
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogenation-induced C–N bond activation enables the synthesis of 1,2-cis C-aryl furanosides via deaminative cyclization†
Wenbo
Wang
a,
Jiawei
Wu
a,
Kaiyu
Jiang
 a,
Maochao
Zhou
a and
Gang
He
*ab
a,
Maochao
Zhou
a and
Gang
He
*ab
aState Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: hegang@nankai.edu.cn
bFrontiers Science Center for New Organic Matter, Nankai University, Tianjin 300071, China
First published on 2nd December 2024
Abstract
1,2-cis C-Aryl furanosides are prevalent in nature and exhibit significant biological activities. The 1,2-cis configuration is less favorable in terms of stereoelectronic and steric effects, making the synthesis of this type of skeleton highly challenging. Traditional methods for the synthesis of 1,2-cis C-aryl furanosides usually require complicated protection manipulations, resulting in lengthy synthetic routes and low overall efficiency. Here, we report a simple and highly applicable procedure for the synthesis of 1,2-cis C-aryl furanosides from unprotected aldoses via Petasis reaction and subsequent deaminative cyclization. Unprotected aldose mediated Petasis reactions yield linear 1,2-trans 1-aryl polyhydroxy amines. Halogenation of the amine motif activates the conventionally inert C–N bond and triggers the key stereoinvertive intramolecular substitution process, affording 1,2-cis C-aryl furanosides with excellent chemo- and diastereoselectivity. This procedure does not require the use of any sensitive reagents, and can be conducted in one-pot without precautions against oxygen or moisture, offering a streamlined approach to 1,2-cis C-aryl furanoside natural products and bioactive agents.
Introduction
1,2-cis C-Aryl furanosides represent a unique class of carbohydrates in which aromatic aglycons are linked to the anomeric position of furanoses via C–C glycosidic bonds from the same side as the C2 hydroxy group (C2-OH). This type of structure has been found in natural products isolated from traditional oriental herbs, the metabolites of bacteria or fungi, and has shown significant biological activity (Scheme 1A).1–5 Notably, gilvocarcin V produced by the genus Streptomyces has shown remarkable antitumor activity and exceptionally low toxicity.6–9 Due to their potent bioactivities, 1,2-cis C-aryl furanosides are attractive targets for total synthesis and medicinal chemistry.10–16 However, the C–C bond linkage and 1,2-cis configuration pose great obstacles for the synthesis of these types of skeletons. | ||
| Scheme 1 Occurrence and synthetic strategies for 1,2-cis C-aryl furanosides. PO: protected hydroxy groups; LG: leaving group; BDE: bond dissociation energy; NBS: N-bromosuccinimide; NCS: N-chlorosuccinimide. | ||
Naturally available unprotected hexoses and pentoses exist in solution as equilibrium mixtures of cyclic hemiacetals and linear glycosyl aldehydes, in which the pyranose forms predominate and the furanose forms are less abundant at equilibrium (Scheme 1B). To achieve the synthesis of 1,2-cis C-aryl furanosides from readily available aldoses, strategies with high selectivity are needed to ensure the construction of the furan skeleton, the C–C bond linkage, and most importantly, the 1,2-cis configuration. Traditional methods for the synthesis of 1,2-cis C-aryl furanosides usually first convert pyranose hemiacetals into highly protected furanosyl donors, and then construct C–C bonds via Lewis-acid-promoted glycosylation reactions, such as the Friedel–Crafts reaction with electron-rich arenes,17 O-to-C rearrangement of O-aryl glycosides18 and C2-OH-directed intramolecular aglycone delivery.19–21 Alternatively, nucleophilic addition to linear glycosyl aldehydes with aryl reagents yields 1-aryl alditol intermediates, and cyclization through an intramolecular substitution can also afford C-aryl furanosides.22–25 However, anionic aryl nucleophiles with high basicity, such as aryl lithium reagents and Grignard reagents, are needed. The addition reactions suffer from low diastereoselectivity. Inevitably, complicated protection manipulations are indispensable in the aforementioned methods, resulting in lengthy synthetic routes and low overall efficiency. Recently, List reported a concise procedure for the synthesis of the C-nucleoside of remdesivir from the reaction of unprotected D-ribose with electron-rich arenes.26 Cyclization of the temporarily protected linear 1,2-trans alditol intermediate gave 1,2-cis C-aryl furanoside as the major product (dr up to 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under acidic conditions. Despite these achievements, the synthesis of 1,2-cis C-aryl furanosides has lagged far behind the synthesis of C-glycosides.27–37 Methods that enable the stereoselective synthesis of 1,2-cis C-aryl furanosides directly from unprotected aldoses are still greatly needed.
:1) under acidic conditions. Despite these achievements, the synthesis of 1,2-cis C-aryl furanosides has lagged far behind the synthesis of C-glycosides.27–37 Methods that enable the stereoselective synthesis of 1,2-cis C-aryl furanosides directly from unprotected aldoses are still greatly needed.
We envision that the C–C bond linkage between unprotected aldoses and aryl motifs can be constructed via a three-component Petasis reaction, wherein aryl-boronic acids or boronates are used as nucleophiles to attack the imines generated in situ from the condensation of aldehydes and amines.38–44 The presence of a hydroxy group at the α position of the aldehyde can accelerate the nucleophilic addition process and control the diastereoselectivity, producing 1,2-trans hydroxy amines.45–51 Then deaminative cyclization of the 1,2-trans 1-aryl-polyhydroxy amines via intramolecular SN2 substitution provides a concise procedure for the synthesis of 1,2-cis C-aryl furanosides (Scheme 1B). However, organic amines are unarguably weak acids and poor leaving groups in the SN2 reaction.52 To increase their nucleofugality (leaving ability), primary amines must be converted to bis-sulfonimides,53 diazo ions54 or pyridinium cations (Katritzky salt),55,56 and the C–N bond of tertiary amines can be activated in the form of quaternary ammonium salts by treatment with stoichiometric amounts of electrophiles, such as methyl iodide, cyanogen bromide (von Braun reaction) or difluorocarbenes (Scheme 1C).57–74 For 1,2-trans 1-aryl-polyhydroxy amines, the inherent multiple free hydroxy groups may also interfere with these electrophiles, making the selective cleavage of C–N bonds a challenging transformation.75–78 Herein, we report an efficient approach for the activation of the C–N bond and triggering of the key stereoinvertive intramolecular substitution process via halogenation of the amine moiety with N-bromosuccinimide or N-chlorosuccinimide (Scheme 1D). Combined with p-toluidine- or indoline-mediated Petasis reactions of unprotected aldoses with aryl trifluoroborates, this procedure provides a general route for the synthesis of 1,2-cis C-aryl furanoside natural products and their analogs from unprotected aldoses in two steps.
Results and discussion
Optimization of the reaction conditions
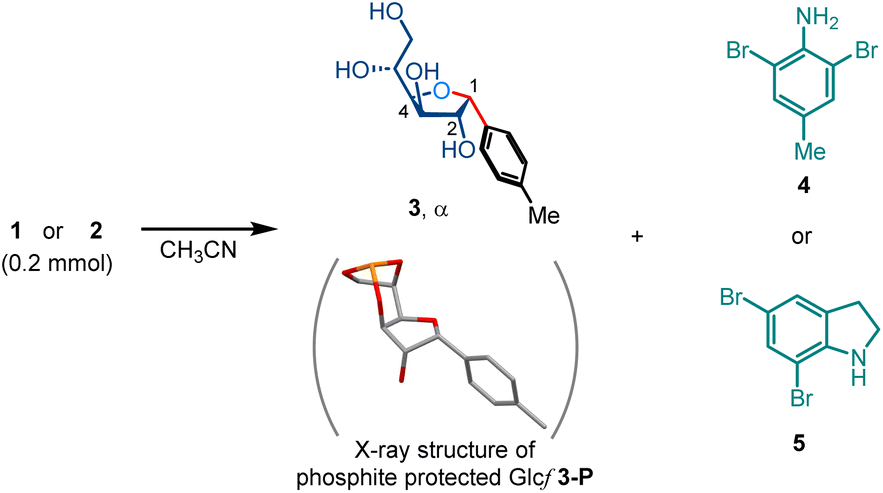
As shown in Scheme 2, p-toluidine and indoline were found to be the optimal amine sources in the Petasis reaction of unprotected glucose with potassium(4-methylphenyl)-trifluoroborate, affording the linear 1-aryl polyhydroxy amines 1 and 2 with excellent 1,2-trans selectivity (see the ESI for the optimization of the Petasis reaction†). Treatment of 1 or 2 with NBS (2.1 equiv.) at elevated temperature in acetonitrile gave 1,2-cis C-aryl furanoside 3 in 85% or 76% isolated yield, respectively, with inversion of the stereochemistry at the C1 position (Table 1, entries 1 & 5). The structure of 3 was confirmed by X-ray crystallographic analysis of its phosphite derivative 3-P.79 In addition to succinimide, 2,6-dibromo-4-methylaniline 4 (91%) and 5,7-dibromoindoline 5 (79%) were identified as byproducts. The deaminative cyclization reaction proceeded smoothly at 60 °C (entries 2 & 6) and even at room temperature (entry 7) with decent yields. The addition of aq. HBr (1.0 equiv.) accelerated the cyclization of 1 at room temperature, giving 3 in 97% NMR yield (entry 3). NCS afforded yields comparable to those of NBS, but required slightly longer reaction times (entries 4 & 8). Notably, the Petasis reaction and deaminative cyclization reactions were conducted under an air atmosphere, and did not require any precautions against moisture. | ||
| Scheme 2 Synthesis of 1,2-trans 1-aryl-polyhydroxy amine via Petasis reaction. Isolated yield on a scale of 0.2 mmol. | ||
|
|
||
|---|---|---|
| Entry | Reagents and conditions | Yield of 3 |
| a All screening reactions were carried out in an 8 mL glass vial on a scale of 0.2 mmol. b Yields were based on 1H NMR analysis of the reaction mixture with 1,1,2,2-tetrachloroethane as internal standard. c Isolated yield. d The amount of 2 remaining. | ||
| 1 | 1, NBS, 80 °C, 5 min | 97% (85% + 4, 91%)c |
| 2 | 1, NBS, 60 °C, 6 h | 70% |
| 3 | 1, NBS, aq. HBr, rt, 6 h | 97% |
| 4 | 1, NCS, 100 °C, 30 min | 95% |
| 5 | 2, NBS, 100 °C, 5 min | 89% (76% + 5, 79%)c |
| 6 | 2, NBS, 60 °C, 8 h | 75% |
| 7 | 2, NBS, rt, 24 h | 61% (20%)d |
| 8 | 2, NCS, 100 °C, 1 h | 85% |
Mechanistic studies
The inverted configuration and brominated arylamine byproducts strongly indicate that the cyclization reaction may have occurred in an intramolecular SN2 substitution fashion. To verify the mechanism of this deaminative cyclization reaction, enantioenriched substrate (R)-6 was prepared and subjected to the halogenation conditions. As the deaminative cyclization proceeded more slowly when NCS was used as the halogenation agent (Table 1, entry 1 vs. entry 4), (R)-6 (85% ee) was treated with NCS to obtain some intermediates that participated in the cyclization reaction.As shown in Scheme 3A, cyclized product (S)-7 was isolated in 28% yield with slightly eroded enantiopurity (78% ee), along with 59% ortho dichlorinated intermediate (R)-8 (84% ee) and 31% 2,6-dichloro-4-methylaniline 9. Reheating (R)-8 in the absence of any additive delivered (S)-7 in only 12% NMR yield, and the addition of succinimide inhibited the cyclization reaction (Scheme 3B). Gratifyingly, the addition of a catalytic amount of NCS (0.1 equiv.) restored the reactivity, affording (S)-7 in 91% yield, albeit with diminished enantiopurity (75% ee). The partial racemization may arise from interference by the SN1 reaction pathway at elevated temperatures. Lowering the reaction temperature to 60 °C led to low conversion and also reduced the likelihood of racemization, giving (S)-7 in 9% yield with 83% ee. These results support the presence of the ortho dichlorinated intermediate during the cyclization reaction, and suggest that NCS plays essential roles beyond halogenating the phenyl skeleton. NCS can transfer the chloronium ion to the nitrogen atom of aromatic amines, generating quaternary ammonium salts,80,81 thus increasing their nucleofugality. To verify this quaternization effect, Brønsted acids that can protonize the amine motif were tested. As shown in Scheme 3B, aqueous hydrobromide (aq. HBr) can promote the cyclization of (R)-8, affording results comparable to those of NCS.
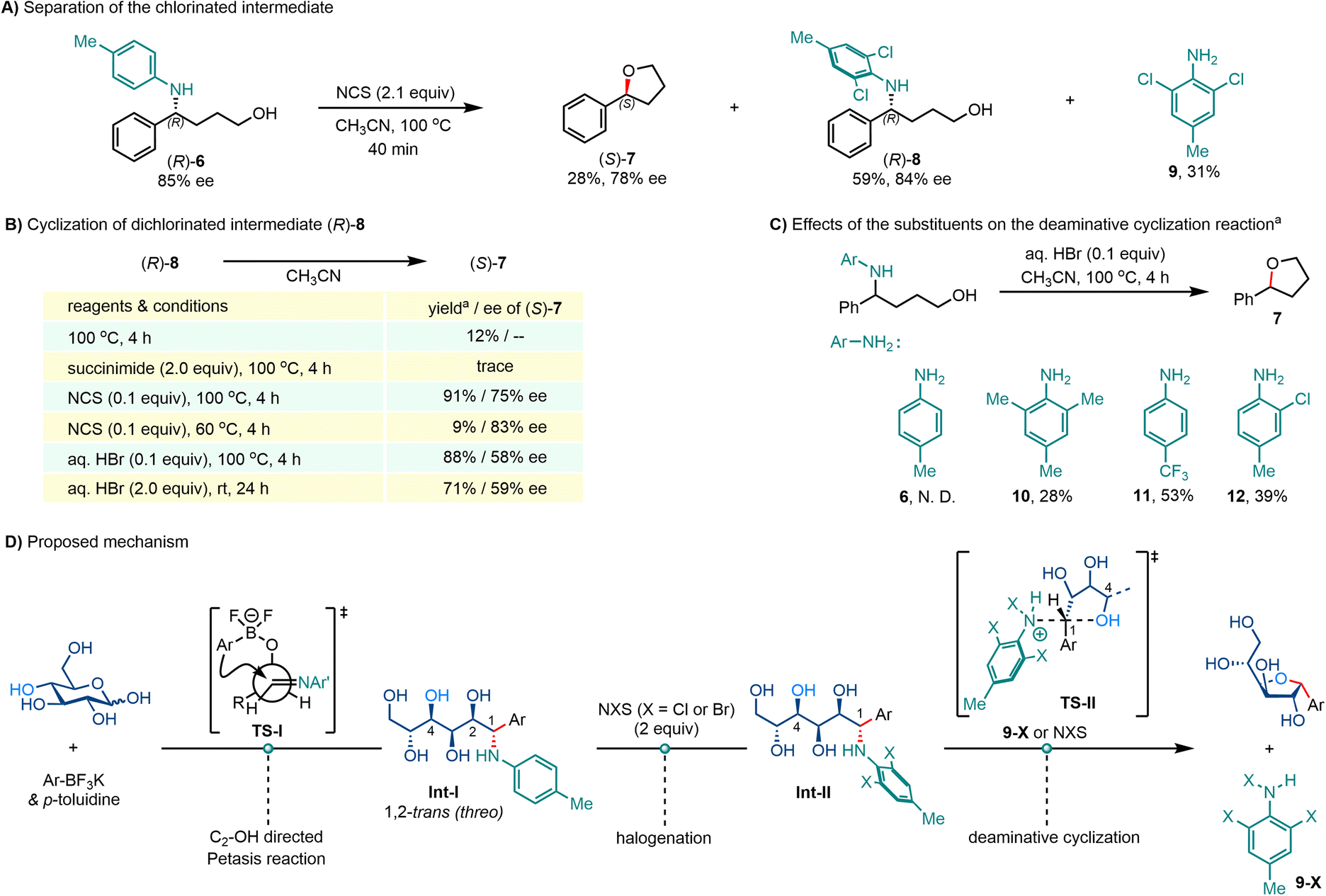 | ||
| Scheme 3 Mechanistic studies. All reactions were carried out in an 8 mL glass vial on a scale of 0.2 mmol. aYields were based on 1H NMR analysis of the reaction mixture with 1,1,2,2-tetrachloroethane as internal standard. N. D.: not detected. | ||
It is evident that quaternization at the nitrogen atom is crucial for inducing selective C–N bond cleavage. Quaternization enhances the acidity of the aryl-amine motif, making it a better leaving group than the neutral form. In addition, chlorination at the ortho position of the amine group may also facilitate C–N bond cleavage by exerting both steric effects and electronic effects. As shown in Scheme 3C, unchlorinated 6 failed to give any cyclized product with a catalytic amount (0.1 equiv.) or excess (2.0 equiv.) of aq. HBr even at 100 °C. By contrast, increasing the steric hindrance by introducing two ortho-methyl groups (10), or increasing the electron deficiency by replacing the para-methyl group with the CF3 strong electron-withdrawing group (11), afforded much improved yields. Cyclization of mono-chlorinated amino-alcohol (12) also afforded 7 in 39% yield. This type of substituent induced weakening of σ bonds was also observed in the C(Me)–O bond cleavage of anisoles and was supported by DFT calculations.82 Although amines 10–12 were amenable for the deaminative cyclization reaction, these types of anilines were unable to participate in the Petasis reaction with unprotected aldoses. Overall, to realize this two-step protocol, arylamines with good nucleophilicity, such as p-toluidine or indoline, are required in the first-step Petasis reaction; then, halogenation converts them to better leaving groups, making the deaminative cyclization reaction feasible.
One the basis of the above results and previous reports, a possible mechanism for this two-step procedure is proposed in Scheme 3D. In the Petasis reaction, p-toluidine reacts with the linear aldehyde to form imine, and at the same time, C2-OH captures the arylborate to give a boronate complex.50,51 Then, intramolecular transfer of the aryl group proceeds through transition state TS-I to minimize the 1,3-allylic strain, giving 1-aryl polyhydroxy amine Int-I with excellent trans selectivity. Halogenation at the aromatic amine skeleton affords Int-II, and quaternization at the nitrogen atom activates the C–N bond and then triggers the intramolecular SN2 reaction with the C4-OH group (TS-II), affording 1,2-cis C-aryl furanoside. N-Halogenated byproduct 9-X (X = Cl or Br) can serve as a halogenation reagent that transfers the halogen cation to succinimide, regenerating NXS, or halogenates Int-II directly to initiate the next round of the deaminative cyclization reaction. The N-chloroaniline intermediates are highly labile and generally not isolated.83,84 Our efforts toward the isolation of 9-Cl also failed. The formation of furanosides has been proven to be kinetically favored over the formation of pyranosides,85,86 which is further verified here, as the presence of C5-OH or C6-OH does not affect the reaction, and no pyranoside or seven-membered product was detected.
 | ||
| Scheme 4 Substrate scope. Isolated yields on a 0.2 mmol scale. aInseparable anomers. | ||
This two-step procedure is compatible with a broad range of unprotected aldoses (Scheme 4B). Free D-galactose (29), L-fucose (30), and even isomaltose (31) with seven unprotected hydroxy groups, reacted smoothly to give 1,2-cis C-aryl furanosides in good yields. In addition to hexoses, unprotected pentoses were competent substrates in this procedure. As exemplified by compounds 32–39, C-aryl furanosides were obtained from reactions of D-arabinose (32), D-xylose (33, 34) and D-ribose (35–39) with an exclusively 1,2-cis configuration.88 This provides a potential method for the preparation of α-C-nucleosides. The Petasis reaction of D-mannose, L-rhamnose and D-lyxose afforded 1-aryl polyhydroxy amines with exclusively 1,2-trans selectivity (see the ESI for details†).89 The subsequent deaminative cyclization reaction delivered the products as mixtures of separable anomers (40–42). The structures of 40α and 40β were confirmed by X-ray crystallographic analysis after being protected with isopropylidene groups (40′α & 40′β).90 Among the expected products (40β, 41β and 42β) generated via the SN2 pathway, all of the substituents on the furannosyl skeleton are located on the same side. This crowded all-cis configuration would cause severe steric repulsion in the transition states of the SN2 pathway, that some α anomers were generated via the SN1 pathway. Nevertheless, β anomers were still obtained as the major products, underscoring the power of the developed synthetic procedure in the synthesis of challenging all-cis C-aryl furanosides.
:1). After the removal of the isopropyl group with BBr3, natural products neopuerarin A (49) and neopuerarin B (50) were obtained in 31% and 31% yields, respectively (Scheme 5C). The structures of 49 and 50 were confirmed by comparison with data reported in the literature1,2 and X-ray crystallographic analysis of the sodium salt of 49.92
 | ||
| Scheme 5 Synthetic utility. aIsolated yields on a 0.2 mmol scale. | ||
Conclusions
In summary, we have developed an efficient two-step method for the synthesis of 1,2-cis C-aryl furanosides from unprotected aldoses. p-Toluidine- or indoline-mediated Petasis reactions with aldoses and potassium aryl-trifluoroborates enable the key C1–C(Ar) bond coupling and covert cyclic pyranoses to linear 1-aryl polyhydroxy amines with excellent 1,2-trans selectivity; subsequent selective halogenation at the aryl-amine motif activates the C–N bond and triggers intramolecular SN2 substitution, furnishing 1,2-cis C-aryl furanosides. This “open-and-close” procedure is compatible with a broad range of free aldoses and aryl-trifluoroborates. The use of readily available reagents, mild reaction conditions and simple operation make this procedure highly practical for accessing 1,2-cis C-aryl furanoside natural products. This halogenation-enabled C–N bond cleavage can inspire additional strategies for the selective transformation of amine-containing substrates.Data availability
Detailed synthetic procedures, compound characterization, NMR spectra, and X-ray crystallographic data are provided in the ESI.† Crystallographic data for 3-P, 34, 39, 40-S, 40′α, 40′β and 49-Na have been deposited at the Cambridge Crystallographic Data Centre.Author contributions
W. W. made the initial discovery of the project, finished most of the experiments and prepared the ESI.† J. W. helped with the structural identification of the products and prepared some starting material. K. J. helped with the mechanistic studies. M. Z. helped with the separation of neopuerarins. G. H. formulated the initial ideas of this work, supervised the project and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (no. 22071119, 22271163, 22221002), Frontiers Science Center for New Organic Matter (63181206), the Fundamental Research Funds for the Central Universities (63243099, 63241203), and Haihe Laboratory of Sustainable Chemical Transformations.Notes and references
- H.-J. Zhang, X.-P. Yang and K.-W. Wang, J. Asian Nat. Prod. Res., 2010, 12, 293–299 CrossRef CAS.
- Q.-W. Hu, H.-Y. Xiang, J.-M. Shan, Q.-Y. Jiao, S. Lv, L.-Y. Li, F. Li, D.-M. Ren and H.-X. Lou, Fitoterapia, 2020, 144, 104594–104599 CrossRef CAS.
- M. A. Asmaey, D. Abatis, A. S. Abdel-Razek, G. Lambrinidis, I. Chinou, N. Fokialakis, N. Tsafantakis, M. Shaaban and N. Aligiannis, Molecules, 2021, 26, 3976–3988 CrossRef CAS.
- F. Li, Z.-S. He and Y. Ye, Acta Pharm. Sin. B, 2017, 7, 527–531 CrossRef PubMed.
- R. Ma, S. Cheng, J.-W. Sun, W.-M. Zhu and P. Fu, J. Nat. Prod., 2022, 85, 2282–2289 CrossRef CAS.
- H. Nakano, Y. Matsuda, K. Ito, S. Ohkubo, M. Morimoto and F. Tomita, J. Antibiot., 1981, 34, 266–270 CrossRef CAS PubMed.
- K. Takahashi, M. Yoshida, F. Tomita and K. Shirahata, J. Antibiot., 1981, 34, 271–275 CrossRef CAS.
- R. K. Elespuru and S. K. Gonda, Science, 1984, 223, 69–71 CrossRef CAS.
- L. R. McGee and R. Misra, J. Am. Chem. Soc., 1990, 112, 2386–2389 CrossRef CAS.
- Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281–12356 CrossRef CAS.
- K. Kitamura, Y. Ando, T. Matsumoto and K. Suzuki, Chem. Rev., 2018, 118, 1495–1598 CrossRef CAS PubMed.
- T. Matsumoto, T. Hosoya and K. Suzuki, J. Am. Chem. Soc., 1992, 114, 3568–3570 CrossRef CAS.
- T. Hosoya, E. Takashiro, T. Matsumoto and K. Suzuki, J. Am. Chem. Soc., 1994, 116, 1004–1015 CrossRef CAS.
- M. Morales-Chamorro, O. Cortezano-Arellano and A. Cordero-Vargas, Synthesis, 2016, 48, 4555–4563 CrossRef CAS.
- F. Wu, J. Zhang, F.-H. Song, S.-S. Wang, H. Guo, Q. Wie, H.-Q. Dai, X.-Y. Chen, X.-K. Xia, X.-T. Liu, L.-X. Zhang, J.-Q. Yu and X.-G. Lei, ACS Cent. Sci., 2020, 6, 928–938 CrossRef CAS.
- A. Cordero-Vargas, B. Quiclet-Sire and S. Z. Zard, Org. Biomol. Chem., 2005, 3, 4432–4443 RSC.
- R. N. Farr, D.-I. Kwok and G. D. Daves, J. Org. Chem., 1992, 57, 2093–2100 CrossRef CAS.
- T. Matsumoto, M. Katsuki and K. Suzuki, Tetrahedron Lett., 1988, 29, 6935–6938 CrossRef CAS.
- I. Singh and O. Seitz, Org. Lett., 2006, 8, 4319–4322 CrossRef CAS.
- A. Nakazaki, J. Usuki and K. Tomooka, Synlett, 2008, 13, 2064–2068 Search PubMed.
- O. R. Martin, S. P. Rao, K. G. Kurz and H. A. El-Shenawy, J. Am. Chem. Soc., 1988, 110, 8698–8700 CrossRef CAS.
- W. Frick and R. R. Schmidt, Liebigs Ann. Chem., 1989, 565–570 CrossRef CAS.
- E. Ravarino, S. K. Jana, R. Frohlich and R. Holl, Carbohydr. Res., 2012, 361, 162–169 CrossRef CAS PubMed.
- R. Cribiù, K. Eszter Borbas and I. Cumpstey, Tetrahedron, 2009, 65, 2022–2031 CrossRef.
- For an efficient method for the synthesis of both 1,2-cis and 1,2-trans C-aryl furanosides via reduction, see: M. Peifer, R. Berger, V. W. Shurtleff, J. C. Conrad and D. W. C. Macmillan, J. Am. Chem. Soc., 2014, 136, 5900–5903 CrossRef CAS PubMed.
- C. Obradors, B. Mitschke, M. H. Aukland, M. Leutzsch, O. Grossmann, S. Brunen, S. A. Schwengers and B. List, Angew. Chem., Int. Ed., 2022, 61, e202114619 CrossRef CAS.
- F. Zhu, S. O'Neill, J. Rodriguez and M. A. Walczak, Chem.–Eur. J., 2019, 25, 3147–3155 CrossRef CAS PubMed.
- F. Li and J. Qu, Chin. J. Org. Chem., 2021, 41, 3949–3964 Search PubMed.
- J. Ghouilem, M. de Robichon, F. Le Bideau, A. Ferry and S. Messaoudi, Chem.–Eur. J., 2021, 27, 491–511 CrossRef CAS.
- H. Gong and M. R. Gagné, J. Am. Chem. Soc., 2008, 130, 12177–12183 CrossRef CAS.
- Q.-Q. Wang, B. C. Lee, N.-X. Song and M. J. Koh, Angew. Chem., Int. Ed., 2023, 62, e202301081 CrossRef CAS PubMed.
- Y. Jiang, Y. Wei, Q.-Y. Zhou, G.-Q. Sun, X.-P. Fu, N. Levin, Y. Zhang, W.-Q. Liu, N. Song, S. Mohammed, B. G. Davis and M. J. Koh, Nature, 2024, 631, 319–327 CrossRef CAS.
- C. Zhang, S.-Y. Xu, H. Zuo, X. Zhang, Q.-D. Dang and D.-W. Niu, Nat. Synth., 2023, 2, 251–260 CrossRef CAS.
- A.-R. Chen, B. Yang, Z.-H. Zhou and F. Zhu, Chem Catal., 2022, 2, 3430–3470 CrossRef CAS.
- M.-L. Liu, Y.-H. Niu, Y.-F. Wu and X.-S. Ye, Org. Lett., 2016, 18, 1836–1839 CrossRef CAS PubMed.
- Q.-Q. Wang, S. An, Z.-Q. Deng, W.-J. Zhu, Z.-Y. Huang, G. He and G. Chen, Nat. Catal., 2019, 2, 793–800 CrossRef CAS.
- Q.-Q. Wang, Y. Fu, W. Zhu, S. An, Q. Zhou, S.-F. Zhu, G. He, P. Liu and G. Chen, CCS Chem., 2020, 2, 1729–1736 Search PubMed.
- N. A. Petasis and I. Akrltopoulou, Tetrahedron Lett., 1993, 34, 583–586 CrossRef CAS.
- N. R. Candeias, F. Montalbano, P. M. S. D. Cal and P. M. P. Gois, Chem. Rev., 2010, 110, 6169–6193 CrossRef CAS.
- P. Wu, M. Givskov and T. E. Nielsen, Chem. Rev., 2019, 119, 11245–11290 CrossRef CAS.
- M. G. Ricardo, D. Llanes, L. A. Wessjohann and D. G. Rivera, Angew. Chem., Int. Ed., 2019, 58, 2700–2704 CrossRef CAS PubMed.
- S. Krajcovicova and D. R. Spring, Angew. Chem., Int. Ed., 2023, 62, e202307782 CrossRef CAS PubMed.
- N. A. Petasis and I. Zavialov, J. Am. Chem. Soc., 1998, 120, 11798–11799 CrossRef CAS.
- N. Kumagai, G. Muncipinto and S. L. Schreiber, Angew. Chem., Int. Ed., 2006, 45, 3635–3638 CrossRef CAS.
- N. A. Petasis, Multicomponent Reactions with Organoboron Compounds in Multicomponent Reactions, Wiley, Weinheim, 2005, pp. 199–223 Search PubMed.
- A. S. Davis, S. G. Pyne, B. W. Skelton and A. H. White, J. Org. Chem., 2004, 69, 3139–3143 CrossRef CAS.
- S. G. Pyne and C. Wentrup, Aust. J. Chem., 2022, 75, 923–944 CrossRef CAS.
- Z.-Y. Hong, L. Liu, C.-C. Hsu and C.-H. Wong, Angew. Chem., Int. Ed., 2006, 45, 7417–7421 CrossRef CAS PubMed.
- Z.-Y. Hong, L. Liu, M. Sugiyama, Y. Fu and C.-H. Wong, J. Am. Chem. Soc., 2009, 131, 8352–8353 CrossRef CAS.
- J. Tao and S. Li, Chin. J. Chem., 2010, 28, 41–49 CrossRef CAS.
- R. Y. Souza, G. A. Bataglion, D. A. C. Ferreira, C. C. Gatto, M. N. Eberlin and B. A. D. Neto, RSC Adv., 2015, 5, 76337–76341 RSC.
- M. B. Smith, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, 8th edn, 2020, p. 466 Search PubMed.
- P. J. DeChristopher, J. P. Adamek, G. D. Lyon, J. J. Galante, H. E. Haffner, R. J. Boggio and R. J. Baumgarten, J. Am. Chem. Soc., 1969, 91, 2384–2385 CrossRef CAS.
- W. Kirmse, Angew. Chem., Int. Ed., 1976, 15, 251–261 CrossRef.
- A. R. Katritzky, Tetrahedron, 1980, 36, 679–699 CrossRef CAS.
- A. R. Katritzky and C. M. Marson, Angew. Chem., Int. Ed., 1984, 23, 420–429 CrossRef.
- D.-W. Yue and R. C. Larock, Org. Lett., 2004, 6, 1037–1040 CrossRef CAS.
- B. Yao, Q. Wang and J.-P. Zhu, Angew. Chem., Int. Ed., 2012, 51, 5170–5174 CrossRef CAS.
- Y. Gui and S.-K. Tian, Org. Lett., 2017, 19, 1554–1557 CrossRef CAS PubMed.
- Q.-M. Zhu, Q.-H. Yuan, M.-W. Chen, M.-P. Guo and H.-M. Huang, Angew. Chem., Int. Ed., 2017, 56, 5101–5105 CrossRef CAS PubMed.
- D. I. Bugaenko, M. A. Yurovskaya and A. V. Karchava, Org. Lett., 2018, 20, 6389–6393 CrossRef CAS.
- C.-X. Cui, H. Li, X.-J. Yang, J. Yang and X.-Q. Li, Org. Lett., 2013, 15, 5944–5947 CrossRef CAS.
- C. Yu, M. A. Shoaib, N. Iqbal, J. S. Kim, H.-J. Ha and E. J. Cho, J. Org. Chem., 2017, 82, 6615–6620 CrossRef CAS PubMed.
- Y. Kim, J. Heo, D. Kim, S. Chang and S. Seo, Nat. Commun., 2020, 11, 4761–4771 CrossRef CAS PubMed.
- J. Scharfbier, B. M. Gross and M. Oestreich, Angew. Chem., Int. Ed., 2020, 59, 1577–1580 CrossRef CAS PubMed.
- H.-Q. Fang, K.-X. Xie, S. Kemper and M. Oestreich, Angew. Chem., Int. Ed., 2021, 60, 8542–8546 CrossRef CAS PubMed.
- J.-K. Su, X.-X. Ma, Z.-L. Ou and Q.-L. Song, ACS Cent. Sci., 2020, 6, 1819–1826 CrossRef CAS PubMed.
- J.-K. Su, C.-B. Li, X.-Y. Hu, Y. Guo and Q.-L. Song, Angew. Chem., Int. Ed., 2022, 61, e202212740 CrossRef CAS PubMed.
- A. Liu, C.-F. Ni, Q.-Q. Xie and J.-B. Hu, Angew. Chem., Int. Ed., 2022, 61, e202115467 CrossRef CAS PubMed.
- H. Lim, S. Seong, Y. Kim, S. Seo and S. Han, J. Am. Chem. Soc., 2021, 143, 19966–19974 CrossRef CAS.
- Q.-Q. Tang, F.-H. Li, H.-Y. Chen, X.-J. Yin, Y. Tang and Q.-L. Zeng, Asian J. Org. Chem., 2021, 10, 1687–1690 CrossRef CAS.
- A. B. Turner, B. I. McBain, R. A. Howie and P. J. Cox, J. Chem. Soc., Perkin Trans. 1, 1986, 1151–1155 RSC.
- A. B. Turner, Chem. Commun., 1968, 1659–1660 RSC.
- W.-Z. Geng, W.-X. Zhang, W. Hao and Z.-F. Xi, J. Am. Chem. Soc., 2012, 134, 20230–20233 CrossRef CAS PubMed.
- K.-B. Ouyang, W. Hao, W.-X. Zhang and Z.-F. Xi, Chem. Rev., 2015, 115, 12045–12090 CrossRef CAS PubMed.
- S. M. Pound and M. P. Watson, Chem. Commun., 2018, 54, 12286–12301 RSC.
- J.-Y. Xu, O. P. Bercher, M. R. Talley and M. P. Watson, ACS Catal., 2021, 11, 1604–1612 CrossRef CAS PubMed.
- K. J. Berger and M. D. Levin, Org. Biomol. Chem., 2021, 19, 11–36 RSC.
- The X-ray data of 3-P has been deposited at the Cambridge Crystallographic Data Centre with a number of CCDC 2297443, DOI: https://doi.org/10.5517/ccdc.csd.cc2h3p2d.
- R. C. Samanta and H. Yamamoto, Chem.–Eur. J., 2015, 21, 11976–11979 CrossRef CAS PubMed.
- X.-D. Xiong and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2016, 55, 16101–16105 CrossRef CAS PubMed.
- M. M. Suryan, S. A. Kafafi and S. E. Stein, J. Am. Chem. Soc., 1989, 111, 4594–4600 CrossRef CAS.
- R. S. Neale, R. G. Schepers and M. R. Walsh, J. Org. Chem., 1964, 29, 3390–3393 CrossRef CAS.
- P. G. Gassman and G. A. Campbell, J. Am. Chem. Soc., 1971, 93, 2567–2569 CrossRef CAS.
- Y. Seo and M. R. Gagné, ACS Catal., 2018, 8, 6993–6999 CrossRef CAS.
- A. Yamaguchi, N. Hiyoshi, O. Sato, K. K. Bando and M. Shirai, Green Chem., 2009, 11, 48–52 RSC.
- Alkenyl boronic acids participated Petasis reaction gave the 1,2-trans products in good yields, however, no desired C-alkenyl furanoside was obtained under the halogenation conditions.
- The X-ray data of 34 has been deposited at the Cambridge Crystallographic Data Centre with a number of CCDC 2387663, DOI: https://doi.org/10.5517/ccdc.csd.cc2l4kdq; the X-ray data of 39 has been deposited at the Cambridge Crystallographic Data Centre with a number of CCDC 2297325, DOI: https://doi.org/10.5517/ccdc.csd.cc2h3k8g.
- The X-ray data has been deposited at the Cambridge Crystallographic Data Centre with a number of CCDC 2297387, DOI: https://doi.org/10.5517/ccdc.csd.cc2h3m8j.
- The X-ray data of 40′α has been deposited at the Cambridge Crystallographic Data Centre with a number of CCDC 2297359, DOI: https://doi.org/10.5517/ccdc.csd.cc2h3lcl; the X-ray data of 40′β has been deposited at the Cambridge Crystallographic Data Centre with a number of CCDC 2297392, DOI: https://doi.org/10.5517/ccdc.csd.cc2h3mfp.
- W. Meng, B. A. Ellsworth, A. A. Nirschl, P. J. McCann, M. Patel, R. N. Girotra, G. Wu, P. M. Sher, E. P. Morrison, S. A. Biller, R. Zahler, P. P. Deshpande, A. Pullockaran, D. L. Hagan, N. Morgan, J. R. Taylor, M. T. Obermeier, W. G. Humphreys, A. Khanna, L. Discenza, J. G. Robertson, A.-Y. Wang, S.-P. Han, J. R. Wetterau, E. B. Janovitz, O. P. Flint, J. M. Whaley and W. N. Washburn, J. Med. Chem., 2008, 51, 1145–1149 CrossRef CAS PubMed.
- The X-ray data has been deposited at the Cambridge Crystallographic Data Centre with a number of CCDC 2337769, DOI: https://doi.org/10.5517/ccdc.csd.cc2jgmxk.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, optimization, characterization of compounds and spectroscopic data. See DOI: https://doi.org/10.1039/d4sc07410f |
| This journal is © The Royal Society of Chemistry 2025 |