This leads to an enhancement of the dipole moment and a redistribution of charge within the retinal. Furthermore, considering that the occurrences at 200 fs involve a change in the configuration of the retinal framework, the increase in dipole moment is related to structural changes and possibly serves as the driving force for subsequent trans-to-cis isomerization.51,52,61,62 The FSRS spectra of the CN stretching mode are satisfactorily fitted including τ1 = 200 fs rising followed by three decay components of τ2 = 500 fs (I → J state), τ3 = 9.1 ps (J → K state), and τ4 = an infinitely long lifetime (K → L state), similar to the transient dynamics observed in the HOOP mode. The findings suggest a strong correlation between the gradual decrease in dipole moment and simultaneous modifications observed in the retinal structure.15,51,52
Conclusion
In this study, we investigate the ultrafast structural dynamics of all-trans-retinal in bR using FSRS and report the previously undiscovered Raman modes at 1820 cm−1, which arise from vibronic coupling between the two-photon allowed 2A−g states and 1A−g ground state. The observation of the vibronic coupling CC stretching mode and CN stretching mode (at 1700 cm−1) as well as the HOOP mode (at 954 cm−1) provides support for the three-state model in elucidating the initial photoisomerization dynamics of retinal in bR (schematic potential energy curves of the three-state model are depicted in Fig. 1a). In our experiment, the I and J states are identified as higher-energy two-photon allowed 2A−g states, in comparison to the 2B+u-like H state. The transition from H (2B+u) to the I (2A−g) state occurs within 200 fs and involves an enhancement in dipole moment resulting from the charge transfer and a small change of the C12–C13C14–C15 dihedral angle in the all-trans retinal structure. The vibronic coupling is subsequently attenuated by twisting the C12–C13C14–C15 dihedral angle, accompanied by a reduction in dipole moment within ∼500 fs, resulting in a transition from I (2A−g) to J (2A−g) states. Afterward, a transition from J (2A−g) to K 13-cis ground states occurs by further twisting the dihedral angle of C12–C13C14–C15, resulting in reduced vibronic coupling strength towards zero due to complete disruption of the C2h symmetry. Eventually, during the K → L transition, there is a further conformation change that results in the formation of a cis-planar configuration. Importantly, our experimental findings are significantly supported by the TR-SFX experimental results,10 providing novel insights into the previously unexplored optical properties of bR and emphasizing the unique utility of FSRS in advancing our comprehension of photoreceptor protein dynamics.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Z. W., Y. C. and W. L. conceived, designed and performed the experiments. Y. C., Z. W. and J. J. analysed the experimental results. Z. W., Y. C., and W. L. co-wrote the manuscript. X. Z. provided the bR samples. All authors participated in discussions and edited the manuscript. W. L. directed the project.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22373068); the Double First-Class Initiative Fund of ShanghaiTech University; ShanghaiTech Start-up Funding (F-0201-16-006).References
- T. Sovdat, G. Bassolino, M. Liebel, C. Schnedermann, S. P. Fletcher and P. Kukura, J. Am. Chem. Soc., 2012, 134, 8318–8320 CrossRef CAS PubMed.
- G. Bassolino, T. Sovdat, A. Soares Duarte, J. M. Lim, C. Schnedermann, M. Liebel, B. Odell, T. D. Claridge, S. P. Fletcher and P. Kukura, J. Am. Chem. Soc., 2015, 137, 12434–12437 CrossRef CAS PubMed.
- C. Schnedermann, X. Yang, M. Liebel, K. Spillane, J. Lugtenburg, I. Fernández, A. Valentini, I. Schapiro, M. Olivucci, P. Kukura and R. Mathies, Nat. Chem., 2018, 10, 449–455 CrossRef CAS PubMed.
- M. Ghosh, K.-H. Jung and M. Sheves, Phys. Chem. Chem. Phys., 2019, 21, 9450–9455 RSC.
- Y. Hontani, M. Broser, M. Luck, J. r. Weißenborn, M. Kloz, P. Hegemann and J. T. Kennis, J. Am. Chem. Soc., 2020, 142, 11464–11473 CrossRef CAS PubMed.
- H. Kandori, Bull. Chem. Soc. Jpn., 2020, 93, 76–85 CrossRef CAS.
- E. Nango, A. Royant, M. Kubo, T. Nakane, C. Wickstrand, T. Kimura, T. Tanaka, K. Tono, C. Song and R. Tanaka, Science, 2016, 354, 1552–1557 CrossRef CAS PubMed.
- K. Schulten, Z. Schulten and P. Tavan, Inf. Energy Transduct. Biol. Member., 1984, 164, 113–131 CAS.
- S. Shim, J. Dasgupta and R. A. Mathies, J. Am. Chem. Soc., 2009, 131, 7592–7597 CrossRef CAS PubMed.
- P. Nogly, T. Weinert, D. James, S. Carbajo, D. Ozerov, A. Furrer, D. Gashi, V. Borin, P. Skopintsev and K. Jaeger, Science, 2018, 361, eaat0094 CrossRef PubMed.
