
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed intramolecular aerobic oxidative cross-coupling of BH/CH between o-carborane and arenes†
Zhen
Wang
ab,
Jiahui
Yu
c,
Jie
Zhang
 d,
Dengsong
Zhang
c,
Zaozao
Qiu
*bc and
Zuowei
Xie
*be
d,
Dengsong
Zhang
c,
Zaozao
Qiu
*bc and
Zuowei
Xie
*be
aState Key Laboratory of Antiviral Drugs, Pingyuan Laboratory, School of Chemistry, and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, China
bShanghai-Hong Kong Joint Laboratory in Chemical Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai 200032, China. E-mail: zxie@cuhk.edu.hk; qiuzz@sioc.ac.cn
cInnovation Institute of Carbon Neutrality, International Joint Laboratory of Catalytic Chemistry, Department of Chemistry, College of Sciences, Institution, Shanghai University, Shanghai 200444, China
dDepartment of Chemistry, The Chinese University of Hong Kong, Shatin, N.T., Hong Kong, China
eShenzhen Grubbs Institute, Department of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China
First published on 23rd January 2025
Abstract
An efficient Pd-catalyzed regioselective intramolecular aerobic oxidative dehydrocoupling of BH/CH between o-carborane and arenes has been achieved with the construction of a series of five-, six- and seven-membered rings under mild reaction conditions. Control experiments indicate that B–H activation proceeds preferentially over the aryl C–H. These new polyarene–carborane conjugates have potential applications in materials as demonstrated by pyrene fused o-carborane that exhibits unique dual-phase emission, intramolecular charge transfer (ICT), and aggregation-induced emission (AIE) properties.
Introduction
Carboranes are a class of polyhedral boron hydride molecular clusters in which one or more of the BH vertices are replaced by CH units.1 Their structural features, such as spherical geometry and three-dimensional electron delocalization, make them valuable components in various applications, ranging from functional materials to pharmaceuticals.2,3 The o-carborane unit is known for its distinct electronic characteristics, including steric electronic conjugation and position-dependent electron-donating and accepting properties.4 Due to the electronic interactions between the carborane unit and the attached π-conjugated substituents, o-carboranes have gained significant interest as a versatile boron element-block for constructing optoelectronic materials, especially for their remarkable ability to suppress aggregation-caused quenching (ACQ). This feature is mainly observed in carbon vertex-substituted derivatives.5 However, the limited functionalization methods for boron vertex-arylated o-carboranes restrict their further applications.6 Among the various methodologies developed for o-carborane B–H activation, transition-metal-catalyzed BH/CH oxidative coupling is regarded as the most direct and efficient method for the formation of C(sp2)–B bonds. For instance, Ir-catalyzed intermolecular oxidative CH/BH cross-coupling of carboranyl carboxylic acid has been demonstrated with thiophene7 and aryl substrates bearing various directing groups including carboxy,8 amide,9 and nitrogen-containing heterocycles (Scheme 1a).10 Additionally, Fe-catalyzed intramolecular CH/BH oxidative cyclization of o-carborane with 8-aminoquinoline-based auxiliaries leads to the formation of C,B-substituted carborane-fused phenanthroline derivatives (Scheme 1b).11 | ||
| Scheme 1 Transition metal-catalyzed dehydrogenative arylation. | ||
These reactions generally require high temperatures and anhydrous/anaerobic reaction conditions for successful B–H activation. As part of an ongoing project in our laboratory, we aim to develop efficient and environmentally friendly methods for cage functionalization to synthesize boron-arylated o-carboranes with potential optoelectronic applications. We report here a Pd-catalyzed intramolecular aerobic oxidative dehydrocoupling of BH/CH between o-carborane and arenes with the construction of a series of five-, six- and seven-membered rings under mild reaction conditions (Scheme 1c).
Results and discussion
Our investigation began by examining the BH/CH oxidative coupling of 1-(2-methoxy-1-naphthyl)-2-benzyl-o-carborane (1aa) in the presence of Pd(OAc)2 catalyst (10 mol%), H2O2 (1.0 equiv.) and TFA (1.0 equiv.) in HOAc at 40 °C in open air, giving acenaphtheno-o-carborane (2aa) with the construction of a boron-containing five-membered ring in 70% NMR yield (Table 1, entry 1). Screening of copper oxidants proved that Cu(OTf)2 was the optimal choice, generating 2aa in 96% yield (Table 1, entries 2–4). Compared with Pd(OAc)2, Pd(TFA)2 and Pd(acac)2 gave slightly lower yield of 2aa, whereas Pd(MeCN)4(BF4)2 and PdCl2 showed poor or no catalytic activity (Table 1, entries 5–8). Solvent was also very crucial for this reaction. The use of toluene, THF, and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) as the solvent gave the cross-coupling product in 16–30% yields. Trifluoroacetic acid (TFA) offered a reduced yield of 78% (Table 1, entries 9–12). While the use of Pd(TFA)2 as catalyst and TFA as solvent led to a 90% yield of 2aa (Table 1, entry 13). Further screening indicated that lowering the catalyst loading or oxidant amount resulted in reduced yields of 2aa (Table 1, entries 14 and 15). Lowering reaction temperature to 25 °C led to poor reaction efficiency (Table 1, entry 16). In view of the yield of 2aa, entry 4 was chosen as the optimal reaction conditions.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | [O] | Solvent | Yieldb (%) |
| a All reactions were carried out on 0.1 mmol scale in 1 mL of solvent. b NMR yield using 1,1,2,2-tetrachloroethane as an internal standard. c 1 equiv. of TFA was added. d 5 mol% Pd(OAc)2. e 0.5 equiv. Cu(OTf)2. f 25 °C, 12 h. | ||||
| 1 | Pd(OAc)2 | H2O2c | HOAc | 70 |
| 2 | Pd(OAc)2 | Cu(OAc)2 | HOAc | 8 |
| 3 | Pd(OAc)2 | Cu(OPiv)2 | HOAc | 7 |
| 4 | Pd(OAc)2 | Cu(OTf)2 | HOAc | 96 |
| 5 | Pd(TFA)2 | Cu(OTf)2 | HOAc | 90 |
| 6 | Pd(acac)2 | Cu(OTf)2 | HOAc | 88 |
| 7 | Pd(MeCN)4(BF4)2 | Cu(OTf)2 | HOAc | 8 |
| 8 | PdCl2 | Cu(OTf)2 | HOAc | N.R. |
| 9 | Pd(OAc)2 | Cu(OTf)2 | Toluene | 16 |
| 10 | Pd(OAc)2 | Cu(OTf)2 | THF | 17 |
| 11 | Pd(OAc)2 | Cu(OTf)2 | HFIP | 30 |
| 12 | Pd(OAc)2 | Cu(OTf)2 | TFA | 78 |
| 13 | Pd(TFA)2 | Cu(OTf)2 | TFA | 90 |
| 14d | Pd(OAc)2 | Cu(OTf)2 | HOAc | 78 |
| 15e | Pd(OAc)2 | Cu(OTf)2 | HOAc | 72 |
| 16f | Pd(OAc)2 | Cu(OTf)2 | HOAc | 44 |
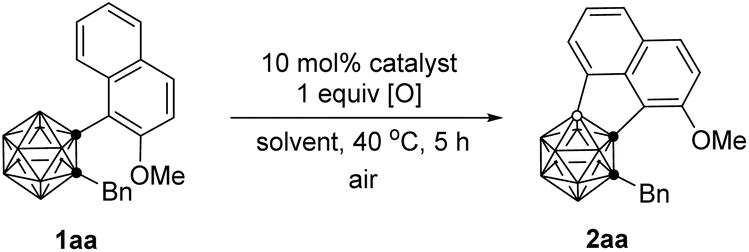
Under such optimized reaction conditions, the scope of this reaction was investigated and the results were compiled in Table 2. We first investigated the substrate generality of 2-methoxy-1-naphthyl group, affording the BH/CH dehydrocoupling products 2aa–2an in 73–92% isolated yields. The mild reaction conditions were tolerant of a variety of functional groups including F, Cl, CO2Me, C(O)Me and CN, and no obvious electronic effect was observed in 2aa–2an. Notably, the reaction of substrate 1aa proceeded smoothly on 1.0 mmol scale with a slightly lower efficiency (86% yield, 334 mg). Moreover, steric hindrance of 2-substitutions on naphthyl group did not affect the reaction efficiency (2ao–2as). 1-(1-Naphthyl)-2-benzyl-o-carborane 1as also worked well, giving acenaphtheno-o-carborane product 2as in 81% isolated yield, while variation of the R3 group at 2-position of naphthyl had little effect on the reactivity for the formation of products 2ao–2ar. 1-(1-Naphthyl)-2-benzyl-o-carboranes (1a) bearing –OMe or –F substituents at different position of the naphthyl group offered the corresponding products (2at–2aw) in 69–86% isolated yields. For 2at–2aw, electron-donating units on naphthyl group generally offered better yields of 2a than those of electron-withdrawing substituents. However, heteroaryl containing substrate 1ax showed poor reactivity towards the BH/CH oxidative coupling between o-carborane and thienyl groups to afford 2ax in only 16% isolated yield due probably to the interactions between Pd and S atoms. On the other hand, different substituents at cage C(2) were also evaluated, leading to the corresponding products 2ba–2ga in moderate to excellent isolated yields. The results indicated that steric factor has an influence on the regioselectivity, as both B(4)- and B(3)-coupling products (2ha and 3ha) were obtained with a molar ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in the reaction of C(2)–H substrate 1ha.
:1 in the reaction of C(2)–H substrate 1ha.

Furthermore, 1,2-di(1-naphthyl)-o-carborane (4) was also compatible with this five-membered ring construction process to afford B(4,7)- and B(4,11)-cross-coupling products 5 and 6 in 11% and 45% yields, respectively. The molecular structures of 2aa, 2ha, 5 and 6 were further confirmed by single-crystal X-ray analyses, supporting the unambiguous assignment of the substituted vertices (Scheme 2).
 | ||
| Scheme 2 Intramolecular dehydrogenative coupling of 1,2-dinaphthyl-o-carborane. | ||
We then examined the feasibility of six-membered ring construction via BH/CH oxidative coupling using 1-(2-biphenyl)-2-benzy-o-carborane (7a) as a model substrate. Treatment of 7a with 1 equiv. of Cu(OTf)2 in the presence of 10 mol% of Pd(OAc)2 catalyst in HOAc at 40 °C for 12 h afforded the desired product 8a in 88% yield (Table 3, entry 1, standard conditions). Screening of other solvents and oxidants did not give better results (Table 3, entries 2–7). Decreasing the catalyst loading to 5 mol% resulted in a decreased yield of 8a (Table 3, entry 8). It is worth noting that lowering the amount of copper salt to 0.5 equiv. gave the product in 91% yield. Finally, using 0.1 equiv. of Cu(OTf)2 as the cocatalyst and air as the oxidant led to 90% yield of 8a at 60 °C (Table 3, entries 9–11).
|
|
||
|---|---|---|
| Entry | Variations from the “standard conditions” | Yieldb (%) |
| a All reactions were carried out on 0.1 mmol scale in 1 mL of HOAc. b NMR yield using 1,1,2,2-tetrachloroethane as the internal standard. | ||
| 1 | None | 88 |
| 2 | HFIP instead of HOAc | 26 |
| 3 | THF instead of HOAc | 13 |
| 4 | Cu(OAc)2 instead of Cu(OTf)2 | 12 |
| 5 | Cu(OPiv)2 instead of Cu(OTf)2 | 9 |
| 6 | 2 equiv. AgOTf instead of Cu(OTf)2 | 20 |
| 7 | 1 equiv. H2O2/TFA instead of Cu(OTf)2 | 8 |
| 8 | 5 mol% Pd(OAc)2 | 59 |
| 9 | 0.5 equiv. Cu(OTf)2 | 91 |
| 10 | 0.1 equiv. Cu(OTf)2 | 75 |
| 11 | 0.1 equiv. Cu(OTf)2, 60 °C, 5 h | 90 |

With the optimized conditions in hand, we investigated the scope and limitation of this 6-membered ring construction (Table 4). The results indicated that both electron-donating and -withdrawing substituents at 4′- and 6′-positions of biphenyl group were well tolerated, leading to the desired products (8a–8l) in moderate to very good isolated yields (68–85%). In addition, the mild reaction conditions were tolerant of various functional groups, including halogens (8e, 8f, 8k, 8l) and ester (8g). For Ccage-connected aromatic ring, biphenyls with substituents at 6- and 4-positions (8m, 8n and 8q–8t) gave very high coupling efficiency (75–88%). However, those with 5-Me, 5-Ph, or 3-OMe substituents delivered the corresponding products (8o, 8p and 8u) in relatively lower yields (61–65%), while the use of 5-F substituent led to no reaction. Excellent yields were observed for C(2)-Me and -Ph substituted o-carborano-phenanthrene products (8v and 8w). With C(2)-H substrate 7x, both 1,4- and 1,3-o-carborane fused phenanthrenes (8x and 9x) were isolated in 77% and 9% yields, respectively, probably for steric reasons.

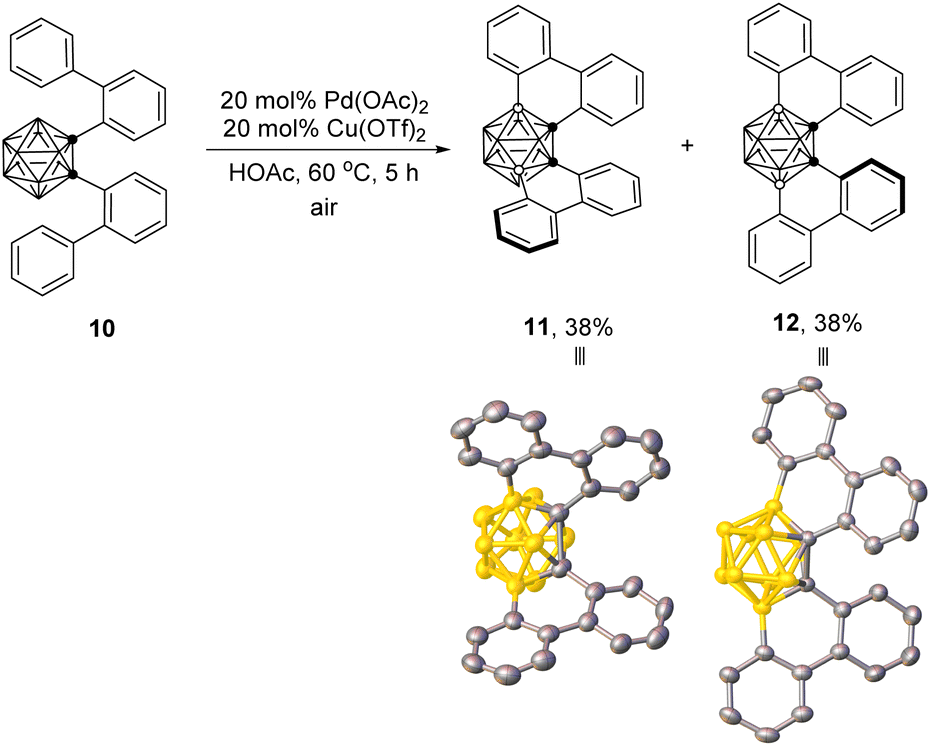
12-Dibiphenyl-o-carborane (10) also worked to afford the desired B(4,7)- and B(4,11)-crosscoupling products 11 and 12, respectively, in the same 38% isolated yields in the presence of 20 mol% Pd(OAc)2 and 20 mol% Cu(OTf)2 in HOAc at 60 °C (Scheme 3). The B(4)-, B(3)-, B(4,7)- and B(4,11)-regioselectivities in 8a, 8x, 9x, 11 and 12 were further confirmed by single-crystal X-ray analyses.
 | ||
| Scheme 3 Intramolecular dehydrogenative coupling of 1,2-dibiphenyl-o-carborane. | ||
This strategy worked also for the synthesis of o-carborane fused seven-membered rings (Scheme 4). In the presence of 10 mol% of Pd(OAc)2 and 1 equiv. of Cu(OTf)2, the intramolecular BH/CH oxidative coupling of 1-(2-(benzyl)phenyl)-o-carboranes 13 afforded the corresponding coupling products 14a and 14b in 25% and 41% yields, respectively. Instead of the methylene unit, substrates with X = O- or NPh-bridging moiety between the two phenyl groups showed poor reactivities for the seven-membered ring formation. In addition, such oxidative cross-coupling was compatible with 1-(2-(1-naphthyl)phenyl)-o-carborane substrate 15 with the employment of 2 equiv. of Cu(OTf)2 to give 16 in 30% yield. Low yields may be related to unfavorable entropy and enthalpy strains in the formation of 7-membered rings.
 | ||
| Scheme 4 Intramolecular dehydrogenative 7-membered ring construction. | ||
To gain some insight into the reaction mechanism, several control experiments were carried out. The oxidative coupling reactions did not occur in the absence of palladium catalyst (Scheme 5a). The yield of 2aa was dramatically dropped to 7% without Cu(OTf)2 (Scheme 5b). In contrast, replacement of air with N2 in the reaction system offered only a 21% yield of 2aa under standard reaction conditions (Scheme 5c), suggesting that oxygen may act as an oxidant with the help of a Cu(II) cocatalyst.12 To gain additional information regarding the initial step of the reaction, treatment of 1aa with 20 mol% Pd(OAc)2 under non-oxidative conditions using DOAc as the solvent led to 19–88% deuteration efficiency on the ten boron vertexes (Scheme 5d). These results indicated that B–H activation is more favorable than the aryl C–H activation in this Pd-catalyzed process, which is calculated [at the B3LYP level of theory in conjunction with the Lanl2dz basis set and the corresponding Hay–Wadt effective core potential (ECP) for Pd and standard 6-31+G** basis set for all remaining atoms] to be thermodynamically favorable with ΔG = −9.9 kcal mol−1 (Fig. 1).
 | ||
| Scheme 5 Control experiments. (a) Reaction without Pd(OAc)2. (b) Reaction without Cu(OTf)2. (c) Reaction under N2 atmosphere. (d) Palladium catalyzed BH deuteration in DOAc. | ||
 | ||
| Fig. 1 Transition states calculated at the B3LYP/6-31+G**/Lanl2dz level of theory. | ||
On the basis of the aforementioned experimental results and literature work,13 a plausible reaction mechanism is proposed in Scheme 6. Electrophilic attack of the Pd(II) center at the B(4)–H affords an intermediate A, which undergoes intramolecular C–H bond activation to give the six-membered palladacycle B. It is noted that this reversible B–Pd bond formation will take place at all the ten BH vertexes of o-carborane cage, which follow the order B(3,6)–H ≪ B(4,5,7,11)–H < B(8,10)–H < B(9,12)–H relied on the differences in vertex charge.14 Among these, only the B(3) and B(4)–Pd can approach the aromatic C–H due to the substrate configuration. The excellent B(4)-regioselectivity can be attributed to the electronic effect of the boron cage as well as the steric effect of C(2)-substituents. Reductive elimination affords the final products 2aa with the release of Pd(0), followed by Cu(II) promoted the aerobic oxidation of Pd(0) to Pd(II).12
 | ||
| Scheme 6 Proposed reaction mechanism. | ||
To further demonstrate the synthetic utility of the above intramolecular BH arylation protocol, 1-pyrenyl-o-carborane 17 was subjected to the palladium-catalyzed aerobic oxidative cross-coupling to afford the corresponding pyrene incorporated 1,4-disubstituted o-carborane 18 in 89% yield (Scheme 7). A single-crystal X-ray structure of 18 is shown in Fig. 2. The dihedral angle between the Ccage–Ccage bond and the pyrene moiety is about 43°. In the crystal packing diagram, there is no intermolecular π⋯π interaction due to the bulkiness of o-carboranyl unit, resulting in the suppressed ACQ effect. Moreover, one-dimensional continuous BcageH⋯π interactions followed by the linear packing structure is observed.15
 | ||
| Scheme 7 Synthesis of carborane-fused polycyclic aromatic compound. | ||
 | ||
| Fig. 2 (a) Molecular structure of 18. Hydrogen atoms are omitted for clarity. (b) Packing structures of 18. | ||
The photophysical properties of 18 were subsequently investigated. The UV/vis absorption spectrum of 18 in solution was recorded and compared to those of pyrene and substrate 17.16 As shown in Fig. 3a, the absorption spectra of 17 and 18 exhibited bathochromic shifts relative to pyrene, which can be attributed to the involvement of the o-carborane unit in an extended π-conjugation.17 On the other hand, the dual-emission bands were observed in the emission spectra of both 17 and 18. While both 17 and 18 displayed emission peaks near 400 nm, their photoluminescence (PL) spectra differed significantly from pyrene, showing broad emission bands with peaks in the range of 570 to 600 nm (Fig. 3b). Furthermore, the B–C bond coupling in 17 caused a red-shifted emission, which was due to the increased electron-withdrawing effect of the bisubstituted o-carborane.18 To explore the dual-emission mechanism in 18, changes in the absorption and PL spectra were monitored in solvents with varying polarities (Fig. 3c and d). No major peak shifts were observed in the absorption spectra, which could be attributed to the minimal solvent effect in the ground state. In contrast, as the solvent polarity increased, substantial bathochromic shifts were observed in the broad emission band at longer wavelengths in the PL spectra, suggesting that the luminescence near 600 nm in 18 is originated from the intramolecular charge transfer (ICT) state.16,18 The fluorescence near 385 nm was largely unaffected by solvent polarity, displaying a mirror-image relationship with the absorption spectrum in the longer-wavelength region, thereby confirming its assignment to the locally excited (LE) state.16
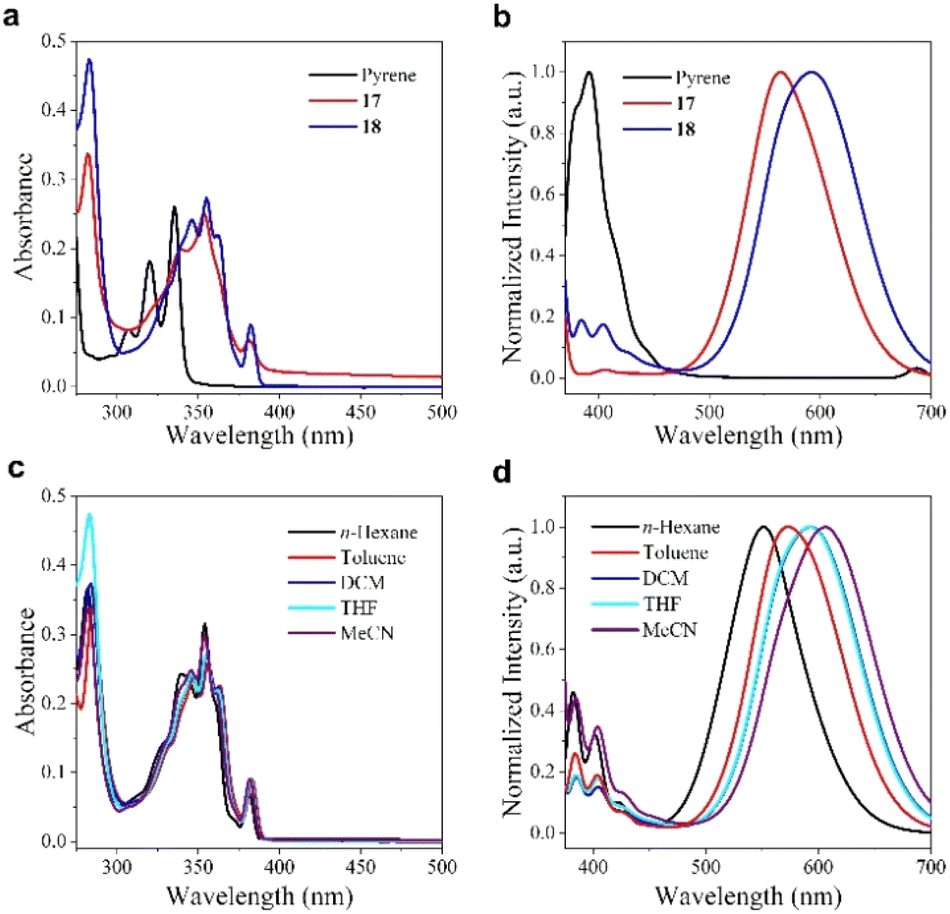 | ||
| Fig. 3 (a) UV-vis absorption spectra of pyrene, 17 and 18 in THF (c = 1.0 × 10−5 M). (b) The normalized PL spectra of pyrene, 17 and 18 in THF (c = 1.0 × 10−5 M), the excitation wavelength is 340, 360 and 360 nm, respectively. (c) UV-vis absorption spectra of 18 in different solvents (c = 1.0 × 10−5 M). (d) The normalized PL spectra of 18 in different solvents (c = 1.0 × 10−5 M, λex = 360 nm). | ||
Subsequently, the aggregation and solid-state emission properties of 18 were examined (Fig. 4 and Table 5). In a study involving THF/water mixtures, a THF solution of 18 emitted yellow light at 590 nm due to its ICT emission. As the water fraction (fw) in the mixture increased from 0% to 60%, the emission intensity gradually decreased, and the spectrum exhibited a continuous red shift, which can be attributed to the ICT effect.19 With further increases in fw, the emission intensity significantly increased again, accompanied by a gradual blue shift. In the aqueous mixture with fw of 99%, the emission shifted to 567 nm. Additionally, a higher quantum efficiency (94%) was observed for the powder sample compared to the THF solution and the aggregated state, indicating that 18 exhibits both aggregation-induced emission (AIE) and AIE enhancement (AIEE) properties. For this arylated o-carborane with a fused structure at the neighboring carbon and boron atoms for fixing molecular conformation, the elongation of the Ccage–Ccage bond in the excited state, followed by nonradiative decay, was proposed as the primary mechanism for emission quenching in solution.20
 | ||
| Fig. 4 (a) Emission spectra of 18 in the binary THF/H2O solution with different volumetric ratios (fw, vol%) of water (c = 100 μM, λex = 360 nm). (b) Dependence of I/I0 ratios of 18 in THF with different water fractions. (c) Photographs of 18 in THF/H2O solution with various fractions of water (fw) taken under UV light (365 nm). | ||
Conclusions
In summary, an efficient intramolecular aerobic Pd-catalyzed BH/CH oxidative coupling has been achieved, leading to the facile synthesis of previously unavailable carborane-fused five-, six- and seven-membered ring structures under mild conditions. A plausible reaction mechanism including sequential electrophilic B–H and C–H activation, and reductive elimination was proposed, which is supported by the deuterium labeling experiment and DFT calculations. The facile synthesis has potential application in carborane-based luminogens.Data availability
The experimental procedures and additional data can be found in the ESI.† Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition number 2406471 (2aa), 2406472 (2ha), 2406473 (5), 2406474 (6), 2406475 (8a), 2406476 (8x), 2406477 (9x), 2406478 (11), 2406479 (12), 2406480 (14b), 2406481 (16), and 2406482 (18). Copies of the data can be obtained free of charge from the CCDC viahttps://www.ccdc.cam.ac.uk/structures/.Author contributions
Z. Q. and Z. X. directed and conceived this project. Z. W. and J. Y. conducted the experiments. J. Z. did the theoretical work. All authors discussed the results and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Project No. 22371290 and 92056106 to Z. Q.; Project No. 22331005 to Z. X.), Shenzhen Science and Technology Program (Project No. KQTD20221101093558015 to Z. X.), and the Shanghai-Hong Kong Joint Laboratory in Chemical Synthesis, CAS.Notes and references
- (a) R. N. Grimes, Carboranes, Academic Press, Amsterdam, The Netherlands, 3rd edn, 2016 Search PubMed; (b) N. S. Hosmane and R. D. Eagling, Handbook of Boron Science, World Scientific, 2018, vol. 2, pp. 1–155 Search PubMed; (c) J. Poater, C. Viñas, M. Sola and F. Teixidor, Nat. Commun., 2022, 13, 3844 CrossRef CAS PubMed; (d) F. Sun, S. Tan, H.-J. Cao, C.-S. Lu, D. Tu, J. Poater, M. Solà and H. Yan, J. Am. Chem. Soc., 2023, 145, 3577–3587 CrossRef CAS PubMed.
- (a) R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307 CrossRef PubMed; (b) S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070–1093 RSC; (c) X. Li, H. Yan and Q. Zhao, Chem.–Eur. J., 2016, 22, 1888–1898 CrossRef CAS PubMed; (d) R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC; (e) B. P. Dash, R. Satapathy, J. A. Maguire and N. S. Hosmane, New J. Chem., 2011, 35, 1955–1972 RSC.
- (a) M. F. Hawthorne and A. Maderna, Chem. Rev., 1999, 99, 3421–3434 CrossRef CAS PubMed; (b) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed; (c) P. Stockmann, M. Gozzi, R. Kuhnert, M. B. Sárosi and E. Hey-Hawkins, Chem. Soc. Rev., 2019, 48, 3497–3512 RSC; (d) K. Fink and M. Uchman, Coord. Chem. Rev., 2021, 431, 213684 CrossRef CAS.
- (a) K. Kokado and Y. Chujo, J. Org. Chem., 2011, 76, 316–319 CrossRef CAS PubMed; (b) A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590–596 CrossRef CAS PubMed.
- (a) K. Kokado and Y. Chujo, Macromolecules, 2009, 42, 1418–1420 CrossRef CAS; (b) K. R. Wee, Y. J. Cho, J. K. Song and S. O. Kang, Angew. Chem., Int. Ed., 2013, 52, 9682–9685 CrossRef CAS PubMed; (c) H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2015, 54, 5084–5087 CrossRef CAS PubMed; (d) D. Tu, P. Leong, S. Guo, H. Yan, C. Lu and Q. Zhao, Angew. Chem., Int. Ed., 2017, 56, 11370–11374 CrossRef CAS PubMed; (e) X. Li, Y. Yin, H. Yan and C. Lu, Chem.–Asian J., 2017, 12, 2207–2210 CrossRef CAS PubMed; (f) R. Huang, H. Liu, K. Liu, G. Wang, Q. Liu, Z. Wang, T. Liu, R. Miao, H. Peng and Y. Fang, Anal. Chem., 2019, 91, 14451–14457 CrossRef CAS PubMed; (g) X. Wei, M. J. Zhu, Z. Cheng, M. Lee, H. Yan, C. Lu and J. J. Xu, Angew. Chem., Int. Ed., 2019, 58, 3162–3166 CrossRef CAS PubMed; (h) J. Ochi, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2020, 59, 9841–9855 CrossRef CAS PubMed; (i) K. Tanaka, M. Gon, S. Ito, J. Ochi and Y. Chujo, Coord. Chem. Rev., 2022, 472, 1–34 CrossRef; (j) Z. Wang, X. Gou, Q. Shi, K. Liu, X. Chang, G. Wang, W. Xu, S. Lin, T. Liu and Y. Fang, Angew. Chem., Int. Ed., 2022, 61, e202207619 CrossRef CAS PubMed; (k) H. Yang, H. Liu, Y. Shen, S. T. Zhang, Q. Zhang, Q. Song, C. Lv, C. Zhang, B. Yang, Y. Ma and Y. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202115551 CrossRef CAS PubMed; (l) R. Chen, J. Liu, C. Lin, Y. Li, Y. Geng and Y. Yuan, Chin. Chem. Lett., 2024, 35, 110074 CrossRef CAS; (m) T. Lee, J. H. Jang, N. N. T. Nguyen, J. Jung, J. H. Lee and M. H. Lee, Adv. Sci., 2024, 11, 2309016 CrossRef CAS PubMed; (n) K. Yuhara and K. Tanaka, Angew. Chem., Int. Ed., 2024, 63, e202319712 CrossRef CAS PubMed; (o) Z. Sun, J. Zong, H. Ren, C. Lu, D. Tu, J. Poater, M. Solà, Z. Shi and H. Yan, Nat. Commun., 2024, 15, 7934 CrossRef CAS PubMed.
- (a) Y. Quan, Z. Qiu and Z. Xie, Chem.–Eur. J., 2018, 24, 2795–2805 CrossRef CAS PubMed; (b) Y. Quan and Z. Xie, Chem. Soc. Rev., 2019, 48, 3660–3673 RSC; (c) Y. K. Au and Z. Xie, Bull. Chem. Soc. Jpn., 2021, 94, 879–899 CrossRef CAS; (d) Z. Qiu and Z. Xie, Acc. Chem. Res., 2021, 54, 4065–4079 CrossRef CAS PubMed.
- Y. Quan, H. Lyu and Z. Xie, Chem. Commun., 2017, 53, 4818–4821 RSC.
- Y. K. Au, H. Lyu, Y. Quan and Z. Xie, Chin. J. Chem., 2020, 38, 383–388 CrossRef CAS.
- (a) Y. K. Au, H. Lyu, Y. Quan and Z. Xie, J. Am. Chem. Soc., 2019, 141, 12855–12862 CrossRef CAS PubMed; (b) K. Cao, J. Wu, C. Y. Zhang, L. F. Ding and J. Yang, ChemistrySelect, 2021, 6, 10178–10181 CrossRef CAS.
- Y. Chen, Y. Quan and Z. Xie, Chem. Commun., 2020, 56, 7001–7004 RSC.
- Y. Chen, H. Lyu, Y. Quan and Z. Xie, Org. Lett., 2021, 23, 4163–4167 CrossRef CAS PubMed.
- (a) J. P. Parrish, Y. C. Jung, S. I. Shin and K. W. Jung, J. Org. Chem., 2002, 67, 7127–7130 CrossRef CAS PubMed; (b) B. J. Li, S. L. Tian, Z. Fang and Z. J. Shi, Angew. Chem., Int. Ed., 2008, 47, 1115–1118 CrossRef CAS PubMed; (c) P. Kannaboina, K. A. Kumar and P. Das, Org. Lett., 2016, 18, 900–903 CrossRef CAS PubMed; (d) D. Wang and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 5704–5707 CrossRef CAS PubMed; (e) S. J. Tereniak, D. L. Bruns and S. S. Stahl, J. Am. Chem. Soc., 2020, 142, 20318–20323 CrossRef CAS PubMed.
- (a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (b) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed; (c) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed.
- F. Teixidor, G. Barberà, A. Vaca, R. Kivekäs, R. Sillanpää, J. Oliva and C. Viñas, J. Am. Chem. Soc., 2005, 127, 10158–10159 CrossRef CAS PubMed.
- X. Zhang, H. Dai, H. Yan, W. Zou and D. Cremer, J. Am. Chem. Soc., 2016, 138, 4334–4337 CrossRef CAS PubMed.
- (a) K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Org. Lett., 2016, 18, 4064–4067 CrossRef CAS PubMed; (b) X. Wu, J. Guo, J. Zhao, Y. Che, D. Jia and Y. Chen, Dyes Pigm., 2018, 154, 44–51 CrossRef CAS; (c) S. Kim, J. H. Lee, H. So, M. Kim, M. S. Mun, H. Hwang, M. H. Park and K. M. Lee, Inorg. Chem. Front., 2020, 7, 2949–2959 RSC.
- B. P. Dash, R. Satapathy, E. R. Gaillard, K. M. Norton, J. A. Maguire, N. Chug and N. S. Hosmane, Inorg. Chem., 2011, 50, 5485–5493 CrossRef CAS PubMed.
- (a) Y. Yin, X. Li, S. Yan, H. Yan and C. Lu, Chem.–Asian J., 2018, 13, 3155–3159 CrossRef CAS PubMed; (b) L. Guo, X. Yu, J. Du, W. Li, V. Bregadze, D. Tu, C. Lu and H. Yan, Chem.–Eur. J., 2022, 28, e202200303 CrossRef CAS PubMed; (c) M. Zhu, Q. Zhou, H. Cheng, Z. Meng, L. Xiang, Y. Sha, H. Yan and X. Li, New J. Chem., 2022, 46, 11382–11388 RSC; (d) W. Fang, K. Liu, G. Wang, Y. Liang, R. Huang, T. Liu, L. Ding, J. Peng, H. Peng and Y. Fang, Anal. Chem., 2021, 93, 8501–8507 CrossRef CAS PubMed; (e) J. Tong, Y. Cao, Y. W. Zhang, P. Wang, P. Wang, X. J. Liao, W. Zhang, Y. Wang, Y. X. Zheng, J. J. Zhu and Y. Pan, Angew. Chem., Int. Ed., 2022, 61, e202209438 CrossRef CAS PubMed.
- (a) R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CrossRef CAS; (b) E. Wang, J. W. Y. Lam, R. Hu, C. Zhang, Y. S. Zhao and B. Z. Tang, J. Mater. Chem. C, 2014, 2, 1801–1807 RSC.
- (a) J. Ochi, K. Tanaka and Y. Chujo, Dalton Trans., 2021, 50, 1025–1033 RSC; (b) J. Ochi, T. Yanagihara, K. Tanaka and Y. Chujo, Phys. Chem. Chem. Phys., 2023, 25, 11839–11844 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures for synthesis of compounds, NMR spectra and X-ray data. CCDC 2406471–2406482. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08496a |
| This journal is © The Royal Society of Chemistry 2025 |