
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iridium trihydride and tetrahydride complexes and their role in catalytic polarisation transfer from parahydrogen to pyruvate†
Ben. J.
Tickner
 a and
Simon B.
Duckett
*b
a and
Simon B.
Duckett
*b
aCentre for Hyperpolarisation in Magnetic Resonance, University of York, Heslington, YO10 5NY, UK
bDepartment of Chemistry, University of York, Heslington, YO10 5DD, UK. E-mail: simon.duckett@york.ac.uk
First published on 12th December 2024
Abstract
This work details how the unusual iridium tetrahydride [Ir(H)4(IMes)(sulfoxide)]Na and trihydride [Ir(H)3(IMes)(sulfoxide)2] can be formed in a solvent dependent reaction of [IrCl(COD)(IMes)] with sulfoxide (dimethyl or methylphenyl), base, and H2. In the case of dimethyl sulfoxide, the four hydride ligands of the tetrahydride are equivalent, and the IMes and sulfoxide ligands mutually trans. However, for phenyl methyl sulfoxide, this isomer of the tetrahydride forms alongside its cis counterpart where the remarkable symmetry breaking effect of the sulfoxide leads to it presenting four chemically distinct hydride ligands. These products and their ligand arrangements are characterised and the reaction pathways leading to their formation probed using NMR spectroscopy and parahydrogen-hyperpolarised methods. Subsequently, they form as previously unidentified low concentration by-products in the important SABRE-catalysed hyperpolarisation of pyruvate, and their concentration should be minimised for efficient polarisation transfer.
Introduction
Metal hydride complexes play a key role in industrial catalysis, fine-chemical manufacture, and materials chemistry.1 Some of the more unusual examples of metal hydrides are reflected in {RuH4} species that commonly exist in dihydride–dihydrogen arrangements,2 and react to form dimers in the absence of coordinating ligands.3,4 These species are readily formed by the reaction of a chloride precursor with H2 and MOH [M = K or Na]. Interestingly, their deprotonation affords anionic trihydride complexes like [RuH3(PPh3)3]M that are useful building blocks for complex structures,5 with 18-crown-6-ether enabling structural studies that confirm their facial hydride ligand arrangement.6 In contrast related [OsH4(PPh3)3] is a classical Os(IV) tetrahydride,7 and while fac-[OsH3(PPh3)3]− is itself highly reactive,8 it can be stabilised by ion pairing effects.9 For rhodium, while many trihydride complexes are known, they are normally highly reactive and their {RhH4} analogues are again cationic dihydride–dihydrogen species.10,11Surprisingly, a wider range of iridium hydride species are known, and these exhibit both classical and non-classical interactions. Notably, pincer ligands have been used to generate neutral Ir dihydrides that now exist in equilibrium with their tetrahydride counterpart.12,13 Now, accessible pentahydride complexes like [Ir(H)5(PiPr3)2], act as weak acids and form anionic tetrahydride Ir(III) species in the presence of KH, with cis and trans isomers forming according to the counterion environment.14,15 More recently, related [IrH4(dppe)]K has been obtained from [IrCl(COD)(dppe)] and KOtBu in isopropanol.16
The bonding and ligand mobility shown by such metal hydride complexes can be readily investigated using nuclear magnetic resonance (NMR) spectroscopy through hydride ligand chemical shifts, H–H and related coupling constants, and relaxation data.17 Furthermore, as hydride ligands are often associated with the activation of molecular H2 metal–dihydrogen complexes that retain a strong H–H bonding interaction have added significantly to our understanding of bond activation processes more generally.17,18
One example of catalysis that relies on these effects is highly topical reversible magnetisation transfer from a parahydrogen (pH2) feedstock. This is typically mediated by a metal complex that activates pH2 to unlock its latent spin order.19–22 Magnetisation can then spread either spontaneously,23,24 or via radiofrequency excitation,25,26 to other sites within the metal complex from the pH2 derived hydride ligands. Ligand dissociation then allows the catalytic build-up of a finite concentration of hyperpolarised ligand, free in solution, with the complex playing a key role in the efficiency of this catalytic process as ligand exchange rates, J-coupling, relaxation, and solubility all affect polarisation transfer.27,28
Typically, such spin polarisation transfer catalysts are iridium-derived dihydride complexes of the form [Ir(H)2(NHC)(substrate)3]Cl or [IrCl(H)2(NHC)(substrate)2], where NHC is an N-heterocyclic carbene.29,30 This process has been termed signal amplification by reversible exchange (SABRE). Significantly, experimentally invisible but theoretically predicted, {Ir(H)2(η2-H2)}-type reaction intermediates are thought to play a crucial role in refreshing the pH2-derived singlet order within the active catalyst that is needed for SABRE,29,31 and hydride complexes derived from this family play a key role in other metal-catalysed pathways such as hydrogenation, hydroformylation, and deuterium isotope exchange.32,33
In this work, we use NMR to rationalise the detection of a series of iridium tri and tetra hydride species that form when the common SABRE catalyst precursor [IrCl(COD)(IMes)] (COD = cis–cis-1,5-cyclooctadiene and IMes = 1,3-bis(2,4,6-trimethyl-phenyl)imidazole-2-ylidene) reacts with sulfoxide, base, and H2. These unstable species are reflective of [Ir(H)3(IMes)(sulfoxide)2] and [Ir(H)4(IMes)(sulfoxide)]Na, and characterised using a combination of detailed NMR studies and parahydrogen-hyperpolarised NMR. We highlight routes to their formation and comment on their reactivity, which leads to the eventual detection of a number of binuclear reaction products. We also rationalise the role these complexes play in the SABRE hyperpolarisation of pyruvate as first reported in 2019 (Fig. 1).34 Given the proven utility of pyruvate as a viable molecular imaging probe of cancer, such studies are highly warranted. Furthermore, since this initial report utilising SABRE, pyruvate polarisation levels have been optimised35–37 to the point in vivo biomedical imaging is possible.38
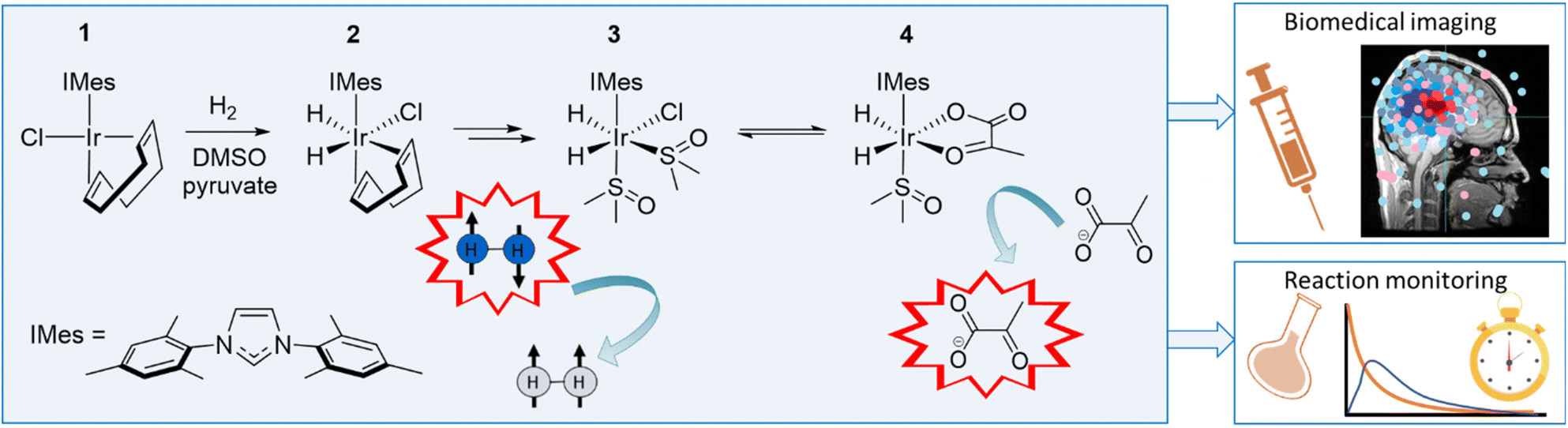 | ||
| Fig. 1 Mechanism leading to the successful hyperpolarisation of pyruvate by SABRE. The reaction of 1 with H2, DMSO and sodium pyruvate in methanol-d4 forms an equilibrium mixture of 3 and 4via2. Reversible exchange of pH2 and pyruvate then dramatically enhance the 13C NMR signals of free pyruvate. Such enhanced signals have been used in applications that include biomedical imaging and reaction monitoring as eluded to in the cartoons shown. | ||
As shown in Fig. 1, the addition of H2 (3 bar) to methanol-d4 solutions containing [IrCl(COD)(IMes)] (1) (5 mM), DMSO (25 mM) and sodium pyruvate (30 mM) is complex and proceeds by the initial formation of [IrCl(H)2(COD)(IMes)] (2), before further reaction leads to an equilibrium mixture of [IrCl(H)2(IMes)(DMSO)2] (3) and [Ir(H)2(κ2-pyruvate)(IMes)(DMSO)] (4).34,394 exists in two dominant isomeric forms, according to the orientation of the pyruvate and hydride ligands, with optimal SABRE hyperpolarisation of pyruvate stemming from the isomer where these ligands lie in the same plane. Highly reactive [IrCl(H)2(IMes)(DMSO)2], 3, is therefore present in these reaction mixtures, and it forms when the reaction is performed without sodium pyruvate.34,393 has been suggested to play a key role in pH2 incorporation during such studies.39
These early reports addressed the identities of the major products, 3 and 4, detected during SABRE.35,37,38,40,41 Other minor hydride-containing species prove visible to NMR during these studies, with X-ray crystal structures resulting for several sulfur-bridged iridium dimers that form at long reaction times. These have been linked to a drop in catalyst activity over time.42,43 However, the identity and the role of the minor reaction products has not yet been resolved due to the fact their NMR signals appear only transiently. This stems from the use of methanol-d4 as the solvent, which leads to the rapid deuteration of any hydride ligands, and this precludes their spectral mapping. Accordingly, we begin this work by studying the reactivity of [IrCl(COD)(IMes)] 1 with sulfoxide, and H2 in methanol-d3 and detail some surprising discoveries.
Results and discussion
Formation of [Ir2(Cl)(μ2-Cl)(H)3(μ2-H)(IMes)2(DMSO)2] (5 and 5′)
When solutions containing 1 and DMSO are reacted with H2 in methanol-d3 they lead to the rapid formation of 3, as expected. 3 yields two very broad hydride ligand NMR signals due to rapid sulfoxide and H2 exchange at 298 K. Its detection when such studies are completed in methanol-d4 at 298 K by NMR is made even more challenging by the fact its hydride ligand sites become rapidly deuterated as CD3OH forms.However, in the methanol-d3 solution the ready detection of a range of other hydride ligand signals besides those of 3 proved possible. The most notable of these signals appear at δ = −15.79, −16.06, −24.80, and −30.66, and possess equivalent signal intensities. They were assigned to the dimer [Ir2(Cl)(μ2-Cl)(H)3(μ2-H)(IMes)2(DMSO)2], 5 of Scheme 1 by a series of complex NMR procedures (see ESI, Section S1†). A second set of much weaker signals appear at δ = −16.90, −18.82, −20.55, and −22.11 were assigned to a minor isomer (5′) of 5 that exhibits a different ligand arrangement (see ESI, Section S1†). 5 and 5′ likely arise from the dimerization of 3, and differ from the literature reported sulfur-bridged dimers that have been reported to form at very long reaction times.42,43
 | ||
| Scheme 1 Reaction pathways revealed through this work build from known 1–3, 6 and 10, with some of these species reported to play a role in iridium-mediated polarisation transfer from pH2 to pyruvate. These complexes were detected here, alongside previously unreported 5, 5′, 7, 8, 8′, and 9. The formation of 6–9 are facilitated by NaOMe, added specifically or formed indirectly when sodium pyruvate is utilised in methanol. | ||
Formation of the tetrahydride [Ir(H)4(DMSO)(IMes)] (7) and trihydride [Ir(H)3(DMSO)2(IMes)] (8 and 8′)
Recently, the unstable trihydride complex [Ir(H)3(COD)(IMes)] (6) was characterised as a minor reaction product alongside dominant [Ir(H)2(IMes)(pyridine)3]Cl when the weak base pyridine and H2 react with 1 in methanol. The proportion of 6 was found to increase when NaOMe was added to methanol in agreement with its formation via deprotonation of the unseen intermediate [Ir(H)2(η2-H2)(COD)(IMes)].44 Consequently, methanol-d3 solutions containing DMSO and a 10-fold excess of NaOMe relative to 1 (without pyruvate) were examined here. Halide substitution was observed to create an equilibrium mixture of 1, [Ir(OCH3)(COD)(IMes)] and [Ir(OH)(COD)(IMes)] (from adventitious water). The displacement of chloride being incomplete, with these complexes existing in a ratio of 2.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.7:1 respectively at 298 K; notably no signal for [Ir(DMSO)(COD)(IMes)]Cl was observed in accordance with the weak nature of the iridium sulfoxide interaction.
:2.7:1 respectively at 298 K; notably no signal for [Ir(DMSO)(COD)(IMes)]Cl was observed in accordance with the weak nature of the iridium sulfoxide interaction.
Surprisingly, the addition of H2 to this solution at 298 K led to the rapid formation of three hydride-containing species. These species yielded paired signals at δ = −9.10 and −13.49, a unique signal at δ = −8.58, and paired signals at δ = −9.49 and −15.24 in the associated 1H NMR spectra. The three species initially appeared to exist in a ratio of 2.7:1.7:1 respectively, although they change with reaction time as detailed in Fig. 2a.
 | ||
| Fig. 2 (a) Reaction time course chemical speciation plot for the changes that take place after H2 (3 bar) is added to 1 (5 mM), DMSO (25 mM) and NaOMe (50 mM) in methanol-d3 at 298 K. Note, the signals for complexes 8′ and 6 disappear after 60 and 100 minutes respectively. The inset chart shows an expansion of the first 25 minutes of this profile for a sample that was prepared in a dry ice/acetone bath prior to placing it into the NMR spectrometer at 298 K where data acquisition was started. This process allows the conversion to 6–9 to be more precisely monitored. (b) Typical hydride spectral regions extracted from the 1H NMR spectra used to create chart (a) at the indicated times after H2 addition. (c) Analogous reaction time course data collected after H2 (3 bar) is added to 1 (5 mM), DMSO (25 mM) and NaOMe (50 mM) in DCM-d2 at 298 K. (d) Typical hydride regions of the corresponding 1H NMR spectra used to assemble chart (c), at the indicated times after H2 addition. | ||
Initially, those for unstable [Ir(H)3(COD)(IMes)] (6) are dominant (Fig. 2a) with the unique hydride ligand signal, at δ = −8.58, being attributed to [Ir(H)4(DMSO)(IMes)]Na, 7 on the basis of its NMR data. This included peak integration and NOE analysis (see ESI, Section S2† for characterisation details). The evolution of the intensity of its hydride signal as a function of reaction time proved complex in this data recorded at 298 K, with its initial high intensity falling, before increasing again and then falling (Fig. 2a). This suggests there are two routes to its formation.
The NMR characterisation of 8 was secured at 243 K as the trihydride [Ir(H)3(DMSO)2(IMes)] (8) of Scheme 1. Its hydride ligand signals appear in a 2:1 ratio, with NOE studies confirming their meridional arrangement, alongside interactions to bound IMes and two inequivalent DMSO ligands (see ESI, Section S2† for characterisation details). Initially, 8 coexists with isomeric 8′, whose three hydrides are arranged in a facial arrangement. Its signals are observed in a 1:2 ratio at δ = −13.85 and −15.45 (see ESI, Section S2† for characterisation details), but their intensity is always low and are no longer evident after 45 minutes of reaction at 298 K. Consequently, it must form separately from 8 and the two species are not in equilibrium. After a few hours at room temperature, the ratio of 7 and 8 remains similar, suggesting that while these two species initially form in different pathways, they equilibrate slowly.
At long reaction times weak signals for 5 and 5′ appear that account for just a few % of the reaction products. However, signals for 9 at δ = −11.56, −13.30, −20.48, −22.61, and −30.11 also appear and these are much more substantial. They are assigned to the pentahydride dimer [Ir2(H)4(μ2-H)(μ2-OCD3)(DMSO)2(IMes)2] (see ESI, Section S3†). This product again likely arises from a reaction of 3, but this time with 8 or 8′ and methoxide rather than with a second molecule of 3. It is worth noting that after 15 hours of reaction the total hydride ligand signal intensity accounts for just 75% of the original iridium concentration, a situation which is not surprising give the long term formation of sulfide derived dimers.44
When the analogous reaction was monitored in DCM-d2 at 298 K (Fig. 2c) the formation of 7, 8, 8′ and 9 was again revealed. 8 now clearly dominates (Fig. 2d) the products formed in this solvent and the hydride ligand signals for 6 are very challenging to discern, with its proportion never exceeding a few %. In contrast, signals for 3 are clearly visible and grow in proportion at longer time points. Furthermore, 9 no longer forms in significant amounts as there is now significantly less methoxide available to create the necessary bridging ligand.
Reaction pathways leading to [Ir(H)4(DMSO)(IMes)] (7) and trihydride [Ir(H)3(DMSO)2(IMes)] (8 and 8′)
In order to probe these reactions in more detail, a series of control measurements were completed. The first of these involved reacting 1 with H2 and DMSO in methanol-d3 to form a mixture of 3 and 5 as detailed earlier. This speciation changed immediately upon adding NaOMe to the solution as 1H NMR signals for 7 and 8 rapidly replaced those of 3 and 5 (see ESI, Section S4†). A second control measurement involved the initial conversion of 1 into 6via NaOMe addition to 1 in methanol-d3 under H2 at 253 K. This reaction also leads to formation of known [Ir2(H)2(μ2-H)2(η2–η2-COD)(IMes)2] (10) as a minor product.44 Once all of the 1 present was consumed, DMSO was added to the solution at 298 K and NMR monitoring commenced. Under these conditions, 6 proved to rapidly convert into both 7 and 8, with the later ultimately dominating.Collectively, these results confirm that 3, 5 and 6 react independently to yield 7 and 8, which have already been stated to equilibrate. Furthermore, the reaction time-course data (Fig. 2a and c) confirm the existence of two pathways to 7 as its hydride ligand signal intensity, both in methanol-d3 and DCM-d2, exhibit two maxima. Literature examples of {IrH3} and {IrH4} species form by H2 addition to {IrH} and {IrH2} precursors respectively, or involve base-driven deprotonation of {IrH4} and {IrH5} species.12–15 Hence, routes to 7via deprotonation of undiscerned {Ir(H)5} and 8via H2 loss from {Ir(H)5}, or deprotonation of hydride dihydrogen intermediates formed from 3 and 6 are possible.
Effect of deuteration on hydride ligand signals of [Ir(H)4(DMSO)(IMes)] (7) and [Ir(H)3(DMSO)2(IMes)] (8)
As indicated earlier, when these reactions are performed in methanol-d4, hydride ligand exchange with the methoxyl deuterium occurs. This is readily evident as when 8 is formed initially in methanol-d4, the H3 form dominates before two regioisomers, 8-d and 8-d′, of its mono 2H-labelled counterpart result depending on which site contains deuterium (Fig. 3). For 8-d, the deuterium label is incorporated into the site trans to hydride, and hence it yields inequivalent hydride ligand signals at essentially the same chemical shifts as those of 8, but now in a 1:1 ratio. In contrast, its regioisomer 8-d′ contains two equivalent hydride ligands as the deuterium label lies trans to DMSO, and yields a single hydride signal at δ = −9.37 (Fig. 3a). Subsequently these signals disappear as 8 becomes fully deuterated in addition to reacting to form deuterated analogues of 9 and its binuclear counterparts.
 | ||
| Fig. 3 Hydride regions taken from 1H NMR spectra recorded at the indicated times after H2 (3 bar) is added to 1 (5 mM), DMSO (25 mM) and NaOMe (50 mM) in methanol-d4 at 298 K. Unassigned signals could result from isotopologues of 9 and other binuclear products. | ||
The effect of deuterium exchange on the hydride ligand signals for 3 and 7 is, however, much less obvious, although these species are clearly deuterated; the resonances for 3 broaden, are less intense, and more challenging to discern. Those of 7 in contrast no longer integrate to four when compared to the IMes ligand signals. Furthermore, the diagnostic signals of 5, 5′, and 9, are much harder to discern due to their formation from these deuterated precursors, and/or the fact that their hydride ligands can become deuterated via exchange with solvent. Consequently, in methanol-d4 after 15 hours reaction, ca. 75% of the starting hydride ligand signal intensity is lost.
Hyperpolarised measurements to rationalise formation of [Ir(H)4(DMSO)(IMes)] (7) and [Ir(H)3(DMSO)2(IMes)] (8)
In order to learn more about these reactions, studies using parahydrogen-enhanced 1H NMR spectroscopy were completed.45–53 Accordingly, a sample containing 1 (5 mM), DMSO (25 mM) and NaOMe (50 mM) in methanol-d3 was exposed to pH2 at 298 K, and then shaken in a magnetic field of 65 G for 10 seconds before being rapidly placed into a 9.4 T NMR spectrometer and examined using a single-scan 45° 1H radio frequency pulse. This process should produce enhanced 1H NMR signals for hydride ligands that were previously located in the pH2 feedstock if the rate of hydrogen addition is faster than the rate of nuclear spin relaxation. Under these conditions, the enhanced hydride ligand signals may display a mixture of ALTADENA and PASADENA effects if the hydrogenation step takes place at both low field and high field respectively.21 However, if the resulting iridium dihydride rapidly converts to a second species then its signals may too be recorded as a hyperpolarised response.31,44Consequently, the 1H NMR spectrum recorded immediately after pH2 addition revealed enhanced hydride ligand signals for both 8 and 8′ with ALTADENA patterns (Fig. 4a). In contrast, those of 6 displayed strong ALTADENA behaviour, with a small PASADENA distortion. Broad enhanced hydride ligand signals for 3 with PASADENA character were also visible (Fig. 4a) that are consistent with its rapid H2 exchange. When further measurements are recorded after this sample is removed and shaken again with fresh pH2 for 10 seconds at 65 G the enhanced signals for 3 are no longer visible, although those of the trihydrides 6 and 8 appear, albeit with reduced intensity (Fig. 4b). Repeating this process sees the intensity of the enhanced 1H NMR signals decrease until they are not visible with enhancement after 1 hour of reaction. It is worth noting that other transient hydride ligand signals of similar intensity to the enhanced signals for 8′ are also discernible in the initial hyperpolarised measurement (Fig. 4a), although their identity could not be confirmed as they are only visible in hyperpolarised experiments. When the aliphatic region of these NMR spectra are examined, a PHIP enhanced signal for hydrogenated COD (cyclooctane) at δ = 1.5 is observed in addition to weak SABRE enhanced IMes ligand signals. These results highlight that trihydrides 6, 8, and 8′ can be created in a hyperpolarised form via PHIP.
 | ||
| Fig. 4 (a) Hyperpolarised single scan 1H NMR spectra recorded using a 45° pulse immediately after the addition of pH2 to a methanol-d3 solution of 1 (5 mM), DMSO (25 mM) and NaOMe (50 mM) at 298 K. Before the spectrum was recorded, the sample was shaken with pH2 for 10 seconds at 65 G to initiate PHIP. The associated time course data detailing the corresponding change in hyperpolarised signal intensity given in (b). Each data point is collected after refilling the tube with fresh pH2 and reshaking as described in (a). | ||
Exchange spectroscopy (EXSY) studies on 8 were then conducted that revealed its H2 loss rate to be negligible (<4% exchange after a mixing time of 1.3 s at 298 K). Hence, it is clear that reversible H2 loss from 8 does not account for the PHIP response it exhibits. Furthermore, this deduction is supported by the fact that the intensity of its hyperpolarised signals decrease over the first ca. 15 minutes of reaction (Fig. 4b), despite the fact its concentration grows during this period (Fig. 2a). If 8 could exchange H2 it would be reflected in a growth of hyperpolarised signal intensity as its concentration increases. As this is not the case, its polarised response must result from the fact that it forms from a hyperpolarised precursor (e.g.3 or 6) with a faster formation rate than the rate of relaxation. This is consistent with the fact that 3 has already been reported to undergo rapid hydrogen exchange,39 while 6 has been reported to form in a hyperpolarised state, via deprotonation of [Ir(H)2(η2-H2)(COD)(IMes)] which forms after pH2 addition to 1 to form hyperpolarised 2.44 This hypothesis is supported by the fact that signals for hyperpolarised 3 (in the early stages of reaction) and 6 are observed throughout the time course of the hyperpolarisation experiment (Fig. 4) and the hyperpolarisation of 8 is no longer observed once 3 and 6 have been consumed. These data in turn support the previous conclusion that that 8 forms from 3 and/or 6.
Tetrahydride 7 does not display any obvious hyperpolarisation, although as its hydrides are magnetically equivalent, any hyperpolarisation is likely to be locked in a singlet state which is invisible to these NMR experiments. EXSY measurements also revealed that DMSO exchange in 7 and 8 does not occur on the NMR timescale, and that 3, 6–9 do not interconvert on this timescale.
When these hyperpolarisation experiments are repeated in DCM-d2, PHIP enhanced 2 and 3 are observed in the first measurement (see ESI, Section S5†). Weakly enhanced signals for 8 can then be detected a few minutes later. Collectively, these experiments confirm that the speed of reaction in DCM-d2 is too slow to produce strongly enhanced signals for 8.
Breaking the symmetry of trihydrides and tetrahydrides using chiral sulfoxides
Trihydride 8 contains two magnetically equivalent hydride ligands when Cs-symmetric DMSO is used. However, when these experiments are repeated using asymmetric methylphenylsulfoxide (MPSO), analogues of 7 and 8 can be created wherein the hydride ligands are magnetically inequivalent. For example, reaction of 1, MPSO, NaOMe and H2 in either methanol-d3 or toluene-d8 yields equilibrium mixtures of [Ir(H)4(MPSO)(IMes)] (11) and [Ir(H)3(MPSO)2(IMes)] (12) (see ESI, Section S6† for details of their characterisation). When the hydride resonances for 12 are examined, two pairs of three distinct signals are now seen. This results from the fact 12 contains two chiral sulfoxides, and forms as two pairs of diastereomers, each with distinct resonances (Fig. 5a). A single hydride resonance was observed at δ = −8.44 for 11, which is analogous in position to that of 7.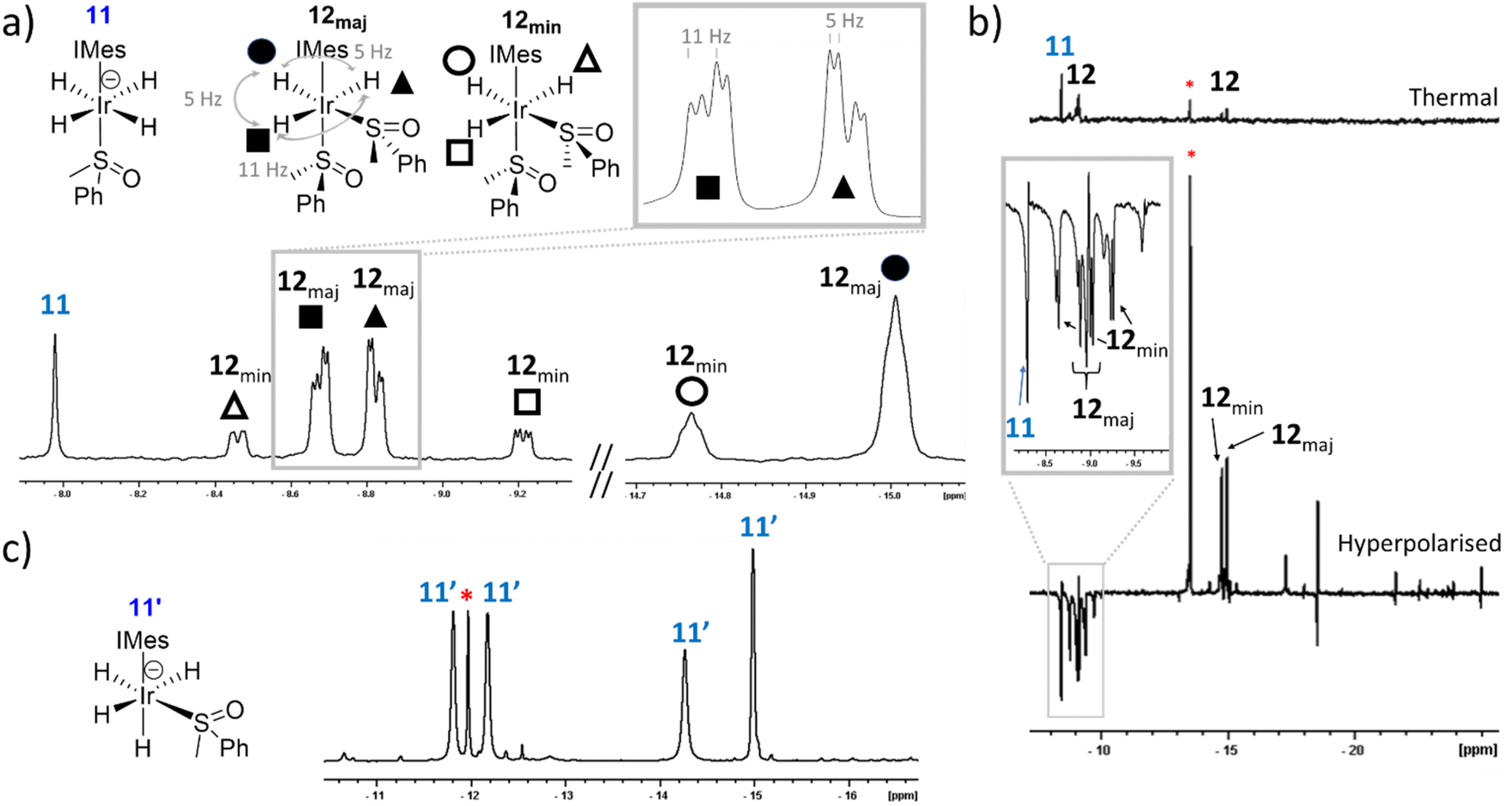 | ||
| Fig. 5 (a) Hydride regions taken from 1H NMR spectra recorded a few minutes after H2 (3 bar) is added to 1 (5 mM), MPSO (25 mM) and NaOMe (50 mM) in methanol-d3 at 298 K. (b) Hydride region of a single scan 1H NMR spectra recorded with a 45° pulse a few minutes after pH2 (3 bar) is added to 1 (5 mM), MPSO (25 mM) and NaOMe (50 mM) in methanol-d3 at 298 K and shaken at 6.5 mT for 10 seconds. A thermally polarised spectrum recorded 30 seconds after this spectrum was recorded presented is shown above on the same vertical scale. The inset trace shows an expansion to illustrate the appearance of the polarised signals for 11 and 12. The singlet marked by the asterisk is tentatively assigned as an analogue of 11. (c) Hydride regions taken from 1H NMR spectra recorded a few days after H2 (3 bar) is added to 1 (5 mM), MPSO (25 mM) and KH (50 mM) with 18-crown-6 (25 mM) in toluene-d8 at 298 K. | ||
Now, hyperpolarisation experiments in methanol-d3 immediately after the addition of pH2 to samples containing MPSO yield transiently enhanced signals for both diastereomers of 12, in analogy to those seen for 8 that were described earlier, although their relative intensity is now weaker. In addition, an enhanced singlet is seen for 11, alongside that of a further singlet at δ = −13.00 in methanol-d3 (Fig. 5b). These observations suggest that MPSO succeeds in breaking the symmetry of the precursors leading to 11. Unfortunately, the low proportion and stability of the species yielding these two singlets precluded their full NMR characterisation, however according to EXSY the two species are in exchange. We suggest therefore that they differ in respect to the location of the cation.
However, when these reactions are repeated in toluene-d8 using KH or KOMe as the base with 18-crown-6, the formation of asymmetric tetrahydride 11′ is achieved. It presents four inequivalent hydride ligands that resonate at δ = −11.75, −12.13, −14.21 and −14.95 (Fig. 5c, ESI S6† for characterisation). This inequivalence reflects the cis relationship between the sulfoxide and IMes ligands. The use of a crown ether to stabilise such facial arrangements has been demonstrated previously.15
Formation and role of 6–9 during hyperpolarisation of pyruvate
The formation of these tri and tetrahydride complexes in the analogous reactions with pyruvate was confirmed through analogous experiments where samples containing H2 (3 bar), 1 (5 mM), DMSO (25 mM) and sodium pyruvate (30 mM) were examined in methanol-d4 (0.6 mL) at 298 K. Now, signals for the hydride ligands of 3, and the two previously reported isomers of 4, are readily seen.34,39 However, when these NMR spectra are examined more closely, extra signals for 6, 7, 8, 8′, 8-d′, and 9 are seen, both in the corresponding thermally polarised and hyperpolarised 1H NMR spectra (Fig. 6). It is notable, that 1H NMR signals for these species are visible even though NaOMe is not specifically added to the samples. This is not surprising as sodium pyruvate equilibrates in methanol with NaOMe and pyruvic acid. | ||
| Fig. 6 (a) 1H NMR spectra 1 min (upper) and 40 min (lower) after H2 (3 bar) addition to a solution of 1 (5 mM), DMSO (25 mM) and sodium pyruvate (30 mM) in methanol-d4 (0.6 mL) at 298 K. (b) Hyperpolarised single scan 1H NMR spectrum (and expansion in blue box with increased vertical scale) recorded using a 90° pulse using the solution for (a) but after shaking with 3 bar of pH2 for 10 seconds at 65 G, 15 minutes after the start of observations. The corresponding NMR spectrum at this time point, where the PHIP effect is absent and the vertical scale increased by a factor of 32 relative to (b) is shown below. For clarity, the peaks have been phased so the largest signals appear in absorption mode. | ||
This hypothesis was confirmed by doping the solutions (1 (5 mM), DMSO (25 mM), and sodium pyruvate (30 mM) in methanol-d4 (0.6 mL)) with NaOMe (50 mM), and this did indeed favour the formation of 6–9. Interestingly, the expected signals for 4 are no longer seen. This agrees with the observation that when 4 is prepared in situ, by H2 addition to 1 (5 mM), DMSO (25 mM), and sodium pyruvate (30 mM) in methanol-d4 (0.6 mL), it rapidly converts into 7, 8 and 8-d′ once NaOMe (50 mM) is added.
In the context of pyruvate hyperpolarisation using SABRE, the presence of 6–9 will serve to hamper the key magnetisation transfer activity as their formation reduces the amount of active catalyst 4. However, as 6–9 appear to exhibit slow/no H2 exchange themselves they are unlikely to play a role in destroying the pH2 singlet order needed for SABRE. Hence, for those seeking to optimise pyruvate hyperpolarisation, the formation of 6–9 should simply be minimised, and thus sample pH controlled as it clearly is a key factor in the production of 7 and 8.
Experimental
All NMR measurements were carried out on a 400 MHz Bruker Avance III spectrometer at 298 K unless otherwise stated. Para-hydrogen (pH2) was produced by passing hydrogen gas over a spin-exchange catalyst (Fe2O3) at 28 K and used for all hyperpolarisation experiments. This method produces constant pH2 with ca. 99% purity. 1H (400 MHz) and 13C (100.6 MHz) NMR spectra were recorded with an internal deuterium lock. Chemical shifts are quoted as parts per million and referenced to residual solvent. All starting compounds were purchased from Sigma Aldrich, Fluorochem, or Alfa-Aesar and used as supplied without further purification. 1 was synthesised according to a literature procedure.54The shake & drop method was employed for recording hyperpolarised SABRE NMR spectra. Samples were prepared in a 5 mm NMR tube that was fitted with a J. Young's tap. NMR samples were subsequently degassed by three freeze–pump–thaw cycles using a Schlenk line before filling the tube with H2 or pH2 at 3 bar overpressure. Once filled with pH2, the tubes were shaken vigorously for 10 seconds at ca. 6.5 mT. Immediately after that, the NMR tubes were put inside the spectrometer for immediate NMR detection. Both hyperpolarised and thermally polarised spectra were recorded on the same sample using the same spectrometer settings.
Conclusions
In this work novel trihydride [Ir(H)3(IMes)(sulfoxide)2] and tetrahydride [Ir(H)4(IMes)(sulfoxide)]M [M = Na and K] complexes result when [IrCl(COD)(IMes)] reacts with sulfoxide, base and H2. Mechanistic and kinetic 1H NMR studies have revealed that these complexes form in two separate pathways: either via [Ir(Cl)(H)2(IMes)(sulfoxide)2], or through replacement of the diene in [Ir(H)3(COD)(IMes)]. At long reaction times, the formation of dinculear complexes like [Ir2(Cl)(μ2-Cl)(H)3(μ2-H)(IMes)2(DMSO)2], and [Ir2(H)4(μ2-H)(μ2-OCD3)(DMSO)2(IMes)2] dominate the reaction products.These reaction products have been characterised using a combination of advanced 2D NMR and parahydrogen hyperpolarised NMR. Notably, they can be detected with hyperpolarised NMR signals when pH2 is used as a reactant. Furthermore, the use of symmetry breaking methylphenylsulfoxide enables analogues of these complexes to be created where their hydride ligands become chemically distinct. This effect is a major benefit when analysing such systems by NMR spectroscopy and PHIP.
This study has also shown that these unusual products are present when related [Ir(H)2(κ2-pyruvate)(IMes)(sulfoxide)] and [IrCl(H)2(IMes)(sulfoxide)2] are used to catalytically transfer spin order from pH2 to pyruvate through SABRE. Significantly, the efficiency of pyruvate hyperpolarisation using SABRE has been dramatically increased from its initial demonstration in 2019 to the point where it has been used to produce probes for in vivo biomedical imaging studies in rodents with promise for clinical translation.37 Our results provide insight into the role these previously unknown species play during SABRE, and suggest their formation should be avoided, by varying sample pH in order to achieve the maximum active catalyst concentration.
Data availability
The raw NMR data collected in this study is available from the University of York data repository.Author contributions
BJT – conceptualisation; investigation; validation; visualisation; writing – original draft, writing – review and editing. SBD – conceptualisation; investigation; validation; writing – review and editing; supervision, funding acquisition.Conflicts of interest
B. J. T. and S. B. D. (and others) are inventors on a patent application filed by the University of York related to this work (patent no. GB1818171.9).Acknowledgements
This work was funded by UK Research and Innovation (UKRI) under the UK government's Horizon Europe funding guarantee [grant number EP/X023672/1]. We are extremely grateful to Dr Victoria Annis for synthesis of the [IrCl(COD)(IMes)] precatalyst and to Dr Peter J. Rayner for synthesis of [IrBr(COD)(IMes)] and [IrCl(13C-COD)(IMes)]. Helpful discussions with Dr Claire Condon and Callum A. Gater are appreciated.References
- J. C. Babón, M. A. Esteruelas and A. M. López, Chem. Soc. Rev., 2022, 51, 9717–9758 Search PubMed.
- N. Sieffert, T. Kendrick, D. Tiana and C. A. Morrison, Dalton Trans., 2015, 44, 4259–4270 Search PubMed.
- L. S. Van der Sluys, G. J. Kubas and K. G. Caulton, Organometallics, 1991, 10, 1033–1038 Search PubMed.
- H. Samouei, F. M. Miloserdov, E. C. Escudero-Adán and V. V. Grushin, Organometallics, 2014, 33, 7279–7283 Search PubMed.
- M. Plois, W. Hujo, S. Grimme, C. Schwickert, E. Bill, B. de Bruin, R. Poettgen and R. Wolf, Angew. Chem., Int. Ed., 2013, 52, 1314–1318 CrossRef CAS PubMed.
- A. S. C. Chan and H.-S. Shieh, Chem. Commun., 1985, 1379–1380 RSC.
- R. H. Crabtree and D. G. Hamilton, J. Am. Chem. Soc., 1986, 108, 3124–3125 CrossRef CAS.
- G. Guilera, G. S. McGrady, J. W. Steed and A. L. Jones, Organometallics, 2006, 25, 122–127 CrossRef CAS.
- D. G. Gusev, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 1998, 120, 13138–13147 Search PubMed.
- J. Ott, L. M. Venanzi, C. A. Ghilardi, S. Midollini and A. Orlandini, J. Organomet. Chem., 1985, 291, 89–100 CrossRef CAS.
- V. I. Bakhmutov, C. Bianchini, M. Peruzzini, F. Vizza and E. V. Vorontsov, Inorg. Chem., 2000, 39, 1655–1660 CrossRef CAS PubMed.
- I. Göttker-Schnetmann, P. S. White and M. Brookhart, Organometallics, 2004, 23, 1766–1776 Search PubMed.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed.
- K. Abdur-Rashid, D. G. Gusev, S. E. Landau, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 1998, 120, 11826–11827 CrossRef CAS.
- S. E. Landau, K. E. Groh, A. J. Lough and R. H. Morris, Inorg. Chem., 2002, 41, 2995–3007 Search PubMed.
- P. Kisten, E. Manoury, A. Lledós, A. C. Whitwood, J. M. Lynam, J. M. Slattery, S. B. Duckett and R. Poli, Dalton Trans., 2023, 52, 2495–2505 RSC.
- G. J. Kubas, J. Organomet. Chem., 2014, 751, 33–49 CrossRef CAS.
- G. J. Kubas, J. Organomet. Chem., 2001, 635, 37–68 CrossRef CAS.
- J. Hövener, A. N. Pravdivtsev, B. Kidd, C. R. Bowers, S. Glöggler, K. V Kovtunov, M. Plaumann, R. Katz-Brull, K. Buckenmaier and A. Jerschow, Angew. Chem., Int. Ed., 2018, 57, 11140–11162 CrossRef PubMed.
- P. J. Rayner and S. Duckett, Angew. Chem., Int. Ed., 2018, 57, 6742–6753 CrossRef CAS PubMed.
- B. J. Tickner and V. V Zhivonitko, Chem. Sci., 2022, 13, 4670–4696 RSC.
- D. A. Barskiy, S. Knecht, A. V Yurkovskaya and K. L. Ivanov, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 114–115, 33–70 Search PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science (1979), 2009, 323, 1708–1711 Search PubMed.
- T. Theis, M. L. Truong, A. M. Coffey, R. V Shchepin, K. W. Waddell, F. Shi, B. M. Goodson, W. S. Warren and E. Y. Chekmenev, J. Am. Chem. Soc., 2015, 137, 1404–1407 Search PubMed.
- T. Theis, M. Truong, A. M. Coffey, E. Y. Chekmenev and W. S. Warren, J. Magn. Reson., 2014, 248, 23–26 Search PubMed.
- A. N. Pravdivtsev, A. V. Yurkovskaya, H.-M. Vieth and K. L. Ivanov, J. Phys. Chem. B, 2015, 119, 13619–13629 CrossRef CAS PubMed.
- P. J. Rayner, P. Norcott, K. M. Appleby, W. Iali, R. O. John, S. J. Hart, A. C. Whitwood and S. B. Duckett, Nat. Commun., 2018, 9, 1–11 CrossRef CAS PubMed.
- P. Pham and C. Hilty, Chem. Commun., 2020, 56, 15466–15469 RSC.
- M. J. Cowley, R. W. Adams, K. D. Atkinson, M. C. R. Cockett, S. B. Duckett, G. G. R. Green, J. A. B. Lohman, R. Kerssebaum, D. Kilgour and R. E. Mewis, J. Am. Chem. Soc., 2011, 133, 6134–6137 CrossRef CAS PubMed.
- B. J. Tickner, M. Dennington, B. G. Collins, C. A. Gater, T. F. N. Tanner, A. C. Whitwood, P. J. Rayner, D. P. Watts and S. B. Duckett, ACS Cat., 2024, 14, 994–1004 CrossRef CAS PubMed.
- B. J. Tickner, R. O. John, S. S. Roy, S. J. Hart, A. C. Whitwood and S. B. Duckett, Chem. Sci., 2019, 10, 5235–5245 RSC.
- G. J. Kubas, Acc. Chem. Res., 1988, 21, 120–128 CrossRef.
- G. J. Kubas, J. Organomet. Chem., 2014, 751, 33–49 CrossRef CAS.
- W. Iali, S. S. Roy, B. J. Tickner, F. Ahwal, A. J. Kennerley and S. B. Duckett, Angew. Chem., Int. Ed. Engl., 2019, 131, 10377–10381 CrossRef.
- B. J. Tickner, O. Semenova, W. Iali, P. J. Rayner, A. C. Whitwood and S. B. Duckett, Cat. Sci. Technol., 2020, 10, 1343–1355 RSC.
- P. TomHon, M. Abdulmojeed, I. Adelabu, S. Nantogma, M. S. H. Kabir, S. Lehmkuhl, E. Y. Chekmenev and T. Theis, J. Am. Chem. Soc., 2022, 144, 282–287 CrossRef CAS PubMed.
- A. B. Schmidt, H. de Maissin, I. Adelabu, S. Nantogma, J. Ettedgui, P. TomHon, B. M. Goodson, T. Theis and E. Y. Chekmenev, ACS Sens., 2022, 7, 3430–3439 CrossRef CAS PubMed.
- H. de Maissin, P. R. Groß, O. Mohiuddin, M. Weigt, L. Nagel, M. Herzog, Z. Wang, R. Willing, W. Reichardt and M. Pichotka, Angew. Chem., Int. Ed., 2023, 62, e202306654 CrossRef CAS PubMed.
- B. J. Tickner, J. S. Lewis, R. O. John, A. C. Whitwood and S. B. Duckett, Dalton Trans., 2019, 48, 15198–15206 RSC.
- C. D. Assaf, X. Gui, A. A. Auer, S. B. Duckett, J.-B. Hövener and A. N. Pravdivtsev, J. Phys. Chem. Lett., 2024, 15, 1195–1203 CrossRef CAS PubMed.
- P. TomHon, M. Abdulmojeed, I. Adelabu, S. Nantogma, M. S. H. Kabir, S. Lehmkuhl, E. Y. Chekmenev and T. Theis, J. Am. Chem. Soc., 2021, 144, 282–287 CrossRef PubMed.
- B. J. Tickner, F. Ahwal, A. C. Whitwood and S. B. Duckett, ChemPhysChem, 2021, 22, 13–17 CrossRef CAS PubMed.
- B. J. Tickner, R. R. Parker, A. C. Whitwood and S. B. Duckett, Organometallics, 2019, 38, 4377–4382 CrossRef CAS PubMed.
- B. J. Tickner, A. C. Whitwood, C. Condon, C. Platas-Iglesias and S. B. Duckett, Eur. J. Inorg. Chem., 2024, e202400397 CrossRef CAS.
- R. Giernoth, P. Huebler and J. Bargon, Angew. Chem., Int. Ed., 1998, 37, 2473–2475 CrossRef CAS PubMed.
- J. Bargon, The Handbook of Homogeneous Hydrogenation, 2006, pp. 313–358 Search PubMed.
- M. Ahlquist, M. Gustafsson, M. Karlsson, M. Thaning, O. Axelsson and O. F. Wendt, Inorg. Chim. Acta, 2007, 360, 1621–1627 Search PubMed.
- C. Godard, S. B. Duckett, S. Polas, R. Tooze and A. C. Whitwood, J. Am. Chem. Soc., 2005, 127, 4994–4995 CrossRef CAS PubMed.
- S. B. Duckett, C. L. Newell and R. Eisenberg, J. Am. Chem. Soc., 1994, 116, 10548–10556 CrossRef CAS.
- N. V Kireev, A. S. Kiryutin, A. A. Pavlov, A. V. Yurkovskaya, E. I. Musina, A. A. Karasik, E. S. Shubina, K. L. Ivanov and N. V Belkova, Eur. J. Inorg. Chem., 2021,(41), 4265–4272 Search PubMed.
- D. Blazina, S. B. Duckett, P. J. Dyson and J. A. B. Lohman, Chem.–Eur. J., 2003, 9, 1045–1061 Search PubMed.
- S. B. Duckett and N. J. Wood, Coord. Chem. Rev., 2008, 252, 2278–2291 Search PubMed.
- R. Gobetto, L. Milone, F. Reineri, L. Salassa, A. Viale and E. Rosenberg, Organometallics, 2002, 21, 1919–1924 Search PubMed.
- L. D. Vazquez-Serrano, B. T. Owens and J. M. Buriak, Inorg. Chim. Acta, 2006, 359, 2786–2797 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06138a and NMR data at DOI: https://doi.org/10.15124/fccecf8d-6d7d-4f76-85e0-7904413f86b5 |
| This journal is © The Royal Society of Chemistry 2025 |