- K. Schulten, W. Humphrey, I. Logunov, M. Sheves and D. Xu, Isr. J. Chem., 1995, 35, 447–464 CrossRef CAS.
- K. Hasson, F. Gai and P. A. Anfinrud, Proc. Natl. Acad. Sci. USA, 1996, 93, 15124–15129 CrossRef CAS PubMed.
- F. Gai, K. Hasson, J. C. McDonald and P. A. Anfinrud, Science, 1998, 279, 1886–1891 CrossRef CAS PubMed.
- W. Humphrey, H. Lu, I. Logunov, H.-J. Werner and K. Schulten, Biophys. J., 1998, 75, 1689–1699 CrossRef CAS PubMed.
- T. Kobayashi, T. Saito and H. Ohtani, Nature, 2001, 414, 531–534 CrossRef CAS PubMed.
- S. Gozem, P. J. Johnson, A. Halpin, H. L. Luk, T. Morizumi, V. I. Prokhorenko, O. P. Ernst, M. Olivucci and R. D. Miller, J. Phys. Chem. Lett., 2020, 11, 3889–3896 CrossRef CAS PubMed.
- R. R. Birge and C. F. Zhang, J. Chem. Phys., 1990, 92, 7178–7195 CrossRef CAS.
- H. Hashimoto and Y. Koyama, Chem. Phys. Lett., 1989, 154, 321–325 CrossRef CAS.
- G. Orlandi, F. Zerbetto and M. Z. Zgierski, Chem. Rev., 1991, 91, 867–891 CrossRef CAS.
- S. Hogiu, W. Werncke, M. Pfeiffer, A. Lau and T. Steinke, Chem. Phys. Lett., 1998, 287, 8–16 CrossRef CAS.
- S. Hogiu, W. Werncke, M. Pfeiffer and A. Lau, Chem. Phys. Lett., 1999, 303, 218–222 CrossRef CAS.
- G. Orlandi and F. Zerbetto, Chem. Phys., 1986, 108, 187–195 CrossRef CAS.
- F. Zerbetto, M. Zgierski, G. Orlandi and G. Marconi, J. Chem. Phys., 1987, 87, 2505–2512 CrossRef CAS.
- S.-Y. Lee, D. Zhang, D. W. McCamant, P. Kukura and R. A. Mathies, J. Chem. Phys., 2004, 121, 3632–3642 CrossRef CAS PubMed.
- P. Kukura, D. W. McCamant, S. Yoon, D. B. Wandschneider and R. A. Mathies, Science, 2005, 310, 1006–1009 CrossRef CAS PubMed.
- J. Dasgupta, R. R. Frontiera, K. C. Taylor, J. C. Lagarias and R. A. Mathies, Proc. Natl. Acad. Sci. USA, 2009, 106, 1784–1789 CrossRef CAS PubMed.
- C. Fang, R. R. Frontiera, R. Tran and R. A. Mathies, Nature, 2009, 462, 200–204 CrossRef CAS PubMed.
- H. Veisi, P. Abassi, P. Mohammadi, T. Tamoradi and B. Karmakar, Sci. Rep., 2021, 11, 2734 CrossRef CAS PubMed.
- J. Dobler, W. Zinth, W. Kaiser and D. Oesterhelt, Chem. Phys. Lett., 1988, 144, 215–220 CrossRef CAS.
- G. Atkinson, T. Brack, D. Blanchard and G. Rumbles, Chem. Phys., 1989, 131, 1–15 CrossRef CAS.
- G. Nass Kovacs, J.-P. Colletier, M. L. Grünbein, Y. Yang, T. Stensitzki, A. Batyuk, S. Carbajo, R. B. Doak, D. Ehrenberg and L. Foucar, Nat. Commun., 2019, 10, 3177 CrossRef PubMed.
- H.-J. Polland, M. Franz, W. Zinth, W. Kaiser and D. Oesterhelt, Biochim. Biophys. Acta, Bioenerg., 1986, 851, 407–415 CrossRef CAS.
- M. Braiman and R. Mathies, Proc. Natl. Acad. Sci. USA, 1982, 79, 403–407 CrossRef CAS PubMed.
- B. Curry, A. Broek, J. Lugtenburg and R. Mathies, J. Am. Chem. Soc., 1982, 104, 5274–5286 CrossRef CAS.
- J. Herbst, K. Heyne and R. Diller, Science, 2002, 297, 822–825 CrossRef CAS PubMed.
- A. Kahan, O. Nahmias, N. Friedman, M. Sheves and S. Ruhman, J. Am. Chem. Soc., 2007, 129, 537–546 CrossRef CAS PubMed.
- P. J. Johnson, A. Halpin, T. Morizumi, V. I. Prokhorenko, O. P. Ernst and R. D. Miller, Nat. Chem., 2015, 7, 980–986 CrossRef CAS PubMed.
- Y. Furutani, Y. Sudo, A. Wada, M. Ito, K. Shimono, N. Kamo and H. Kandori, Biochemistry, 2006, 45, 11836–11843 CrossRef CAS PubMed.
