
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of exciton delocalization in chiroptical properties of benzothiadiazole carbon nanohoops†
Kovida
Kovida
a,
Juraj
Malinčík
ac,
Carlos M.
Cruz
 b,
Araceli G.
Campaña
b and
Tomáš
Šolomek
*ac
b,
Araceli G.
Campaña
b and
Tomáš
Šolomek
*ac
aVan't Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam, PO Box 94157, 1090 GD Amsterdam, The Netherlands. E-mail: t.solomek@uva.nl
bDepartment of Organic Chemistry, University of Granada, Avda Fuentenueva, s/n, 18071 Granada, Spain
cPrievidza Chemical Society, M. Hodžu 10/16, 971 01 Prievidza, Slovakia
First published on 20th December 2024
Abstract
Development of chiral organic materials with a strong chiroptical response is crucial to advance technologies based on circularly polarized luminescence, enantioselective sensing, or unique optical signatures in anti-counterfeiting. The progress in the field is hampered by the lack of structure–property relationships that would help designing new chiral molecules. Here, we address this challenge by synthesis and investigation of two chiral macrocycles that integrate in their structure a pseudo-meta [2.2]paracyclophane with planar chirality and a highly fluorescent benzothiadiazole. Both compounds display remarkably red-shifted fluorescence with high quantum yields and large Stokes shifts. They differ in the extent of π-electron conjugation that allowed, for the first time, systematic examination of the effect of exciton delocalization on the absorption and luminescence of circularly polarized light. By a combination of steady-state spectroscopy and quantum chemical calculations, we constructed a unique structure–property relationship offering critical insights that will aid and abet the development of robust design guidelines for materials with strong electronic circular dichroism or circularly polarized luminescence of exceptional brightness.
Introduction
Organic materials with strong electronic circular dichroism (ECD) and circularly polarized luminescence (CPL) are attractive synthetic targets for their potential applications in chiroptical devices.1–5 The quality of such chiroptical materials is determined by a few key metrics: the dissymmetry factor (g), the molar absorptivity (ε), and the fluorescence quantum yield (ϕF).6,7 Combined, they define the overall brightness of CPL, BCPL.8 The typical values of the luminescence dissymmetry factor (glum) in small organic molecules are relatively low. The dissymmetry factor critically depends on the size and mutual orientation of the electric (μ) and magnetic (m) transition dipole moments and, as a result, it is relatively difficult to tune the dissymmetry factor systematically by precision organic synthesis due to the absence of robust molecular design guidelines. For example, among the few known approaches to improve glum is imposing the Dn symmetry9,10 to a chiral fluorophore, as demonstrated in a figure-eight-shaped [5]helicene dimer by Matsuda and Hirose.11 Here, the electric and magnetic transition dipole moments adopt ideal parallel orientation providing |glum| = 1.5 × 10−2. However, this system suffers from a relatively low ϕF, i.e., low μ. Similarly, the high absorption dissymmetry factor (gabs) in ECD is often associated with lower molar absorptivity, with the latter further impacting BCPL. Although the underlying theory is well established,7,10,12 systematic studies on how the nature of the excited state transitions in CPL-active compounds depends on the molecular structure are relatively scarce.13–16 Recently, some of us proposed a rationalization for enhancing |m| in helically chiral compounds to achieve high values of g in ECD or CPL by finding a direct relationship with the area of the inner cavity of the helix.17In recent years, chiral carbon nanohoops, π-conjugated macrocycles derived from cycloparaphenylenes ([n]CPPs) have emerged as a promising class of CPL-active molecules. The curved architecture of CPPs endows them with visible-light fluorescence with relatively high ϕF that can be tuned by controlling their size with synthesis.18–21 But CPPs are achiral. Therefore, chirality in carbon nanohoops is typically induced by incorporation of a chiral unit into CPPs, such as biphenyl, binaphthyl, helicene or others.22–30 Although they provide chiral nanohoops with a relatively high configurational stability, they can in principle racemize. Racemization can be fully prevented by creating topologically chiral carbon nanohoops as reported for example by Stępień or Jasti.31,32 Recently, Jiang et al. used planar chiral pseudo-meta [2.2]paracyclophane (PCP) to induce stable chirality and CPL response in carbon nanohoops, such as 1 (Fig. 1),33 and also used tetrasubstituted PCP to synthesize a topologically chiral carbon nanohoop.34
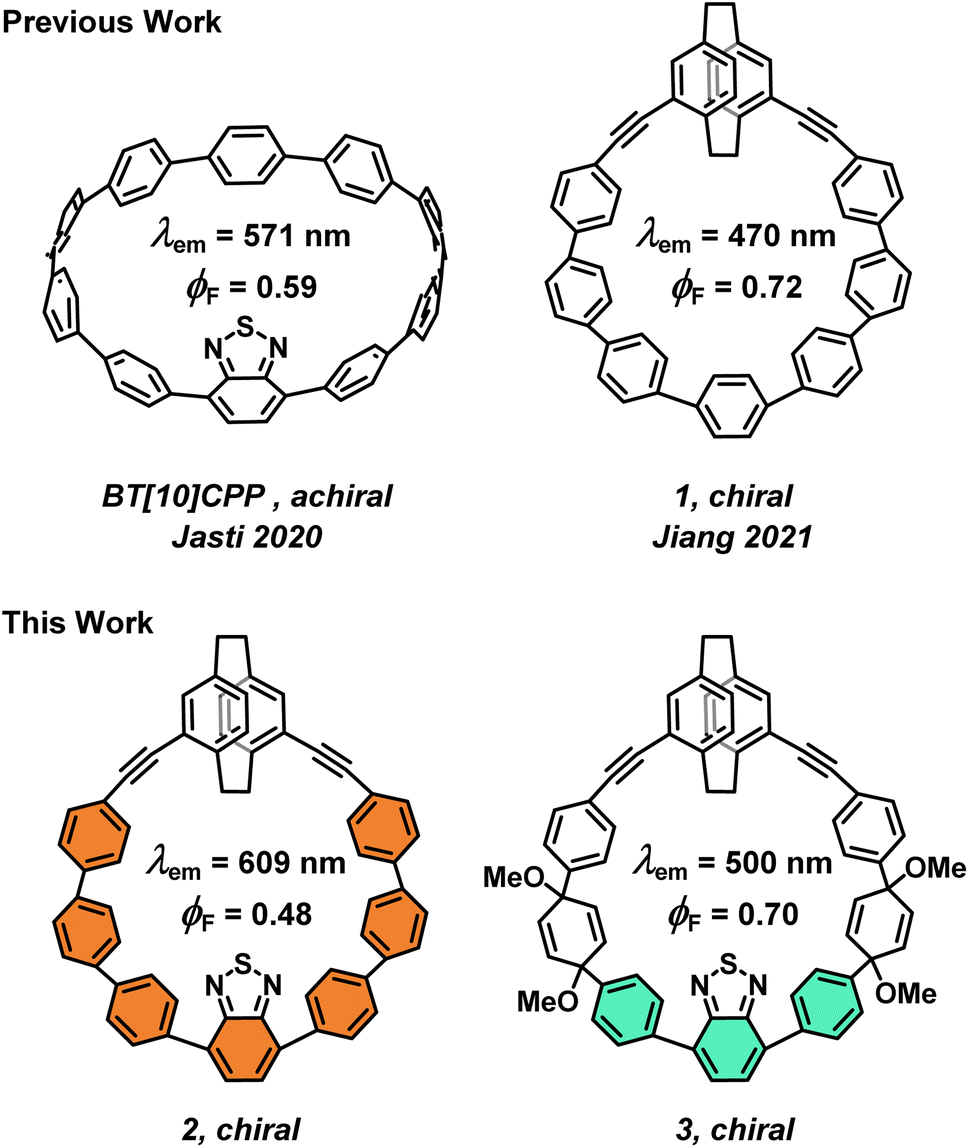 | ||
| Fig. 1 Previous and concept of the present work. | ||
CPPs emitting light above 500 nm display a low ϕF,21 a key metric of a good fluorophore. In 2020, Jasti et al. reported an achiral benzothiadiazole (BT) nanohoop BT[10]CPP (Fig. 1) with λmax at 571 nm, i.e., with >100 nm red-shift compared to [10]CPP, and impressive ϕF = 0.59.35 However, chiral nanohoops with CPL with λmax > 550 nm and high ϕF are only rarely reported.36
Here, we report on simultaneous integration of pseudo-meta PCP and BT into a carbon nanohoop (2, Fig. 1) accomplishing a CPL beyond 600 nm with a good ϕF. In addition, we achieved a turn-off of the π–electron conjugation in 2 using the pro-aromatic units in its equally luminous chiral precursor 3. Compound 3 displayed a bright fluorescence at 500 nm, the first such nanohoop precursor to do so. Ultimately, the chiroptical properties of 1, 2, and 3 allowed us to explore, for the first time, the effect of the extent of exciton delocalization in this important class of molecules on their chiroptical response in ECD and CPL. The structure–property relationship constructed in this work together with previous studies allowed us to formulate guidelines to aid the synthesis of new molecular systems with outstanding chiroptical properties.
Results and discussion
Synthesis
The target chiral carbon nanohoop 2 was synthesized according to the strategy developed by Jasti,18i.e., via aromatization of macrocycle 3 with pro-aromatic cyclohexa-2,5-dienyl units prepared according to Scheme 1. First, Sonogashira cross coupling between 4,15-bis(ethynyl)[2.2]paracyclophane 4 and building block 5 provided the C-shape intermediate 6 (ref. 33) in 75% yield. The Suzuki cross-coupling of 6 with 7 gave chiral macrocycle 3 in 23% yield.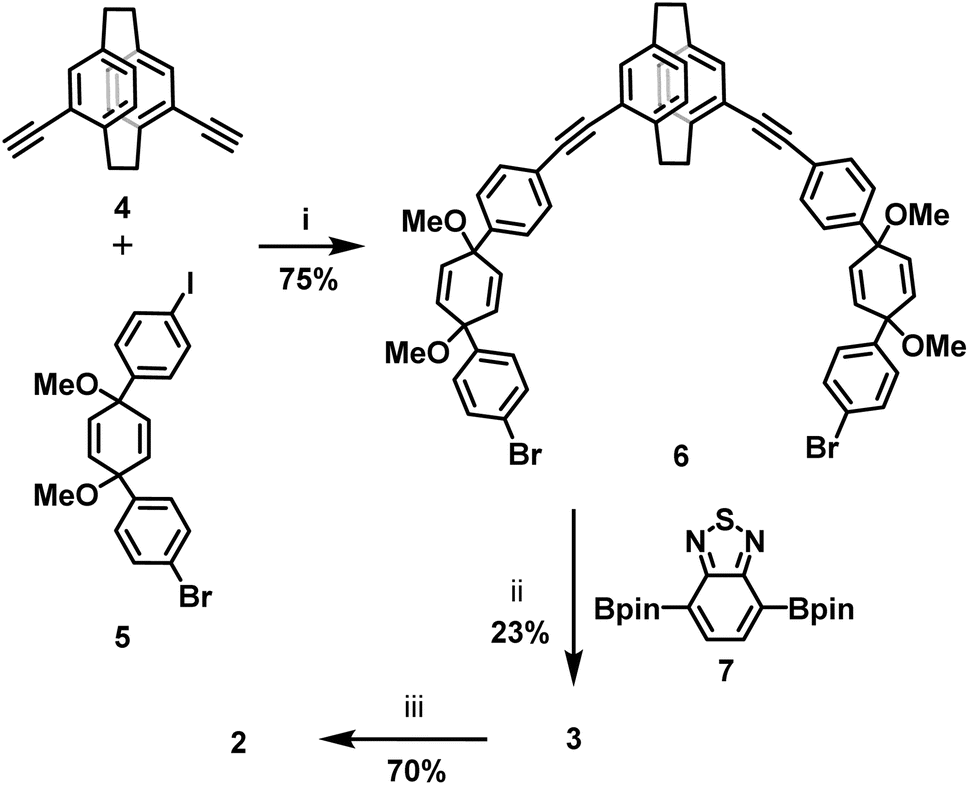 | ||
| Scheme 1 Synthesis of BT containing macrocycles 2 and 3. Reaction conditions: (i) Pd2dba3, PPh3, CuI, THF/TEA, RT, 4 h. (ii) K3PO4, SPhos Pd Gen 3, dioxane, 80 °C, 18 h (iii) A: SnCl2, HCl, THF, RT, 2 h or B: Na, naphthalene, THF, −78 °C, 10 min. | ||
Reductive aromatization of 3 using SnCl2/HCl leads to the desired nanohoop 2 alongside several side products, a result of acid-catalysed rearrangement reactions. Under these conditions, 2 was obtained as a bright fluorescent orange solid in 60% yield. We also conducted the aromatization using sodium naphthalenide as the reducing agent. In this case, nanohoop 2 was formed in a markedly cleaner process in 70% yield after purification by column chromatography. All compounds were fully characterized by 1H, 13C NMR and high-resolution mass spectrometry (see the ESI†).
Photophysical properties
We then investigated the optoelectronic properties of 2 and 3. Note that the pro-aromatic cyclohexa-2,5-dienyls in 3 interrupt the π–electron conjugation between PCP and BT in the macrocycle, thereby preventing the unique optoelectronic properties of fully conjugated carbon nanohoops to emerge. Nevertheless, 3 displays a bright green fluorescence, which is highly unusual in macrocyclic precursors of carbon nanohoops. Here, the observed green luminescence in 3 allowed us to investigate how the conjugation affects the absorption and emission in these macrocycles (Fig. 2a and Table 1). The absorption of 2 is similar to that of [n]CPPs with a characteristic S0 → S2 transition at 324 nm, blue-shifted by 4 nm compared to 1,33 showing a minor influence of replacing a phenylene by BT on the dominant absorption maximum. The absorption profile of 3 at ∼300 nm is not well resolved due to the presence of multiple electronic transitions of similar energy (Table S2†). The absorption band for the S0 → S1 transition is observed for 2 and 3 at 450 nm (ε = 0.3 × 104 M−1 cm−1) and 389 nm (0.9 × 104 M−1 cm−1), respectively. Both the bathochromic shift and the decrease in the ε with increased conjugation in 2 may suggest an enhancement of the charge-transfer character in this transition.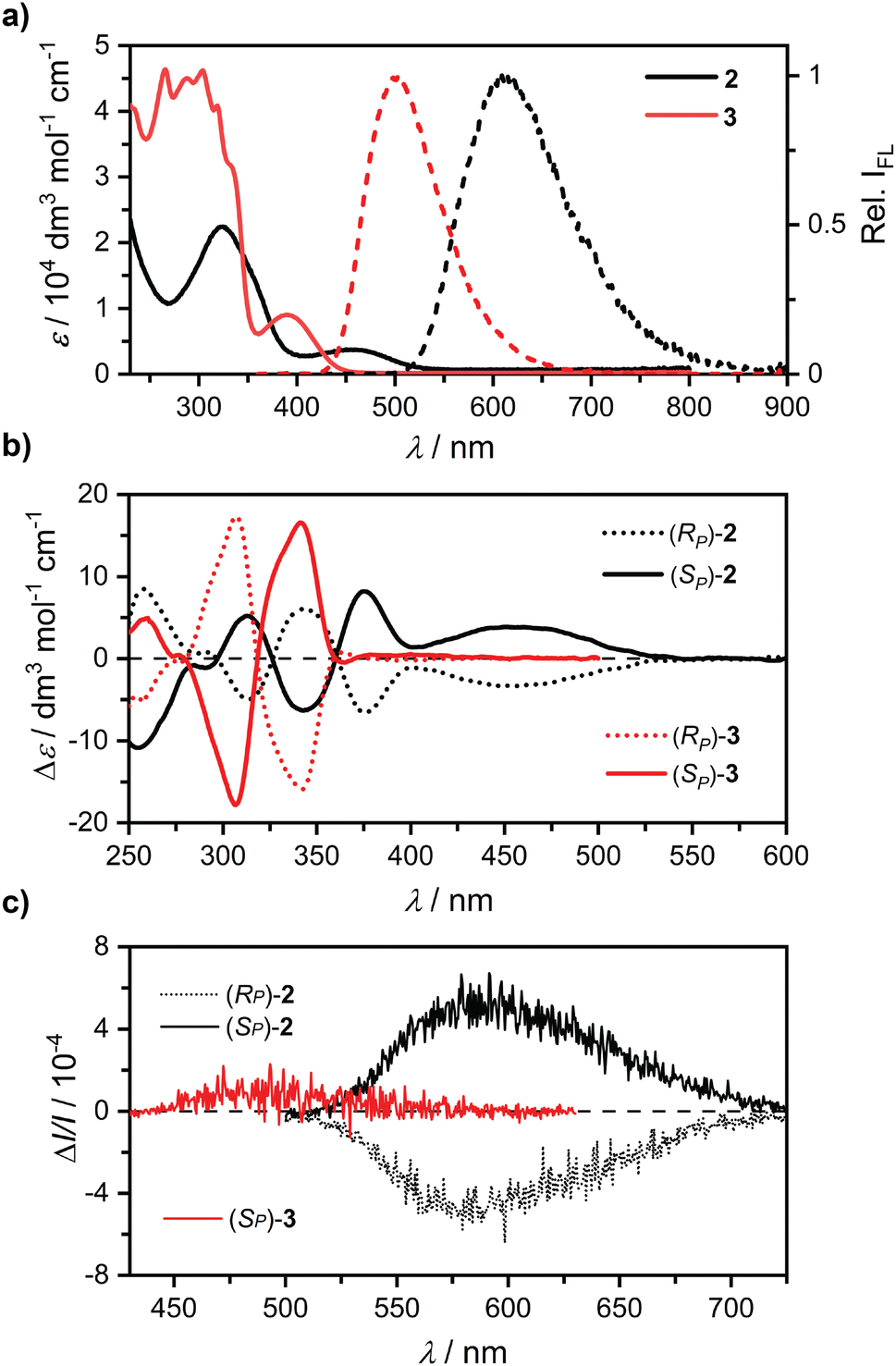 | ||
| Fig. 2 (a) UV-Vis absorption (solid line) and fluorescence (dashed line) spectra of 2 and 3. (b) ECD of 2 and 3 (Δε of 3 scaled by 0.1) and (c) CPL spectra of 2 and 3. All samples were measured in dichloromethane (c ∼5 × 10−5 M). | ||
| Compound | λ abs (nm) | λ em (nm) | ϕ F | ε (104 M−1 cm−1) | |gabs|c (10−3) | |glum|d (10−3) | Stokes shift (cm−1) | B CPL (M−1 cm−1) |
|---|---|---|---|---|---|---|---|---|
| a Dichloromethane solutions at room temperature (∼10−5 M). b The values for the first and the second absorption maximum, respectively. c At 450 and 390 nm for 2 and 3, respectively. d At 600 and 500 nm for 2 and 3, respectively. e Brightness of CPL (BCPL = ε × ϕF × |glum|/2). f From ref. 33. g Estimated for the S0 → S1 transition from data in ref. 33. h TD-CAM-B3LYP/6-31g(d) value. i Value cannot be determined because the S0 → S1 transition is not resolved. j Calculated for a model 2′ with interrupted conjugation (see text and Fig. 3). k From ref. 35. l Not determined because of the absence of the characteristic S0 → S2 transition found in carbon nanohoops used to calculate the brightness from ref. 33. | ||||||||
| 1 | <400, 328 | 470 | 0.72 | —, 3.95 | ∼11g (20.1h) | 2.7(2.3h) | —i | 36.8 |
| 2 | 450, 324 | 609(596h) | 0.48 | 0.3, 2.2 | 1.3 (1.2h; 0.27j) | 0.5(0.7h) | 5800 | 2.6 |
| 3 | 389, ∼320 | 500(473h) | 0.70 | 0.9, 3.5 | 0.3 (0.22h) | <0.1(0.17h) | 5700 | —l |
| BT[10]CPP | 445, 334 | 571 | 0.59 | —, 5.4 | — | — | 4960 | — |
However, we observe only a minor and comparable solvatochromism in 2 and 3 (Fig. S12–15†). Our TD-DFT calculations (Tables S1, S2 and Fig. S35–38†) confirm that the lowest-energy absorption in 2 corresponds to the HOMO → LUMO transition, while it is characterized by the HOMO−2 → LUMO transition in 3. The HOMO in 2 involves BT and all six phenylenes and the LUMO is centred mainly in the BT moiety, while both MOs are dominantly localised in the BT moiety with adjacent phenylenes in 3. The MO analysis and the computed excited state dipole moments thus show a marginal charge-transfer character in the lowest-energy transition in 2, and the changes in the absorption can be attributed solely to the increase in conjugation.
The same effect is also clearly visible upon comparison of the photoluminescence spectra (Fig. 2a). Both macrocycles are strongly luminescent with the emission maxima at λem ≥ 500 nm (Table 1). The maximum in 2 resides at impressive 609 nm with the band onset reaching the near IR region (∼850 nm) establishing this molecule as the most red-shifted chiral nanohoop reported to date with a good ϕF (see below).
In addition, macrocycle 3 itself represents the first chiral nanohoop precursor with bright green fluorescence at 500 nm. The red shift in 2 compared to 3 is 3580 cm−1 (109 nm) and can again, be attributed to the increase in the π-conjugation length.
Interestingly, the Stokes shifts in 2 and 3 are significant at 5800 cm−1 and 5700 cm−1, respectively, yet comparable despite the different π-conjugation. Clearly, mostly the energy of the S1 state but not the reorganization from the Franck-Condon point (ΔE2,3 ∼ 100 cm−1) is affected by the extra conjugation. Both 2 and 3 display high ϕF values of 0.48 and 0.70, respectively, despite the significant shift in the emission, particularly in 2. Both nanohoops are also fluorescent in the solid state (Fig. S57†). Comparison of 2 and 1, which have the same size, shows that the fluorescence maximum is red-shifted by striking 139 nm (4860 cm−1). Clearly, incorporation of BT into carbon nanohoops proposed by Jasti35 is a very effective strategy to achieve a significant red-shift. The λem in 2 is even red-shifted by 38 nm compared to BT[10]CPP without a notable drop in ϕF.
Chiroptical properties and theoretical calculations
The three compounds, 1, 2, and 3, form a series that shares the same critical structural features (Fig. 1): (i) a PCP unit with planar chirality, (ii) a central phenylene or BT unit attached to PCP via (iii) two arms with three p-phenylenes each, although masked as cyclohexa-2,5-dienyls in 3. Nevertheless, these chiral compounds have similar size and geometry, and are all luminescent (Table 1). This provided us with a rare opportunity to use their chiroptical properties to systematically investigate the effect of exciton delocalization on the chiroptical properties of these PCP-based nanohoops. To tackle the proposed challenge, we successfully separated the enantiomers of 2 and 3 using recycling HPLC equipped with a chiral stationary phase (see ESI†). We characterized the individual enantiomers by ECD and CPL (Fig. 2b–d, Table 1 and Fig. S23–30†). We fully assigned the recorded ECD and CPL spectra to each individual enantiomer by TD-DFT calculations (Fig. S31–34†). The observed CPL response for the isolated enantiomer of 3 was very weak, reaching the detection limit of our instrument. Nevertheless, the obtained experimental and calculated values of gabs and glum agree well for both 2 and 3. Both factors for 1 can be derived from data in ref. 33 and we additionally calculated them at the same level of theory as for 2 and 3 (see the ESI†).The measured dissymmetry factors for 2 and 3 are markedly lower compared to those obtained for 1, and obey the following trend: gabs,lum(1) > gabs,lum(2) > gabs,lum(3). The mutual differences in the series reach roughly an order of magnitude and so decrease the BCPL. To allow for a balanced comparison between the three compounds, we examined the individual lowest-energy transitions by computing the corresponding natural transition orbitals (NTOs, S37, S38 and S49†) and electronic transition densities (Fig. 3). Inspection of the S0 → S1 transition densities and NTOs shows that the exciton in 1 reaches the PCP unit in the nanohoop and mixes some of its MOs. The situation changes in 2, where both the occupied and virtual NTOs are localized to the half of the nanohoop with the BT unit. Clearly, replacing a p-phenylene for a strong electron acceptor, such as BT, localizes the electron and the hole density away from the PCP unit upon excitation. The turn-off of the conjugation in 3 further exacerbates the effect. The same observation can be made for the S1 → S0 transition (Fig. S53–56†), reflecting the trend in the CPL response, although the effect is more subtle as the result of exciton localization upon relaxation from the Franck-Condon point (see below). To fully exclude that the observed outcome emerged from the change in the geometry, such as between 2 and 3, where the rehybridization in the latter does affect the PCP and BT distance and orientation (Fig. 3), we designed a model compound 2′ (structure in Fig. S43†) for a computational experiment to effectively turn off the conjugation in 2. This was achieved by addition of four methoxy groups to the central p-phenylenes as in 3 and relaxing their coordinates, while keeping all others frozen in the process. As a result, the distance and orientation of BT and PCP in 2′ remained identical to those in 2 (Fig. S43†). The partial rehybridization of the central p-phenylenes in our computational model had a dramatic effect on the NTOs involved in the S0 → S1 transition (2′, Fig. S46†). The partially delocalized NTOs in 2 collapsed to localized NTOs similar in shape to those in 3. Essentially, the nature of the transition in 2′ became the same as in 3 despite the minimal effect on the PCP and BT geometry found in 2. Accordingly, the calculated gabs dropped to a value commensurate to that found for 3.
 | ||
| Fig. 3 The S0 → S1 transition densities and gabs values (multiplied by 103) calculated for 1, 1′, 2, 3 and (TD-CAM-B3LYP/6-31g(d)), isosurface value = 0.004). See the ESI† for the involved natural transition orbitals. | ||
Clearly, the lack of conjugation that allows the exciton to couple BT and the PCP has a detrimental effect on the size of the dissymmetry factor. Nevertheless, the values of gabs and glum in all three compounds are relatively small and even those in 1 do not exceed the values of typical small organic chromophores. We argue that the reason is the connection of PCP to the remainder chromophore via the pseudo-meta positions, which do not permit an effective exciton delocalization to the chiral unit according to the structure–property relationship that we constructed here. We thus decided to test its predictive ability and examine if using a pseudo-para instead of pseudo-meta PCP in 1 could provide a chiral carbon nanohoop37 (1′, Fig. 3 and S50†) with a larger exciton delocalization and, therefore, a larger dissymmetry factor.‡ The internal area of this model analogue would also be slightly enhanced compared to 1, affecting |m| and subsequently gabs. We calculated the parameters of the S0 → S1 transition in hypothetical enantiomer 1′ and investigated the corresponding transition density and NTOs (Fig. 3, S52 and Tables S8 and S9†). Indeed, the individual MOs of pseudo-para PCP are now mixed with those from the curved para-phenylenes and the PCP is thus strongly involved in the transition. The predicted dissymmetry factor for 1′ reached a value of |gabs| = 0.114, an order of magnitude higher than that in 1. Note that this is an extraordinary gabs for a molecule that lacks any symmetry. Comparable values of gabs are typically achieved in macrocycles that belong to high symmetry point groups.9,35,36
Extending the delocalization directly affects the value of the angle (θ) between μ and m, which is nearly 90° in 3 and increases steadily in the series to a value of 110° in 1′ (Tables S3–S10†). Despite the transition density that spans the entire 1′, the |cos(θ)| = 0.342 highlights the importance of approaching a uniform exciton delocalization that could be reached for chromophores with Dn symmetry.11,38 Adopting such high symmetry is, however, beyond reach for carbon nanohoops with a single chiral element. An alternative strategy that provides a higher flexibility in molecular design with a slight sacrifice in the gabs factor is imposing structures to a Cn point group of symmetry,38 in which the individual components of μ and m transform under the same irreducible representations, i.e., they can even be parallel. Lifting the symmetry further could still lead to acceptable values of θ (≥135°) if the exciton was sufficiently delocalized. A similar trend is observed for |m| that is slightly enhanced in 1′ as a result of an extended exciton delocalization, which would ultimately be related to the slight increase in the internal area of the helical circuit.
However, excited state reorganization in carbon nanohoops is known to lead to exciton localization,39,40 which often decreases θ close to 90° compromising the value of glum in CPL similarly to what we observe in the compounds investigated here. Preventing exciton localization is challenging, in particular in carbon nanohoops that undergo extensive excited state geometry relaxation. For this reason, bright chiral fluorophores with or close to C2 symmetry and small Stokes shifts could preserve sufficiently delocalized excited states in non-polar environments, representing interesting targets to accomplish CPL materials with high BCPL.
Conclusions
In summary, we synthesized two macrocycles incorporating planar chiral pseudo-meta [2.2]paracyclophane and benzothiadiazole that allowed for a significant red-shift of their CPL, while displaying high fluorescence quantum yields and large Stokes shifts. These molecules allowed for unprecedent, systematic investigation of the role of exciton delocalization in the chiroptical properties in carbon nanohoops. Thereby, we constructed a unique structure–property relationship that allowed for a critical insight into the factors limiting the desired chiroptical response. We believe that this work will thus aid and abet the development of robust guidelines to design materials with strong electronic circular dichroism or circularly polarized luminescence with high brightness.Data availability
The data underlying this study are available in the published article and its ESI,† or available from the authors on request.Author contributions
TS and KK conceived the idea. KK carried out the synthesis of all compounds and their spectroscopic investigation. JM performed the DFT calculations. CMC and AGC performed the CD and CPL experiments. All authors analyzed and discussed the data. TS and KK wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Program (grant agreement no. 949397, TS) and from grant PID2021-127521NB-I00 funded by MICIU/AEI/10.13039/501100011033 and by ERDF/EU.Notes and references
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem.–Eur. J., 2015, 21, 13488–13500 CrossRef PubMed.
- N. Chen and B. Yan, Molecules, 2018, 23, 3376 CrossRef PubMed.
- X. Li, Y. Xie and Z. Li, Adv. Photonics Res., 2021, 2, 2000136 CrossRef CAS.
- C. Zhang, X. Wang and L. Qiu, Front. Chem., 2021, 9, 711488 CrossRef CAS PubMed.
- Y. Zhang, S. Yu, B. Han, Y. Zhou, X. Zhang, X. Gao and Z. Tang, Matter, 2022, 5, 837–875 CrossRef CAS.
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
- Y. Nagata and T. Mori, Front. Chem., 2020, 8, 448 CrossRef CAS PubMed.
- L. Arrico, L. Di Bari and F. Zinna, Chem.–Eur. J., 2021, 27, 2920–2934 CrossRef CAS PubMed.
- P. Rivera-Fuentes, J. L. Alonso-Gómez, A. G. Petrovic, P. Seiler, F. Santoro, N. Harada, N. Berova, H. S. Rzepa and F. Diederich, Chem.–Eur. J., 2010, 16, 9796–9807 CrossRef CAS PubMed.
- M. Hasegawa, Y. Nojima and Y. Mazaki, ChemPhotoChem, 2021, 5, 1042–1058 CrossRef CAS.
- H. Kubo, D. Shimizu, T. Hirose and K. Matsuda, Org. Lett., 2020, 22, 9276–9281 CrossRef CAS PubMed.
- I. Warnke and F. Furche, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 150–166 CAS.
- K. Takaishi, K. Iwachido, R. Takehana, M. Uchiyama and T. Ema, J. Am. Chem. Soc., 2019, 141, 6185–6190 CrossRef CAS PubMed.
- K. Dhbaibi, L. Favereau, M. Srebro-Hooper, C. Quinton, N. Vanthuyne, L. Arrico, T. Roisnel, B. Jamoussi, C. Poriel, C. Cabanetos, J. Autschbach and J. Crassous, Chem. Sci., 2020, 11, 567–576 RSC.
- K. Dhbaibi, L. Abella, S. Meunier-Della-Gatta, T. Roisnel, N. Vanthuyne, B. Jamoussi, G. Pieters, B. Racine, E. Quesnel, J. Autschbach, J. Crassous and L. Favereau, Chem. Sci., 2021, 12, 5522–5533 RSC.
- K. Takaishi, S. Murakami, K. Iwachido and T. Ema, Chem. Sci., 2021, 12, 14570–14576 RSC.
- R. G. Uceda, C. M. Cruz, S. Míguez-Lago, L. Á. de Cienfuegos, G. Longhi, D. A. Pelta, P. Novoa, A. J. Mota, J. M. Cuerva and D. Miguel, Angew. Chem., Int. Ed., 2024, 63, e202316696 CrossRef CAS PubMed.
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 17646–17647 CrossRef CAS PubMed.
- M. Fujitsuka, D. W. Cho, T. Iwamoto, S. Yamago and T. Majima, Phys. Chem. Chem. Phys., 2012, 14, 14585 RSC.
- Y. Segawa, A. Fukazawa, S. Matsuura, H. Omachi, S. Yamaguchi, S. Irle and K. Itami, Org. Biomol. Chem., 2012, 10, 5979–5984 RSC.
- E. R. Darzi and R. Jasti, Chem. Soc. Rev., 2015, 44, 6401–6410 RSC.
- W. Xu, X.-D. Yang, X.-B. Fan, X. Wang, C.-H. Tung, L.-Z. Wu and H. Cong, Angew. Chem., Int. Ed., 2019, 58, 3943–3947 CrossRef CAS PubMed.
- L.-H. Wang, N. Hayase, H. Sugiyama, J. Nogami, H. Uekusa and K. Tanaka, Angew. Chem., Int. Ed., 2020, 59, 17951–17957 CrossRef CAS PubMed.
- K. Sato, M. Hasegawa, Y. Nojima, N. Hara, T. Nishiuchi, Y. Imai and Y. Mazaki, Chem.–Eur. J., 2021, 27, 1323–1329 CrossRef CAS PubMed.
- J. Nogami, Y. Nagashima, K. Miyamoto, A. Muranaka, M. Uchiyama and K. Tanaka, Chem. Sci., 2021, 12, 7858–7865 RSC.
- D. Wassy, M. Hermann, J. S. Wössner, L. Frédéric, G. Pieters and B. Esser, Chem. Sci., 2021, 12, 10150–10158 RSC.
- J. Malinčík, S. Gaikwad, J. P. Mora-Fuentes, M.-A. Boillat, A. Prescimone, D. Häussinger, A. G. Campaña and T. Šolomek, Angew. Chem., Int. Ed., 2022, 61, e202208591 CrossRef PubMed.
- Y. Xu, F. Steudel, M.-Y. Leung, B. Xia, M. von Delius and V. W.-W. Yam, Angew. Chem., Int. Ed., 2023, 62, e202302978 CrossRef CAS PubMed.
- Y. Zhou, X. Zhang, B. Yuan, D. Lu, G.-L. Zhuang and P. Du, Org. Lett., 2024, 26, 5635–5639 CrossRef CAS PubMed.
- X. Kong, X. Zhang, B. Yuan, W. Zhang, D. Lu and P. Du, J. Org. Chem., 2024, 89, 8255–8261 CrossRef CAS PubMed.
- K. Senthilkumar, M. Kondratowicz, T. Lis, P. J. Chmielewski, J. Cybińska, J. L. Zafra, J. Casado, T. Vives, J. Crassous, L. Favereau and M. Stępień, J. Am. Chem. Soc., 2019, 141, 7421–7427 CrossRef CAS PubMed.
- T. A. Schaub, E. A. Prantl, J. Kohn, M. Bursch, C. R. Marshall, E. J. Leonhardt, T. C. Lovell, L. N. Zakharov, C. K. Brozek, S. R. Waldvogel, S. Grimme and R. Jasti, J. Am. Chem. Soc., 2020, 142, 8763–8775 CrossRef PubMed.
- J. He, M. Yu, M. Pang, Y. Fan, Z. Lian, Y. Wang, W. Wang, Y. Liu and H. Jiang, Chem.–Eur. J., 2022, 28, e202103832 CrossRef CAS PubMed.
- J. He, M.-H. Yu, Z. Lian, Y.-Q. Fan, S.-Z. Guo, X.-N. Li, Y. Wang, W.-G. Wang, Z.-Y. Cheng and H. Jiang, Chem. Sci., 2023, 14, 4426–4433 RSC.
- T. C. Lovell, Z. R. Garrison and R. Jasti, Angew. Chem., Int. Ed., 2020, 59, 14363–14367 CrossRef CAS PubMed.
- Y. Xue, Y. Shi and P. Chen, Adv. Opt. Mater., 2024, 12, 2303322 CrossRef CAS.
- Y. Wu, G. Zhuang, S. Cui, Y. Zhou, J. Wang, Q. Huang and P. Du, Chem. Commun., 2019, 55, 14617–14620 RSC.
- S. Sato, A. Yoshii, S. Takahashi, S. Furumi, M. Takeuchi and H. Isobe, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13097–13101 CrossRef CAS PubMed.
- J. Xia and R. Jasti, Angew. Chem., Int. Ed., 2012, 51, 2474–2476 CrossRef CAS PubMed.
- T. C. Lovell, C. E. Colwell, L. N. Zakharov and R. Jasti, Chem. Sci., 2019, 10, 3786–3790 RSC.
- K. J. Weiland, T. Brandl, K. Atz, A. Prescimone, D. Häussinger, T. Šolomek and M. Mayor, J. Am. Chem. Soc., 2019, 141, 2104–2110 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07333a |
| ‡ Racemization of enantiomers of 1′ is rapid. A strategy to kinetically stabilize enantiomers of such macrocycles has already been reported by one of us.41 |
| This journal is © The Royal Society of Chemistry 2025 |