
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lattice oxygen insertion mechanism in CeO2-catalyzed reactions in water: nitrile hydration reaction†
Takaaki
Endo
 a,
Tatsushi
Ikeda
a,
Koki
Muraoka
a,
Yusuke
Kita
b,
Masazumi
Tamura
*b and
Akira
Nakayama
*a
a,
Tatsushi
Ikeda
a,
Koki
Muraoka
a,
Yusuke
Kita
b,
Masazumi
Tamura
*b and
Akira
Nakayama
*a
aDepartment of Chemical System Engineering, The University of Tokyo, Tokyo 113-8656, Japan. E-mail: nakayama@chemsys.t.u-tokyo.ac.jp
bDepartment of Chemistry and Bioengineering, School of Engineering, Osaka Metropolitan University, 3-3-138, Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: mtamura@omu.ac.jp
First published on 2nd December 2024
Abstract
Cerium oxide (CeO2) exhibits prominent catalytic activity in various organic reactions owing to its unique acid–base and redox properties. One of the most interesting applications of pure CeO2-catalyzed organic reactions is the hydration of nitriles in water. The experimental results showed that the hydration of 2-cyanopyridine to picolinamide in water using CeO2 catalysts proceeds readily at low temperatures (30–100 °C) in high yields and that this reaction occurs exclusively on CeO2 among various metal-oxide catalysts. To elucidate the unique catalytic activity of CeO2, the reaction mechanism is dissected using the density functional theory-based molecular dynamics (DFT-MD) simulations. Based on the free energy analysis, it is demonstrated that the reaction proceeds with the involvement of the surface lattice oxygen, where the lattice oxygen atom is inserted into picolinamide. The involvement of the surface lattice oxygen is notably uncommon given the low temperatures of the reaction, and this computational prediction is verified by the two experiments using H218O solvent and 18O-exchanged CeO2 catalyst, where the introduction of surface lattice oxygen into picolinamide is confirmed. The inherent flexibility of the surface lattice oxygen and the unique acid–base properties of CeO2, which can favorably bind and activate both nitrile and water molecules, are key factors in the high reactivity for various organic reactions, which characterizes the outstanding catalytic activity of CeO2.
1. Introduction
Among various metal-oxides, cerium oxide (CeO2) has received considerable attention because of its immense scientific and technological importance in fields such as catalysis, glass polishing, biomedical technology, and sensors. In particular, the number of catalytic applications of CeO2 is continuously increasing, and CeO2 is extensively used in crude oil refining, solid oxide fuel cells (SOFC), vehicle emission control, and so on. The wide applicability of CeO2 is attributed to its unique redox and acid–base properties, oxygen storage capacity, and high thermal stability and durability (see ref. 1–6 for the recent reviews). In the catalytic applications of CeO2, its high reducibility and basicity promote the activation and dispersion of metal nanoparticles via strong metal-oxide electronic interactions,7–9 and CeO2-supported metal nanoparticles have a wide range of catalytic applications.10–14 Even without supported metal nanoparticles, pure CeO2 is also quite effective for various organic reactions (see the reviews in ref. 15–17), which characterizes CeO2 as a highly unique metal-oxide.In one of the most interesting applications of pure CeO2-catalyzed organic reactions, Tamura et al. reported that the hydration of 2-cyanopyridine to picolinamide over CeO2 proceeds very smoothly at low temperatures (30–100 °C) with high yields (Fig. 1).18,19 It was also found that this reaction proceeds exclusively on CeO2 among various metal-oxide catalysts and that the activity is two orders of magnitude higher than that of other metal-oxides. Based on this finding, 2-cyanopyridine was later used as a dehydration agent for the direct conversion of CO2 and methanol to dimethyl carbonate on CeO2 catalysts, where 94% yield and 99% selectivity were reported.20 Also, it was further utilized in CO2 conversion with alcohols and amines into carbonates, ureas, carbamates over CeO2.21,22 In our previous works on the adsorption of 2-cyanopyridine over CeO2(111),23–26 we found a unique adsorption structure of 2-cyanopyridine, involving a covalent interaction between the C atom in the CN group of 2-cyanopyridine and the surface lattice O atom along with the acid–base interaction between the N atoms in the pyridine ring and the surface Ce atom. This unique adsorption structure creates a strong base site at the position of the N atom in the CN group, and it has been demonstrated that base-catalyzed reactions proceed effectively and that the pKa (CH3CN) is estimated to be ∼21.23 In another related study, we examined the adsorption structure of picolinic acid over CeO2(111) and found a similar adsorption structure that involves a covalent bond between the C atom in the carboxylic group and the surface O atom.27 Interestingly, when we further performed the DFT-based molecular dynamics simulations (DFT-MD) to gain insight into the adsorption process in water, the spontaneous dissociation of a hydroxide ion from the carboxylic group was observed, which implies the rich chemistry at the interface of water and CeO2 surface.
 | ||
| Fig. 1 Hydration of 2-cyanopyridine to picolinamide over CeO2 catalyst. | ||
These observations motivated us to investigate the reaction mechanism of the hydration of 2-cyanopyridine over CeO2 by employing DFT-MD simulations with explicit solvent water molecules. The DFT-MD simulations are now widely used as an invaluable tool to obtain a bottom-up picture of the liquid/metal-oxide interface and thus provide useful information on the microscopic properties and dynamical behavior of molecules at the interfaces. In the previous studies by the DFT-MD simulations at the water/CeO2 interface, high activity at the interface involving proton hopping was reported.28–31 In this study, the reaction mechanisms of nitrile hydration on the CeO2(111) surface are examined by computing the free energy landscape, and we decipher the role of CeO2 in these unique catalytic reactions. Based on the free energy analysis, we find that the reaction proceeds with the involvement of the surface lattice oxygen, and this computational prediction is further verified by the two experiments using H218O solvent and 18O-exchanged CeO2 catalyst.
2. Computational details
2.1. DFT calculations
All DFT calculations were performed under the periodic-boundary condition with the mixed Gaussian and plane-waves (GPW) approach implemented in the CP2K program package.32 The short-range variants of the double-ζ valence plus polarization (DZVP) basis sets of the MOLOPT type33 were employed for H, C, N, and O atoms to represent the valence electrons, and the norm-conserving Goedecker–Teter–Hutter pseudopotentials34,35 were used to describe the interactions between the valence and core electrons. For Ce atoms, we employed the basis sets and pseudopotential generated by Wang and coworkers.36 The energy cutoff of 400 Ry was taken for the auxiliary plane wave expansion of the density. The generalized-gradient approximation by the Perdew–Burke–Ernzerhof (PBE) functional models was employed as the exchange and correlation potential, and the DFT+U approach was taken in order to correctly represent the nature of 4f orbitals of Ce atoms, where the occupancies of 4f orbitals were calculated based on the Mulliken population analysis.37,38 The U value was set to 7.0 eV following the previous works.36 The Brillouin zone integration was performed with a reciprocal space mesh consisting of only the Γ-point in the GPW approach. The convergence criteria for the energy in the SCF calculation were set to 1 × 10−6 hartree.Cerium oxide exhibits fluorite structure in the low-temperature regime. It has been determined theoretically39 and experimentally40 that the CeO2(111) surface is the most stable low-index surface among single crystal terminations of CeO2, and therefore it represents a large portion of the polycrystalline CeO2 surface.39,41 CeO2-catalyzed organic reactions are often reported to be influenced by the exposed facets.42 In our catalyst system in the nitrile hydration, the activity per surface area does not change significantly even after high-temperature calcination,18 suggesting that the main active facet is the (111) facet. Furthermore, a recent study has demonstrated that the (111) facet exhibits high activity for the hydration of 2-cyanopyridine.43 Therefore, the surface of the CeO2(111) facet was employed in this study. The CeO2(111) surface was modeled as a periodic p(3 × 3) hexagonal slab of 27 CeO2 units with three O–Ce–O trilayers. The dimensions of a simulation cell were set to a = b = 11.56 Å, c = 30.0 Å and α = β = 90°, γ = 60°, and this slab was separated by ∼16 Å of vacuum space in the direction perpendicular to the surface. The lattice parameters of the CeO2(111) slab were determined by the cell optimization of the bulk CeO2, and the optimized lattice constant of 5.450 Å was in good agreement with the experimental value of 5.411 Å,44,45 where the deviation from the experimental value was less than 1%. For geometry optimization, the forces on all atoms are minimized to less than 0.02 eV Å−1 (4.5 × 10−4 hartree bohr−1). The bottom O–Ce–O tri-layers were fixed at the bulk positions during geometry optimization and DFT-MD simulations. In calculating the energies of gas-phase molecules, the simulation cell of a = b = c = 15.0 Å in the cubic box is used.
The adsorption energy of a molecule EA to the CeO2(111) surface is calculated according to
| EA = Emol+surf − Emol − Esurf | (1) |
2.2. Molecular dynamics simulations with umbrella sampling
For the investigation of chemical reactions that occur on a much longer timescale than that allowed in DFT-MD simulations (so-called rare events), free energy calculations are generally carried out. Computing the free energy landscape and finding the lowest free energy pathway on this landscape provide valuable information on the mechanism and kinetics of the reaction.46 The free energy landscape is generally represented by a few selected coarse-grained coordinates or collective variables (CVs), and several advanced sampling techniques have been proposed to explore the free energy landscape along these CVs: these include metadynamics,47,48 umbrella sampling,49 and several others.50–52In this work, we employ the umbrella sampling techniques for constructing free energy landscape. Two types of CVs are used in this work. The first one is the distance between atoms i and j, and it is given as
dij ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) = |Ri − Rj| = |Ri − Rj| | (2) |
The second one is a coordination number, and the coordination number between two atom types (X and Y) is defined as
 | (3) |
 | (4) |
In the following simulations, 32 water molecules were placed over the CeO2(111) surface in addition to 2-cyanopyridine. The initial configurations of water molecules were generated by the GROMACS package.53 The DFT-MD simulations were performed at a system temperature of T = 360 K with the umbrella potential, where quadratic potentials centered at predefined points were applied. The temperature was controlled by the Nosé–Hoover thermostat. The mass of hydrogen was replaced with that of deuterium, allowing for a larger time step of 1.0 fs. For each umbrella window, 10 ps simulations were performed for equilibrium and subsequent 10 ps simulations were used for analysis, which is sufficient to ensure the dissociative equilibrium of water molecules.28,29,31 Around the transition state region, additional umbrella sampling was performed using the configuration of adjacent window that is closer to the transition state region as an initial configuration, and the convergence of probability distributions was confirmed. The biased probability distributions from different umbrella windows were reweighted and patched by the weighted histogram analysis method (WHAM)54 using the implementation by Grossfield.55 The PLUMED package56 interfaced with CP2K was used for the umbrella sampling. Free energy barrier heights were estimated using multidimensional lowest energy (MULE) algorithm code implemented by Fu et al.57
3. Results and discussions
In this section, the reaction mechanism of hydration of 2-cyanopyridine over CeO2 is investigated. Experimentally, the following reaction mechanism has been proposed:18 (1) dissociation of water molecules on CeO2 giving rise to Hδ+ and OHδ− pair species, (2) formation of adsorption complex between 2-cyanopyridine and CeO2, (3) addition of OHδ− to the C atom in the CN group via an amide anion intermediate, and (4) desorption of the amide product (picolinamide) from the CeO2 surface accompanying regeneration of the adsorption site. Since no kinetic isotopic effect was observed (kH/kD = 0.95), meaning that the water dissociation is fast on CeO2, the third step is considered to be the rate-determining step. As already reported in the literature,28–31 water molecules are activated on CeO2 and about 20–33% of the surface oxygen sites are hydroxylated (degree of hydroxylation). Since adsorption energy of a water molecule in the molecular and dissociative states are quite similar, proton hopping is quite active at the water/CeO2(111) interface.28–31 Therefore, the presence of surface hydroxy groups does not impede the approach and adsorption of 2-cyanopyridine on the surface. Consequently, we consider steps (2) and (3) in the following analysis.3.1. Adsorption process of 2-cyanopyridine over CeO2
The adsorption structures of 2-cyanopyridine over CeO2(111) have already been reported,23 and there are three types of adsorption states, which are shown in Fig. 2. The strongest adsorption involves the covalent bond between the C atom in the CN group (CCN) and the surface lattice O atom (OS), along with the acid–base interaction between the N atom in the pyridine ring (Npy) and the surface Ce atom (denoted by St-I). The adsorption energy of St-I was calculated to be −68 kJ mol−1. It has been known that this adsorption structure forms a strong base site in the CN position of 2-cyanopyridine.23 Another adsorption structure (denoted by St-II) involves molecular adsorption where 2-cyanopyridine interacts with the surface Ce atoms through the two N atoms in the pyridine ring and the CN group with a distance of ∼3.0 Å, where the adsorption energy was −47 kJ mol−1. St-III involves the acid–base interaction only between the N atom in the CN group and the surface Ce atom with the adsorption energy of −33 kJ mol−1. | ||
| Fig. 2 Adsorption structures of 2-cyanopyridine on CeO2(111) (a) St-I, (b) St-II, and (c) St-III. | ||
The adsorption process of 2-cyanopyridine from the molecular adsorption structure, which corresponds to St-II, to the structure with the CCN–OS covalent bond (corresponding to St-I) is then investigated in water environment by DFT-MD simulations with umbrella sampling (hereinafter referred to as Step 1-(II)). When we performed the standard DFT-MD simulation starting from St-I with surrounding water molecules, a proton was spontaneously transferred to the N atom in the CN group (NCN) from an adjacent water molecule due to the strong basicity of the NCN atom formed by this unique adsorption structure of St-I.23 The resulting hydroxide ion then accepts a proton from a nearby water molecule, and subsequently, a hydroxide ion is transferred to the Ce atom after a couple of proton hopping events via the Grotthuss-like diffusion mechanism. This implies that during the adsorption process from St-II to St-I in water environment, a proton is transferred to the N atom in the CN group. The schematic representation is given in Fig. 3. For the sake of simplicity, a proton transferred to the N atom is depicted as originating from surface hydroxy group, although in DFT-MD simulations it originates from a nearby water molecule, as mentioned above. To obtain the free energy profile for this process, two CVs were introduced. The first CV (CV1) was set as the distance between CCN and one of the surface OS atoms, d(CCN–OS), and the second CV (CV2) was chosen as c(NCN–HW), where HW is hydrogen atoms of water molecules, to promote proton transfer events between NCN and the surrounding water molecules. For CV2, the cutoff bond distance was set to R0(NCN–HW) = 1.2 Å, and p = 6 and q = 6 were used to define the shape of the switching function. The umbrella potentials were placed between 1.3 Å and 3.1 Å for CV1, and those of CV2 were chosen between 0.1 and 0.9. The force constants of CV1 and CV2 were set to 20.7 eV Å−2 and 31.1 eV, respectively. For CV2, a larger force constant of 41.5 eV was used near the transition state region.
 | ||
| Fig. 3 Schematics of reaction mechanisms. | ||
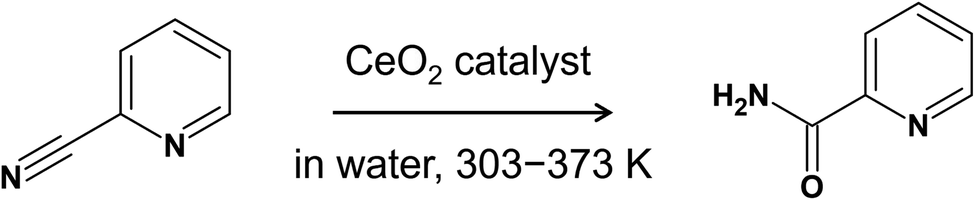
The free energy surface was constructed using CV1 and CV2, and the two-dimensional contour plot of the free energy surface along CV1 = d(CCN–OS) and CV2 = c(NCN–HW) is shown in Fig. 4(a). Starting from the region corresponding to the molecular adsorption state at d(CCN–OS) ≈ 3.0 Å (A1, the snapshot is given in Fig. 4(b)), the molecule approaches the surface by surmounting a small free energy barrier of 25 kJ mol−1 at d(CCN–OS) ≈ 2.1 Å. After passing through this barrier, the free energy surface reveals a metastable state at d(CCN–OS) ≈ 1.4 Å and c(NCN–HW) ≈ 0.1 (A2), representing the adsorption state with a covalent CCN–OS bond. This structure corresponds to St-II in Fig. 2. Then, there is a small barrier for a proton transfer from a neighboring water molecule, which is not an adsorbed one, to the NCN atom, leading to the stable protonated adsorption state (A3) with d(CCN–OS) ≈ 1.4 Å and c(NCN–HW) ≈ 0.75. The free energy difference between the molecular adsorption state (A1) and protonated adsorption state (A3) is 76 kJ mol−1, which indicates a strong adsorption of protonated 2-cyanopyridine. Considering the difference in adsorption energy between St-I and St-II given in Fig. 2 (21 kJ mol−1), the large stabilization of A3 is mainly due to the protonation of 2-cyanopyridine. The barrier height for the adsorption process is close to the values obtained in a similar simulation for the adsorption of picolinic acid to the CeO2(111) surface (∼30 kJ mol−1),27 which also involves the CCN–OS covalent bond formation.
 | ||
| Fig. 4 (a) Contour plot of the free energy surface (given in kJ mol−1) for adsorption process of 2-cyanopyridine from the molecular adsorption state (Step 1-(II)). The minimum free energy pathway, which is determined by the MULE algorithm, is shown in white. The inset shows the free energy profile along the minimum free energy pathway. (b) Snapshots of representative configuration near A1, A2, and A3. | ||
Here we also examine the adsorption process of 4-cyanopyridine where it is experimentally shown that the reaction rate of hydration of 4-cyanopyridine is 10−7-order of magnitude smaller than that of 2-cyanopyridine. The free energy surface of the adsorption process was constructed in the same way as above (the details are provided in ESI†), and it was found that the free energy barrier leading to the product state was 67 kJ mol−1, which is much higher than that of 2-cyanopyridine. This is mainly due to the steric hindrance between the pyridine ring and the surface, and the unfavorable adsorption of 4-cyanopyridine is related to the very low activity of the hydration reaction.
3.2. Hydration of 2-cyanopyridine on CeO2
The hydration reaction is next investigated from the above adsorption state as a protonated 2-cyanopyridine species (A3). Two reaction mechanisms are considered. The first mechanism involves the nucleophilic attack of hydroxide ion that is formed on the nearby Ce atom to the CCN atom (Step 2-(I)), while the second one involves the surface OS atom where the surface OS atom is released from the surface and introduced into picolinamide (Step 2-(II)). The second mechanism has not been considered because the surface OS atoms have not been thought to be involved in the reaction at low temperatures (below room temperature). The two reaction mechanisms are schematically shown in Fig. 3.In the first mechanism (Step 2-(I)), we consider the reaction mechanism where a hydroxide ion that is formed on the Ce atom nucleophilically attacks the CCN atom. Here, the following two CVs were introduced for the free energy analysis: CV1 = d(CCN–OW) and CV2 = c(OW–HW). CV1 corresponds to the distance between the CCN atom and one of the O atoms deriving from water molecules, which promotes the nucleophilic attack of the hydroxide ion. CV2 was introduced to accelerate the deprotonation of the attacking water on the surface, where the cutoff bond distance was set to R0(OW–HW) = 1.2 Å with p = 6 and q = 6. The umbrella potentials were placed between 1.3 Å and 3.4 Å for CV1, and those of CV2 were chosen between 0.1 and 1.7, and the force constants of CV1 and CV2 were set to 20.7 eV Å−2 and 31.1 eV, respectively.
Fig. 5(a) shows the contour plot of the reconstructed free energy surface along CV1 = d(CCN–OW) and CV2 = c(OW–HW), where the reactant region consists of the two stable states, B1-I (d(CCN–OW) ≈ 3.0 Å, c(OW–HW) ≈ 1.6) and B1-II (d(CCN–OW) ≈ 3.2 Å, c(OW–HW) ≈ 0.9). B1-I corresponds to the configuration where the attacking water molecule is coordinated to the nearby Ce atom (snapshot is given in Fig. 5(b)), while B1-II represents the configuration where the attacking water molecule is dissociatively adsorbed on the surface, being a hydroxide ion. The free energy profile between B1-I and B1-II simply represents the free energy difference between the water molecule and hydroxide ion on the nearby Ce atom. Free energy difference and activation energy between B1-I and B1-II are small, which indicates the facile proton hopping between the hydroxide ion and surface hydroxy group or adjacent water molecule, which has been already reported for the simulation of the water/CeO2(111) interface.31
 | ||
| Fig. 5 (a) Contour plot of the free energy surface (given in kJ mol−1) for the nucleophilic attack of a hydroxide ion to the CCN atom (Step 2-(I)). The minimum free energy pathway, which is determined by the MULE algorithm, is shown in white. The inset shows the free energy profile along the minimum free energy pathway. (b) Snapshots of representative configuration near B1-I, B1-II, and B2. | ||
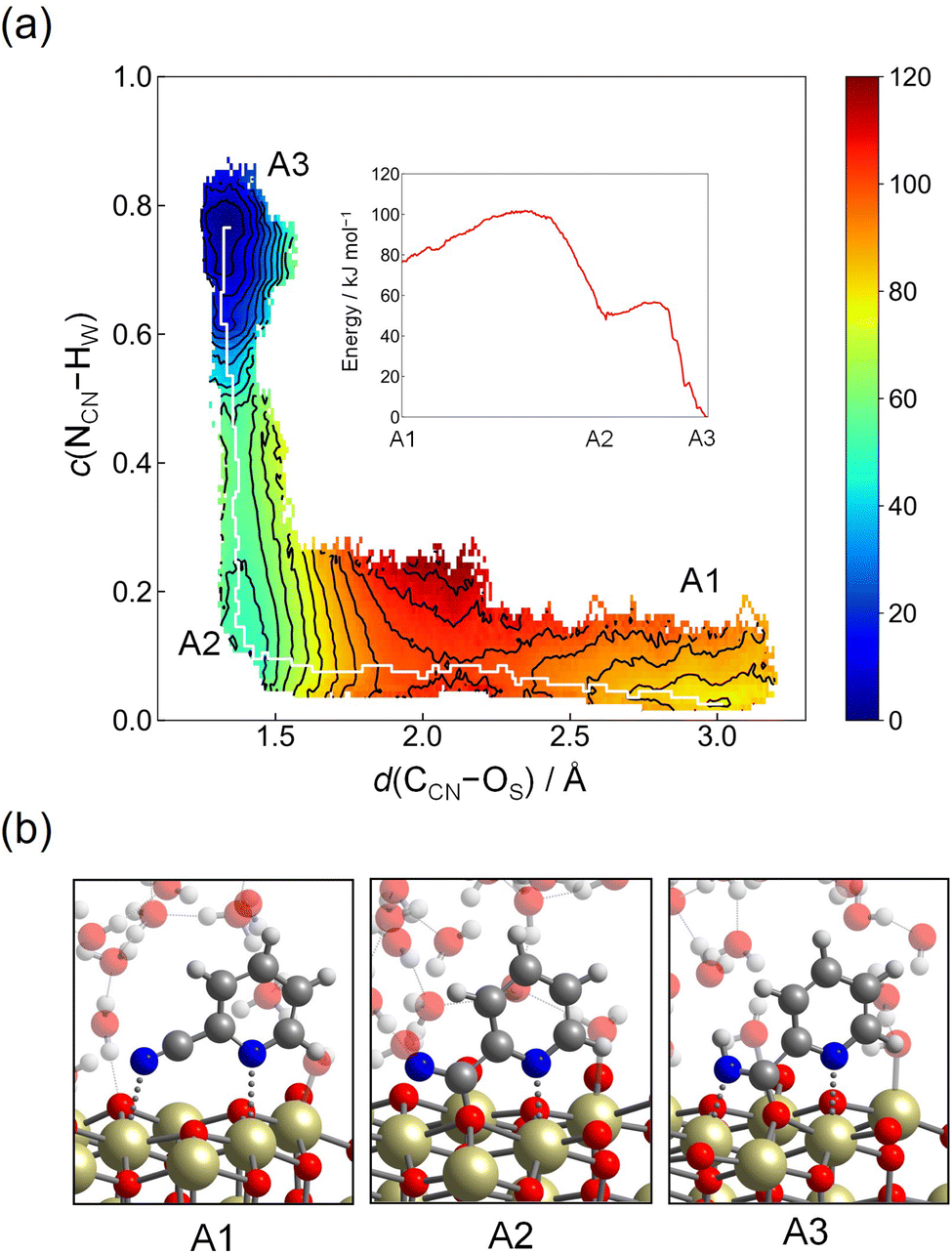
Hydration reaction starts from B1-II, and the transition state region is found at d(CCN–OW) ≈ 1.8 Å and c(OW–HW) ≈ 0.8, representing the conformation of the nucleophilic attack of a hydroxide ion residing on the Ce atom to the CCN atom. The free energy barrier is estimated as 70 kJ mol−1. After passing through this region, tautomerization readily occurs by a proton relay through the hydrogen bond network, leading to the amide product (picolinamide) (B2). The free energy barrier of 70 kJ mol−1 is appreciably lower than the value estimated in the hydration reaction in bulk water (120 kJ mol−1, see below and ESI†). This is rationalized by the unique adsorption structure of 2-cyanopyridine, where the CCN atom is more positively charged due to the formation of protonated 2-cyanopyridine on the surface. At B2, there still exits the CCN–OS bond, and the free energy profile for the breaking of the CCN–OS bond was obtained by umbrella sampling using only one CV, d(CCN–OS). Fig. 6 shows the free energy profile for this step, and the free energy barrier of 53 kJ mol−1 was obtained. This barrier height is smaller than that of the nucleophilic attack of a hydroxide ion (B1-II → B2).
 | ||
| Fig. 6 Free energy profile for the breaking of the CCN–OS bond as a function of d(CCN–OS) starting from B2 in Step 2-(I). | ||
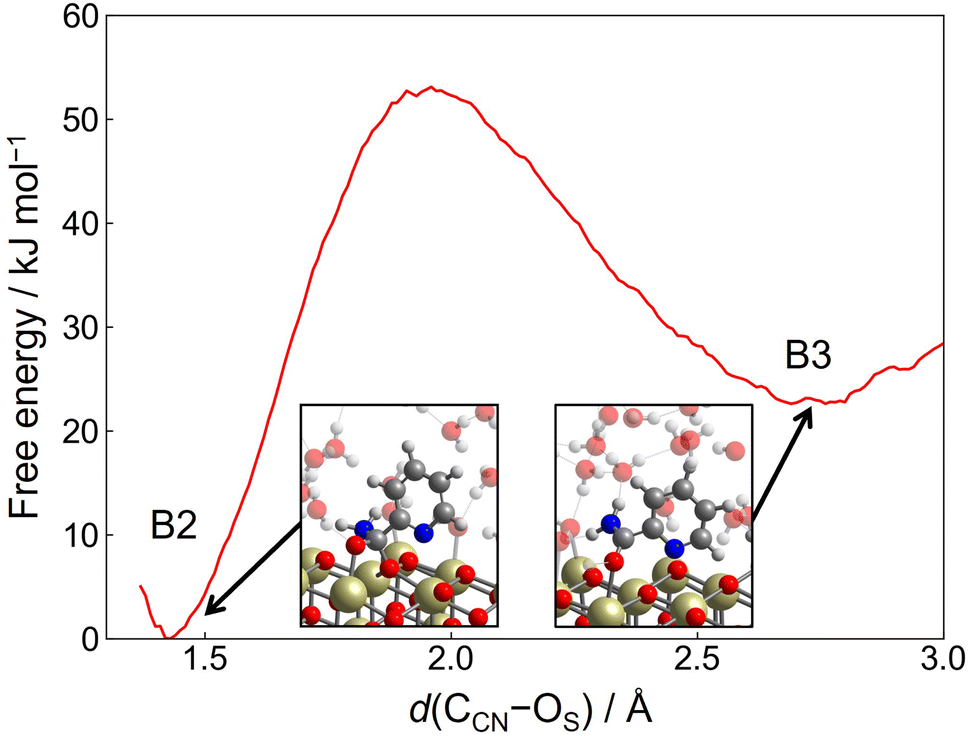
Next, we consider the second mechanism (Step 2-(II)), where the surface OS atom is involved. In this step, the surface OS atom of the CCN–OS bond is introduced into 2-cyanopyridine, leading to the formation of picolinamide anion on the surface and an oxygen vacancy. Then, the resulting oxygen vacancy is replenished by a hydroxide ion on the surface. To investigate this reaction mechanism, the free energy surface is constructed by two CVs, CV1 and CV2. CV1 is the distance between the surface OS atom of the CCN–OS bond and the vacancy site VO that is defined by the center of mass of the three surrounding surfaces Ce atoms, d(VO–OS). CV2 is defined as the difference between two coordination numbers, one of which is the coordination number between the vacancy site VO and O atoms of solvent water molecules, c(VO–OW). The introduction of c(VO–OW) promotes the replenishment of the oxygen vacancy after the formation of picolinamide anion on the surface. The other one is defined as c(VO–NCN). We introduce this coordination number by noting that as the distance d(VO–OS) (=CV1) increases, the NCN atom tends to fill the oxygen vacancy site due to the charge-negativity of the NCN atom (see snapshot of C2 in Fig. 7(b)). The introduction of c(VO–NCN) is essential to promote the replenishment of the oxygen vacancy by a hydroxide ion rather than the NCN atom. The cutoff bond distances of these coordination numbers were set to R0(VO–OW) = 2.0 Å with p = 6 and q = 4 and R0(VO–NCN) = 2.0 Å with p = 6 and q = 2, respectively.
 | ||
| Fig. 7 (a) Contour plot of the free energy surface (given in kJ mol−1) for the mechanism involving surface lattice oxygen atom (Step 2-(II)). The minimum free energy pathway, which is determined by the MULE algorithm, is shown in white. The inset shows the free energy profile along the minimum free energy pathway. (b) Snapshots of representative configuration near C1, C2, and C3. OS represents the surface lattice oxygen involving the CCN–OS bond and OW represents an oxygen atom originating from solvent water molecule. | ||
Fig. 7(a) shows the contour plot of the free energy surface along CV1 = d(VO–OS) and CV2 = c(VO–OW) – c(VO–NCN). Starting from the minimum located at d(VO–OS) ≈ 1.2 Å and c(VO–OW) − c(VO–NCN) ≈ −0.1 (C1), which corresponds to the protonated adsorption structure of A3, the free energy increases slightly as the distance d(VO–OS) is increased until d(VO–OS) ≈ 2.6 Å. At d(VO–OS) ≈ 2.6 Å, the OS atom is detached from the original lattice position, and the resulting configuration can be viewed as an adsorbed picolinamide anion near the oxygen vacancy site. From this structure, there are two pathways leading to C2 or C3. C2 is a metastable adsorption state of picolinamide anion with a shallow minimum at d(VO–OS) ≈ 2.7 Å and c(VO–OW) – c(VO–NCN) ≈ −0.5, where the NCN atom of picolinamide anion partially fills the vacancy. When d(VO–OS) is increased further, there exists the barrier at d(VO–OS) ≈ 3.0 Å and c(VO–OW) – c(VO–N) ≈ 0.4 to accommodate a hydroxide ion at the vacancy site, which leads to C3. The barrier height associated with this process is estimated as 54 kJ mol−1, and it is lower than the value obtained in Step 2-(I) (70 kJ mol−1). After the vacancy site is filled by a hydroxide ion, the picolinamide anion readily accepts a proton from the surface hydroxy group that occupies the vacancy site, leading to the picolinamide product. The formed picolinamide is adsorbed on the surface by acid–base interaction between the O atom of the amide group and the surface Ce atom (see snapshot of C3 in Fig. 7(b)).
This reaction mechanism using surface lattice oxygen atom is quite unique in that the reaction occurs at room temperature. In CeO2-catalyzed reactions, it is generally accepted that the surface oxygen is involved in the redox reaction and that these reactions occur at much higher temperatures of ∼600 K.4,58 As an exception, it was reported that CeO2 works as an oxidation catalyst of alcohols at low temperatures around room temperature, where the surface lattice oxygen of CeO2 is used for the oxidation.59 When we monitor the oxidation state of Ce atoms for representative snapshots along the hydration reaction in Step 2-(II), the magnetic moment of all Ce atoms is almost zero, implying that the oxidation state of all Ce atoms is IV and the oxygen vacancy is accompanied by Ce(IV). It is suggested that Ce(IV) is maintained by the coordination of picolinamide anion and hydroxide ion, which are also stabilized by surrounding water molecules. The remarkable reactivity of CeO2 could be attributed to the flexibility of surface lattice oxygen atoms.
As another possible reaction mechanism, hydration of 2-cyanopyridine from the molecular adsorption structure, which corresponds to St-II (see Fig. 2), is also examined in a similar manner to Step 2-(I), and this reaction mechanism is denoted as Step 2-(III) hereafter (see Fig. 3 for schematics). This mechanism is intuitively more plausible for nitrile hydration reaction on metal-oxide surface, where the CN group coordinated to the Lewis acid site of Ce atoms is nucleophilically attacked by a hydroxide ion on the surface, which is closely related to the reaction mechanism of nitrile hydration catalyzed by metal-complexes such as Ru-complexes.60,61 The starting configuration is the same as Step 1-(II) (corresponding to A1), and two CVs, d(CCN–OW) and c(OW–HW), are employed to obtain the free energy surface as has been chosen in Step 2-(I). The force constants of umbrella potential are set to be the same values as in Step 2-(I). The contour plot of the free energy surface spanned by d(CCN–OW) and c(OW–HW) is shown in Fig. 8(a). The reactant corresponds to D1, which is located at d(CCN–OW) ≈ 3.4 Å and c(OW–HW) ≈ 1.0, where an attacking hydroxide ion is coordinated on the nearby Ce atom. Here, the dissociation equilibrium of the attacking hydroxide ion and water molecule on the Ce atom is not considered as in Step 2-(I), which corresponds to the region of c(OW–HW) >1.0. As d(CCN–OW) becomes shorter, the molecule reaches the transition state region at d(CCN–OW) ≈ 1.7 Å and c(OW–HW) ≈ 0.8, where the free energy barrier is calculated to be 69 kJ mol−1. After passing through the transition state region, the shallow minimum is seen at d(CCN–OW) ≈ 1.3 Å and c(OW–HW) ≈ 0.8 (D2), which corresponds to an imidic acid. Then, picolinamide (D3) is formed by tautomerization through a proton relay. The free energy barrier of 69 kJ mol−1 is comparable to that of Step 2-(I) (70 kJ mol−1), but the high stability of the protonated adsorption state of A3, compared to molecular adsorption state of A1 (difference in free energy is 76 kJ mol−1), would eliminate the possibility of taking this reaction pathway. The hydration of 2-cyanopyridine in the bulk water is also examined for reference (the details are provided in the ESI†), and in this case, the free energy barrier was estimated as 120 kJ mol−1, which is significantly high compared to the reaction occurring at the interface of water/CeO2. In the experiments of CeO2-catalyzed reaction, the activation energy was estimated to be 81.7 kJ mol−1 and also the activation Gibbs energy was reported to be 79.0 kJ mol−1 by the Eyring plot.19 These values are slightly higher than the barrier heights of Step 2-(I) (70 kJ mol−1) and Step 2-(II) (54 kJ mol−1) estimated in the simulation. One reason would be the simulation temperature of 360 K in our simulations. We employ this temperature because it is a good compromise for describing the structural properties of ambient liquid water for PBE and its derived functionals in DFT calculations.62,63
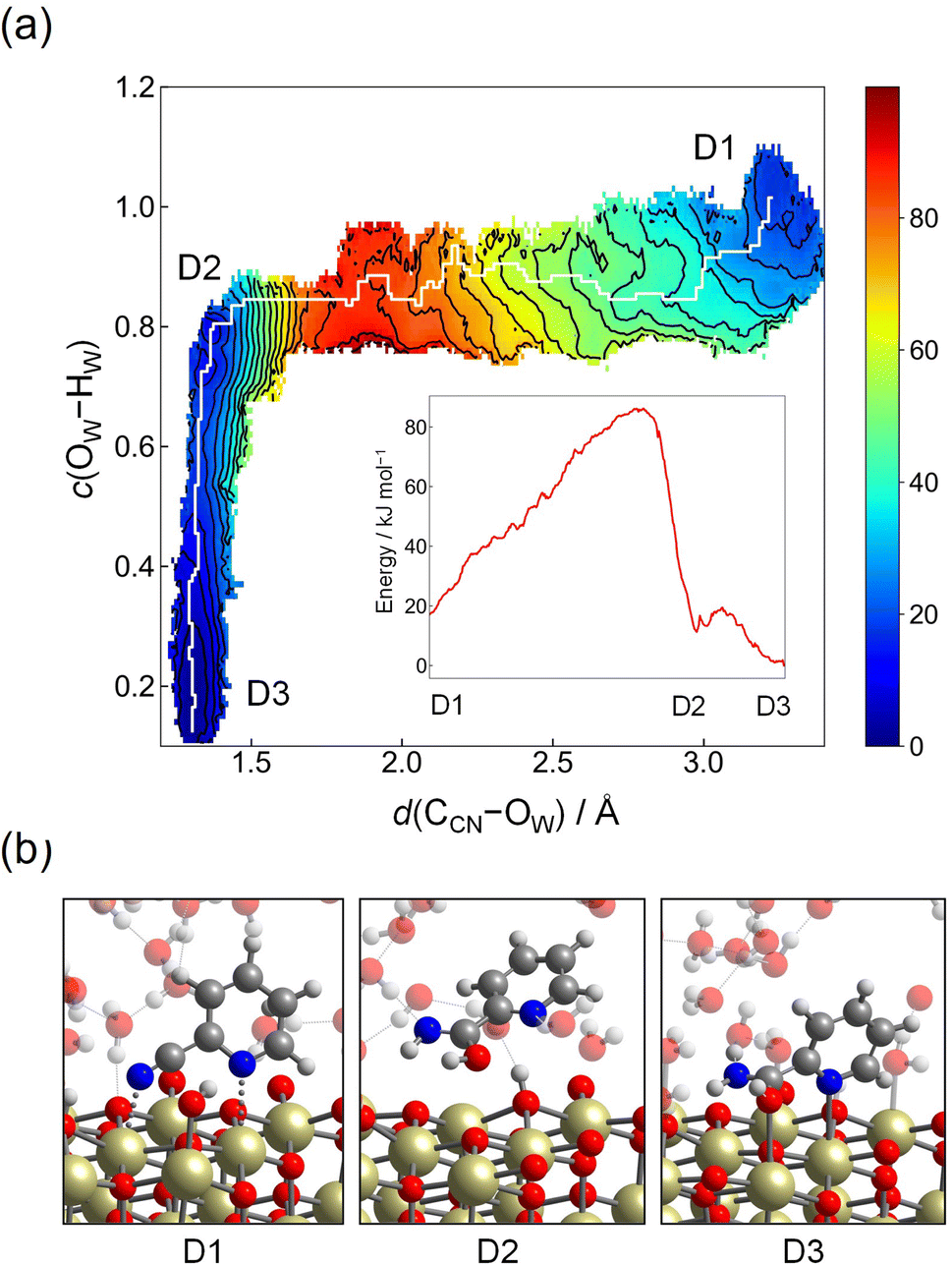 | ||
| Fig. 8 (a) Contour plot of the free energy surface (given in kJ mol−1) for the nucleophilic attack of a hydroxide ion to the CCN atom from molecular adsorption structure (Step 2-(III)). The minimum free energy pathway, which is determined by the MULE algorithm, is shown in white. The inset shows the free energy profile along the minimum free energy pathway. (b) Snapshots of representative configuration near D1, D2, and D3. | ||
Fig. 9 depicts the free energy diagram for the reaction pathways investigated in this work. Starting from the molecular adsorption state (A1), a protonated species with the CCN–OS covalent bond is formed (A3). From this strong adsorption state, two reaction pathways are possible. Based on the difference in the free energy barrier, the reaction involving surface lattice oxygen is energetically more favorable.
 | ||
| Fig. 9 Free energy diagram starting from molecular adsorption state of A1 and D1, where zero of the free energy is set to this state. | ||
3.3. Experimental verification
If the reaction mechanism involving surface lattice oxygen (Step 2-(II)) is the main reaction pathway, the lattice oxygen will be introduced into 2-cyanopyridine to form picolinamide in the hydration of 2-cyanopyridine. If not, the oxygen atoms of water molecules will be introduced into 2-cyanopyridine. We conducted two experiments: using (i) CeO2 with lattice oxygen partially substituted with 18O (18O-substituted CeO2) and (ii) 18O-substituted water (H218O) solvent. The introduction of 18O in picolinamide can be confirmed by the difference in the mass number of picolinamide; The mass number of the product with 16O and 18O are 122 and 124, respectively. The experimental details are provided in ESI.†In the first experiment, we prepared 18O-substituted CeO2 by introducing 18O2 after hydrogen reduction at 873 K, and the substituted ratio of 18O/16O in 18O-introduced CeO2 was determined to be 1.42% by O2 adsorption of the reduced CeO2 (Fig. S4†). Hydration of 2-cyanopyridine was conducted at 274 K and 279 K by using the 18O-introduced CeO2, and the product was confirmed by mass spectroscopy. Fig. 10 shows the time course of the hydration of 2-cyanopyridine with CeO2 and 18O-substituted CeO2. In the initial stage of the reaction, the mass intensity ratio of 124/122 in picolinamide with 18O-substituted CeO2 was significantly higher (about five times) compared to that with CeO2, confirming that 18O from 18O-substituted CeO2 was introduced into the picolinamide. The relationship between the 18O/16O in picolinamide and conversion is shown in Fig. 11. As a result, the 18O/16O was around 1.4% at low conversions, which is in good agreement with the 18O/16O in 18O-substituted CeO2 (1.42%), and the ratio decreased as the conversion increased. This result can be interpreted as follows. In the initial stage of the reaction, picolinamide was produced with the surface lattice oxygen of 18O-substituted CeO2, and therefore, the 18O/16O in picolinamide (∼1.41%) is almost equal to that of the 18O-substituted CeO2 (1.42%). As the reaction consumes 18O in the 18O-substituted CeO2, 16O is replenished from water and the surface 18O/16O of the 18O-substituted CeO2 decreased, resulting in the decrease in the 18O/16O in picolinamide. If the reaction mechanism of Step 2-(I) is involved, 16O of H2O will be introduced into the product and 18O/16O in picolinamide would be lower than 1.42%.
 | ||
| Fig. 10 Conversion and mass intensity ratio of 124/122 in hydration of 2-cyanopyridine with 18O-substituted CeO2 and CeO2 catalysts at (a) 279 K and (b) 274 K. Marks of 18O-substituted CeO2. Conversion: white circle, mass intensity ratio of 124/122: white square. Marks of CeO2. Conversion: black circle, mass intensity ratio of 124/122: black square. Reaction conditions (a): CeO2 or 18O-substituted CeO2 100 mg (0.58 mmol), 2-cyanopyridine 104 mg (1.0 mmol), H2O 5 g, 279 K. Reaction conditions (b): CeO2 or 18O-substituted CeO2 100 mg (0.58 mmol), 2-cyanopyridine 104 mg (1.0 mmol), H2O 5 g, 274 K. The detailed data are shown in Table S1.† | ||
 | ||
| Fig. 11 The relationship between the 18O/16O in picolinamide and the conversion in hydration of 2-cyanopyridine over 18O-substituted CeO2. The detailed data are shown in Table S2.† | ||
In the second experiment, we conducted the hydration of 2-cyanopyridine with H218O solvent and CeO2. If picolinamide is produced by the surface lattice oxygen of CeO2, the mass number of the produced picolinamide is 122. On the other hand, if picolinamide is produced directly by water molecules, the mass number of the produced picolinamide is 124. At first, we checked the reactivity of the hydration of 2-cyanopyridine with H2O and H218O (Fig. S5 and Table S3†), showing no significant difference between the reactions. A typical example of the mass spectrum of the produced picolinamide is shown in Fig. S6,† and the results are shown in Fig. 12 and Table S3.† The mass intensity ratio of 122/124 increased with decreasing the reaction time, and the mass number ratio at 0 min is expected to be quite high, indicating that under the conditions where TON is 1 for the surface lattice oxygen of CeO2, a quantitative amount of the amide with 16O, namely with a molecular weight of 122, was formed. Therefore, the hydration of 2-cyanopyridine proceeds with the involvement of surface lattice oxygen, and these two experimental results strongly support the reaction mechanism suggested by the DFT-MD simulations.
 | ||
| Fig. 12 Mass intensity ratio of 122/124 in hydration of 2-cyanopyridine in H218O over CeO2. Reaction conditions: CeO2 100 mg (0.58 mmol), 2-cyanopyridine 104 mg (1.0 mmol), H2O or H218O 3 g, 278 K. | ||
4. Conclusions
The detailed mechanism of the hydration of 2-cyanopyridine over CeO2 catalysts was investigated by the DFT-MD simulations. The possible reaction mechanism was discussed by depicting the free energy landscape. The adsorption of 2-cyanopyridine onto CeO2(111) is facile, leading to the stable adsorption structure of the protonated species, which is covalently bonded to the surface lattice oxygen atom. Two reaction schemes were compared from this adsorption structure, and the free energy calculations reveal that the reaction mechanism involving the surface lattice oxygen was energetically preferable with the energy barrier associated with this process being 54 kJ mol−1. The other possible mechanism is the nucleophilic attack of a hydroxide ion formed on the surface Ce atom to the C atom of the protonated nitrile species. The free energy barrier associated with this process is calculated to be 70 kJ mol−1, which is higher than the former process. The former lattice oxygen insertion mechanism is quite unique considering that this reaction proceeds readily at room temperature or below. The contribution of the surface lattice oxygen to the hydration reaction was also investigated by the two experiments using H218O solvent and 18O-exchanged CeO2 catalyst. It was confirmed that the lattice oxygen-introduced product was obtained by both experiments, which strongly supports theoretical prediction. In a related study, the oxygen vacancy-assisted mechanism over CeO2 catalyst was reported in the direct dimethyl carbonate synthesis from CO2 and methanol.64 The flexibility of surface oxygen and the unique acid–base properties of CeO2, which can accommodate substrate with the covalent bond along with acid–base interactions and activate water molecules on the surface, are the key factors in its high reactivity for various organic reactions. These unique characteristics of CeO2 will be further utilized in the development of more efficient and environmentally friendly catalysts for various organic reactions at low temperatures. In particular, this unique reactivity of the lattice oxygen of CeO2 in water is believed to be effective for hydrolysis and hydration reactions using water, making it promising not only for green reaction systems that employ water as a green solvent, but also for applications in the low-temperature hydrolysis of plastics such as polyesters and polyamides, which have recently garnered attention.Data availability
Additional data supporting this article are included in the ESI.†Author contributions
T. E. and A. N. performed calculations. Y. K. and M. T. conducted experiments. M. T. and A. N. conceived of the work. All authors contributed to the writing and proofing of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support by JSPS KAKENHI (Grant Numbers 23H05404 and 23K13071), MEXT program: Data Creation and Utilization-Type Material Research and Development Project (Grant Number JPMXP1122712807), and JST PRESTO (JPMJPR2378). Part of this work was supported by Green Innovation Fund Project (Grant Number 21502377-0) from the New Energy and Industrial Technology Development Organization (NEDO). Part of the calculations were performed on supercomputers at RIIT (Kyushu Univ.), ACCMS (Kyoto Univ.), and CCMS, IMR (Tohoku Univ., Proposal No. 202312-SCKXX-0006).References
- S. Park, J. M. Vohs and R. J. Gorte, Direct Oxidation of Hydrocarbons in a Solid-Oxide Fuel Cell, Nature, 2000, 404(6775), 265–267, DOI:10.1038/35005040.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Active Nonmetallic Au and Pt Species on Ceria-Based Water-Gas Shift Catalysts, Science, 2003, 301(5635), 935–938, DOI:10.1126/science.1085721.
- G. A. Deluga, J. R. Salge, L. D. Schmidt and X. E. Verykios, Renewable Hydrogen from Ethanol by Autothermal Reforming, Science, 2004, 303(5660), 993–997, DOI:10.1126/science.1093045.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Fundamentals and Catalytic Applications of CeO2-Based Materials, Chem. Rev., 2016, 116(10), 5987–6041, DOI:10.1021/acs.chemrev.5b00603.
- D. R. Mullins, The Surface Chemistry of Cerium Oxide, Surf. Sci. Rep., 2015, 70(1), 42–85, DOI:10.1016/j.surfrep.2014.12.001.
- M. V. Ganduglia-Pirovano, The Non-Innocent Role of Cerium Oxide in Heterogeneous Catalysis: A Theoretical Perspective, Catal. Today, 2015, 253, 20–32, DOI:10.1016/j.cattod.2015.01.049.
- C. T. Campbell, Electronic Perturbations, Nat. Chem., 2012, 4(8), 597–598, DOI:10.1038/nchem.1412.
- A. Bruix, J. A. Rodriguez, P. J. Ramírez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, A New Type of Strong Metal–Support Interaction and the Production of H2 through the Transformation of Water on Pt/CeO2(111) and Pt/CeOx/TiO2(110) Catalysts, J. Am. Chem. Soc., 2012, 134(21), 8968–8974, DOI:10.1021/ja302070k.
- Y. Lykhach, S. M. Kozlov, T. Skála, A. Tovt, V. Stetsovych, N. Tsud, F. Dvořák, V. Johánek, A. Neitzel, J. Mysliveček, S. Fabris, V. Matolín, K. M. Neyman and J. Libuda, Counting Electrons on Supported Nanoparticles, Nat. Mater., 2016, 15(3), 284–288, DOI:10.1038/nmat4500.
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Hydrodeoxygenation of Vicinal OH Groups over Heterogeneous Rhenium Catalyst Promoted by Palladium and Ceria Support, Angew. Chem., Int. Ed., 2015, 54(6), 1897–1900, DOI:10.1002/anie.201410352.
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Performance, Structure, and Mechanism of ReOx–Pd/CeO2 Catalyst for Simultaneous Removal of Vicinal OH Groups with H2, ACS Catal., 2016, 6(5), 3213–3226, DOI:10.1021/acscatal.6b00491.
- S. Tazawa, N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Deoxydehydration with Molecular Hydrogen over Ceria-Supported Rhenium Catalyst with Gold Promoter, ACS Catal., 2016, 6(10), 6393–6397, DOI:10.1021/acscatal.6b01864.
- J. Cao, M. Tamura, R. Hosaka, A. Nakayama, J. Hasegawa, Y. Nakagawa and K. Tomishige, Mechanistic Study on Deoxydehydration and Hydrogenation of Methyl Glycosides to Dideoxy Sugars over a ReOx–Pd/CeO2 Catalyst, ACS Catal., 2020, 10(20), 12040–12051, DOI:10.1021/acscatal.0c02309.
- M. Tamura, N. Hayashigami, A. Nakayama, Y. Nakagawa and K. Tomishige, Heterogeneous Enantioselective Hydrogenation of Ketones by 2-Amino-2’-Hydroxy-1,1’binaphthyl-Modified CeO2-Supported Ir Nanoclusters, ACS Catal., 2022, 12(2), 868–876, DOI:10.1021/acscatal.1c04427.
- L. Vivier and D. Duprez, Ceria-Based Solid Catalysts for Organic Chemistry, ChemSusChem, 2010, 3(6), 654–678, DOI:10.1002/cssc.201000054.
- S. Sato, F. Sato, H. Gotoh and Y. Yamada, Selective Dehydration of Alkanediols into Unsaturated Alcohols over Rare Earth Oxide Catalysts, ACS Catal., 2013, 3(4), 721–734, DOI:10.1021/cs300781v.
- M. Tamura, K. Shimizu and A. Satsuma, CeO2-Catalyzed Transformations of Nitriles and Amides, Chem. Lett., 2012, 41(11), 1397–1405, DOI:10.1246/cl.2012.1397.
- M. Tamura, A. Satsuma and K. Shimizu, CeO2-Catalyzed Nitrile Hydration to Amide: reaction Mechanism and Active Sites, Catal. Sci. Technol., 2013, 3(5), 1386–1393, 10.1039/c3cy00033h.
- M. Tamura, K. Sawabe, K. Tomishige, A. Satsuma and K. Shimizu, Substrate-Specific Heterogeneous Catalysis of CeO2 by Entropic Effects via Multiple Interactions, ACS Catal., 2015, 5(1), 20–26, DOI:10.1021/cs501448n.
- M. Honda, M. Tamura, Y. Nakagawa, S. Sonehara, K. Suzuki, K. Fujimoto and K. Tomishige, Ceria-Catalyzed Conversion of Carbon Dioxide into Dimethyl Carbonate with 2-Cyanopyridine, ChemSusChem, 2013, 6(8), 1341–1344, DOI:10.1002/cssc.201300229.
- K. Tomishige, M. Tamura and Y. Nakagawa, CO2 Conversion with Alcohols and Amines into Carbonates, Ureas, and Carbamates over CeO2 Catalyst in the Presence and Absence of 2-Cyanopyridine, Chem. Rec., 2019, 19(7), 1354–1379, DOI:10.1002/tcr.201800117.
- M. Tamura, Y. Nakagawa and K. Tomishige, Direct CO2 Transformation to Aliphatic Polycarbonates, Asian J. Org. Chem., 2022, 11(11), e202200445, DOI:10.1002/ajoc.202200445.
- M. Tamura, R. Kishi, A. Nakayama, Y. Nakagawa, J. Hasegawa and K. Tomishige, Formation of a New, Strongly Basic Nitrogen Anion by Metal Oxide Modification, J. Am. Chem. Soc., 2017, 139(34), 11857–11867, DOI:10.1021/jacs.7b05227.
- M. Tamura, D. Hiwatashi, Y. Gu, A. Nakayama, Y. Nakagawa and K. Tomishige, Organic Compound Modification of CeO2 and 2-Cyanopyridine Hybrid Catalyst in Carbonate Synthesis from CO2 and Alcohols, J. CO2 Util., 2021, 54, 101744, DOI:10.1016/j.jcou.2021.101744.
- M. Tamura, E. Sagawa, A. Nakayama, Y. Nakagawa and K. Tomishige, Hydrogen Atom Abstraction by Heterogeneous–Homogeneous Hybrid Catalyst of CeO2 and 2-Cyanopyridine via Redox of CeO2 for C–H Bond Oxidation with Air, ACS Catal., 2021, 11(19), 11867–11872, DOI:10.1021/acscatal.1c03757.
- W. Sun, P. Li, M. Yabushita, Y. Nakagawa, Y. Wang, A. Nakayama and K. Tomishige, Comparative Study between 2-Furonitrile and 2-Cyanopyridine as Dehydrants in Direct Synthesis of Dialkyl Carbonates from CO2 and Alcohols over Cerium Oxide Catalyst, ChemSusChem, 2023, 16(24), e202300768, DOI:10.1002/cssc.202300768.
- T. Seki, K. Takeda and A. Nakayama, Ab Initio Molecular Dynamics Simulations for Adsorption Structures of Picolinic Acid at the Water/CeO2 Interface, J. Phys. Chem. C, 2022, 126(7), 3404–3410, DOI:10.1021/acs.jpcc.1c10005.
- M. Farnesi Camellone, F. Negreiros Ribeiro, L. Szabová, Y. Tateyama and S. Fabris, Catalytic Proton Dynamics at the Water/Solid Interface of Ceria-Supported Pt Clusters, J. Am. Chem. Soc., 2016, 138(36), 11560–11567, DOI:10.1021/jacs.6b03446.
- Z. Ren, N. Liu, B. Chen, J. Li and D. Mei, Theoretical Investigation of the Structural Stabilities of Ceria Surfaces and Supported Metal Nanocluster in Vapor and Aqueous Phases, J. Phys. Chem. C, 2018, 122(9), 4828–4840, DOI:10.1021/acs.jpcc.7b10208.
- L. Agosta, D. Arismendi-Arrieta, M. Dzugutov and K. Hermansson, Origin of the Hydrophobic Behaviour of Hydrophilic CeO2, Angew. Chem., Int. Ed., 2023, 62(35), e202303910, DOI:10.1002/anie.202303910.
- T. Kobayashi, T. Ikeda and A. Nakayama, Long-Range Proton and Hydroxide Ion Transfer Dynamics at the Water/CeO2 Interface in the Nanosecond Regime: Reactive Molecular Dynamics Simulations and Kinetic Analysis, Chem. Sci., 2024, 15, 6816–6832, 10.1039/D4SC01422G.
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, CP2K: An Electronic Structure and Molecular Dynamics Software Package - Quickstep: Efficient and Accurate Electronic Structure Calculations, J. Chem. Phys., 2020, 152(19), 194103, DOI:10.1063/5.0007045.
- J. VandeVondele and J. Hutter, Gaussian Basis Sets for Accurate Calculations on Molecular Systems in Gas and Condensed Phases, J. Chem. Phys., 2007, 127(11), 114105, DOI:10.1063/1.2770708.
- S. Goedecker, M. Teter and J. Hutter, Separable Dual-Space Gaussian Pseudopotentials, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54(3), 1703–1710, DOI:10.1103/PhysRevB.54.1703.
- C. Hartwigsen, S. Goedecker and J. Hutter, Relativistic Separable Dual-Space Gaussian Pseudopotentials from H to Rn, Phys. Rev. B:Condens. Matter Mater. Phys., 1998, 58(7), 3641–3662, DOI:10.1103/PhysRevB.58.3641.
- Y.-G. Wang, D. Mei, J. Li and R. Rousseau, DFT + U Study on the Localized Electronic States and Their Potential Role During H2O Dissociation and CO Oxidation Processes on CeO2(111) Surface, J. Phys. Chem. C, 2013, 117(44), 23082–23089, DOI:10.1021/jp409953u.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Electron Localization Determines Defect Formation on Ceria Substrates, Science, 2005, 309(5735), 752–755, DOI:10.1126/science.1111568.
- J. Paier, C. Penschke and J. Sauer, Oxygen Defects and Surface Chemistry of Ceria: Quantum Chemical Studies Compared to Experiment, Chem. Rev., 2013, 113(6), 3949–3985, DOI:10.1021/cr3004949.
- M. Nolan, V. S. Verdugo and H. Metiu, Vacancy Formation and CO Adsorption on Gold-Doped Ceria Surfaces, Surf. Sci., 2008, 602(16), 2734–2742, DOI:10.1016/j.susc.2008.06.028.
- H. Nörenberg and G. A. D. Briggs, Defect Formation on CeO2(111) Surfaces after Annealing Studied by STM, Surf. Sci., 1999, 424(2–3), L352–L355, DOI:10.1016/S0039-6028(99)00212-5.
- M. Nolan, S. C. Parker and G. W. Watson, The Electronic Structure of Oxygen Vacancy Defects at the Low Index Surfaces of Ceria, Surf. Sci., 2005, 595(1–3), 223–232, DOI:10.1016/j.susc.2005.08.015.
- X. Huang, K. Zhang, B. Peng, G. Wang, M. Muhler and F. Wang, Ceria-Based Materials for Thermocatalytic and Photocatalytic Organic Synthesis, ACS Catal., 2021, 11(15), 9618–9678, DOI:10.1021/acscatal.1c02443.
- L. Tian, Y.-S. Liao, Z. Xiao, G. Sun, J.-P. Chou, C.-Y. Wong, J. C. Ho, Y. Zhao, P.-T. Chou and Y.-K. Peng, Unveiling the Pivotal Role of Ce Coordination Structures and Their Surface Arrangements in Governing 2-Cyanopyridine Hydrolysis for Direct Dimethyl Carbonate Synthesis from CO2 and Methanol, ACS Catal., 2024, 14(22), 16861–16871, DOI:10.1021/acscatal.4c04639.
- L. Gerward and J. S. Olsen, Powder Diffraction Analysis of Cerium Dioxide at High Pressure, Powder Diffr., 1993, 8(2), 127–129, DOI:10.1017/S0885715600017966.
- A. Nakajima, A. Yoshihara and M. Ishigame, Defect-Induced Raman Spectra in Doped CeO2, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50(18), 13297–13307, DOI:10.1103/PhysRevB.50.13297.
- M. E. Tuckerman, Statistical Mechanics, Oxford Graduate Texts, Oxford University Press, Incorporated, Oxford, 2nd edn, 2023 Search PubMed.
- A. Laio and M. Parrinello, Escaping Free-Energy Minima, Proc. Natl. Acad. Sci. U.S.A., 2002, 99(20), 12562–12566, DOI:10.1073/pnas.202427399.
- A. Barducci, G. Bussi and M. Parrinello, Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method, Phys. Rev. Lett., 2008, 100(2), 020603, DOI:10.1103/PhysRevLett.100.020603.
- G. M. Torrie and J. P. Valleau, Monte Carlo Free Energy Estimates Using Non-Boltzmann Sampling: Application to the Sub-Critical Lennard-Jones Fluid, Chem. Phys. Lett., 1974, 28(4), 578–581, DOI:10.1016/0009-2614(74)80109-0.
- F. Wang and D. P. Landau, Efficient, Multiple-Range Random Walk Algorithm to Calculate the Density of States, Phys. Rev. Lett., 2001, 86(10), 2050–2053, DOI:10.1103/PhysRevLett.86.2050.
- Y. I. Yang, J. Zhang, X. Che, L. Yang and Y. Q. Gao, Efficient Sampling over Rough Energy Landscapes with High Barriers: A Combination of Metadynamics with Integrated Tempering Sampling, J. Chem. Phys., 2016, 144(9), 094105, DOI:10.1063/1.4943004.
- J. Comer, J. C. Gumbart, J. Hénin, T. Lelièvre, A. Pohorille and C. Chipot, The Adaptive Biasing Force Method: Everything You Always Wanted To Know but Were Afraid To Ask, J. Phys. Chem. B, 2015, 119(3), 1129–1151, DOI:10.1021/jp506633n.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers, SoftwareX, 2015, 1–2, 19–25, DOI:10.1016/j.softx.2015.06.001.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, The Weighted Histogram Analysis Method for Free-energy Calculations on Biomolecules. I. The Method, J. Comput. Chem., 1992, 13(8), 1011–1021, DOI:10.1002/jcc.540130812.
- A. Grossfield, WHAM: The Weighted Histogram Analysis Method, Version 2.0.11, http://membrane.urmc.rochester.edu/?page_id=126.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, PLUMED 2: New Feathers for an Old Bird, Comput. Phys. Commun., 2014, 185(2), 604–613, DOI:10.1016/j.cpc.2013.09.018.
- H. Fu, H. Chen, X. Wang, H. Chai, X. Shao, W. Cai and C. Chipot, Finding an Optimal Pathway on a Multidimensional Free-Energy Landscape, J. Chem. Inf. Model., 2020, 60(11), 5366–5374, DOI:10.1021/acs.jcim.0c00279.
- J. L. Vincent and P. A. Crozier, Atomic Level Fluxional Behavior and Activity of CeO2-Supported Pt Catalysts for CO Oxidation, Nat. Commun., 2021, 12(1), 5789, DOI:10.1038/s41467-021-26047-8.
- M. Tamura and K. Tomishige, Redox Properties of CeO2 at Low Temperature: The Direct Synthesis of Imines from Alcohol and Amine, Angew. Chem., Int. Ed., 2015, 54(3), 864–867, DOI:10.1002/anie.201409601.
- B. Guo, J. G. De Vries and E. Otten, Hydration of Nitriles Using a Metal–Ligand Cooperative Ruthenium Pincer Catalyst, Chem. Sci., 2019, 10(45), 10647–10652, 10.1039/C9SC04624K.
- S. Yadav and R. Gupta, Hydration of Nitriles Catalyzed by Ruthenium Complexes: Role of Dihydrogen Bonding Interactions in Promoting Base-Free Catalysis, Inorg. Chem., 2022, 61(39), 15463–15474, DOI:10.1021/acs.inorgchem.2c02058.
- D. Prendergast, J. C. Grossman and G. Galli, The Electronic Structure of Liquid Water within Density-Functional Theory, J. Chem. Phys., 2005, 123(1), 014501, DOI:10.1063/1.1940612.
- M. J. Gillan, D. Alfè and A. Michaelides, Perspective: How Good Is DFT for Water?, J. Chem. Phys., 2016, 144(13), 130901, DOI:10.1063/1.4944633.
- D. Stoian, T. Sugiyama, A. Bansode, F. Medina, W. Van Beek, J. Hasegawa, A. Nakayama and A. Urakawa, Dimethyl Carbonate Synthesis from CO2 and Methanol over CeO2: Elucidating the Surface Intermediates and Oxygen Vacancy-Assisted Reaction Mechanism, Chem. Sci., 2023, 14(47), 13908–13914, 10.1039/D3SC04466A.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison with 4-cyanopyridine; hydration of 2-cyanopyridine in the bulk water; experimental details. See DOI: https://doi.org/10.1039/d4sc06294a |
| This journal is © The Royal Society of Chemistry 2025 |