- J. P. Kraack, T. Buckup and M. Motzkus, J. Phys. Chem. Lett., 2013, 4, 383–387 CrossRef CAS PubMed.
- D. W. McCamant, P. Kukura and R. A. Mathies, J. Phys. Chem. B, 2005, 109, 10449–10457 CrossRef CAS PubMed.
- K. Niu, B. Zhao, Z. Sun and S.-Y. Lee, J. Chem. Phys., 2010, 132, 084510 CrossRef PubMed.
- M. Garavelli, P. Celani, F. Bernardi, M. A. Robb and M. Olivucci, J. Am. Chem. Soc., 1997, 119, 11487–11494 CrossRef CAS.
- D. W. McCamant, P. Kukura and R. A. Mathies, J. Phys. Chem. A, 2003, 107, 8208–8214 CrossRef CAS PubMed.
- R. L. Christensen, M. G. I. Galinato, E. F. Chu, J. N. Howard, R. D. Broene and H. A. Frank, J. Phys. Chem. A, 2008, 112, 12629–12636 CrossRef CAS PubMed.
- R. L. Christensen, M. M. Enriquez, N. L. Wagner, A. Y. Peacock-Villada, C. Scriban, R. R. Schrock, T. s. Polívka, H. A. Frank and R. R. Birge, J. Phys. Chem. A, 2013, 117, 1449–1465 CrossRef CAS PubMed.
- Y. Mukai, H. Hashimoto, Y. Koyama, S. Kuroda, Y. Hirata and N. Mataga, J. Phys. Chem., 1991, 95, 10586–10592 CrossRef CAS.
- J. M. Artes Vivancos, I. H. Van Stokkum, F. Saccon, Y. Hontani, M. Kloz, A. Ruban, R. van Grondelle and J. T. Kennis, J. Am. Chem. Soc., 2020, 142, 17346–17355 CrossRef CAS PubMed.
- Z. Wang, Y. Zhang, C. Chen, R. Zhu, J. Jiang, T. C. Weng, Q. Ji, Y. Huang, C. Fang and W. Liu, Angew. Chem., Int. Ed., 2023, 62, e202212209 CrossRef CAS PubMed.
- L. Nagy, P. Maróti and M. Terazima, FEBS Lett., 2008, 582, 3657–3662 CrossRef CAS PubMed.
- A. Petrone, D. B. Williams-Young, D. B. Lingerfelt and X. Li, J. Phys. Chem. A, 2017, 121, 3958–3965 CrossRef CAS PubMed.
- R. González-Luque, M. Garavelli, F. Bernardi, M. Merchán, M. A. Robb and M. Olivucci, Proc. Natl. Acad. Sci. USA, 2000, 97, 9379–9384 CrossRef PubMed.
- S. v. Schenkl, F. Van Mourik, G. Van der Zwan, S. Haacke and M. Chergui, Science, 2005, 309, 917–920 CrossRef CAS PubMed.
- A. Cembran, F. Bernardi, M. Olivucci and M. Garavelli, Proc. Natl. Acad. Sci. USA, 2005, 102, 6255–6260 CrossRef CAS PubMed.
- P. E. Konold, E. Arik, J. Weißenborn, J. C. Arents, K. J. Hellingwerf, I. H. van Stokkum, J. T. Kennis and M. L. Groot, Nat. Commun., 2020, 11, 4248 CrossRef CAS PubMed.
- H. Nagae, M. Kuki, J.-P. Zhang, T. Sashima, Y. Mukai and Y. Koyama, J. Phys. Chem. A, 2000, 104, 4155–4166 CrossRef CAS.
- I. Schapiro, M. N. Ryazantsev, L. M. Frutos, N. Ferré, R. Lindh and M. Olivucci, J. Am. Chem. Soc., 2011, 133, 3354–3364 CrossRef CAS PubMed.
- J. P. Kraack, T. Buckup, N. Hampp and M. Motzkus, ChemPhysChem, 2011, 12, 1851–1859 CrossRef CAS PubMed.
- C. Wickstrand, P. Nogly, E. Nango, S. Iwata, J. Standfuss and R. Neutze, Annu. Rev. Biochem., 2019, 88, 59–83 CrossRef CAS PubMed.
- S. J. Doig, P. J. Reid and R. A. Mathies, J. Phys. Chem., 1991, 95, 6372–6379 CrossRef CAS.
- Y. Hontani, K. Inoue, M. Kloz, Y. Kato, H. Kandori and J. T. Kennis, Phys. Chem. Chem. Phys., 2016, 18, 24729–24736 RSC.
- S. Schenkl, F. Van Mourik, N. Friedman, M. Sheves, R. Schlesinger, S. Haacke and M. Chergui, Proc. Natl. Acad. Sci. USA, 2006, 103, 4101–4106 CrossRef CAS PubMed.
- A. Yabushita and T. Kobayashi, Biophys. J., 2009, 96, 1447–1461 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07540d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |