
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of oxygen vacancies on hydrogenation efficiency by spillover in catalysts
Lijuan
Xie
a,
Jinshan
Liang
a,
Lizhi
Jiang
 *a and
Wei
Huang
*abc
*a and
Wei
Huang
*abc
aStrait Institute of Flexible Electronics (SIFE, Future Technologies), Fujian Normal University and Strait Laboratory of Flexible Electronics (SLoFE), Fuzhou, Fujian 350117, China. E-mail: ifelzhjiang@fjnu.edu.cn
bKey Laboratory of Flexible Electronics & Institute of Advanced Materials, Nanjing Tech University, Nanjing, 211816, China. E-mail: vc@nwpu.edu.cn
cFrontiers Science Center for Flexible Electronics, Shaanxi Institute of Flexible Electronics (SIFE), Northwestern Polytechnical University (NPU), 127 West Youyi Road, Xi'an, 710072, China
First published on 28th January 2025
Abstract
Hydrogen spillover is crucial for hydrogenation reactions on supported catalysts. The properties of supports have been reported to be very important for affecting hydrogen spillover and the subsequent hydrogenation process. The introduction of oxygen vacancies offers a promising strategy to enhance efficiency of catalysts. Recent advanced characterization and theoretical modeling techniques have provided us with increasing new insights for understanding hydrogen spillover effects. However, a comprehensive understanding of oxygen vacancy effects on hydrogen spillover and hydrogenation efficiency of catalysts is still lacking. This review focuses on the recent advances in support effects especially oxygen vacancy effects on improving the efficiency of catalysts from three process aspects including hydrogen dissociation, active hydrogen spillover, and hydrogenation by spillover. The challenges in studying the effects on hydrogenations by spillover on the supported catalysts are highlighted at the end of the review. It aims to provide valuable strategies for the development of high-performance catalytic hydrogenation materials.
 Lijuan Xie | Lijuan Xie received her B.S. degree in environmental engineering from Fujian Agriculture and Forestry University in 2022, and is currently pursuing her MS degree at the Strait Institute of Flexible Electronics of Fujian Normal University. Her research interests focus on the mechanisms of hydrogen spillover and hydrogenation catalysis. |
 Lizhi Jiang | Lizhi Jiang received her PhD degree from Kyushu University in 2013. In 2021, she joined the Strait Institute of Flexible Electronics, Fujian Normal University, and became an associate professor. Her research interests focus on electronic properties of materials and mechanisms of catalysis on surfaces and interfaces of materials. |
 Wei Huang | Wei Huang received his PhD from Peking University in 1992. In 1993, he began his postdoctoral research at the National University of Singapore. In 2001, he joined Fudan University, and then founded the Institute of Advanced Materials. In 2006, he was appointed as the Deputy President of the Nanjing University of Posts and Telecommunications. Then he was appointed as the President of Nanjing Tech University in 2012. Then he was appointed as the Deputy President of Northwestern Polytechnical University in 2017. His research interests include organic electronics and flexible electronics. |
1. Introduction
The development of effective catalysts is urgently demanded for the practical application of chemical synthesis, energy storage, and conversion technologies. It is known as a smart strategy to create new active sites to enhance efficiency for heterogeneous catalysts by spillover.1–3 In catalysis, only the activated species spillover is important. Recent studies have highlighted the multifaceted role of metal oxide supports in catalytic hydrogenation. These supports not only facilitate the dissociation of H2 but also act as “hydrogen reservoirs”, absorbing and storing hydrogen atoms and releasing them under favorable conditions to sustain catalytic reactions.A critical feature of metal oxide supports contributing to this process is the presence of oxygen vacancies. These vacancies have attracted significant attention due to their ability to modulate the electronic properties of metal oxide supports, thereby influencing their catalytic behavior. Cao et al. investigated the role of oxygen vacancies in CeO2 using carbon dioxide hydrogenation as a model reaction.4 Their findings provide direct evidence that surface oxygen atoms are integral to the activation of H2, while oxygen vacancies accelerate the dissociation of CO2, underscoring the unique catalytic roles of both surface oxygen and oxygen vacancies. Similarly, recent work by Wang et al. has demonstrated that oxygen-deficient tungsten oxide is a versatile and efficient catalyst for the hydrogenation of nitroaromatic hydrocarbons.5,6 The study revealed that the catalytic activity of tungsten oxide is closely linked to the concentration of oxygen vacancies, with higher vacancy concentrations correlating with enhanced catalytic performance. Furthermore, oxygen vacancies alter the dissociation pathway of H2, with one dissociated hydrogen atom bonding to a surface metal atom and the other to an adjacent oxygen vacancy site, thereby optimizing catalytic efficiency.5 Despite these promising advancements, challenges such as slow hydrogen spillover rates, limited spillover distances, and insufficient adsorption capacities of the support materials continue to impede their industrial application, challenges that become especially acute when scaling these technologies for broader industrial deployment, where maintaining consistent performance and efficiency is paramount.7,8
The process of hydrogenation by spillover involves three primary processes: the dissociation of H2 on the active metal, the migration of dissociated H on the “inert” support9–13 and hydrogenation. Recent advancements in both experimental and computational methods have provided us with more evidence for deeper understanding of the process of hydrogenation by spillover, as the advent of advanced surface characterization techniques such as in situ spectroscopy and high-resolution electron microscopy has enabled researchers to observe, with unprecedented clarity, the dissociation of H2 on active metal sites in real time, thereby providing direct and compelling evidence for the spillover phenomenon.14 Furthermore, computational methods, particularly density functional theory (DFT) and molecular dynamics simulations, have proven indispensable for elucidating the migration pathways of dissociated hydrogen on inert supports while accurately predicting the energetics and kinetics of these processes.15 These techniques have not only confirmed the existence of spillover hydrogen but have also yielded critical insights into the factors influencing their efficiency, including the properties of the support material, particle size, and the nature of the active metal. Despite these significant advancements, a conspicuous gap remains in the literature—a comprehensive and systematic review that summarizes these findings is still absent, a shortfall that, if addressed, would be invaluable for consolidating the current state of knowledge, identifying persistent challenges.
This review highlights recent progress in understanding the oxygen vacancy effects on hydrogenations by spillover on catalysts through the three important step processes (Fig. 1). We start to discuss the different types of hydrogen dissociation, and then briefly introduce the migration and reaction of active hydrogen species on various supports, and clearly summarize the typical factors affecting hydrogenation including oxygen vacancies. In conclusion, this review provides a comprehensive summary of the current research status and prospects of hydrogen spillover, aiming to offer new insights for the design and application of this phenomenon in catalytic processes.
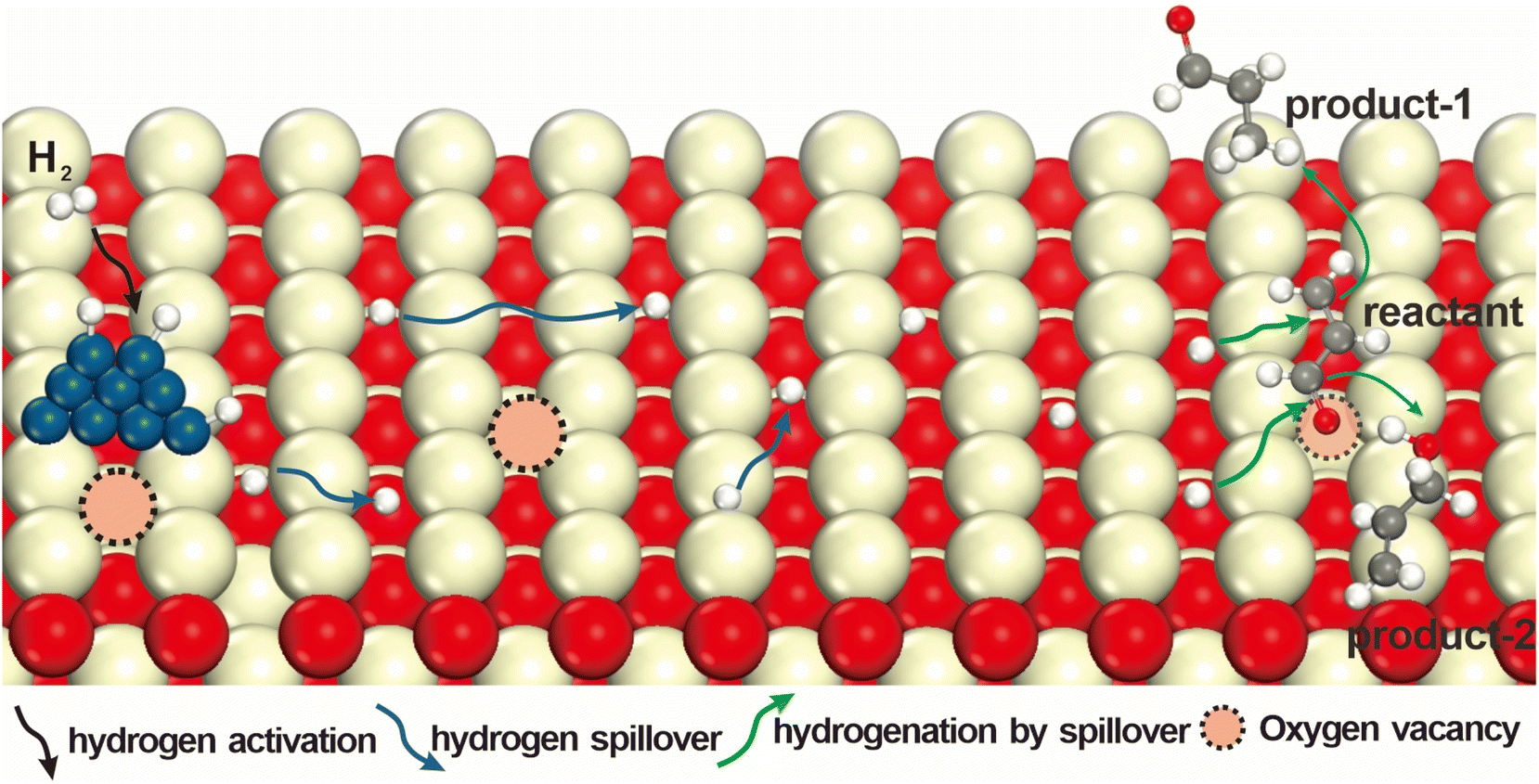 | ||
| Fig. 1 Schematic diagram of the effects of oxygen vacancies on the hydrogenation efficiency of an unsaturated substrate in the case of acrolein. The elementary reactions typically occur in the following sequence: activation of H2; transfer of active hydrogen and various hydrogenation reactions with unsaturated substrates. The presence of oxygen vacancies in surfaces facilitates effective hydrogenations. | ||
2. Oxygen vacancies in metal oxides supports
Supported catalysts used in hydrogenation reactions typically consist of two main components: a metal phase and a support material. The metal phase, often composed of noble metals such as Pt, Pd, Rh, or Ru, is responsible for the adsorption and dissociation of hydrogen molecules, and is also as the active site for hydrogenation. The supports can usually be divided into reducible and irreducible materials. The dissociated hydrogens could then spill to the reducible support materials such as CeO2 and TiO2, which provide a platform for hydrogen atom migration and stabilize the active hydrogen species through various interactions.10,12Metal oxide catalysts exhibit excellent activity, selectivity and stability in various redox reactions.16–18 The redox performance of these catalysts is strongly influenced by the distribution and concentration of oxygen vacancies, which are considered key active sites within the oxide matrix. Oxygen vacancies can exist at both the surface and subsurface regions of the support material (Fig. 2), and they are critical for the activation and stabilization of hydrogen atoms. Surface oxygen vacancies are particularly significant due to their higher reactivity, as they directly influence the adsorption and activation of reactant molecules. In contrast, subsurface oxygen vacancies, situated several atomic layers beneath the surface, do not directly engage in reactions. However, they play a vital role in modulating the electronic structure of the catalyst by redistributing electrons, which can indirectly impact the overall catalytic performance.19
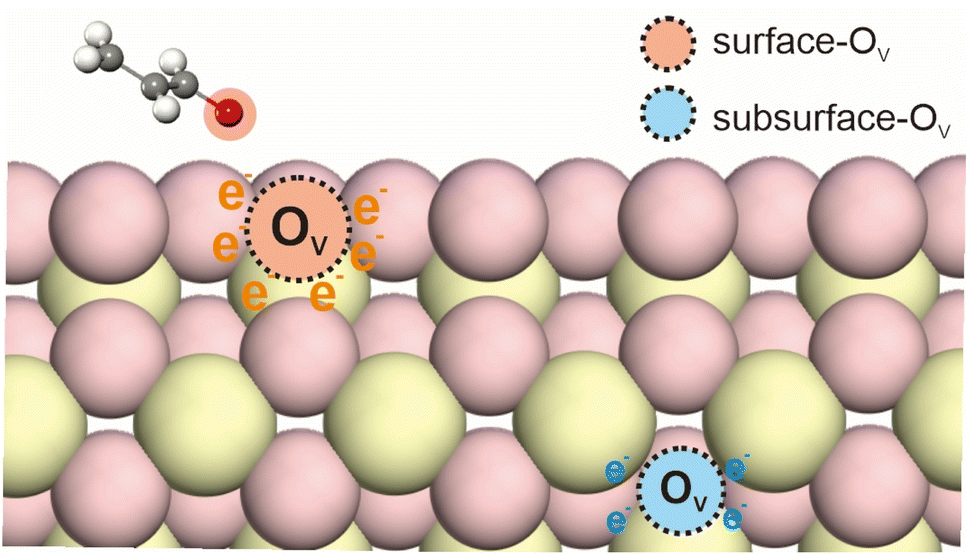 | ||
| Fig. 2 Diagram of oxygen vacancy species on a metal oxide catalyst. | ||
These oxygen vacancies not only act as active sites for hydrogen migration from the metal phase to the support but also influence the adsorption and activation of reactant molecules. The coordination environment around these vacancies, such as the number and distribution of neighboring metal cations, greatly affects their reactivity. Moreover, the migration of oxygen vacancies within the support materials is essential for maintaining catalytic activity, as it influences the electronic structure of the catalyst and its overall stability. The following sections will discuss their coordination environments, and the mechanisms governing their migration. Understanding these factors is crucial for optimizing the efficiency of hydrogenation reactions.
2.1 Coordination environment of oxygen vacancies
In catalytic systems, particularly those involving metal oxides like CeO2, the reactivity of oxygen vacancies is crucial for efficient catalytic processes. A highly reactive oxygen vacancy must balance the strong adsorption of reactant molecules with the effective desorption of product molecules. This delicate balance is significantly influenced by the local atomic and electronic environment surrounding the vacancy. For instance, in the fluorite structure of CeO2, oxygen ions are typically tetrahedrally coordinated with Ce4+ cations. Introducing structural asymmetry by doping with other metal cations can disrupt this symmetry (Fig. 3a), leading to a redistribution of electron density around the oxygen vacancies.20 This redistribution may enhance the activity of the oxygen vacancy, allowing it to participate more efficiently in catalytic reactions and providing a new dimension for catalyst design.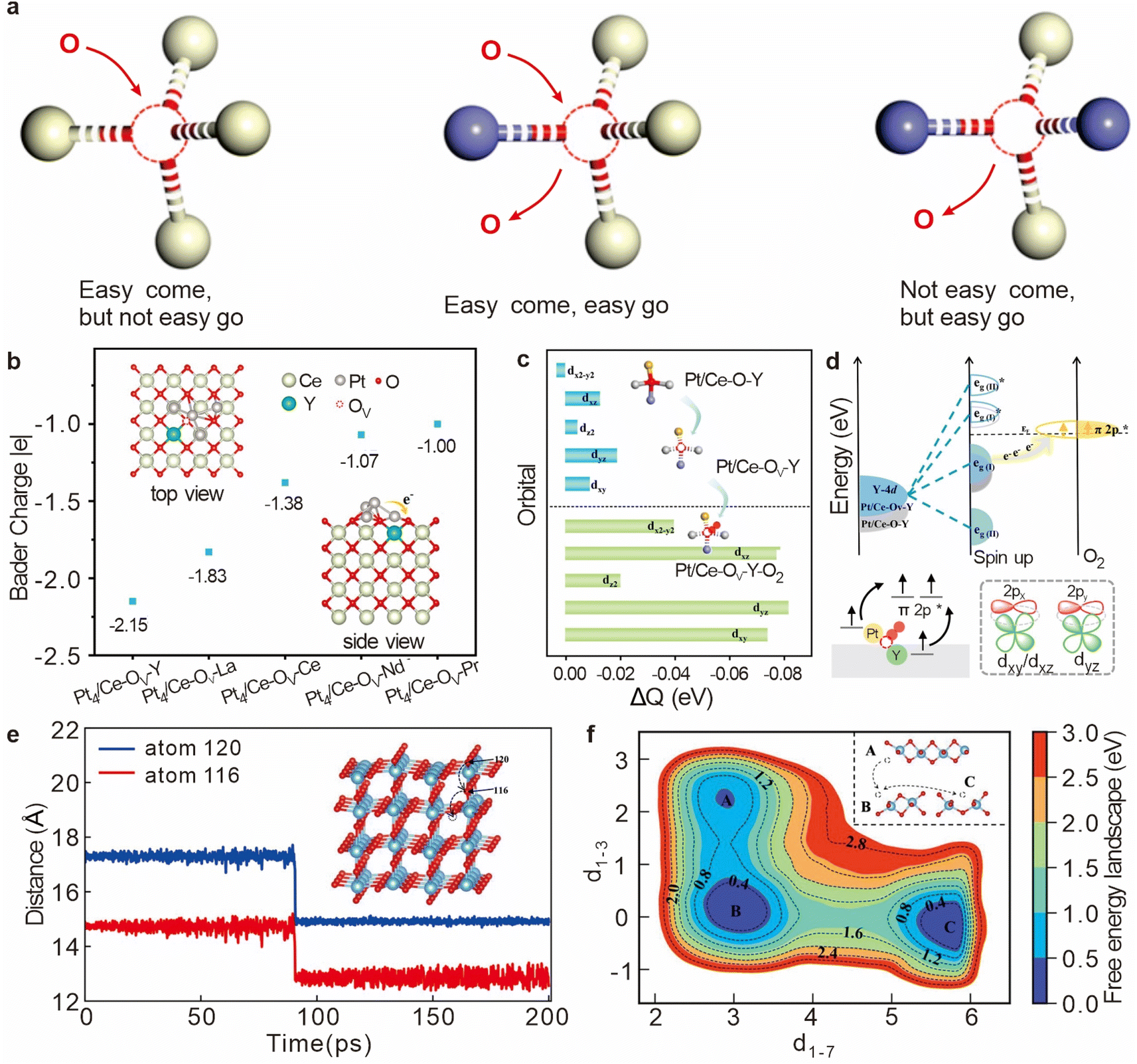 | ||
| Fig. 3 (a) Schematic diagram of the relationship between the microstructure of an oxygen vacancy and its redox properties. Reproduced with permission. Reproduced from ref. 20. Copyright 2019 Wiley-VCH GmbH. (b) Bader charge of the Pt4 cluster deposited onto different oxygen vacancy sites. (c) Variation of charge density due to oxygen vacancy formation and oxygen adsorption in Pt4/Ce–Ov–Y (d) Electronic modulation effect of Y doping in Pt4/Ce–OV–Y on O2 activation. Reproduced from ref. 21. Copyright 2024 American Chemical Society. (e) Vertical diffusion process of OV–5 to the surface. (f) Free energy surface of OV–1 diffusion at 330 K as a function of collective variables d1–3 and d1–7. Reproduced from ref. 22. Copyright 2023 American Chemical Society. | ||
CeO2 and its derivatives are regarded as excellent active supports due to their abundant oxygen vacancies.23 Recent advancements in the field have demonstrated that doping CeO2 with various transition metals can strategically modify the electronic properties of these vacancies, thereby significantly enhancing the material's catalytic efficiency. In a study conducted by Yu et al., low-cost doping ions with distinct d-band centers were introduced into Pt/CeO2(100) to create the Pt/Ce–OV–M interface.21 This strategic doping alters the charge densities within the oxygen vacancies, as the doped ions influence the electronic effects at these sites (Fig. 3b). Notably, the transfer of electrons from Pt4 clusters to the asymmetric Ce–OV–Y and Ce–OV–La sites was observed, with doping by Y and La demonstrating a marked enhancement in interfacial charge transfer. Further investigations revealed that when O2 was adsorbed at the Pt4/Ce–OV–Y site, there was significant electron depletion in the Y-4d orbital (Fig. 3c and d), which was corroborated by Bader charge analysis. This highlights the critical role of doping in modifying the electronic environment of oxygen vacancies, thus improving their ability to participate in catalytic reactions.
2.2 Migration of oxygen vacancies
The migration behavior of oxygen vacancies is a pivotal aspect of catalytic science, as the diffusion of these vacancies during catalytic reactions intricately affects the activity and stability of the catalyst.24 In the context of transition metal oxides like TiO2, oxygen vacancies play a crucial role in modulating the material's electronic and structural properties, which in turn influences its photocatalytic performance.The introduction and precise control of oxygen vacancies in TiO2 have been shown to enhance its photocatalytic efficiency, making it a subject of intense research. Studies such as those conducted by Wu et al. have employed advanced computational methods, including Deep Potential Molecular Dynamics (DPMD) and enhanced sampling techniques, to model the diffusion behavior of oxygen vacancies in TiO2, particularly focusing on the rutile (110) surface.22 By constructing a detailed Deep Potential (DP) model, the researchers were able to simulate the diffusion of oxygen vacancies both within the atomic layers and on the surface of TiO2 (Fig. 3e). To further elucidate the diffusion mechanism of surface oxygen vacancies, Wu et al. utilized the Well-Tempered Metadynamics (WT-MetaD) method to reconstruct the free energy surface associated with the diffusion process at 330 K (Fig. 3f).22 The analysis employed key coordinate variables (cv d1–3 and d1–7) to model the migration pathways, revealing distinct states (B and C) that correspond to oxygen vacancies at different locations on the surface. The findings showed that the diffusion of an oxygen vacancy from state B to state C in the [001] direction encounters a free energy barrier of approximately 1.43 eV. This barrier is indicative of the kinetic challenges involved in vacancy migration, which can influence the overall reactivity and efficiency of the catalytic process.
These insights into the kinetic and thermodynamic behavior of oxygen vacancies on TiO2 surfaces contribute to a deeper understanding of how vacancy migration impacts catalytic activity. By manipulating the concentration, distribution, and mobility of oxygen vacancies, researchers can optimize the performance of TiO2-based catalysts, making them highly adaptable for specific applications in photocatalysis and other catalytic processes. The ability to fine-tune these factors offers significant potential for improving the selectivity and efficiency of catalytic reactions, thus enhancing the industrial applicability of TiO2 and related materials.
3. Hydrogen activation
3.1 Hydrogen activation mechanism
The dissociation of H2 on metal surfaces, a crucial step in catalytic hydrogenation, occurs through two primary pathways: homolytic and heterolytic dissociation.25 Homolytic dissociation involves the symmetrical cleavage of the H–H bond, where each hydrogen atom binds to a metal center, typically favored in environments where metal atoms are closely spaced, as observed in transition metals like platinum and palladium (Fig. 4a). In contrast, heterolytic dissociation involves an asymmetric cleavage, with one hydrogen atom binding to the Lewis acidic metal center and the other associating with a nearby Lewis base, commonly seen in metal–support systems with electron-donating species (Fig. 4b). | ||
| Fig. 4 (a) Illustration of H2 homolytic dissociation pathways. (b) Illustration of H2 heterolytic dissociation pathways. | ||
The choice of dissociation pathway significantly impacts catalytic performance, influencing the overall activity, selectivity, and stability of the catalyst. Homolytic dissociation typically produces highly reactive metal-hydride species that, although effective in hydrogenation reactions, may also lead to side reactions if not carefully controlled. In contrast, heterolytic dissociation, by involving a base, generally enables more controlled and selective hydrogenation processes, especially when multifunctional catalysts leverage the base's properties to direct the reaction towards the desired products. To gain a deeper understanding of the dissociation mechanism, theoretical calculations including DFT are widely used to model the energy barriers and reaction pathways of different dissociation mechanisms. Computational methods provide insights into the microscopic mechanisms of the reactions, uncovering details that may not be fully understood through experimental observations alone.
It has been widely believed that hydrogen molecules dissociate into hydrogen atoms with a partial negative charge (Hδ−) through homolytic cleavage on Pd particle catalysts.26 However, when Pd atoms are individually dispersed in Pd1/TiO2 catalysts, no Pd–Pd pairs are available for the homolytic cleavage of H2. DFT calculations conducted by Zheng et al. showed that H2 adsorbed on Pd1 readily splits into two H atoms, and one H atom migrates to a nearby oxygen atom forming an O–Hδ+ bond, while the other H atom remains on Pd forming an Hδ− species.27 This discovery not only challenges traditional understanding but also underscores the importance of studying hydrogen dissociation mechanisms in catalytic hydrogenation and hydrogen storage processes.
3.2 Oxygen vacancy effects on hydrogen activation
The dissociation mode of hydrogen significantly influences the type and behavior of active hydrogen species, which in turn dictates the catalytic reaction pathway and product distribution. Recent studies have highlighted the potential of oxide catalysts in promoting heterolytic dissociation of hydrogen, and especially those located on the surface or at the metal–support interface, can shift the dissociation process, thereby enhancing the generation of active hydrogen atoms (Fig. 5a). Gong et al. reported that on the CeO2(110) surface, homolytic dissociation of H2 into two OH groups is thermodynamically favorable, but becomes unstable as the concentration of surface oxygen vacancies increases. Meanwhile, on the CeO2(110) surface with moderate defects, heterolytic dissociation of H2 into an H− atom at the oxygen vacancy site and an OH group is thermodynamically permissible. This is consistent with previous DFT calculations, which suggest that surface oxygen vacancies can enhance the stability of H− species.28 Yang et al. discovered that the homolytic cleavage of hydrogen on α-Ga2O3 occurs at the coordination-unsaturated Ga3+ sites formed during the initial heterolytic dissociation process.29 As shown in Fig. 5c, Ga–H and Ga–OH rapidly form and intensify simultaneously when α-Ga2O3 is exposed to H2, but the concentration of Ga–H continues to increase at a rate of 0.9 nm−2 min−1 after Ga–OH stabilization, with the final hydride-to-hydroxyl ratio reaching 5.6. Further DFT calculations revealed the dissociation behavior of H2 on the oxygen-deficient α-Ga2O3(001) surface (Fig. 5d), the heterolytic dissociation of H2 initially releases a significant amount of energy (1.65 eV), and the resulting hydrides migrate to neighboring O atoms to form hydroxyl groups, releasing an additional 0.58 eV of energy. Interestingly, the average dissociation energy of H2 in the presence of adjacent hydroxyl groups is −0.46 eV (Fig. 5d), whereas the dissociation energy of H2 without prior heterolytic cleavage is +0.28 eV, which indicates that heterolytic dissociation is a prerequisite for homolytic cleavage. This dual dissociation mechanism can significantly enhance the overall catalytic activity. This highlights the importance of designing catalysts that can selectively control the hydrogen dissociation pathway to optimize the catalytic process for specific reactions. | ||
| Fig. 5 (a) Mechanism of oxygen vacancies affecting hydrogen dissociation. (b) Calculated energy profiles of adsorption and dissociation of H2. Reproduced from ref. 30. Copyright 2022 Springer Nature. (c) The evolution of surface Ga–H and Ga–OH over α-Ga2O3 samples with contact time of H2 at 350 °C. (d) H2 adsorption energy by homolytic dissociation over α-Ga2O3 (001). Reproduced from ref. 29. Copyright 2024 Springer Nature. | ||
The dissociation capacity of hydrogen is a critical factor for determining the production rate of active hydrogen species in catalytic hydrogenation processes. This capacity is influenced not only by the inherent properties of the metal at the dissociation site but also by the characteristics of the support material. The interaction between metal particles and the support can significantly alter the electronic environment and the distribution of active sites, thereby enhancing or hindering hydrogen dissociation. Recent studies have shown that by carefully designing and engineering the catalyst's structure, researchers can significantly improve hydrogen dissociation efficiency. By combining spectral experiments and computational models, Wang et al. elucidated the promoting effect of a CoO shell with oxygen vacancies on the H2 dissociation process.30 They found that the heterolytic dissociation of H2 on the CoO(100) surface required overcoming a potential barrier of 0.60 eV, but the presence of oxygen vacancies on the CoO(100)–OV surface made the adsorption and dissociation process (Fig. 5b) of H2 more favorable; the heat absorption was reduced (0.36 eV) and the barrier was lower at 0.56 eV.
4. Hydrogen spillover process
4.1 Hydrogen spillover mechanism
The phenomenon of hydrogen spillover, initially discovered by Khoobiar during the hydrogen reduction of WO3, marks a pivotal development in catalysis science.31 Initially, hydrogen molecules are adsorbed onto the metal surface, where they undergo dissociation to form active hydrogen atoms. These hydrogen atoms then migrate from the metal surface to the metal–support interface, where they can diffuse across the support material, as hydrogen dissociation does not occur on the support itself. Importantly, hydrogen spillover is not confined to the support surface immediately adjacent to the activated metal; it can extend to other areas of the support. In cases where the composition of a secondary accepting surface differs from that of the initial surface, secondary spillover may take place, further facilitating hydrogen migration.32,33 Despite decades of research, the underlying mechanisms of hydrogen spillover remain incompletely understood. An in-depth exploration of the spillover mechanism on various supports is essential for effectively leveraging this phenomenon to enhance catalytic performance. For supported metal catalysts, it is widely accepted that hydrogen spillover can efficiently occur on easily reducible oxide supports such as WO3 and TiO2.15,34 These oxides facilitate the migration of hydrogen atoms across their surfaces, enhancing the catalytic activity of the supported metal particles. However, the possibility of hydrogen spillover on less reducible oxide supports, such as SiO2 and Al2O3, has been a contentious topic in the academic community. This controversy stems from the inherent difficulty in experimentally observing hydrogen spillover on such supports due to their low reducibility and the challenges associated with visualizing this phenomenon.9,35–38Traditional methods for studying hydrogen spillover have encountered significant limitations, particularly in the development of model systems with clearly separated catalytic functions. These challenges have hindered the direct observation and quantification of hydrogen spillover, complicating the understanding of its mechanisms. To address these challenges, Waiz Karim et al. developed an innovative approach by placing pairs of iron oxide and platinum nanoparticles on different supports, creating a model system with a distance gradient to directly observe the chemical transformations induced by hydrogen spillover (Fig. 6a).10 This experimental setup provided direct and intuitive evidence for hydrogen spillover behavior on various supports, shedding light on the complex interplay between support materials and spillover phenomena. Additionally, the adsorption and migration mechanisms of hydrogen on TiO2 and γ-Al2O3 were explored through first-principles simulations.39,40 These computational studies revealed that the spillover barrier of hydrogen atoms (H*) to TiO2 after separation is approximately 0.45 eV, indicating the feasibility of hydrogen spillover in this system (Fig. 6b). On the γ-Al2O3 support, the transfer of H* between adjacent alumina sites exhibited an activation energy barrier ranging from 1.15 eV to 1.63 eV, depending on the degree of surface hydration (Fig. 6c).
 | ||
| Fig. 6 (a) Scheme of hydrogen spillover from platinum to an iron oxide particle over a titanium oxide or aluminium oxide support. (b) Hydrogen dissociation on the platinum cluster and spillover to anatase titanium oxide. (c) Hydride mobility on γ-aluminium oxide. Reproduced from ref. 10. Copyright 2017 Springer Nature. (d) Schematic diagram of H2 dissociation and hydrogen spillover on the Pt1−x/Cu(111) surface. (e) Diffusion barrier of H over the catalyst surface. Reproduced from ref. 41. Copyright 2024 American Chemical Society. (f) Proposed mechanism for the hydrogen spillover. Reproduced from ref. 9. Copyright 2014 Springer Nature. | ||
These findings align with those of previous theoretical and experimental work,42,43 confirming that the mobility of hydrogen on the surface of γ-Al2O3 is more restricted than on TiO2. On irreducible oxide catalysts, such as silica–alumina (SiO2–Al2O3), early research has identified Brønsted acid sites (BAS) and defect sites produced by surface hydroxyl groups as potential stabilizers for atomic hydrogen.44,45 These sites are significant because they may stabilize atomic hydrogen, facilitating its migration across the catalyst surface. However, the mechanism by which atomic hydrogen migrates at these sites has not been systematically explored, leaving gaps in the understanding of this process.
Juhwan Im et al. made notable strides in this area by developing Pt/NaA zeolites with Pt clusters selectively embedded within the zeolite's micropore structure.9 This novel material design provides an excellent platform for investigating the migration mechanism of atomic hydrogen in zeolites. As shown in Fig. 6f, at the bridge site of two oxygen atoms in the [AlO4]− framework, the hydrogen radical (H˙) is stabilized through the formation of a three-center bond (O–H–O). This three-center bond formation is driven by the presence of spatially localized unpaired electrons, also known as polarons, which induce a local structural distortion at the original BAS site [AlO4]−H+. The unpaired electrons at the [AlO4]−H˙ site facilitate the migration of H˙ by converting the conventional O–H bond into a three-center O–H–O bond. This polaron conduction mechanism enables the H˙ to migrate more easily to adjacent BAS sites. Through multiple migrations, the unpaired electron can eventually reach a cation defect site, where it recombines with a proton to reform H˙. At this stage, the hydrogen radical can be readily transferred to organic molecules adsorbed on external Al sites, thereby participating in hydrogenation reactions. This discovery not only elucidates the stabilization mechanism of atomic hydrogen on irreducible oxides but also provides insight into the polaron-assisted migration of hydrogen across these surfaces.
4.2 Mechanism of sustainable hydrogen spillover
The continuity of hydrogen activation and spillover processes plays a critical role in catalytic reactions, heavily influenced by both kinetic and thermodynamic factors. A deep understanding of these processes at the atomic scale is essential for improving catalytic efficiency, as it provides insight into the driving forces behind hydrogen activation and migration.To probe the atomic-scale dynamics of hydrogen activation, researchers have employed various theoretical approaches, Busnengo et al. discovered that during the dissociation of H2 on the surface of Pd1/Cu(111), one hydrogen atom tends to remain bound to the active Pd site, highlighting the site-specific nature of hydrogen interactions on SAAs.46 This finding was later supported by Lin's group, who observed a similar behavior on the Pt1/Cu(111) surface at a temperature of 300 K.47 They identified two distinct hydrogen adsorption states at the Pd site during H2 activation on Pd1/Cu(111) through XPS,48 but the precise nature of these states remains unclear, which may influence the reactivity and sustainability of H2 activation. Busnengo et al. employed ab initio molecular dynamics (AIMD) simulations to explore the H2 dissociation process on Pd1/Cu(111).46 These simulations provided detailed insights into the atomic-scale dynamics of hydrogen adsorption, dissociation, and spillover on such surfaces, complementing experimental findings and deepening our understanding of the catalytic behavior of SAAs. The surface structure represented by Pt1−x/Cu(111) serves as an ideal model system for such studies (Fig. 6d). In this model, Cu atoms adjacent to Pt (referred to as Cu1) and other Cu atoms (referred to as Cu2) exhibit distinct coordination environments,41 which could lead to different catalytic behaviors. As shown in Fig. 6e, substituting subsurface Cu atoms with early transition metals like vanadium has been shown to lower the diffusion barrier of H* from site 1 to site 2, thereby enhancing the efficiency of the spillover process. However, the diffusion barrier from site 2 to site 3 remains consistent across different surfaces, approximately 0.06 eV, indicating a universal characteristic of these sites in promoting hydrogen spillover.
Further investigations by Lin et al. revealed that the dissociation energy barrier of H2 on Pt/Cu(111) surfaces increases from 0.047 eV to 0.189 eV (for 1H-hcp), and further to 0.785 eV (for 2H-hcp&fcc), indicating that the initial hydrogen adsorption can partially or completely deactivate Pt atoms (Fig. 7a).49 To gain a better understanding of this process, they developed a neural network potential energy surface (PES) to describe the hydrogen spillover mechanism on Pt/Cu(111). Extensive molecular dynamics simulations incorporating adiabatic effects were performed, which showed that the collision between H2 molecules and the pre-adsorbed H atom at the Pt site is crucial for continuous hydrogen spillover (Fig. 7b and c). At an incident kinetic energy of 0.25 eV, the horizontal kinetic energy of the H2 molecule is initially too low to overcome the diffusion barrier. However, at 95 fs, the distance between the H atom in the dissociated H2 and the closest H atom reaches a minimum of 1.43 Å, indicating the strongest repulsive force (Fig. 7b). This force enhances the horizontal kinetic energy of the H atom, allowing it to overcome the barrier at the Cu–Cu bridge site and spillover onto the host Cu surface. For the H2 + 2H-adsorbed Pt/Cu(111) system (Fig. 7c), the strongest collision at 105 fs provides sufficient energy for the H atom to spillover onto the Cu surface, ensuring the continuity of the spillover process.
 | ||
| Fig. 7 (a) Energy profiles for H2 dissociation on the bare-Pt/Cu(111) surface. (b) The distance between the atom closest to the spilled H atom in the H2 molecule and the spilled H atom. (c) Total kinetic energy and kinetic energy components along x + y and z. Reproduced from ref. 49. Copyright 2023 Wiley-VCH GmbH. (d) DFT calculations for H* adsorbed on fully hydroxylated rutile (110) at 0.1 H* nm−2. (e) Temperature effects on H* adsorption. (f) Entropic contributions to hydrogen spillover energetics. Reproduced from ref. 50. Copyright 2023 Springer Nature. | ||
Hydrogen activation is a key step that governs the availability of active hydrogen species while hydrogen spillover plays a pivotal role in determining the quantity and distribution of these active hydrogen species across the surface of the catalyst. Recent studies by Akbar Mahdavi-Shakib et al. have advanced the understanding of the complex process of hydrogen spillover.50 By investigating weakly reversible H2 adsorption on Au/TiO2 catalysts (Fig. 7d), the researchers quantified the surface concentration of spilled hydrogen, providing new insights into this intricate phenomenon. Their findings reveal a notably unconventional behavior: contrary to typical gas adsorption systems, where adsorption generally decreases with increasing temperature, the adsorption of H* on the Au/TiO2 system exhibits a positive temperature dependence (Fig. 7e). This counterintuitive trend can be attributed to the high proton mobility on the catalyst surface, combined with configurational surface entropy, which creates favorable adsorption conditions at elevated temperatures (Fig. 7f). Additionally, the increase in temperature elevates the hydroxyl acid–base equilibrium constant on the TiO2 surface, thereby increasing the number of active spillover sites. This rise in spillover points is associated with an increased concentration of zwitterions on the titanium dioxide surface, which further enhances the H* adsorption observed at higher temperatures.
4.3 Oxygen vacancy effects on hydrogen spillover
Metal oxides are critical components in catalytic hydrogenation reactions, and are extensively utilized in the production of both fine and bulk chemicals.51–53 The efficiency of these reactions is heavily dependent on the ability of hydrogen atoms to diffuse across the surface of metal oxides, as this diffusion directly impacts the concentration of surface-active hydrogen species and, consequently, the overall catalytic activity.32,54,55 However, the process of hydrogen diffusion on metal oxide surfaces is often impeded by the requirement to overcome significant energy barriers (55–236 kJ mol−1). These barriers not only limit the rate of hydrogenation reactions but also reduce the effectiveness of the catalyst. To address this challenge, introducing oxygen vacancies into the metal oxide lattice has emerged as a potent strategy for enhancing the kinetic efficiency of hydrogen spillover. Oxygen vacancies on supports play a critical role in the spillover process not only by providing sites for the migration and stabilization of hydrogen atoms, but also by manipulating the coordination of the metal environments around them, which may directly affect the hydrogen migration barriers.56 These vacancies not only facilitate the movement of hydrogen atoms across the catalyst surface, but also affect their stability through electron redistribution (Fig. 8).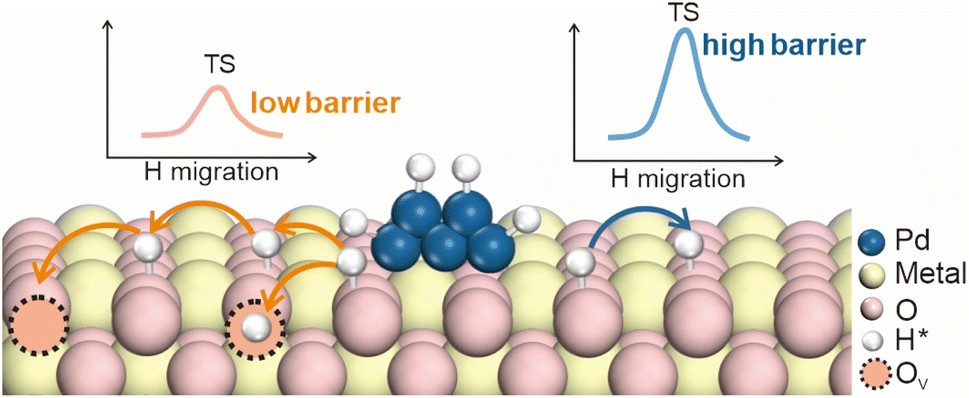 | ||
| Fig. 8 Mechanism of oxygen vacancies affecting hydrogen spillover. | ||
Oxygen vacancies have a profound impact on hydrogen migration by modifying the local electronic environment. The formation of these vacancies, which involves the removal of oxygen atoms from the lattice, alters the coordination of adjacent metal atoms. It leads to a rearrangement of the local electronic structure, often resulting in an increased electron density at specific metal sites. Sun et al. proposed a mechanism in which oxygen vacancies create a built-in electric field (BIEF) that facilitates hydrogen spillover.57 In their study, the Mo–Co diatomic catalyst underwent structural reconstruction in a high-temperature H2 atmosphere, leading to the formation of oxygen vacancies. These vacancies caused electron accumulation near the Co atom, generating a weak BIEF between the Mo and Co atoms (Fig. 9a). Further DFT calculations confirmed that the formation of oxygen vacancies induces a differential Bader charge distribution between the Mo and Co atoms, promoting the diffusion of positively charged hydrogen atoms (+0.10 e−) through the localized electric field (Fig. 9b).
 | ||
| Fig. 9 (a) XPS comparison of MoCo DAC/C before and after the reaction. (b) Two alternative minimum energy paths for the first hydrogenation step of adsorbed DBT on the MoCo DACs/C surface and the corresponding structures. Reproduced from ref. 57. Copyright 2024 American Chemical Society. (c) The charge density difference analysis of Pt/TiO2 and VO-Pt/TiO2. (d) Optimized geometric structures of Pt NCs anchored on TiO2 supports with/without oxygen vacancies and the corresponding DFT calculation of the free energy. Reproduced from ref. 58. Copyright 2021 Wiley-VCH GmbH. (e) The schematic diagram of WAPH with oxygen vacancies. DFT calculations of WAPH for the (f) pristine MoO3 surface and water and (g) MoO3−x surface and water. Reproduced from ref. 59. Copyright 2023 American Chemical Society. | ||
Oxygen vacancies also play a regulatory role in charge transfer processes between the support and the catalytically active sites, influencing hydrogen migration either positively or negatively. Pt known for its unique electronic properties and surface reactivity, is highly effective in hydrogen dissociation.60–62 Pt's low electronegativity (2.28 compared to O's 3.44) tends to result in a higher oxidation state for Pt atoms on metal oxide supports, which can hinder hydrogen desorption. In contrast, Pt nanoclusters (NCs), which contain Pt–Pt bonds, are better at preventing electron transfer from Pt to the support, and under specific conditions, they can even reverse the direction of charge transfer. This reversal can lower the d-band center of Pt, thereby facilitating hydrogen desorption. Zheng et al. demonstrated that oxygen vacancies in the support material can induce unique dual effects of electron-rich Pt in Pt/TiO2 catalysts, including reverse charge transfer and enhanced hydrogen spillover.58 In a Pt/TiO2 system, approximately 0.367 electrons are transferred from Pt nanoclusters to TiO2, but in a VO-Pt/TiO2 system, about 0.406 electrons are transferred from TiO2 to Pt nanoclusters, indicating that oxygen vacancies can reverse the charge transfer direction, enriching the Pt nanoclusters with electrons (Fig. 9c). Further free energy calculations revealed significant differences in the hydrogen adsorption free energy (ΔGH) at different sites on the Pt/TiO2 surface, with a variance of up to 0.70 eV between site-C and site-D (Fig. 9d). In contrast, the ΔGH difference between the two sites on VO-Pt/TiO2 was reduced to 0.54 eV, suggesting that oxygen vacancies substantially enhance the hydrogen spillover effect.
The role of acidic hydroxyl groups on the surface of oxides, or defect sites introduced during synthesis, is well-documented in promoting hydrogen transfer processes.9,63,64 Recent studies have highlighted the significance of these groups in facilitating the migration of hydrogen atoms, especially when synergized with other molecules or structural features of the catalyst. Wang et al. demonstrated that gaseous organic molecules containing carbonyl functional groups could facilitate the migration of hydrogen from Pt to Fe sites on an oxide surface.65 This mechanism is reminiscent of molecular coenzymes in enzymatic reactions, where the coenzyme assists in the transfer of reactive groups between sites, thereby enhancing the overall catalytic process.66 Metal–organic frameworks (MOFs) have also been recognized for their ability to absorb water through their clusters or ligands.67,68 Gu et al. emphasized this understanding by constructing a water-assisted spillover pathway in a Pt@MOF-801 system.69 Their calculations revealed that the energy barrier for hydrogen migration via this water-assisted pathway is lower than that of the traditional ligand-based spillover route, underscoring the potential of water molecules in facilitating hydrogen diffusion.
On the surface of metal oxides, the presence of water can further enhance the diffusion of hydrogen atoms through a mechanism known as proton-coupled electron transfer (PCET). This process, also referred to as the water-assisted proton jump (WAPH),70,71 involves the temporary formation of hydronium ions (H3O+) as water molecules absorb hydrogen atoms. The electrons from these hydrogen atoms are then transferred to the d-orbitals of the metal oxide, promoting subsequent hydrogenation reactions on metal oxide-supported catalysts.72,73 Wang et al. showed that the presence of oxygen vacancies can accelerate the adsorption of water molecules (Fig. 9e),59 thereby reducing the energy barrier associated with WAPH. Their findings indicate that on a defect-free surface, the barrier energy for proton jumping is approximately 0.30 eV (Fig. 9f), but this barrier decreases to 0.25 eV when oxygen vacancies are present (Fig. 9g). The impact of oxygen vacancies on proton jumping is further elucidated by charge density distribution studies, which reveal how these vacancies alter the electronic states of surrounding atoms, particularly oxygen and neighboring metal atoms.
5. Hydrogenations by spillover
5.1 Hydrogenation by spillover mechanism
The hydrogenation process involves two key steps: the formation of adsorbed H* on a catalyst's surface and the subsequent desorption of these species to drive the hydrogenation reaction.74,75 Traditionally, these steps are managed by a single catalytic component, but it often leads to a balance in optimizing both formation and desorption. To overcome this limitation, researchers have used hydrogen spillover which decouples these steps by employing distinct catalytic components specialized for each function.76,77Following the hydrogen spillover, the active hydrogen atoms which have migrated to the support surface may participate in subsequent catalytic reactions under mild conditions depending on the properties of supports. These hydrogen atoms interact with reactant molecules, particularly in systems where the support material, such as metal oxides, provides active sites like oxygen vacancies. These sites not only stabilize the hydrogen atoms but also facilitate the activation and adsorption of reactants. The spillover process effectively increases the availability of hydrogen atoms on the support surface, which can significantly enhance the overall reaction rate and improve the efficiency of the hydrogenation process.12,78 Such a separation strategy not only maximizes the overall efficiency of hydrogen utilization but also significantly enhances the selectivity and precision of the hydrogenation process, leading to improved catalytic performance.
 | ||
Fig. 10 (a) Adsorption energies and configurations of CAL and COL on PtCo(111) and CoBOx-6OH surfaces. (b) Catalytic performance and (c) TOF values of the CAL hydrogenation. Reproduced from ref. 79. Copyright 2021 Elsevier. (d) Energy barrier for water dissociation on Co1–TiOx/Ti. (e) ELF analysis of CAP adsorption on Co1–TiOx/Ti. (f) Dechlorination ratio on Co1–TiOx/Ti and other control samples. Reproduced from ref. 81. Copyright 2024 Wiley-VCH GmbH. (g) Adsorption configurations of 4-NS on Ni(111) and RuNi SAA(111) surfaces. (h) Product distribution in the presence of monometallic Ni, Ru, and RuNi catalysts with various Ru loadings (0.1–2 wt%). (i) Potential energy profiles and the corresponding optimized structures for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogenation and N–O scission in 4-NS over the RuNi SAA(111) surface. Reproduced from ref. 82. Copyright 2022 Springer Nature. C hydrogenation and N–O scission in 4-NS over the RuNi SAA(111) surface. Reproduced from ref. 82. Copyright 2022 Springer Nature. | ||
Electrochemical dechlorination is of critical importance for the environmental remediation of organochlorine compounds, which are notorious for their “teratogenic, carcinogenic, and mutagenic” properties, posing severe risks to both ecosystems and human health. An exemplary advancement in this field is the CoPc/CNT catalyst developed by Wang et al., which demonstrated the efficient conversion of dichloroacetic acid (DCA) into ethylene and chloride ions in aqueous solutions.83
For effective electrochemical dechlorination, it is essential to optimize the dissociation of water molecules into reactive H*, while concurrently inhibiting the undesirable recombination of H* into H2. This balance is crucial because, while noble metal electrodes are highly effective at promoting water dissociation,84 they also tend to enhance the recombination of H*, thereby reducing its availability for dechlorination reactions.85,86 Consequently, there is a pressing need to develop innovative electrode materials that can efficiently promote water dissociation while minimizing H* recombination to maximize dechlorination efficiency. To address the challenge of optimizing the generation, transfer, and utilization of atomic hydrogen in electrochemical processes, Zhang et al. developed an innovative electrocatalytic system based on a reverse hydrogen spillover effect,81 using a Co1–TiOx/Ti electrode. Individual cobalt atoms are anchored onto a titanium oxide substrate, leveraging the excellent hydrolytic ionization and hydrogen adsorption properties of the TiOx support. This configuration allows for the rapid migration of hydrogen atoms to the cobalt sites, where they actively participate in the dechlorination reaction via the reverse hydrogen spillover mechanism (Fig. 10d and e). The electrochemical performance of the Co1–TiOx/Ti electrode was rigorously evaluated, demonstrating a markedly higher dechlorination efficiency (99%) compared to the TiOx/Ti electrode alone (Fig. 10f). These findings highlight the potential of hydrogen spillover systems for advancing the effectiveness of catalytic processes, particularly in applications requiring precise control over hydrogen atom dynamics.
The activated adsorption of specific functional groups is a crucial step in the selective hydrogenation of substrates, as it directly influences both the efficiency of catalytic reactions and the selectivity of the resulting products. While an increase in catalytic activity can sometimes lead to a decrease in the selectivity of the target product,90–92 the PtCo/CoBOx catalyst demonstrates a remarkable exception, maintaining a selectivity of 94.5% (Fig. 10b). Through DFT calculations, Zhang et al. discovered that this high selectivity is due to the interaction between the carbonyl group (–CO) and three surface hydroxyl groups on the catalyst.81 This interaction causes CAL to adopt an inclined adsorption configuration on the catalyst surface, which positions the CC bond away from the surface while elongating the CO bond. Furthermore, the adsorption energy of the corresponding alcohol product on the CoBOx-6OH surface is +0.61 eV, indicating that desorption is energetically favorable (Fig. 10a).
In catalyst design, researchers must carefully consider surface properties to achieve the activated adsorption of specific functional groups. This may involve fine-tuning the composition, structure, and surface modifications of the catalyst to optimize its ability to adsorb and transform particular substrates. Liu et al. reported a RuNi single-atom alloy catalyst where hydrogen dissociates at an adjacent Ni site and then undergoes hydrogenation at the Ruδ− site (Fig. 10g).82 This catalyst demonstrated excellent performance in the selective hydrogenation of 4-nitrostyrene to produce 4-aminostyrene (Fig. 10h). In situ experimental studies, supported by DFT calculations (Fig. 10i), revealed that the Ru–Ni interface site, as the characteristic active center, facilitated the activation and adsorption of the nitro group with a lower energy barrier (0.46 eV) compared to the monometallic Ni catalyst (0.74 eV), ultimately leading to the successful production of the target compound.
5.2 Oxygen vacancy effects on hydrogenation by spillover
Although the concept of hydrogenation by spillover has been recognized in the field of catalytic hydrogenation for many years, the precise structural characteristics of the support required to facilitate effective hydrogen spillover remain inadequately understood.9–13 The efficiency of hydrogenation by spillover is not solely dependent on the catalyst's structural properties, but the reactant's functional groups and substituents also determine its affinity for the catalyst surface and its susceptibility to hydrogenation. Additionally, the nature of the active hydrogen species and the presence of promoters or dopants on the catalyst can further modify these interactions, ultimately affecting the overall catalytic performance.C bond, reducing selectivity for CALA (Fig. 11a and b).
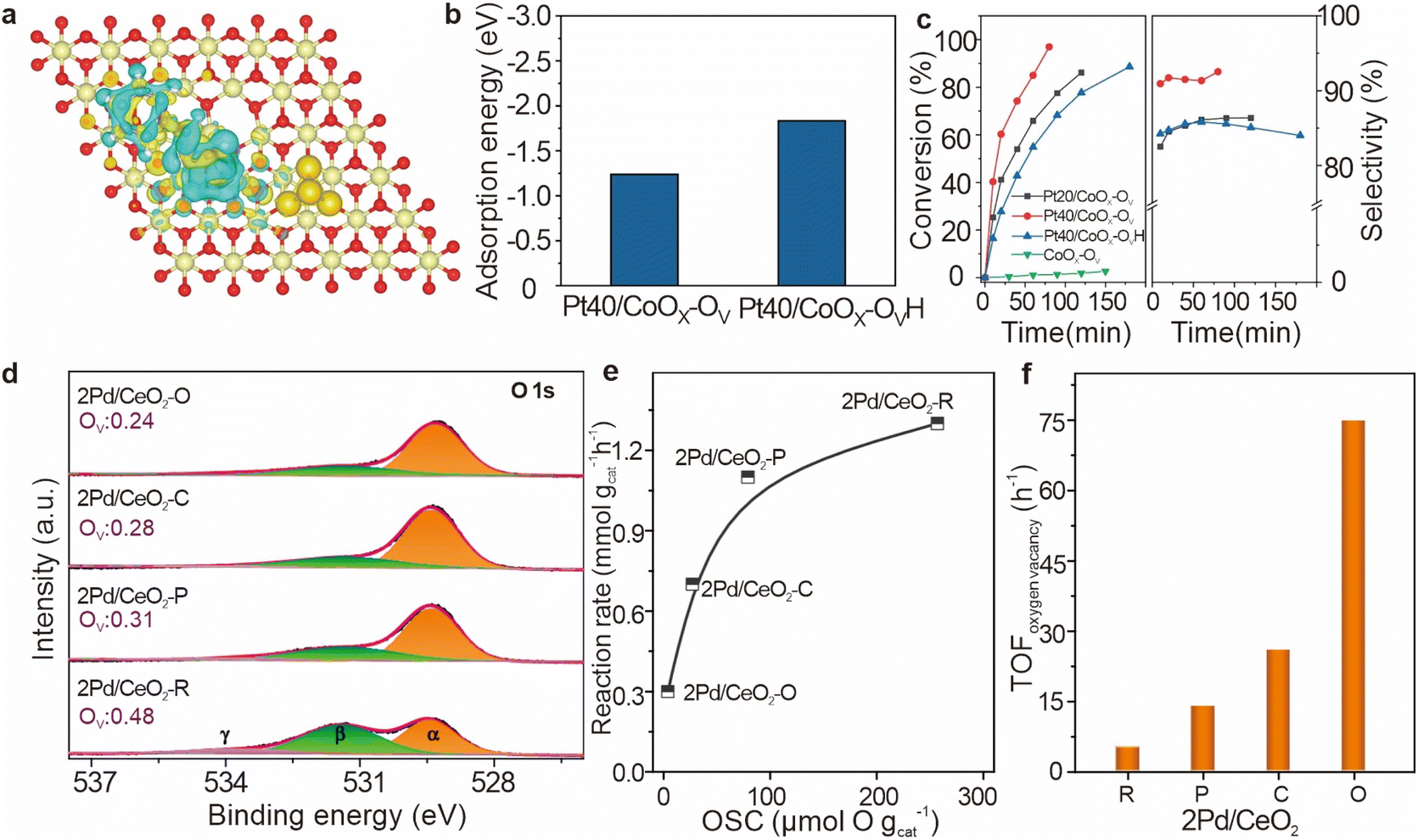 | ||
| Fig. 11 (a) The charge density differences of a CALD molecule adsorbed on Pt40/CoOx–OVH catalysts. (b) Adsorption energies of CALD on Pt40/CoOx–OV and Pt40/CoOx–OVH catalysts. (c) Catalytic performance of different catalysts. Reproduced from ref. 100. Copyright 2024 Elsevier. (d) In situ XPS curves of O 1s for the reduced Pd/CeO2 catalysts. (e) Reaction rate as a function of the number of surface oxygen vacancies (OSCs). (f) TOFoxygen vacancy over various Pd/CeO2 catalysts. Reproduced from ref. 23. Copyright 2020 American Chemical Society. | ||
In the context of hydrogenation reactions, the density of oxygen vacancies on catalyst surfaces plays a crucial role in determining catalytic activity. Liu et al. confirmed a strong correlation between the formation energy of oxygen vacancies and the hydrogenation performance of CO2.23 For Pd/CeO2 catalysts with different morphologies (Fig. 11d), the oxygen vacancy density follows the order: 2Pd/CeO2–R (0.48) > 2Pd/CeO2–P (0.31) > 2Pd/CeO2–C (0.28) > 2Pd/CeO2–O (0.24), as indicated by O 1s spectra and supported by Ce 3d spectral analysis. As the oxygen storage capacity (OSC) increases, the rate of CO2 adsorption and activation on the oxygen vacancies also increases, resulting in higher reaction rates. However, this increase becomes less pronounced at higher OSC values, showing diminishing returns at elevated oxygen vacancy concentrations (Fig. 11e). To further assess the intrinsic activity of oxygen vacancies, the turnover frequency for oxygen vacancy-related reactions was calculated. The 2Pd/CeO2–R catalyst, which exposes the (110) and (111) crystal facets, exhibits the lowest TOF, whereas 2Pd/CeO2–O, which exposes the (111) facet, demonstrates the highest TOF (Fig. 11f). These findings suggest that the formation and reactivity of oxygen vacancies are influenced by both the crystal structure and the ease with which vacancies form. On CeO2–R, the formation of oxygen vacancies is facilitated by the high oxygen mobility and low vacancy formation energy of the CeO2(110) facet, but this leads to lower reactivity. In contrast, CeO2–O, with a lower density of oxygen vacancies and a higher energy barrier for vacancy formation on the (111) facet, shows stronger interaction with CO2 and higher reactivity for hydrogenation. This difference in reactivity can be explained by the balance between vacancy formation and filling.
O group in the glycol molecule increases in the D-configuration (Fig. 12c), and there is a notable difference in hydride transfer barriers between the D- and U-configurations (Fig. 12d), which can be attributed to minor geometric reconstructions of the copper surface during hydrogen transfer. These observations underscore the critical influence of oxygen vacancies on the adsorption structures, which in turn play a vital role in modulating the subsequent hydrogenation processes.
 | ||
| Fig. 12 (a) Diagram of oxygen vacancies affecting hydrogenation by spillover. (b) Comparison of Eads in two configurations. (c) Change in the distance of CO compared to the free gas. (d) Kinetic barrier diagram for glycolyl hydrogenation via the D- and U-configurations. Reproduced from ref. 106. Copyright 2024 American Chemical Society. (e) Scheme of the adsorption configuration of 2-nitrotoluene and 4-nitrotoluene on the oxygen vacancy of WO3. (f) Reaction rate of 2-nitrotoluene/4-nitrotoluene hydrogenation by using the PdHD/WO3-350 and PdNP/WO3-350 catalysts. Reproduced from ref. 107. Copyright 2023 American Chemical Society. | ||
Additionally, in the selective hydrogenation of butenal, metal-free CeO2 nanorods with a high concentration of surface oxygen vacancies exhibit superior selectivity for CO hydrogenation compared to CC hydrogenation, achieving turnover frequencies comparable to those of precious metal-based catalysts, along with very high selectivity for butanol. This enhanced selectivity is attributed to the interaction between the oxygen vacancies and the CO group, which stabilizes the group and promotes its preferential hydrogenation.28 Similarly, another study on selective hydrogenation of α,β-unsaturated hydrocarbons using transition metal photocatalysts supported by LDH (layered double hydroxide) surfaces found that oxygen vacancies act as active adsorption centers, favoring the adsorption of CO groups while hindering the adsorption of CC groups. This selective adsorption pathway ensures high cinnamyl alcohol yield.108
The importance of oxygen vacancies extends beyond simple reactant adsorption and is particularly relevant in the selective hydrogenation of functionalized nitroaromatics. The selective reduction of nitro groups to amines is a key reaction in the synthesis of various fine chemicals, pharmaceuticals, and agrochemicals; the adsorption configuration of the nitro group at an oxygen vacancy is highly sensitive to the presence and properties of adjacent substituents on the aromatic ring.109–115Ortho-substituents that are positioned near the nitro group can exert both electronic and spatial effects that influence the interaction between the nitro group and the catalyst surface (Fig. 12e).107 When the ortho-position on the benzene ring is occupied by a larger substituent, steric effects can weaken the adsorption of the reactant molecule at the oxygen vacancy, thereby reducing catalytic activity. Under identical reaction conditions, the hydrogenation rate of 4-nitrotoluene by PdHD/WO3-350 is observed to be 19 times greater than that of 2-nitrotoluene (Fig. 12f). This significant difference in catalytic activity highlights the impact of steric effects, where the presence of an ortho-methyl group at the oxygen vacancy significantly diminishes the efficiency of p-nitro-hydrogenation.
Oxygen vacancies promote catalytic hydrogenation primarily by regulating the electronic structure and surface properties of the catalyst. Ongoing research will continue to explore the mechanisms by which oxygen vacancies influence different catalytic systems and how these active sites can be optimized through material design to achieve more efficient and environmentally friendly catalytic processes.
5.3 Other effects on hydrogenation by spillover
In the study of hydrogenation by spillover reactions, introducing lattice defects in the support material can lead to additional active sites or alter the charge distribution to optimize catalytic performance. Additionally, controlling the morphology, regulating the crystal surface, and modifying the catalyst surface can provide more favorable adsorption geometries and reduce activation barriers, thereby promoting catalytic reactions. For instance, selectively exposing specific crystal faces can change the density and distribution of active sites on the catalyst.116 Jiang et al. dispersed Pd atoms onto Cu nanomaterials with distinct crystallographic surfaces.117 Three types of potential catalytic sites were identified: Pd-related sites, interfacial Cu sites, and Cu sites (Fig. 13a and b). Both Pd1/Cu catalysts demonstrated excellent catalytic performance and high durability during the semi-hydrogenation of phenylacetylene to styrene (PhCHCH2) under mild conditions (Fig. 13c). Theoretical calculations revealed that hydrogenation on Cu using spilled hydrogen atoms is highly dependent on the specific surface structure of copper. The Cu(100) plane exhibited a lower hydrogenation barrier and higher surface coverage of phenylacetylene (PA) compared to the Cu(111) plane, facilitating PA conversion on Cu(100) (Fig. 13d). This enhanced catalytic activity on Cu(100) is attributed to more effective rehybridization of the s–p–d orbitals, leading to greater stabilization of the transition state (TS1). The energy barrier for TS1 at the Cu site of Pd1/Cu(111) is significantly higher than at the Pd site or the Cu site at the interface (Fig. 13e). The superior catalytic activity of Cu(100) compared to Cu(111) underscores its potential as an effective support material for enhancing catalytic performance.
 | ||
Fig. 13 (a) Three kinds of possible reactive sites on the Pd1/Cu surface. (b) Top and side views of the Pd1/Cu(111) and Pd1/Cu(100) surface models. (c) Catalytic durability of Pd1/Cu(111) and Pd1/Cu(100) in the semi-hydrogenation of PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH. (d) Calculated barriers for stepwise hydrogenation of PhCCH on the two low-Miller-index Cu surfaces. (e) Calculated barriers for TS1 over different active sites on different Pd1/Cu catalysts. Reproduced from ref. 117. Copyright 2020 Springer Nature. (f) Pt encapsulated in a dense support matrix that allows selective diffusion of hydrogen over organic reactants. (g) Turnover rates in benzene hydrogenation. Reproduced from ref. 9. Copyright 2014 Springer Nature. (h) Structure of PMo11O39Pd1-H16. (i) Reaction kinetics for the hydrogenation of VA with the addition of 2 equivalents benzyl mercaptan after different reaction times. Reproduced from ref. 118. Copyright 2022 Wiley-VCH GmbH. (j) AFM image of the TiO2 nanoparticle monolayer assembled on a smooth Pt film (TiO2/Pt). (k) In situ SERS spectra of the selective hydrogenation of R–NO2 in the presence of R–NC on Au/Pt and Au/30 nm-TiO2/Pt, respectively. Reproduced from ref. 53. Copyright 2020 Wiley-VCH GmbH. CH. (d) Calculated barriers for stepwise hydrogenation of PhCCH on the two low-Miller-index Cu surfaces. (e) Calculated barriers for TS1 over different active sites on different Pd1/Cu catalysts. Reproduced from ref. 117. Copyright 2020 Springer Nature. (f) Pt encapsulated in a dense support matrix that allows selective diffusion of hydrogen over organic reactants. (g) Turnover rates in benzene hydrogenation. Reproduced from ref. 9. Copyright 2014 Springer Nature. (h) Structure of PMo11O39Pd1-H16. (i) Reaction kinetics for the hydrogenation of VA with the addition of 2 equivalents benzyl mercaptan after different reaction times. Reproduced from ref. 118. Copyright 2022 Wiley-VCH GmbH. (j) AFM image of the TiO2 nanoparticle monolayer assembled on a smooth Pt film (TiO2/Pt). (k) In situ SERS spectra of the selective hydrogenation of R–NO2 in the presence of R–NC on Au/Pt and Au/30 nm-TiO2/Pt, respectively. Reproduced from ref. 53. Copyright 2020 Wiley-VCH GmbH. | ||
The influence of the support's particle size is equally important, as it directly impacts the specific surface area and the number of active sites. A study by Juhwan Im et al. demonstrated that increasing the surface area of an aluminosilicate matrix significantly enhances catalytic activity for benzene hydrogenation.9 The Pt/NaHA-0.95 sample, characterized by a large surface area and a highly amorphous structure, exhibited the highest catalytic activity, surpassing even Pt/SiO2 samples with larger accessible metal surface areas (Fig. 13f and g). With careful catalyst nanostructure design, it is possible to achieve high hydrogenation and dehydrogenation catalytic activity. This principle was further confirmed by Wei et al., who enhanced the selectivity of hydrogen spillover by carefully controlling the chemical potential of H*.53 They found that as H* spilled over from TiO2, the coverage of active hydrogen at the Au site decreased with increasing spillover distance due to the presence of a reaction barrier (Fig. 13j), thereby reducing the chemical potential of active hydrogen. As the chemical potential decreases, the available hydrogen preferentially follows hydrogenation pathways that require lower energy, thus enhancing selectivity (Fig. 13k). This work provides valuable molecular-level insights into the mechanism of hydrogen activation, offering a deeper understanding of the rational design of selective hydrogenation catalysts.
While the hypothesis that spillover H* directly contributes to hydrogenation reactions has been proposed, it is important to note that for certain reactants requiring direct H transfer from the metal active site, spillover H* may not participate in the hydrogenation process.12,119 In the structure of PMo11O39Pd1-H16 (Fig. 13h), the introduction of Pd poison (benzyl mercaptan) prevents the conversion of vinyl acetate, which contains CC bonds, causing the reaction rate to drop to zero (Fig. 13i). This indicates that the hydrogen involved in the hydrogenation of the CC bond originates directly from the Pd active site, rather than from spilled H*.118
6. Conclusions and perspective
Enhancing the efficiency of active sites has long been a significant challenge for supported catalysts, and the spillover effects offer a promising strategy to activate more sites on the supports. The advent of advanced surface characterization techniques such as in situ spectroscopy and high-resolution electron microscopy has enabled researchers to observe the dissociation of H2 on active metal sites, thereby providing direct and compelling evidence of the spillover phenomenon. The integration of machine learning with DFT calculations has illuminated the underlying mechanisms of hydrogen spillover. The hydrogenation efficiency of spilled hydrogen can be significantly enhanced by manipulating various factors, including the introduction of oxygen vacancies, the modification of metal crystal surfaces, and so on. Recent studies provide compelling evidence of the catalytic functions of hydrogen spillover, highlighting its potential in the design and development of advanced catalysts. The support effects especially the effects of the oxygen vacancies on hydrogen dissociation, hydrogen spillover, and the hydrogenation process are introduced in detail.The hydrogen dissociation process is sometimes the rate-limiting step in hydrogenation reactions. The dissociation mode of hydrogen significantly influences the type and behavior of active hydrogen species, which in turn dictates the catalytic reaction pathway and product distribution. Recent studies have shown that by carefully designing and engineering the catalyst's structure, the presence of oxygen vacancies on the CoO(100)–OV surface made the adsorption and dissociation process of H2 more favorable. In the hydrogen spillover process, oxygen vacancies have a profound impact on hydrogen migration by modifying the local electronic environment. The formation of these vacancies, which involves the removal of oxygen atoms from the lattice, alters the coordination of adjacent metal atoms. It leads to a rearrangement of the local electronic structure, resulting in an increased electron density at specific metal sites. In the hydrogenation process, oxygen vacancies enhance hydrogenation activity by creating a local charge imbalance that alters the adsorption properties of the metal oxide surface. These vacancies create stronger and more selective binding sites, which profoundly influence the orientation and reactivity of the adsorbed species.
Although significant progress has been made in hydrogen spillover and its application to hydrogenation, several challenges remain for practical implementation. First, developing accurate imaging and characterization techniques: the characterization of active sites is still a challenge for the reaction process. It is difficult to separate the hydrogenation reactions occurring on the metal catalyst from those on the support to accurately assess the spillover efficiency of active hydrogen species on the support. Secondly, developing experimental methods: catalysts with oxygen vacancies are usually not stable, and thus how to equilibrate the stability and reactivity of materials is still a challenge. Finally, developing theoretical calculations with machine learning methods: the combination of dynamics and thermodynamics is necessary to be considered for the migration rate of spillover species, which is essential for understanding the impact of spillover on enhancing hydrogenation activity. The influence of oxygen vacancies in supports on the hydrogenation reaction varies depending on their location. A deeper understanding of oxygen vacancy migration behavior can inform the design of novel catalyst systems for highly selective chemical reactions. Therefore, a broader investigation of the dynamic activities of species behaviors on various types of supports, by ab initio molecular dynamics, machine learning, and other more accurate methods, is strongly recommended. In summary, the spillover effect plays a crucial role in enhancing catalytic performance, offering a pathway to more efficient and selective hydrogenation processes. The deep understating of support effects especially oxygen vacancy effects presents significant opportunities and challenges for chemical synthesis, energy storage, and conversion technologies, requiring a multidisciplinary approach that combines experimental insights with advanced computational techniques.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Lijuan Xie: conceptualization; resources; writing – original draft; writing – review & editing. Jinshan Liang: resources; writing – review & editing. Lizhi Jiang: conceptualization; funding acquisition; supervision; writing – review & editing. Wei Huang: supervision; project administration. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 22102027), Fujian Province Natural Science Foundation of China (Grant No. 2022J01653), and start-up research funding of Fujian Normal University (Grant No. Y0720309K13).References
- Y. Y. Geng and H. Li, Hydrogen spillover-enhanced heterogeneously catalyzed hydrodeoxygenation for biomass upgrading, ChemSusChem, 2022, 15, e202102495 CrossRef CAS PubMed.
- J. Li, H. Liu, W. Gou, M. Zhang, Z. Xia, S. Zhang, C. Chang, Y. Ma and Y. Qu, Ethylene-glycol ligand environment facilitates highly efficient hydrogen evolution of Pt/CoP through proton concentration and hydrogen spillover, Energy Environ. Sci., 2019, 12, 2298–2304 RSC.
- J. Cho, M. Kim, H. Seok, G. H. Choi, S. S. Yoo, N. C. Sagaya Selvam, P. J. Yoo and T. Kim, Patchwork-structured heterointerface of 1T-WS2/a-WO3 with sustained hydrogen spillover as a highly efficient hydrogen evolution reaction electrocatalyst, ACS Appl. Mater. Interfaces, 2022, 14, 24008–24019 CrossRef CAS.
- F. Cao, Y. Xiao, Z. Zhang, J. Li, Z. Xia, X. Hu, Y. Ma and Y. Qu, Influence of oxygen vacancies of CeO2 on reverse water gas shift reaction, J. Catal., 2022, 414, 25–32 CrossRef CAS.
- J. Song, Z. Huang, L. Pan, J.-J. Zou, X. Zhang and L. Wang, Oxygen-deficient tungsten oxide as versatile and efficient hydrogenation catalyst, ACS Catal., 2015, 5, 6594–6599 CrossRef CAS.
- N. Zhao, J. Li, X. Chang, W. Zheng, J. Zhang and X. Liu, Synthesis and application of graphdiyne-based materials for advanced chemical sensors, Coord. Chem. Rev., 2024, 521, 216171 CrossRef CAS.
- J. Li, J. Hu, M. Zhang, W. Gou, S. Zhang, Z. Chen, Y. Qu and Y. Ma, A fundamental viewpoint on the hydrogen spillover phenomenon of electrocatalytic hydrogen evolution, Nat. Commun., 2021, 12, 3502 CrossRef CAS PubMed.
- L. Wang and R. T. Yang, New sorbents for hydrogen storage by hydrogen spillover – a review, Energy Environ. Sci., 2008, 1, 268–279 RSC.
- J. Im, H. Shin, H. Jang, H. Kim and M. Choi, Maximizing the catalytic function of hydrogen spillover in platinum-encapsulated aluminosilicates with controlled nanostructures, Nat. Commun., 2014, 5, 3370 CrossRef.
- W. Karim, C. Spreafico, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci and J. A. van Bokhoven, Catalyst support effects on hydrogen spillover, Nature, 2017, 541, 68–71 CrossRef CAS PubMed.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations, Science, 2012, 335, 1209–1212 CrossRef CAS.
- R. Prins, Hydrogen spillover. Facts and fiction, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS.
- F. Zaera, The long and winding road to catalysis, Nature, 2017, 541, 37–38 CrossRef CAS PubMed.
- E. Lalik, S. F. Parker, G. Irvine, I. da Silva, M. J. Gutmann, G. Romanelli, K. Druzbicki, R. Kosydar and M. Krzystyniak, Hydrogen spillover in tungsten oxide bronzes as observed by broadband neutron spectroscopy, Energies, 2023, 16, 5496 CrossRef CAS.
- K. Shun, K. Mori, S. Masuda, N. Hashimoto, Y. Hinuma, H. Kobayashi and H. Yamashita, Revealing hydrogen spillover pathways in reducible metal oxides, Chem. Sci., 2022, 13, 8137–8147 RSC.
- M. Cargnello, V. V. T. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Control of metal nanocrystal size reveals metal–support interface role for ceria catalysts, Science, 2013, 341, 771–773 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- T. Boningari, P. R. Ettireddy, A. Somogyvari, Y. Liu, A. Vorontsov, C. A. McDonald and P. G. Smirniotis, Influence of elevated surface texture hydrated titania on Ce-doped Mn/TiO2 catalysts for the low-temperature SCR of NOx under oxygen-rich conditions, J. Catal., 2015, 325, 145–155 CrossRef CAS.
- G. X. Zhuang, Y. W. Chen, Z. Y. Zhuang, Y. Yu and J. G. Yu, Oxygen vacancies in metal oxides: recent progress towards advanced catalyst design, Sci. China Mater., 2020, 63, 2089–2118 CrossRef CAS.
- K. Yu, L. Lou, S. Liu and W. Zhou, Asymmetric oxygen vacancies: the intrinsic redox active sites in metal oxide catalysts, Adv. Sci., 2020, 7, 1901970 CrossRef CAS.
- W. Yang, F. Qi, W. An, H. Yu, S. Liu, P. Ma, R. Chen, S. Liu, L. Lou and K. Yu, Local electronic structure modulation of interfacial oxygen vacancies promotes the oxygen activation capacity of Pt/Ce1−xMxO2−δ, ACS Catal., 2024, 14, 5936–5948 CrossRef CAS.
- Z. Wu, W. Yin, B. Wen, D. Ma and L. Liu, Oxygen vacancy diffusion in rutile TiO2: insight from deep neural network potential simulations, J. Phys. Chem. Lett., 2023, 14, 2208–2214 CrossRef CAS PubMed.
- F. Jiang, S. S. Wang, B. Liu, J. Liu, L. Wang, Y. Xiao, Y. B. Xu and X. H. Liu, Insights into the Influence of CeO2 crystal facet on CO2 hydrogenation to methanol over Pd/CeO2 catalysts, ACS Catal., 2020, 10, 11493–11509 CrossRef CAS.
- Y. E. Kim, B. Kim, W. Lee, Y. N. Ko, M. H. Youn, S. K. Jeong, K. T. Park and J. Oh, Highly tunable syngas production by electrocatalytic reduction of CO2 using Ag/TiO2 catalysts, Chem. Eng. J., 2021, 413, 127448 CrossRef CAS.
- D. R. Aireddy and K. Ding, Heterolytic dissociation of H2 in heterogeneous catalysis, ACS Catal., 2022, 12, 4707–4723 CrossRef CAS.
- S. Syrenova, C. Wadell, F. A. A. Nugroho, T. A. Gschneidtner, Y. A. D. Fernandez, G. Nalin, D. Switlik, F. Westerlund, T. J. Antosiewicz, V. P. Zhdanov, K. Moth-Poulsen and C. Langhammer, Hydride formation thermodynamics and hysteresis in individual Pd nanocrystals with different size and shape, Nat. Mater., 2015, 14, 1236–1244 CrossRef CAS.
- P. X. Liu, Y. Zhao, R. X. Qin, S. G. Mo, G. X. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. D. Zang, B. H. Wu, G. Fu and N. F. Zheng, Photochemical route for synthesizing atomically dispersed palladium catalysts, Science, 2016, 352, 797–801 CrossRef CAS.
- Z. H. Zhang, Z. Q. Wang, Z. R. Li, W. B. Zheng, L. P. Fan, J. Zhang, Y. M. Hu, M. F. Luo, X. P. Wu, X. Q. Gong, W. X. Huang and J. Q. Lu, Metal-free ceria catalysis for selective hydrogenation of crotonaldehyde, ACS Catal., 2020, 10, 14560–14566 CrossRef CAS.
- C. Yang, S. Ma, Y. Liu, L. Wang, D. Yuan, W. Shao, L. Zhang, F. Yang, T. Lin, H. Ding, H. He, Z. Liu, Y. Cao, Y. Zhu and X. Bao, Homolytic H2 dissociation for enhanced hydrogenation catalysis on oxides, Nat. Commun., 2024, 15, 540 CrossRef CAS.
- S. Xiang, L. Dong, Z. Wang, X. Han, L. L. Daemen, J. Li, Y. Cheng, Y. Guo, X. Liu, Y. Hu, A. J. Ramirez-Cuesta, S. Yang, X. Gong and Y. Wang, A unique Co@CoO catalyst for hydrogenolysis of biomass-derived 5-hydroxymethylfurfural to 2,5-dimethylfuran, Nat. Commun., 2022, 13, 3657 CrossRef CAS PubMed.
- S. Khoobiar, Particle to particle migration of hydrogen atoms on platinum—alumina catalysts from particle to neighboring particles, J. Phys. Chem., 1964, 68, 411–412 CrossRef CAS.
- M. Xiong, Z. Gao and Y. Qin, Spillover in heterogeneous catalysis: new insights and opportunities, ACS Catal., 2021, 11, 3159–3172 CrossRef CAS.
- M. Z. Li, W. A. Yin, J. A. Pan, Y. W. Zhu, N. Sun, X. Y. Zhang, Y. T. Wan, Z. Z. Luo, L. H. Yi and L. L. Wang, Hydrogen spillover as a promising strategy for boosting heterogeneous catalysis and hydrogen storage, Chem. Eng. J., 2023, 471, 144691 CrossRef CAS.
- S. Masuda, K. Shun, K. Mori, Y. Kuwahara and H. Yamashita, Synthesis of a binary alloy nanoparticle catalyst with an immiscible combination of Rh and Cu assisted by hydrogen spillover on a TiO2 support, Chem. Sci., 2020, 11, 4194–4203 RSC.
- M. D. Marcinkowski, A. D. Jewell, M. Stamatakis, M. B. Boucher, E. A. Lewis, C. J. Murphy, G. Kyriakou and E. C. H. Sykes, Controlling a spillover pathway with the molecular cork effect, Nat. Mater., 2013, 12, 523–528 CrossRef CAS PubMed.
- S. S. E. Collins, M. Cittadini, C. Pecharromán, A. Martucci and P. Mulvaney, Hydrogen spillover between single gold nanorods and metal oxide supports: a surface plasmon spectroscopy study, ACS Nano, 2015, 9, 7846–7856 CrossRef CAS PubMed.
- R. A. Bennett, P. Stone and M. Bowker, Pd nanoparticle enhanced re-oxidation of non-stoichiometric TiO2: STM imaging of spillover and a new form of SMSI, Catal. Lett., 1999, 59, 99–105 CrossRef CAS.
- Y. Lykhach, T. Staudt, M. Vorokhta, T. Skála, V. Johánek, K. C. Prince, V. Matolín and J. Libuda, Hydrogen spillover monitored by resonant photoemission spectroscopy, J. Catal., 2012, 285, 6–9 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, QUICKSTEP: fast and accurate density functional calculations using a mixed Gaussian and plane waves approach, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- C. Spreafico and J. VandeVondele, The nature of excess electrons in anatase and rutile from hybrid DFT and RPA, PCCP, 2014, 16, 26144–26152 RSC.
- Z. Tan, J. Chen and S. Lin, Theoretical insights into H2 activation and hydrogen spillover on near-surface alloys with embedded single Pt atoms, ACS Catal., 2024, 14, 2194–2201 CrossRef CAS.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, Use of DFT to achieve a rational understanding of acid–basic properties of γ-alumina surfaces, J. Catal., 2004, 226, 54–68 CrossRef CAS.
- R. Wischert, P. Laurent, C. Copéret, F. Delbecq and P. Sautet, γ-Alumina: the essential and unexpected role of water for the structure, stability, and reactivity of “defect” sites, J. Am. Chem. Soc., 2012, 134, 14430–14449 CrossRef CAS.
- M. J. Nash, A. M. Shough, D. W. Fickel, D. J. Doren and R. F. Lobo, High-temperature dehydrogenation of Bronsted acid sites in zeolites, J. Am. Chem. Soc., 2008, 130, 2460–2462 CrossRef CAS.
- M. Vitiello, N. Lopez, F. Illas and G. Pacchioni, H2 cracking at SiO2 defect centers, J. Phys. Chem. A, 2000, 104, 4674–4684 CrossRef CAS.
- M. Ramos, A. E. Martínez and H. F. Busnengo, H2 dissociation on individual Pd atoms deposited on Cu(111), PCCP, 2012, 14, 303–310 RSC.
- K. X. Gu, F. F. Wei, Y. H. Cai, S. Lin and H. Guo, Dynamics of initial hydrogen spillover from a single atom platinum active site to the Cu(111) host surface: the impact of substrate electron-hole pairs, J. Phys. Chem. Lett., 2021, 12, 8423–8429 CrossRef CAS PubMed.
- W. Osada, S. Tanaka, K. Mukai, M. Kawamura, Y. Choi, F. Ozaki, T. Ozaki and J. Yoshinobu, Elucidation of the atomic-scale processes of dissociative adsorption and spillover of hydrogen on the single atom alloy catalyst Pd/Cu(111), PCCP, 2022, 24, 21705–21713 RSC.
- K. Gu and S. Lin, Sustained hydrogen spillover on Pt/Cu(111) single-atom alloy: dynamic insights into gas-induced chemical processes, Angew. Chem., Int. Ed., 2023, 62, e202312796 CrossRef CAS PubMed.
- A. Mahdavi-Shakib, T. N. Whittaker, T. Y. Yun, K. B. Sravan Kumar, L. C. Rich, S. Wang, R. M. Rioux, L. C. Grabow and B. D. Chandler, The role of surface hydroxyls in the entropy-driven adsorption and spillover of H2 on Au/TiO2 catalysts, Nat. Catal., 2023, 6, 710–719 CrossRef CAS.
- S. Campisi, C. E. Chan-Thaw, L. E. Chinchilla, A. Chutia, G. A. Botton, K. M. H. Mohammed, N. Dimitratos, P. P. Wells and A. Villa, Dual-site-mediated hydrogenation catalysis on Pd/NiO: selective biomass transformation and maintenance of catalytic activity at low Pd loading, ACS Catal., 2020, 10, 5483–5492 CrossRef CAS.
- T. C. Xiao, X. H. Liu, G. Y. Xu and Y. Zhang, Phase tuning of ZrO2 supported cobalt catalysts for hydrodeoxygenation of 5-hydroxymethylfurfural to 2,5-dimethylfuran under mild conditions, Appl. Catal., B, 2021, 295, 120270 CrossRef CAS.
- J. Wei, S. N. Qin, J. L. Liu, X. Y. Ruan, Z. Q. Guan, H. Yan, D. Y. Wei, H. Zhang, J. Cheng, H. X. Xu, Z. Q. Tian and J. F. Li, In Situ raman monitoring and manipulating of interfacial hydrogen spillover by precise fabrication of Au/TiO2/Pt sandwich structures, Angew. Chem., Int. Ed., 2020, 59, 10343–10347 CrossRef CAS.
- T. Ioannides and X. E. Verykios, The interaction of benzene and toluene with Rh dispersed on SiO2, Al2O3, and TiO2 carriers, J. Catal., 1993, 143, 175–186 CrossRef CAS.
- G. J. Zhang, F. Y. Tang, X. Wang, L. Q. Wang and Y. N. Liu, Atomically dispersed Co–S–N active sites anchored on hierarchically porous carbon for efficient catalytic hydrogenation of nitro compounds, ACS Catal., 2022, 12, 5786–5794 CrossRef CAS.
- C. L. Mao, J. X. Wang, Y. J. Zou, G. D. Qi, J. Y. Y. Loh, T. H. Zhang, M. K. Xia, J. Xu, F. Deng, M. Ghoussoub, N. P. Kherani, L. Wang, H. Shang, M. Q. Li, J. Li, X. Liu, Z. H. Ai, G. A. Ozin, J. C. Zhao and L. Z. Zhang, Hydrogen spillover to oxygen vacancy of TiO2-xHy/Fe: breaking the scaling relationship of ammonia synthesis, J. Am. Chem. Soc., 2020, 142, 17403–17412 CrossRef CAS PubMed.
- G. Sun, D. Liu, H. Shi, J. Li, L. Yang, F. Tian, Y. Cui, C. Wang, F. Li, T. Zhao, H. Zhu, B. Liu, Y. Chai, Y. Liu and Y. Pan, Oxygen-vacancy-induced built-in electric field across MoCo dual-atomic site catalyst for promoting hydrogen spillover in hydrocracking and hydrodesulfurization, ACS Catal., 2024, 14, 3208–3217 CrossRef CAS.
- Z. W. Wei, H. J. Wang, C. Zhang, K. Xu, X. L. Lu and T. B. Lu, Reversed charge transfer and enhanced hydrogen spillover in platinum nanoclusters anchored on titanium oxide with rich oxygen vacancies boost hydrogen evolution reaction, Angew. Chem., Int. Ed., 2021, 60, 16622–16627 CrossRef CAS PubMed.
- X. Zhao, J. Wang, L. Lian, G. Zhang, P. An, K. Zeng, H. He, T. Yuan, J. Huang, L. Wang and Y. Liu, Oxygen vacancy-reinforced water-assisted proton hopping for enhanced catalytic hydrogenation, ACS Catal., 2023, 13, 2326–2334 CrossRef CAS.
- N. B. A. Jr, H. Kasai, W. A. Diño and H. Nakanishi, Potential energy of H2 dissociation and adsorption on Pt(111) surface: first-principles calculation, Jpn. J. Appl. Phys., 2007, 46, 4233 CrossRef.
- F. P. Netzer and G. Kneringer, The adsorption of hydrogen and the reaction of hydrogen with oxygen on Pt (100), Surf. Sci., 1975, 51, 526–538 CrossRef CAS.
- B. Poelsema, K. Lenz and G. Comsa, The dissociative adsorption of hydrogen on defect-‘free’ Pt(111), J. Phys.: Condens. Matter, 2010, 22, 304006 CrossRef.
- M. M. Bettahar, The hydrogen spillover effect. A misunderstanding story, Catal. Rev. Sci. Eng., 2022, 64, 87–125 CrossRef CAS.
- S. Lee, K. Lee, J. Im, H. Kim and M. Choi, Revisiting hydrogen spillover in Pt/LTA: effects of physical diluents having different acid site distributions, J. Catal., 2015, 325, 26–34 CrossRef CAS.
- M. Tan, Y. Yang, Y. Yang, J. Chen, Z. Zhang, G. Fu, J. Lin, S. Wan, S. Wang and Y. Wang, Hydrogen spillover assisted by oxygenate molecules over nonreducible oxides, Nat. Commun., 2022, 13, 1457 CrossRef CAS.
- N. M. Zou, X. C. Zhou, G. Q. Chen, N. M. Andoy, W. Jung, G. K. Liu and P. Chen, Cooperative communication within and between single nanocatalysts, Nat. Chem., 2018, 10, 607–614 CrossRef CAS PubMed.
- M. F. de Lange, K. J. F. M. Verouden, T. J. H. Vlugt, J. Gascon and F. Kapteijn, Adsorption-driven heat pumps: the potential of metal–organic frameworks, Chem. Rev., 2015, 115, 12205–12250 CrossRef CAS PubMed.
- N. C. Burtch, I. M. Walton, J. T. Hungerford, C. R. Morelock, Y. Jiao, J. Heinen, Y. S. Chen, A. A. Yakovenko, W. Q. Xu, D. Dubbeldam and K. S. Walton, In situ visualization of loading-dependent water effects in a stable metal–organic framework, Nat. Chem., 2020, 12, 186–192 CrossRef CAS.
- Z. Gu, M. Li, C. Chen, X. Zhang, C. Luo, Y. Yin, R. Su, S. Zhang, Y. Shen, Y. Fu, W. Zhang and F. Huo, Water-assisted hydrogen spillover in Pt nanoparticle-based metal–organic framework composites, Nat. Commun., 2023, 14, 5836 CrossRef CAS.
- R. I. Cukier and D. G. Nocera, Proton-coupled electron transfer, Annu. Rev. Phys. Chem., 1998, 49, 337–369 CrossRef CAS PubMed.
- X. Li, X. M. Ren, M. Guo, W. J. Li and Q. H. Yang, Water accelerated activity of Ru NPs in sequential hydrogenation of nitrobenzene to cyclohexylamine, J. Catal., 2022, 413, 546–553 CrossRef CAS.
- Z. Zhao, R. Bababrik, W. H. Xue, Y. P. Li, N. M. Briggs, D. T. Nguyen, U. Nguyen, S. P. Crossley, S. W. Wang, B. Wang and D. E. Resasco, Solvent-mediated charge separation drives alternative hydrogenation path of furanics in liquid water, Nat. Catal., 2019, 2, 431–436 CrossRef CAS.
- Y. H. Dai, X. F. Chu, J. J. Gu, X. Gao, M. Xu, D. Lu, X. Y. Wan, W. Qi, B. S. Zhang and Y. H. Yang, Water-enhanced selective hydrogenation of cinnamaldehyde to cinnamyl alcohol on RuSnB/CeO2 catalysts, Appl. Catal., A, 2019, 582, 117098 CrossRef CAS.
- R. Samanta, B. K. Manna, R. Trivedi, B. Chakraborty and S. Barman, Hydrogen spillover enhances alkaline hydrogen electrocatalysis on interface-rich metallic Pt-supported MoO, Chem. Sci., 2023, 15, 364–378 RSC.
- Y. An, P. Chatterjee, P. Naik, S. Banerjee, W. Y. Huang, I. I. Slowing and V. Venditti, Hydrogen spillover and substrate-support hydrogen bonding mediate hydrogenation of phenol catalyzed by palladium on reducible metal oxides, Chem. Sci., 2023, 14, 14166–14175 RSC.
- P. X. Liu, R. X. Qin, G. Fu and N. F. Zheng, Surface coordination chemistry of metal nanomaterials, J. Am. Chem. Soc., 2017, 139, 2122–2131 CrossRef CAS PubMed.
- N. Kaeffer, D. Mance and C. Copéret, N-Heterocyclic carbene coordination to surface copper sites in selective semihydrogenation catalysts from solid-state NMR spectroscopy, Angew. Chem., Int. Ed., 2020, 59, 19999–20007 CrossRef CAS PubMed.
- F. Zaera, CHEMISTRY the long and winding road to catalysis, Nature, 2017, 541, 37–38 CrossRef CAS PubMed.
- S. Zhang, Z. Xia, M. Zhang, Y. Zou, H. Shen, J. Li, X. Chen and Y. Qu, Boosting selective hydrogenation through hydrogen spillover on supported-metal catalysts at room temperature, Appl. Catal., B, 2021, 297, 120418 CrossRef CAS.
- S. C. Tsang, N. Cailuo, W. Oduro, A. T. S. Kong, L. Clifton, K. M. K. Yu, B. Thiebaut, J. Cookson and P. Bishop, Engineering preformed cobalt-doped platinum nanocatalysts for ultraselective hydrogenation, ACS Nano, 2008, 2, 2547–2553 CrossRef CAS.
- Q. Zheng, H. Xu, Y. Yao, J. Dai, J. Wang, W. Hou, L. Zhao, X. Zou, G. Zhan, R. Wang, K. Wang and L. Zhang, Cobalt single-atom reverse hydrogen spillover for efficient electrochemical water dissociation and dechlorination, Angew. Chem., Int. Ed., 2024, 63, e202401386 CrossRef CAS PubMed.
- W. Liu, H. Feng, Y. Yang, Y. Niu, L. Wang, P. Yin, S. Hong, B. Zhang, X. Zhang and M. Wei, Highly-efficient RuNi single-atom alloy catalysts toward chemoselective hydrogenation of nitroarenes, Nat. Commun., 2022, 13, 3188 CrossRef CAS.
- C. Choi, X. Wang, S. Kwon, J. L. Hart, C. L. Rooney, N. J. Harmon, Q. P. Sam, J. J. Cha, W. A. Goddard, M. Elimelech and H. Wang, Efficient electrocatalytic valorization of chlorinated organic water pollutant to ethylene, Nat. Nanotechnol., 2023, 18, 160–167 CrossRef CAS PubMed.
- C. Han, J. Zenner, J. Johny, N. Kaeffer, A. Bordet and W. Leitner, Electrocatalytic hydrogenation of alkenes with Pd/carbon nanotubes at an oil–water interface, Nat. Catal., 2022, 5, 1110–1119 CrossRef CAS.
- G. Han, G. Li and Y. Sun, Electrocatalytic dual hydrogenation of organic substrates with a Faradaic efficiency approaching 200%, Nat. Catal., 2023, 6, 224–233 CrossRef CAS.
- D. Huang, D. J. Kim, K. Rigby, X. Zhou, X. Wu, A. Meese, J. Niu, E. Stavitski and J.-H. Kim, Elucidating the role of single-atom Pd for electrocatalytic hydrodechlorination, Environ. Sci. Technol., 2021, 55, 13306–13316 CAS.
- M. Li and J. Shen, Microcalorimetric studies of O2 and C2H4 adsorption on Pd/SiO2 catalysts modified by Cu and Ag, Thermochim. Acta, 2001, 379, 45–50 CrossRef CAS.
- H. Zhou, X. Yang, L. Li, X. Liu, Y. Huang, X. Pan, A. Wang, J. Li and T. Zhang, PdZn intermetallic nanostructure with Pd–Zn–Pd ensembles for highly active and chemoselective semi-hydrogenation of acetylene, ACS Catal., 2016, 6, 1054–1061 CrossRef CAS.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Identification of non-precious metal alloy catalysts for selective hydrogenation of acetylene, Science, 2008, 320, 1320–1322 CrossRef CAS PubMed.
- K. R. Kahsar, D. K. Schwartz and J. W. Medlin, Control of metal catalyst selectivity through specific noncovalent molecular interactions, J. Am. Chem. Soc., 2014, 136, 520–526 CrossRef CAS PubMed.
- K.-i. Shimizu, Y. Miyamoto, T. Kawasaki, T. Tanji, Y. Tai and A. Satsuma, Chemoselective hydrogenation of nitroaromatics by supported gold catalysts: mechanistic reasons of size-and support-dependent activity and selectivity, J. Phys. Chem. C, 2009, 113, 17803–17810 CrossRef CAS.
- B. Wu, H. Huang, J. Yang, N. Zheng and G. Fu, Selective hydrogenation of α,β-unsaturated aldehydes catalyzed by amine-capped platinum-cobalt nanocrystals, Angew. Chem., Int. Ed., 2012, 51, 3440–3443 CrossRef CAS PubMed.
- Y. Li, Y. Zhang, K. Qian and W. Huang, Metal–support interactions in metal/oxide catalysts and oxide–metal interactions in oxide/metal inverse catalysts, ACS Catal., 2022, 12, 1268–1287 CrossRef CAS.
- D. X. Ji, L. Fan, L. Tao, Y. J. Sun, M. G. Li, G. R. Yang, T. Q. Tran, S. Ramakrishna and S. J. Guo, The kirkendall effect for engineering oxygen vacancy of hollow CoO nanoparticles toward high-performance portable zinc-air batteries, Angew. Chem., Int. Ed., 2019, 58, 13840–13844 CrossRef CAS PubMed.
- C. D. Lv, C. Lee, L. X. Zhong, H. J. Liu, J. W. Liu, L. Yang, C. S. Yan, W. Yu, H. H. Hng, Z. M. Qi, L. Song, S. Z. Li, K. P. Loh, Q. Y. Yan and G. H. Yu, A defect engineered electrocatalyst that promotes high-efficiency urea synthesis under ambient conditions, ACS Nano, 2022, 16, 8213–8222 CrossRef CAS.
- C. Xie, D. F. Yan, H. Li, S. Q. Du, W. Chen, Y. Y. Wang, Y. Q. Zou, R. Chen and S. Y. Wang, Defect chemistry in heterogeneous catalysis: recognition, understanding, and utilization, ACS Catal., 2020, 10, 11082–11098 CrossRef CAS.
- Y. Lu, T. Liu, C.-L. Dong, C. Yang, L. Zhou, Y. Huang, Y. Li, B. Zhou, Y. Zou and S. Wang, Tailoring competitive adsorption sites by oxygen-vacancy on cobalt oxides to enhance the electrooxidation of biomass, Adv. Mater., 2022, 34, 2107185 CrossRef CAS.
- F. Zhang, B. Li, X. Quan, K. Wang, J. Xu, T. Wu, Z. Li, M. Yan, S. Liu, Y. He, Y. Shi, Y. Su and P. Xie, Revealing the dynamics of oxygen vacancy in ZnO1−x/Cu during robust methanol synthesis from CO2, ACS Catal., 2024, 14, 7136–7148 CrossRef CAS.
- G. Xiang, H. Chen, C. Yi, Z. Liu and S. Dai, Oxygen vacancy-regulated atomic dispersed Au on CeFeOx for preferential oxidation of CO in H2-rich stream, Chem. Eng. J., 2024, 479, 147775 CrossRef CAS.
- H. Xu, H. Zhang, L. Cui, X. Zhao, J. Xiao, J. Zhang, Y. Qin and J. Zhang, Unravelling the synergy of platinum-oxygen vacancy in CoOx for modulating hydrogenation performance, Chem. Eng. J., 2024, 488, 150841 CrossRef CAS.
- F. X. Zhang, B. Y. Li, X. Quan, K. Wang, J. Y. Xu, T. T. Wu, Z. L. Li, M. Yan, S. J. Liu, Y. He, Y. Shi, Y. Q. Su and P. F. Xie, Revealing the dynamics of oxygen vacancy in ZnO1−x/Cu during robust methanol synthesis from CO2, ACS Catal., 2024, 14, 7136–7148 CrossRef CAS.
- X. X. Wei, X. J. Wen, Y. Y. Liu, C. Chen, C. Xie, D. D. Wang, M. Y. Qiu, N. H. He, P. Zhou, W. Chen, J. Cheng, H. Z. Lin, J. F. Jia, X. Z. Fu and S. Y. Wang, Oxygen vacancy-mediated selective C–N coupling toward electrocatalytic urea synthesis, J. Am. Chem. Soc., 2022, 144, 11530–11535 CrossRef CAS PubMed.
- S. N. Jiang, N. Ji, X. Y. Diao, H. Y. Li, Y. Rong, Y. X. Lei and Z. H. Yu, Vacancy engineering in transition metal sulfide and oxide catalysts for hydrodeoxygenation of lignin-derived oxygenates, ChemSusChem, 2021, 14, 4377–4396 CrossRef CAS PubMed.
- M. Yazdanpanah, M. Fereidooni, V. Marquez, C. V. Paz, T. Saelee, M. S. Villanueva, M. Rittiruam, P. Khajondetchairit, S. Praserthdam and P. Praserthdam, The underlying catalytic role of oxygen vacancies in fatty acid methyl esters ketonization over TiO2 catalysts, ChemSusChem, 2024, 17, e202301033 CrossRef CAS.
- P. Nikacevic, F. S. Hegner, J. R. Galán-Mascarós and N. López, Influence of oxygen vacancies and surface facets on water oxidation selectivity toward oxygen or hydrogen peroxide with BiVO4, ACS Catal., 2021, 11, 13416–13422 CrossRef CAS.
- Q. Wang, R. Duan, Z. Feng, Y. Zhang, P. Luan, Z. Feng, J. Wang and C. Li, Understanding the synergistic catalysis in hydrogenation of carbonyl groups on Cu-based catalysts, ACS Catal., 2024, 14, 1620–1628 CrossRef CAS.
- Y. Sun, B. Du, Y. Wang, M. Zhang and S. Zhang, Hydrogen spillover-accelerated selective hydrogenation on WO3 with ppm-level Pd, ACS Appl. Mater. Interfaces, 2023, 15, 20474–20482 CrossRef CAS PubMed.
- J. Zhang, M. Gao, R. Wang, X. Li, P. Zhu, Y. Wang and Z. Zheng, Oxygen vacancy regulated selective hydrogenation of α,β-unsaturated aldehydes over LDH surface group coordinated transition metal photocatalysts, Catal. Sci. Technol., 2022, 12, 6163–6173 RSC.
- R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Nanoscale Fe2O3-based catalysts for selective hydrogenation of nitroarenes to anilines, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed.
- H. S. Wei, X. Y. Liu, A. Q. Wang, L. L. Zhang, B. T. Qiao, X. F. Yang, Y. Q. Huang, S. Miao, J. Y. Liu and T. Zhang, FeOx-supported platinum single-atom and pseudo-single-atom catalysts for chemoselective hydrogenation of functionalized nitroarenes, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed.
- T. Schwob and R. Kempe, A reusable Co catalyst for the selective hydrogenation of functionalized nitroarenes and the direct synthesis of imines and benzimidazoles from nitroarenes and aldehydes, Angew. Chem., Int. Ed., 2016, 55, 15175–15179 CrossRef CAS PubMed.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Selective hydrogenation over supported metal catalysts: from nanoparticles to single atoms, Chem. Rev., 2020, 120, 683–733 CrossRef CAS.
- A. Deshpande, F. Figueras, M. Lakshmi Kantam, K. Jeeva Ratnam, R. Sudarshan Reddy and N. S. Sekhar, Environmentally friendly hydrogenation of nitrobenzene to p-aminophenol using heterogeneous catalysts, J. Catal., 2010, 275, 250–256 CrossRef CAS.
- W. C. Cheong, W. J. Yang, J. Zhang, Y. Li, D. Zhao, S. J. Liu, K. L. Wu, Q. G. Liu, C. Zhang, D. S. Wang, Q. Peng, C. Chen and Y. D. Li, Isolated iron single-atomic site-catalyzed chemoselective transfer hydrogenation of nitroarenes to arylamines, ACS Appl. Mater. Interfaces, 2019, 11, 33819–33824 CrossRef CAS PubMed.
- V. K. Das, S. Mazhar, L. Gregor, B. D. Stein, D. G. Morgan, N. A. Maciulis, M. Pink, Y. Losovyj and L. M. Bronstein, Graphene derivative in magnetically recoverable catalyst determines catalytic properties in transfer hydrogenation of nitroarenes to anilines with 2-propanol, ACS Appl. Mater. Interfaces, 2018, 10, 21356–21364 CrossRef CAS.
- Y. Guo, M. Wang, Q. Zhu, D. Xiao and D. Ma, Ensemble effect for single-atom, small cluster and nanoparticle catalysts, Nat. Catal., 2022, 5, 766–776 CrossRef CAS.
- L. Jiang, K. Liu, S. Hung, L. Zhou, R. Qin, Q. Zhang, P. Liu, L. Gu, H. M. Chen, G. Fu and N. Zheng, Facet engineering accelerates spillover hydrogenation on highly diluted metal nanocatalysts, Nat. Nanotechnol., 2020, 15, 848–853 CrossRef CAS.
- M. J. Hülsey, V. Fung, X. Hou, J. Wu and N. Yan, Hydrogen spillover and its relation to hydrogenation: observations on structurally defined single-atom sites, Angew. Chem., Int. Ed., 2022, 61, e202208237 CrossRef.
- V. Fung, G. Hu, Z. Wu and D.-e. Jiang, Hydrogen in nanocatalysis, J. Phys. Chem. Lett., 2020, 11, 7049–7057 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |