
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible formation of tetraphenylpentalene, a room temperature stable antiaromatic hydrocarbon†
Hugh J.
Sanderson
 a,
Andreas
Helbig
b,
Gabriele
Kociok-Köhn
c,
Holger
Helten
*b and
Ulrich
Hintermair
*a
a,
Andreas
Helbig
b,
Gabriele
Kociok-Köhn
c,
Holger
Helten
*b and
Ulrich
Hintermair
*a
aDepartment of Chemistry and Institute for Sustainability, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: u.hintermair@bath.ac.uk
bJulius-Maximilians-Universität Würzburg, Institute of Inorganic Chemistry and Institute for Sustainable Chemistry & Catalysis with Boron, Am Hubland, 97074 Würzburg, Germany. E-mail: holger.helten@uni-wuerzburg.de
cChemical Characterisation Facility, University of Bath, Claverton Down, Bath BA2 7AY, UK
First published on 25th November 2024
Abstract
1,3,4,6-Tetraphenylpentalene (Ph4Pn) has been synthesised by chemical oxidation of the corresponding pentalenide complex Mg[Ph4Pn] with iodine. Ph4Pn is a rare example of a room-temperature stable hydrocarbon that is antiaromatic by Hückel's rule and has been fully characterised by NMR and UV-vis spectroscopy, mass spectrometry as well as single-crystal X-ray diffraction. Quantum chemical studies including nucleus-independent chemical shift (NICS) and anisotropy of the induced current density (ACID) calculations showed the existence of an 8π antiaromatic core decorated with four independent 6π aromatic substituents. The formation of Ph4Pn is reversible and it can be reduced back to the 10π aromatic Ph4Pn2− with potassium.
1. Introduction
Aromaticity, and by extension antiaromaticity, are core concepts in chemistry with different classifications.1 The most familiar is Hückel aromaticity, embodied by the 4n + 2 π-electron rule developed by Doering and Detert,2 which is used for planar closed-shell monocyclic conjugated systems.3 The opposite, Hückel antiaromaticity, is termed for molecules that have a 4n π-electron count yet satisfy the rest of the criteria.4 Whereas aromaticity serves to provide a molecule with increased stability, antiaromaticity imparts destabilisation to the molecule along with other contrasting spectroscopic and magnetic properties (such as paratropic ring currents).5,6 The concept of (anti)aromaticity was later extended to open-shell systems by Baird,7 who showed that molecules with a 4n π-electron count display aromaticity in the triplet state whilst those with a formal 4n + 2 π-electron count exhibit antiaromatic behaviour in the triplet state.8 Other types of (anti)aromaticity have also been reported,1 and mechanisms for electron delocalisation are known beyond π-conjugation.9–13Compared to the interest in the different forms of aromaticity,5 less attention has been paid to antiaromatic molecules and the properties they can possess. Hückel antiaromaticity can result in a lowering of the HOMO–LUMO gap14 and the presence of a low-lying triplet state which may exhibit Baird aromaticity.4,7,8,15,16 Thus, molecules possessing antiaromaticity have found use in areas such as optoelectronics, for example.17–20 However, the destabilisation that arises from a fully conjugated 4n π-electron system often results in highly unstable molecules that react readily to relieve the antiaromatic strain – e.g. through dimerisation or by adoption of non-planar conformations.21 The idea of concealed antiaromaticity has been proposed as a framework with which to identify the structural motifs used to stabilise antiaromatic systems, such that upon oxidation/reduction or photoexcitation aromatic systems may be formed.22 Such motifs presented include sharing of 2π or 4π-electron fragments by a locally aromatic group to the 4n π-system and internal connections (e.g. C–C bonds) within the 4n π-system itself.
A classic example of a 4n π-system that alleviates, or conceals, its antiaromaticity is cyclooctatetraene (C8H8, COT) which adopts a non-planar boat conformation thus avoiding π-conjugation and formation of antiaromatic ring currents (Scheme 1). Pentalene (C8H6, Pn) is an 8 π-electron hydrocarbon related to COTvia a transannular ring closure23 that forces it to adopt a coplanar structure and thus yields a Hückel antiaromatic system. As a consequence Pn dimerises via a [2 + 2] cycloaddition pathway (c.f. cyclobutadiene) above −196 °C to form a non-aromatic pentafulvene-type system.24,25 This process indicates the presence of a low-lying triplet state since such a reaction is believed to proceed via diradical intermediates.26 The increased rigidity imparted by the transannular bond also prevents conformational rearrangements that are often seen when switching between aromatic and antiaromatic states of larger molecules.27–34 However, to date no crystallographic data of a pentalene and its corresponding pentalenide have been reported to compare the extent of any changes.
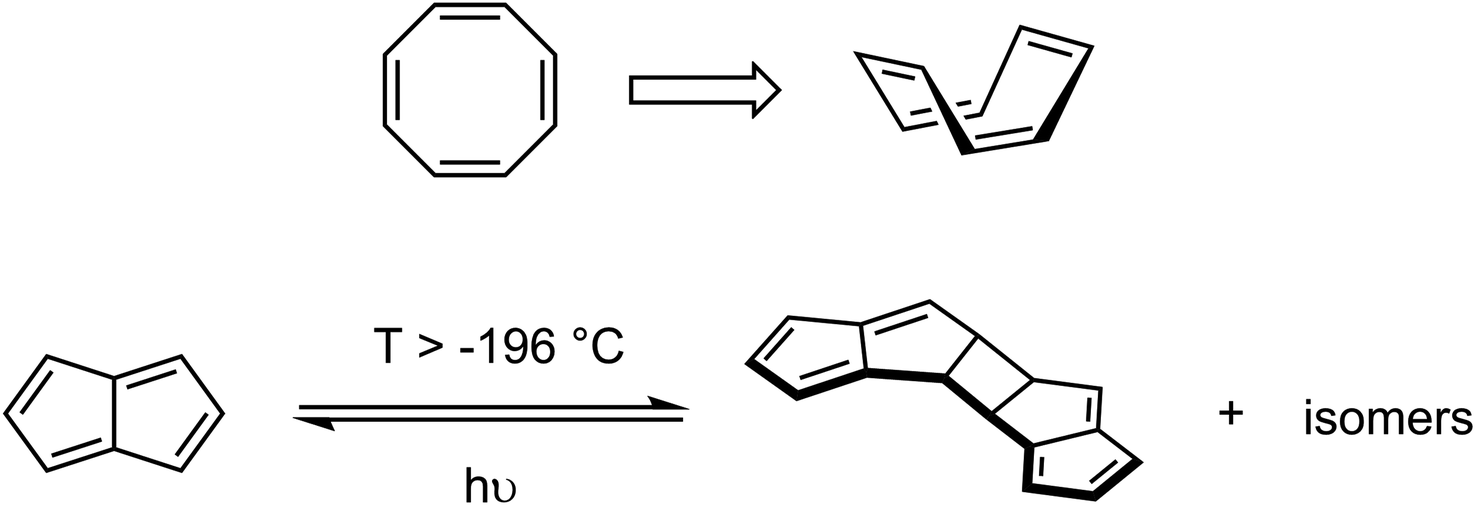 | ||
| Scheme 1 Examples of carbocyclic 8π systems: non-planar conformation of COT (top) and dimerization of Pn (bottom). | ||
The most common method to stabilise (or conceal) the antiaromatic character of pentalene is through annelation with other π systems such as in benzopentalenes, with the first dibenzopentalene reported in 1912, nearly 50 years prior to the first report of an isolable Pn.35,36 Conjugation of the pentalene to locally aromatic benzene groups imparts stability onto the antiaromatic core and renders dibenzopentalenes to be bench-stable despite being formally Hückel antiaromatic 4n π-electron systems.16,18,37–39 Annelation can also be used to vary the degree of (anti)aromaticity, as shown for dinaphtho[2,1,a,f]pentalene,40 an effect which is useful for tuning the electronics of polycyclic pentalene derivatives for potential use in organic semiconductors.41–43
However, reports of stable Pn derivatives that do not rely on annelation with aromatic groups are sparse (Fig. 1).44–50 1,3,5-tBu3Pn was the first Pn derivative to be characterised by single crystal XRD which confirmed the coplanarity of the two rings in the Pn core and showed bond alternation consistent with localised C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.51 The stabilisation of 1,3,5-tBu3Pn arises from the steric bulk of the tBu substituents providing a high kinetic barrier that prevents the [2 + 2] cycloaddition from occurring; the less sterically hindered derivatives 1,3-tBu2-5-RPn (R = CO2Me, CHO or CN) were found to exist in equilibrium with their [2 + 2] dimers.49 1,2,3,4,5,6-hexamethylpentalene (Pn*) avoids forming an antiaromatic π-system through an exocyclic CC bond which results in the Pn core not being fully sp2 hybridised.44 The corresponding [2 + 2] dimer could be accessed through oxidation of the dianion Pn*2− by FeCl2.52 Another hexasubstituted Pn, an aminopentalenecarbonitrile, was reported by Hartke where the opposing push–pull effects of the amino and nitrile groups were proposed to electronically stabilise the antiaromatic core.45 A thienylpentalene has been reported where the aromatic heterocycle stabilises the antiaromaticity of the core as in benzopentalenes. In the same report a hexaarylated Pn derivative bearing p-tolyl substituents was described.50 The hexasubstitution pattern is likely to impart a significant steric barrier to the [2 + 2] dimerisation, however, the electronic influence of the aromatic tolyl-substituents on the Pn core is unknown. More recently a tetra-substituted Pn with cyclopropyl groups was described, however, the cyclo-propyl groups did not impart enough steric or electronic stabilisation to prevent [2 + 2] dimerisation.53 Le Goff reported the synthesis of hexaphenylpentalene from the base-catalysed condensation of a cyclopentadiene with an α,β-unsaturated carbonyl compound followed by oxidation of the resultant dihydropentalene.36 Whilst the synthetic rationale of forming substituted PnH2 from cyclopentadienes and α,β-unsaturated carbonyls is a well-established pathway,54–58 the subsequent oxidation of PnH2 to Pn is less well known. Le Goff also noted that a sample of Li2[Ph6Pn] could be oxidised by addition of I2 to yield Ph6Pn,36 a route that in theory should allow facile formation of Pn derivatives where the only by-product would be an inorganic salt. Recently we reported the synthesis of the first alkaline earth pentalenide Mg[Ph4Pn].59 Herein we report the remarkably facile and fully reversible oxidation of Mg[Ph4Pn] to Ph4Pn which has been fully characterised by NMR and UV-vis spectroscopy as well as X-ray diffraction. The antiaromaticity of the Pn core and the steric and electronic influence of the phenyl substituents have been investigated by computational studies.
C bonds.51 The stabilisation of 1,3,5-tBu3Pn arises from the steric bulk of the tBu substituents providing a high kinetic barrier that prevents the [2 + 2] cycloaddition from occurring; the less sterically hindered derivatives 1,3-tBu2-5-RPn (R = CO2Me, CHO or CN) were found to exist in equilibrium with their [2 + 2] dimers.49 1,2,3,4,5,6-hexamethylpentalene (Pn*) avoids forming an antiaromatic π-system through an exocyclic CC bond which results in the Pn core not being fully sp2 hybridised.44 The corresponding [2 + 2] dimer could be accessed through oxidation of the dianion Pn*2− by FeCl2.52 Another hexasubstituted Pn, an aminopentalenecarbonitrile, was reported by Hartke where the opposing push–pull effects of the amino and nitrile groups were proposed to electronically stabilise the antiaromatic core.45 A thienylpentalene has been reported where the aromatic heterocycle stabilises the antiaromaticity of the core as in benzopentalenes. In the same report a hexaarylated Pn derivative bearing p-tolyl substituents was described.50 The hexasubstitution pattern is likely to impart a significant steric barrier to the [2 + 2] dimerisation, however, the electronic influence of the aromatic tolyl-substituents on the Pn core is unknown. More recently a tetra-substituted Pn with cyclopropyl groups was described, however, the cyclo-propyl groups did not impart enough steric or electronic stabilisation to prevent [2 + 2] dimerisation.53 Le Goff reported the synthesis of hexaphenylpentalene from the base-catalysed condensation of a cyclopentadiene with an α,β-unsaturated carbonyl compound followed by oxidation of the resultant dihydropentalene.36 Whilst the synthetic rationale of forming substituted PnH2 from cyclopentadienes and α,β-unsaturated carbonyls is a well-established pathway,54–58 the subsequent oxidation of PnH2 to Pn is less well known. Le Goff also noted that a sample of Li2[Ph6Pn] could be oxidised by addition of I2 to yield Ph6Pn,36 a route that in theory should allow facile formation of Pn derivatives where the only by-product would be an inorganic salt. Recently we reported the synthesis of the first alkaline earth pentalenide Mg[Ph4Pn].59 Herein we report the remarkably facile and fully reversible oxidation of Mg[Ph4Pn] to Ph4Pn which has been fully characterised by NMR and UV-vis spectroscopy as well as X-ray diffraction. The antiaromaticity of the Pn core and the steric and electronic influence of the phenyl substituents have been investigated by computational studies.
 | ||
| Fig. 1 Previously reported stable pentalene derivatives and their means of characterisation. | ||
2. Results and discussion
The addition of one equivalent of iodine to a THF solution of Mg[Ph4Pn] at room temperature led to an immediate colour change from bright orange to dark yellow, with a precipitate of MgI2 forming after a few minutes. NMR spectroscopic analysis of the filtered solution showed the complete and selective formation of Ph4Pn (Fig. 2). When either an excess of iodine or an equivalent of bromine was used instead, an unidentifiable mixture of products originating from unselective (over)oxidation was observed. This difference in reactivity can be attributed to the increased oxidation power of Br2 over I2 (E0 = 1.07 V for Br2; E0 = 0.54 V for I2)60 and its propensity to undergo bromination reactions. Previous attempts to oxidise Ph4PnH2 or Ph4Pn2− to Ph4Pn with N-bromosuccinimide were unsuccessful,61 although Cu(II), which has a lower oxidation potential (E0 = 0.34 V) than iodine, has previously been reported to facilitate the oxidative coupling of Li2[Pn] to afford [Pn]2.25,62 | ||
| Fig. 2 Synthesis of 1,3,4,6-Ph4Pn (top) and its 500 MHz room temperature 1H NMR spectrum in THF-H8 (bottom). | ||
The room temperature 1H NMR spectrum of Ph4Pn showed a C2h symmetrical molecule with three well-resolved, diamagnetic signals observed at 7.14, 7.05 and 5.44 ppm (Fig. 2). The chemical shift of the two equivalent wingtip protons Hw in Ph4Pn was found to be 1.5 ppm upfield relative to Ph4Pn2− (Table 1), and the corresponding wingtip carbons Cw had a resonance at 134.9 ppm, a 20 ppm downfield shift relative to the respective signal in Ph4Pn2−. These shifts are consistent with formation of a localised olefinic π-system arising from an 8π antiaromatic pentalene and indicate full oxidation of the core of Ph4Pn2− to Ph4Pn, with the four equivalent phenyl groups remaining largely unperturbed. The cleanliness of the crude 1H NMR spectrum also indicates that in solution there is no significant [2 + 2] dimer formation. Variable temperature NMR studies from −80 °C to 60 °C in THF showed no discernible changes (Fig. S4†), indicating monomeric Ph4Pn to be remarkably stable under inert conditions, although exposure to air quickly led to decomposition.
| δ 1Hwa (13Cw)/ppm | C–C perimeter/Å | C–C bridge/Å | Aryl twist angle/° | |
|---|---|---|---|---|
| a Data for Ph4PnH2 in acetone-d6, Mg[Ph4Pn] and Ph4Pn in THF-H8. | ||||
| Ph4PnH2 | 7.45 (139.4) | 1.357(6)–1.512(8) | 1.448(7) | 43.3–47.0 |
| Mg[Ph4Pn] | 6.80 (115.5) | 1.406(3)–1.455(3) | 1.451(3) | 17.1–35.2 |
| Ph4Pn | 5.42 (134.9) | 1.388(2)–1.464(3) | 1.450(3) | 31.7–42.3 |
| [Ph4Pn]2 | n/a | 1.357(2)–1.517(2) | 1.471(2), 1.476(2) | 11.9–47.3 |
XRD analysis of single crystals grown from THF at −35 °C confirmed the structure of Ph4Pn with retention of a bicyclic, co-planar Pn core (Fig. 3). Bond alternation within the sp2 perimeter was evident by C–C distances within the Pn ring ranging from 1.388(2)–1.464(3) Å. For comparison, the average C–C bond length in Mg[Ph4Pn] was found to range between 1.406(3)–1.455(3) Å (Table 1).59 The CC bond lengths in Ph4Pn were longer than in the non-aromatic Ph4PnH2 (1.36(1)–1.37(1) Å)57 in reflection of its antiaromatic character. The C–C bridgehead bond length of 1.450(3) Å was identical to the equivalent distance in Mg[Ph4Pn] (1.451(3) Å), suggesting the electronics of the Pn core do not significantly influence this bond. The C–C/CC bond alternation found in Ph4Pn was less pronounced than in 1,3,5-tBu3Pn where the C–C bonds ranged from 1.46–1.54 Å and the CC bonds between 1.28–1.41 Å, which is likely a consequence of the different electronic nature of the substituents on the pentalene core (alkyl versus aryl).51 No stacking of Ph4Pn was observed in the solid or solution state, ruling out the formation of a so-called “3D aromatic” system as a means of alleviating the antiaromatic 8π count.64–66
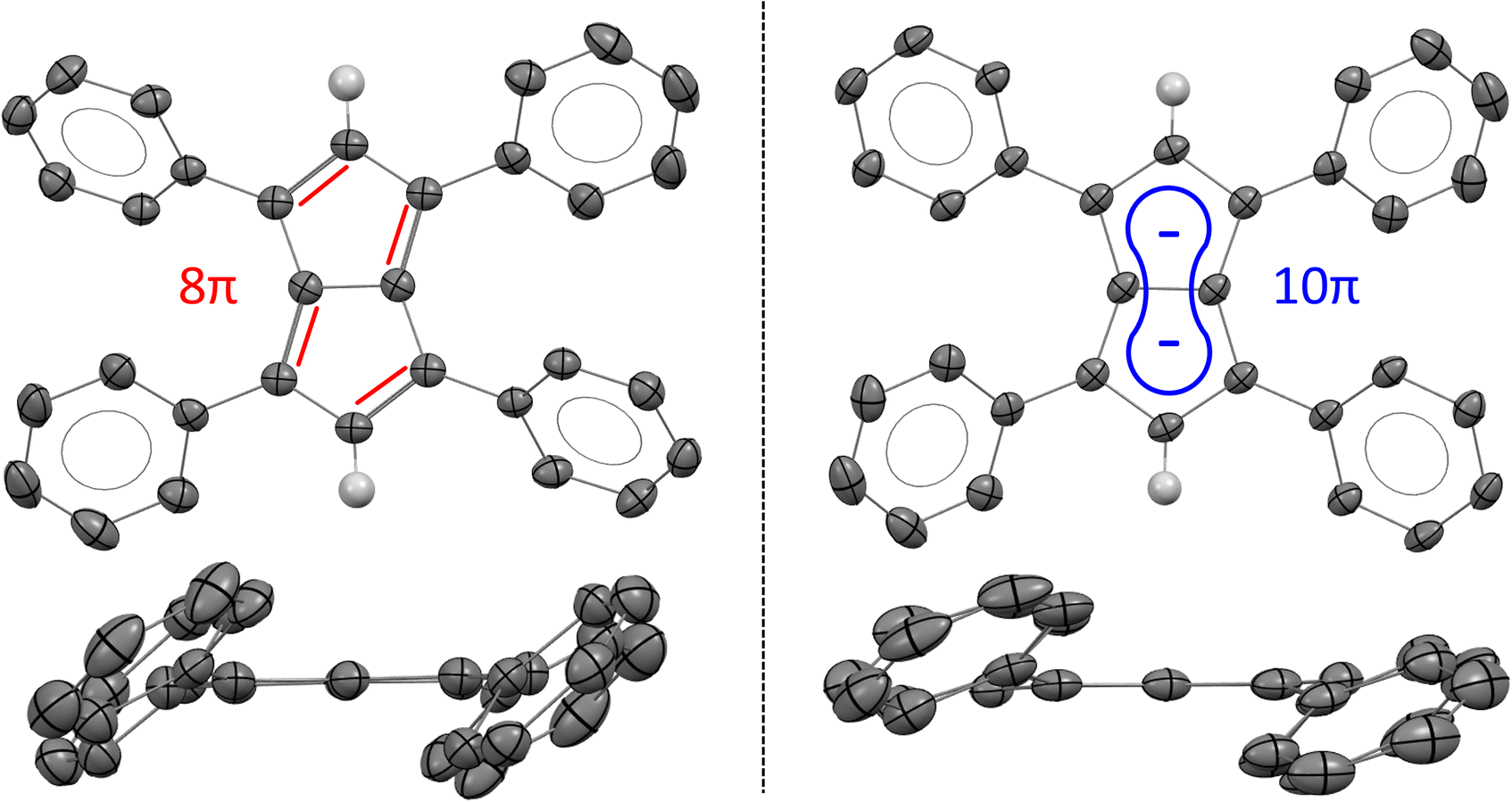 | ||
| Fig. 3 X-ray crystal structures of 1,3,4,6-Ph4Pn (left; top and side views) and 1,3,4,6-Ph4Pn2− (right; top and side views)63 with thermal ellipsoids at the 50% probability level. Phenyl hydrogen atoms omitted for clarity, and double bonds and aromatic conjugation added for illustration. | ||
As in Mg[Ph4Pn] and Ph4PnH2, the phenyl substituents in Ph4Pn were twisted versus the plane of the Pn core by 31.7(3)–42.3(3) Å.57,59 This value was larger than in Mg[Ph4Pn] but marginally less than the non-aromatic Ph4PnH2 (Table 1). In Mg[Ph4Pn] the phenyl groups strive to adopt a co-planar arrangement to delocalise the negative charge as much as sterically possible,67 whilst in Ph4Pn and Ph4PnH2 there is no charge to distribute and so the phenyl groups likely relax in a slightly larger twist angle to minimise steric clash.
Serendipitously, the [2 + 2] cycloaddition dimer [Ph4Pn]2 was found to co-crystallise with Ph4Pn in the same unit cell. In this structure two Pn units were connected via a cyclobutane bridge in an extended chair-like conformation (Fig. 4). The [2 + 2] cycloaddition means that [Ph4Pn]2 is formally non-aromatic with each 6π pentafulvene units containing two adjacent sp3 centres. The CC bond lengths in [Ph4Pn]2 ranged between 1.357(2)–1.374(2) Å and are contracted relative to the anti-aromatic Ph4Pn (Table 1), more reminiscent of those found for the non-aromatic Ph4PnH2 which [Ph4Pn]2 is most closely related to.57 The sp3 bonds within the cyclobutane unit were significantly longer than the other sp2 bonds in the dimer, with bond lengths of 1.567(2)–1.675(2) Å. Sun and co-workers reported similar, but marginally shorter, bond lengths of the cyclobutane linkage (1.56–1.60 Å) for their [cPr4Pn]2.53 O'Hare and co-workers reported that reaction of cis-(Me3Sn)2Pn* with FeCl2 resulted in oxidation of the pentalenide followed by dimerisation, however, no crystallographic data are available for [Pn*]2.52
 | ||
| Fig. 4 X-ray crystal structure of [1,3,4,6-Ph4Pn]2 with thermal ellipsoids at the 50% probability level (phenyl hydrogen atoms omitted for clarity). | ||
Geometry optimisation of Ph4Pn at the B3LYP/6-311++g(d,p) level of theory in the gas phase gave good agreement with the crystallographic data, including the 39.2–44.5° twist angle of the phenyl groups. This suggests the origin of the rotation not to be due to packing effects in the solid state, but to arise so as to avoid overlap of the aromatic (phenyl groups) and antiaromatic (pentalene core) ring currents. The calculated C–C perimeter range of 1.37–1.48 Å for the pentalene core and 1.45 Å for the C–C bridge also matched well with the crystallographic data.
To investigate the extent of antiaromaticity in Ph4Pn, harmonic oscillator model of aromaticity (HOMA) calculations were performed using the experimental X-ray data, alongside DFT calculations of the anisotropy of the induced current density (ACID) and calculations on the nucleus independent chemical shift (NICS) along different axes of the molecule (Fig. 5). A HOMAB value of 0.28 was found for the pentalene core, with the phenyl groups returning a value of 0.99 indicating no overlap of the two π-systems. The lack of overlap is also reflected in the NICS scan along the Z-axis through the centre of one phenyl group. This returned a value for the out-of-plane component of the chemical shift of −26.34 ppm at Z = 0.7 Å, which is very close to that of unsubstituted benzene (Fig. S6†).68 In contrast to the dianionic tetraphenylpentalenide,67 the ESP map of neutral Ph4Pn showed an equally distributed charge density over the pentalene core and the flanking phenyl groups (Fig. S10†). The aromatic pentalenide core of Ph4Pn2− gave a HOMAB value of 0.63, showing a clear change in aromaticity between the 8π and 10π versions of the molecule. The ACID plot of Ph4Pn showed a strong paratropic ring current about the perimeter of the pentalene core with a significant contribution from the transannular C–C bridge. The diatropic ring currents of the four aromatic phenyl substituents were clearly separated and essentially unperturbed by the presence of the anti-aromatic core, as indicated by the corresponding HOMAB values. The NICS scan along the Z-axis, starting from the centre of one 5-membered ring running perpendicular to the pentalene plane, showed features characteristic of an antiaromatic π system (Fig. 5). The isotropic shift values were positive throughout the scan, with a maximum of 24.15 ppm at Z = 0.4 Å, and the shape of the curve was strongly dominated by the out-of-plane component of the chemical shift. The NICS-Y scan, running parallel to the pentalene plane at a height of 1.7 Å revealed two maxima at Y = −0.9 Å (20.28 ppm) and at Y = 1 Å (22.67 ppm) close to the centres of each 5-membered ring, consistent with the paratropic current found in the ACID plot, and mirrors that of the strongly aromatic dianion Ph4Pn2−.67 These maxima were separated by a local minimum at the centre of the molecule around the transannular C–C bridge (Y = 0 Å, 15.50 ppm) as a result of two contributions: a paratropic current about the perimeter of the pentalene system combined with two local paratropic currents in each 5-membered ring. As observed by Stanger et al. for unsubstituted pentalene, the latter contribution typically dominates in these systems,69 although retaining some global antiaromaticity. This strong influence of local currents in the two 5-membered rings can also be seen by the turbulence around the transannular C–C bridge in the ACID plot of Ph4Pn (Fig. 5). The NICS and ACID calculations for the [2 + 2] cycloaddition product [Ph4Pn]2 showed a complete loss of the antiaromatic current (Fig. S12;† with only the flanking phenyl rings retaining their aromatic character), consistent with the notion that the dimerisation of pentalenes is driven by a relief of antiaromatic destabilisation.
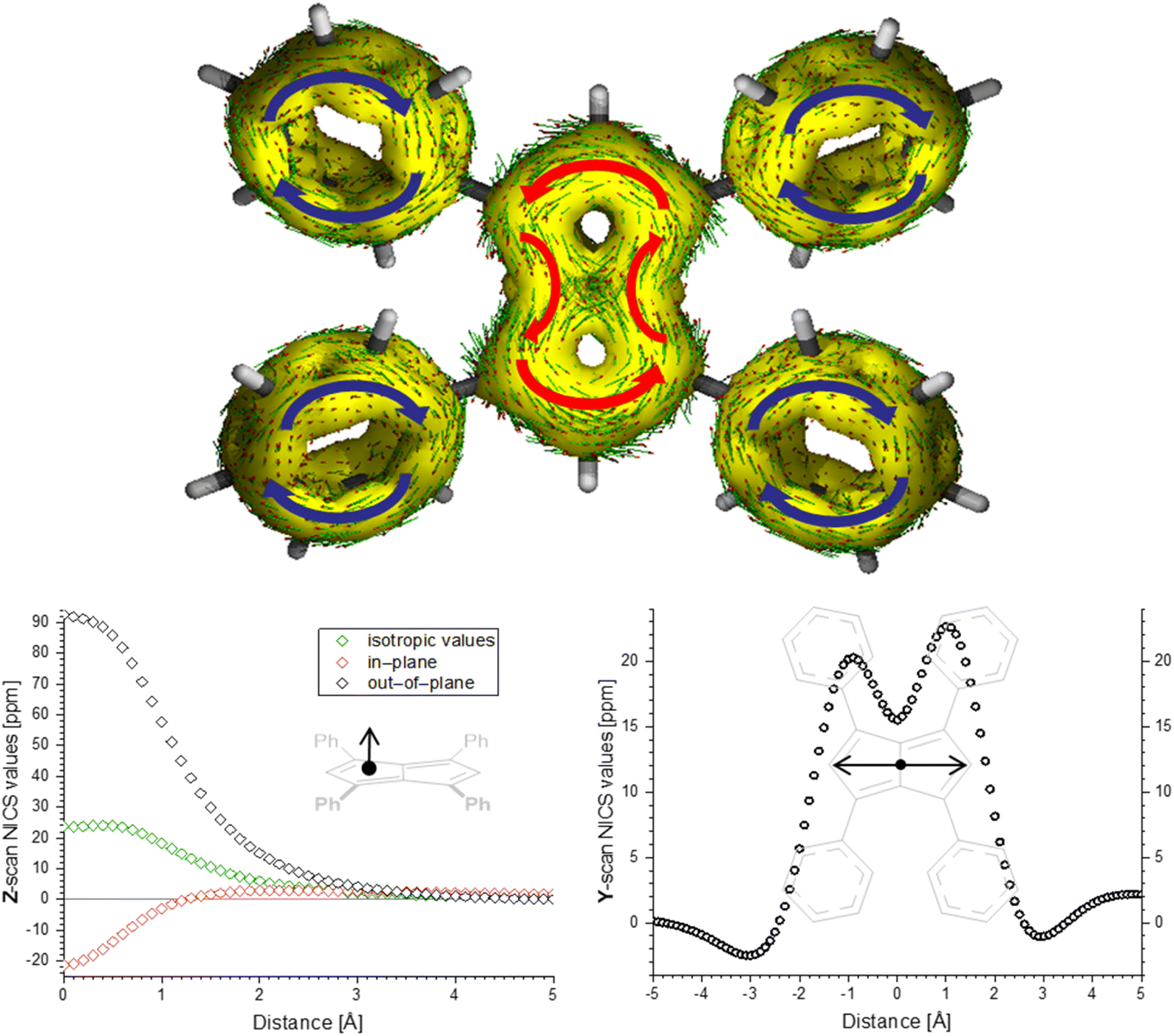 | ||
| Fig. 5 ACID plot ((top); iso-value = 0.03) and NICS scans ((bottom); Z scan [left] and Y scan [right], at a Z height of 1.7 Å; NICS probe BQ shown as •) of Ph4Pn. | ||
The computed frontier orbitals of Ph4Pn (Fig. 6) agree well with those calculated for unsubstituted Pn.70 Both the HOMO and the LUMO are characterised as π orbitals of the pentalene system, with minimal contribution from the substituents. The HOMO showed major contributions from the localised trisubstituted CC bond in each C5-ring, polarised towards the wingtip carbon, as well as a noticeable contribution from the transannular C–C bond. In contrast, in the HOMO of Ph4Pn2− the largest contributions were found to be localised on the 1,3,4,6-positions.67 The LUMO of Ph4Pn showed increased delocalisation across the transannular bond into the 1,4-ipso-carbons but (unlike in Ph4Pn2−) without any delocalisation into the phenyl substituents. The HOMO–LUMO gap for Ph4Pn of 2.11 eV was about 1 eV smaller than that of Ph4Pn2− (3.15 eV), indicating a more reactive system due to the antiaromatic core. Pn has been calculated to have a HOMO–LUMO gap of 1.12 eV,70 1 eV smaller than for Ph4Pn emphasising the stabilising influence of the four phenyl substituents. A singlet-triplet gap of 41.1 kJ mol−1 (0.43 eV) was calculated for Ph4Pn, about four times greater than that computed for Pn (0.1 eV),71 highlighting the decreased propensity of Ph4Pn to dimerise through increased kinetic stabilisation of the singlet state.
 | ||
| Fig. 6 Frontier molecular orbitals of Ph4Pn. | ||
The UV-vis spectrum of Ph4Pn in THF gave rise to two broad absorptions (Fig. 7). The first band at λmax = 312 nm (ε = 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) was nearly three times more intense than comparable absorptions observed for Ph4PnH2 (λ = 301, ε = 31500 M−1 cm−1 and λ = 339 nm, ε = 26800 M−1 cm−1) and are attributed to electronic transitions from the HOMO−1 into the LUMO+2 and LUMO+3 on the basis of TD-DFT (Fig. S16†). The second maximum at λmax = 460 nm (ε = 40000 M−1 cm−1) is assigned as a HOMO−1 → LUMO transition. A similar absorption was seen for Ph4PnH2 at 468 nm, though an order of magnitude less intense (ε = 3600 M−1 cm−1), and was assigned to the fulvene-like double bond in the molecule.57 The UV-vis spectrum of Ph4Pn in benzene largely retained the same features as in THF, indicating there is no significant change in the solution structure of Ph4Pn between the two solvents, consistent with the NMR data. The main band at λmax = 275 nm was of slightly lower intensity (ε = 90000 M−1 cm−1) and blue shifted by 50 nm compared to the same absorption in THF. A marginally more pronounced shoulder at λmax = 350 nm was seen in the benzene spectrum and accounted for by TD-DFT (Fig. S16†). Dilute solutions of Ph4Pn in THF and benzene were both pale yellow due to the absorption at 460 nm, even though in benzene this band was only about half the intensity than in THF. 1,3,5-tBu3Pn has been reported to be dark blue in colour with a visible absorption at 598 nm, and 1,3-(NMe2)2Pn, 1,4-Me2-2,5-(CN)2-3,6-(NH2)2Pn and Ph6Pn were also reported to have absorptions >600 nm. Similar low energy absorptions were found computationally for Ph4Pn but with much lower intensities (Fig. S16 and S17†).
000 M−1 cm−1) was nearly three times more intense than comparable absorptions observed for Ph4PnH2 (λ = 301, ε = 31500 M−1 cm−1 and λ = 339 nm, ε = 26800 M−1 cm−1) and are attributed to electronic transitions from the HOMO−1 into the LUMO+2 and LUMO+3 on the basis of TD-DFT (Fig. S16†). The second maximum at λmax = 460 nm (ε = 40000 M−1 cm−1) is assigned as a HOMO−1 → LUMO transition. A similar absorption was seen for Ph4PnH2 at 468 nm, though an order of magnitude less intense (ε = 3600 M−1 cm−1), and was assigned to the fulvene-like double bond in the molecule.57 The UV-vis spectrum of Ph4Pn in benzene largely retained the same features as in THF, indicating there is no significant change in the solution structure of Ph4Pn between the two solvents, consistent with the NMR data. The main band at λmax = 275 nm was of slightly lower intensity (ε = 90000 M−1 cm−1) and blue shifted by 50 nm compared to the same absorption in THF. A marginally more pronounced shoulder at λmax = 350 nm was seen in the benzene spectrum and accounted for by TD-DFT (Fig. S16†). Dilute solutions of Ph4Pn in THF and benzene were both pale yellow due to the absorption at 460 nm, even though in benzene this band was only about half the intensity than in THF. 1,3,5-tBu3Pn has been reported to be dark blue in colour with a visible absorption at 598 nm, and 1,3-(NMe2)2Pn, 1,4-Me2-2,5-(CN)2-3,6-(NH2)2Pn and Ph6Pn were also reported to have absorptions >600 nm. Similar low energy absorptions were found computationally for Ph4Pn but with much lower intensities (Fig. S16 and S17†).
 | ||
| Fig. 7 UV-vis spectra of 1,3,4,6-Ph4Pn (6 μM), Mg[Ph4Pn] (15 μM) and Ph4PnH2 (11 μM) in THF at 298 K. | ||
In line with the electrochemistry of Ph4Pn showing a reduction peak ∼0.3 V vs. NHE in THF (Fig. S18†), Ph4Pn can be reduced back to Ph4Pn2− by contacting a THF solution of Ph4Pn with a freshly prepared potassium mirror, causing an immediate colour change from dark yellow to bright red. The 1H NMR spectrum of the solution showed full consumption of Ph4Pn and the spectroscopically quantitative formation of K2[Ph4Pn] (Fig. 8) with a characteristic Hw shift of 6.76 ppm.57 The ease of the reduction back to the aromatic Ph4Pn2− is supported by DFT where the enthalpy of Ph4Pn was found to be 72.8 kJ mol−1 higher than Ph4Pn2−.
 | ||
| Fig. 8 500 MHz 1H NMR spectra of K2[Ph4Pn] (top) and 1,3,4,6-Ph4Pn (bottom). | ||
3. Conclusion
A stable pentalene has been synthesised through the facile oxidation of its corresponding pentalenide using elemental iodine. Unlike previous reports of pentalenes, Ph4Pn crystallised as a mixture of both the monomer and dimer in the same unit cell. However, NMR spectroscopy showed that in solution the monomeric form dominates, and UV-vis spectroscopy indicated no solvatochromism or change in the solution structure in THF and benzene. Ph4Pn is a rare example of a small molecule featuring both Hückel aromatic and antiaromatic subunits in close proximity, albeit electronically separated from each other as shown by quantum chemical calculations. Therefore, the stabilisation of the anti-aromatic pentalene core imparted by the four aromatic phenyl substituents is largely steric in nature. The reduction of Ph4Pn back to Ph4Pn2− was found to be reversible within a potential range of ∼1 V, representing a unique example of a small hydrocarbon that can easily switch between an aromatic and anti-aromatic state without any conformational or skeletal rearrangements. These properties alongside its structural simplicity and ease of synthesis58,67 suggest that Ph4Pn and its derivatives could find applications similar to those of dibenzopentalenes in electrochemical sensing or optoelectronics. In the context of organometallic chemistry, the facile oxidation of Ph4Pn2− by iodine provides a useful threshold for the redox potential of metal ions bound to it to guide the design and synthesis of novel pentalenide complexes.Data availability
Crystallographic datasets are available from the CCDC deposition number 2335451, and other analytical data can be found in the ESI.†Author contributions
HJS carried out all synthetic work, spectroscopic analyses, electrochemistry, and drafted the manuscript. GKK conducted all XRD analyses. AH carried out all calculations with guidance and supervision by HH. UH led the project, and all authors contributed to refining the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
UH and HJS thank the Royal Society (award UF160458) and the University of Bath for funding this work. HH and AH thank the German Research Foundation (DFG) for funding through the Heisenberg Grant HE 6171/9-1 (468457264). Kathryn Proctor (University of Bath) is acknowledged for assistance with conducting the mass spectrometry measurements, and we thank Simon Lewis and Dan Pantos (University of Bath) for stimulating discussions about aromaticity.References
- M. Solà, Aromaticity Rules, Nat. Chem., 2022, 14(6), 585–590, DOI:10.1038/s41557-022-00961-w.
- W. E. von Doering and F. L. Detert, Cycloheptatrienylium Oxide, J. Am. Chem. Soc., 1951, 73(2), 876–877, DOI:10.1021/JA01146A537.
- E. Hückel, Quantentheoretische Beiträge Zum Benzolproblem - I. Die Elektronenkonfiguration Des Benzols Und Verwandter Verbindungen, Z. Phys., 1931, 70(3–4), 204–286, DOI:10.1007/BF01339530.
- R. Breslow, Antiaromaticity, Acc. Chem. Res., 1973, 6(12), 393–398, DOI:10.1021/AR50072A001.
- G. Merino, M. Solà, I. Fernández, C. Foroutan-Nejad, P. Lazzeretti, G. Frenking, H. L. Anderson, D. Sundholm, F. P. Cossío, M. A. Petrukhina, J. Wu, J. I. Wu and A. Restrepo, Aromaticity: Quo Vadis, Chem. Sci., 2023, 14(21), 5569–5576, 10.1039/D2SC04998H.
- P. Lazzeretti, Ring Currents, Prog. Nucl. Magn. Reson. Spectrosc., 2000, 36(1), 1–88, DOI:10.1016/S0079-6565(99)00021-7.
- N. C. Baird, Quantum Organic Photochemistry. II. Resonance and Aromaticity in the Lowest 3ππ* State of Cyclic Hydrocarbons, J. Am. Chem. Soc., 1972, 94(14), 4941–4948, DOI:10.1021/JA00769A025.
- H. Ottosson, Exciting Excited-State Aromaticity, Nat. Chem., 2012, 4(12), 969–971, DOI:10.1038/nchem.1518.
- C. S. Wannere, C. Corminboeuf, Z. X. Wang, M. D. Wodrich, R. B. King and P. V. R. Schleyer, Evidence for d Orbital Aromaticity in Square Planar Coinage Metal Clusters, J. Am. Chem. Soc., 2005, 127(15), 5701–5705, DOI:10.1021/JA042716Q.
- A. C. Tsipis, C. E. Kefalidis and C. A. Tsipis, The Role of the 5f Orbitals in Bonding, Aromaticity, and Reactivity of Planar Isocyclic and Heterocyclic Uranium Clusters, J. Am. Chem. Soc., 2008, 130(28), 9144–9155, DOI:10.1021/JA802344Z.
- I. A. Popov, A. A. Starikova, D. V. Steglenko and A. I. Boldyrev, Usefulness of the σ-Aromaticity and σ-Antiaromaticity Concepts for Clusters and Solid-State Compounds, Chem.–Eur. J., 2018, 24(2), 292–305, DOI:10.1002/CHEM.201702035.
- J. T. Boronski, J. A. Seed, D. Hunger, A. W. Woodward, J. van Slageren, A. J. Wooles, L. S. Natrajan, N. Kaltsoyannis and S. T. Liddle, A Crystalline Tri-Thorium Cluster with σ-Aromatic Metal–Metal Bonding, Nature, 2021, 598(7879), 72–75, DOI:10.1038/s41586-021-03888-3.
- B. Peerless, A. Schmidt, Y. J. Franzke and S. Dehnen, φ-Aromaticity in Prismatic {Bi6}-Based Clusters, Nat. Chem., 2022, 15(3), 347–356, DOI:10.1038/s41557-022-01099-5.
- A. Minsky, A. Y. Meyer and M. Rabinovitz, Paratropicity and Antiaromaticity: Role of the Homo-Lumo Energy Gap, Tetrahedron, 1985, 41(4), 785–791, DOI:10.1016/S0040-4020(01)96458-0.
- C. Gellini and P. R. Salvi, Structures of Annulenes and Model Annulene Systems in the Ground and Lowest Excited States, Symmetry, 2010, 2(4), 1846–1924, DOI:10.3390/SYM2041846.
- J. Sprachmann, T. Wachsmuth, M. Bhosale, D. Burmeister, G. J. Smales, M. Schmidt, Z. Kochovski, N. Grabicki, R. Wessling, E. J. W. List-Kratochvil, B. Esser and O. Dumele, Antiaromatic Covalent Organic Frameworks Based on Dibenzopentalenes, J. Am. Chem. Soc., 2023, 145, 2840–2851, DOI:10.1021/JACS.2C10501.
- D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, 6,12-Diarylindeno[1,2-b]Fluorenes: Syntheses, Photophysics, and Ambipolar OFETs, J. Am. Chem. Soc., 2012, 134(25), 10349–10352, DOI:10.1021/JA303402P.
- H. Hopf, Pentalenes—From Highly Reactive Antiaromatics to Substrates for Material Science, Angew. Chem., Int. Ed., 2013, 52(47), 12224–12226, DOI:10.1002/ANIE.201307162.
- Z. Zeng, X. Shi, C. Chi, J. T. López Navarrete, J. Casado and J. Wu, Pro-Aromatic and Anti-Aromatic π-Conjugated Molecules: An Irresistible Wish to Be Diradicals, Chem. Soc. Rev., 2015, 44(18), 6578–6596, 10.1039/C5CS00051C.
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. Ponce Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Diindeno-Fusion of an Anthracene as a Design Strategy for Stable Organic Biradicals, Nat. Chem., 2016, 8(8), 753–759, DOI:10.1038/nchem.2518.
- L. J. Karas and J. I.-C. Wu, Antiaromatic Compounds: A Brief History, Applications, and the Many Ways They Escape Antiaromaticity, in Aromaticity: Modern Computational Methods and Applications, ed. I. Fernandez, Elsevier, 2021, pp 319–338, DOI:10.1016/B978-0-12-822723-7.00010-8.
- F. Glöcklhofer, Concealed Antiaromaticity, ChemRxiv, 2023 DOI:10.26434/CHEMRXIV-2023-HNL0W-V2.
- S. A. R. Knox, F. Gordon and A. Stone, Approaches to the Synthesis of Pentalene via Metal Complexes, Acc. Chem. Res., 1974, 7(10), 321–328 CrossRef CAS.
- R. Bloch, R. A. Marty and P. de Mayo, 1-Methylpentalene, J. Am. Chem. Soc., 1971, 93(12), 3071–3072, DOI:10.1021/ja00741a056.
- T. Bally, S. Chai, M. Neuenschwander and Z. Zhu, Pentalene: Formation, Electronic, and Vibrational Structure, J. Am. Chem. Soc., 1997, 119(8), 1869–1875, DOI:10.1021/ja963439t.
- S. Poplata, A. Tröster, Y. Q. Zou and T. Bach, Recent Advances in the Synthesis of Cyclobutanes by Olefin [2+2] Photocycloaddition Reactions, Chem. Rev., 2016, 116(17), 9748–9815, DOI:10.1021/ACS.CHEMREV.5B00723.
- W. Stawski, Y. Zhu, Z. Wei, M. A. Petrukhina and H. L. Anderson, Crystallographic Evidence for Global Aromaticity in the Di-Anion and Tetra-Anion of a Cyclophane Hydrocarbon, Chem. Sci., 2023, 14(48), 14109–14114, 10.1039/D3SC04251K.
- W. Stawski, Y. Zhu, I. Rončević, Z. Wei, M. A. Petrukhina and H. L. Anderson, The Anti-Aromatic Dianion and Aromatic Tetraanion of [18]Annulene, Nat. Chem., 2024, 1–5, DOI:10.1038/s41557-024-01469-1.
- T. Y. Gopalakrishna and V. G. Anand, Reversible Redox Reaction Between Antiaromatic and Aromatic States of 32π-Expanded Isophlorins, Angew. Chem., Int. Ed., 2014, 53(26), 6678–6682, DOI:10.1002/ANIE.201403372.
- S. Eder, D. J. Yoo, W. Nogala, M. Pletzer, A. Santana Bonilla, A. J. P. White, K. E. Jelfs, M. Heeney, J. W. Choi and F. Glöcklhofer, Switching between Local and Global Aromaticity in a Conjugated Macrocycle for High-Performance Organic Sodium-Ion Battery Anodes, Angew. Chem., Int. Ed., 2020, 59(31), 12958–12964, DOI:10.1002/ANIE.202003386.
- W. Y. Cha, T. Soya, T. Tanaka, H. Mori, Y. Hong, S. Lee, K. H. Park, A. Osuka and D. Kim, Multifaceted [36]Octaphyrin(1.1.1.1.1.1.1.1): Deprotonation-Induced Switching among Nonaromatic, Möbius Aromatic, and Hückel Antiaromatic Species, Chem. Commun., 2016, 52(36), 6076–6078, 10.1039/C6CC02051H.
- T. Y. Gopalakrishna, J. S. Reddy and V. G. Anand, An Amphoteric Switch to Aromatic and Antiaromatic States of a Neutral Air-Stable 25π Radical, Angew. Chem., Int. Ed., 2014, 53(41), 10984–10987, DOI:10.1002/ANIE.201406893.
- Y. Zhu, Z. Zhou, Z. Wei, A. Tsybizova, R. Gershoni-Poranne and M. A. Petrukhina, What a Difference an Electron Makes: Structural Response of Saddle-Shaped Tetraphenylene to One and Two Electron Uptake, ChemRxiv, 2024, 2(5), e202400055, DOI:10.1002/CEUR.202400055.
- X. Yin, Y. Zang, L. Zhu, J. Z. Low, Z. F. Liu, J. Cui, J. B. Neaton, L. Venkataraman and L. M. Campos, A Reversible Single-Molecule Switch Based on Activated Antiaromaticity, Sci. Adv., 2017, 3(10) DOI:10.1126/SCIADV.AAO2615.
- K. Brand, Über Gefärbte Kohlenwasserstoffe Der Diphensuccinden-Reihe. I, Ber. Dtsch. Chem. Ges., 1912, 45(3), 3071–3077, DOI:10.1002/CBER.19120450334.
- E. L. Goff, The Synthesis of Hexaphenylpentalene, J. Am. Chem. Soc., 1962, 84(20), 3975–3976, DOI:10.1021/ja00879a044.
- M. Saito, M. Nakamura and T. Tajima, New Reactions of a Dibenzo[a,e]Pentalene, Chem.–Eur. J., 2008, 14(20), 6062–6068, DOI:10.1002/CHEM.200800451.
- A. Konishi, Y. Okada, M. Nakano, K. Sugisaki, K. Sato, T. Takui and M. Yasuda, Synthesis and Characterization of Dibenzo[a,f]Pentalene: Harmonization of the Antiaromatic and Singlet Biradical Character, J. Am. Chem. Soc., 2017, 139(43), 15284–15287, DOI:10.1021/JACS.7B05709.
- P. J. Mayer, O. El Bakouri, T. Holczbauer, G. F. Samu, C. Janáky, H. Ottosson and G. London, Structure-Property Relationships in Unsymmetric Bis(Antiaromatics): Who Wins the Battle between Pentalene and Benzocyclobutadiene?, J. Org. Chem., 2020, 85(8), 5158–5172, DOI:10.1021/ACS.JOC.9B03119.
- A. Konishi, Y. Okada, R. Kishi, M. Nakano and M. Yasuda, Enhancement of Antiaromatic Character via Additional Benzoannulation into Dibenzo[a,f]Pentalene: Syntheses and Properties of Benzo[a]Naphtho[2,1-f]Pentalene and Dinaphtho[2,1-a,f]Pentalene, J. Am. Chem. Soc., 2019, 141(1), 560–571, DOI:10.1021/JACS.8B11530.
- C. K. Frederickson, L. N. Zakharov and M. M. Haley, Modulating Paratropicity Strength in Diareno-Fused Antiaromatics, J. Am. Chem. Soc., 2016, 138(51), 16827–16838, DOI:10.1021/JACS.6B11397.
- S. Jalife, A. Tsybizova, R. Gershoni-Poranne and J. I. Wu, Modulating Paratropicity in Heteroarene-Fused Expanded Pentalenes, Org. Lett., 2024, 26(6), 1293–1298, DOI:10.1021/ACS.ORGLETT.4C00188.
- H. Oshima, A. Fukazawa and S. Yamaguchi, Facile Synthesis of Polycyclic Pentalenes with Enhanced Hückel Antiaromaticity, Angew. Chem., Int. Ed., 2017, 56(12), 3270–3274, DOI:10.1002/ANIE.201611344.
- A. E. Ashley, A. R. Cowley and D. O'Hare, Permethylpentalene Chemistry, Eur. J. Org. Chem., 2007, 14, 2239–2242, DOI:10.1002/ejoc.200700033.
- K. Hartke and R. Matusch, Aminopentalenecarbonitriles, a Group of Stable Pentalenes, Angew. Chem., Int. Ed. Engl., 1972, 11(1), 50–51, DOI:10.1002/anie.197200501.
- K. Hafner, K. F. Bangert and V. Orfanos, 1,3-Bis(Dimethylamino)Pentalene, Angew. Chem., Int. Ed. Engl., 1967, 475(5), 451–452 CrossRef.
- K. Hafner and H. U. Süss, 1,3,5-Tri-tert-Butylpentalene. A Stabilized Planar 8π-Electron System, Angew. Chem., Int. Ed. Engl., 1973, 12(7), 575–577, DOI:10.1002/anie.197305751.
- K. Hafner and M. Suda, [6+2] Cycloadditions of Pentafulvene. A Facile Pentalene Synthesis, Angew. Chem., Int. Ed. Engl., 1976, 15(5), 314–315 CrossRef.
- M. Suda and K. Hafner, Synthesis of 4.6-Di-Tert.-Butyl-Pentalene Derivatives and Their Reversible Dimerization, Tetrahedron Lett., 1977, 18(28), 2449–2452, DOI:10.1016/S0040-4039(01)83790-4.
- Z. U. Levi and T. D. Tilley, Versatile Synthesis of Pentalene Derivatives via the Pd-Catalyzed Homocoupling of Haloenynes, J. Am. Chem. Soc., 2009, 131(8), 2796–2797, DOI:10.1021/JA809930F.
- B. Kitschke and H. J. Lindner, The Crystal and Molecular Structures of Two Substituted Pentalenes, Tetrahedron Lett., 1977, 18(29), 2511–2514, DOI:10.1016/S0040-4039(01)83806-5.
- A. E. Ashley, R. T. Cooper, G. G. Wildgoose, J. C. Green and D. O'Hare, Homoleptic Permethylpentalene Complexes: “Double Metallocenes” of the First-Row Transition Metals, J. Am. Chem. Soc., 2008, 130, 15662–15677, DOI:10.1021/ja8057138.
- H. Zhao, R. K. Gupta, W. Zhang, J. Jia, Q. Yu, Z. Gao, G. Zhuang, D. Li, X. Wang, C. H. Tung and D. Sun, Facile One-Pot Synthesis of a Novel All-Carbon Stair Containing Dimerized Pentalene Core from Alkyne, Chin. Chem. Lett., 2022, 33(4), 2047–2051, DOI:10.1016/J.CCLET.2021.10.036.
- A. G. Griesbeck, Pyrrolidine-Catalyzed Reactions between α,β-Unsaturated Carbonyl Compounds and Cyclopentadiene: A Convenient Approach to 1,2-and 1,5-Dihydropentalenes, J. Org. Chem., 1989, 54(2), 4981–4982 CrossRef CAS.
- A. G. Griesbeck, Ein Effizienter Zugang Zu 1,5-Dihydropentalen Und 2-Methyl-1,5-Dihydropentalen Aus Acrolein Und Methacrolein, Synthesis, 1990, 2, 144–147 CrossRef.
- A. G. Griesbeck, Notizen/Notes: Synthesis of 1-Phenyl,1,2- and 4-Phenyl-1,5-Dihydropentalenes, Chem. Ber., 1991, 124(2), 403–405 CAS.
- S. M. Boyt, N. A. Jenek, H. J. Sanderson, G. Kociok-Köhn and U. Hintermair, Synthesis of a Tetraphenyl-Substituted Dihydropentalene and Its Alkali Metal Hydropentalenide and Pentalenide Complexes, Organometallics, 2022, 41(3), 211–225, DOI:10.1021/acs.organomet.1c00495.
- N. A. Jenek, M. Balschun, S. M. Boyt and U. Hintermair, Connect Four: Tetraarylated Dihydropentalenes and Triarylated Monocyclic Pentafulvenes from Cyclopentadienes and Enones, J. Org. Chem., 2022, 87(21), 13790–13802, DOI:10.1021/ACS.JOC.2C01507.
- H. J. Sanderson, G. Kociok-Köhn and U. Hintermair, Synthesis, Structure, and Reactivity of Magnesium Pentalenides, Inorg. Chem., 2023, 62(39), 15983–15991, DOI:10.1021/ACS.INORGCHEM.3C02087.
- Handbook of Chemistry and Physics, ed. J. R. Rumble, CRC Press, 104th edn, 2023 Search PubMed.
- S. M. Boyt, Synthesis of Novel Dihydropentalenes , Pentalenides and Hydropentalenides, PhD Thesis, Bath, 2019 Search PubMed.
- S. You and M. Neuenschwander, New Pathways to Precursors of Pentalene, Chimia, 1996, 50(1–2), 26, DOI:10.2533/chimia.1996.24.
- H. J. Sanderson, G. Kociok-Köhn, C. L. McMullin and U. Hintermair, Twinned versus Linked Organometallics - Bimetallic “Half-Baguette” Pentalenide Complexes of Rh(I), Dalton Trans., 2024, 53, 5881–5899, 10.1039/D3DT04325H.
- R. Nozawa, J. Kim, J. Oh, A. Lamping, Y. Wang, S. Shimizu, I. Hisaki, T. Kowalczyk, H. Fliegl, D. Kim and H. Shinokubo, Three-Dimensional Aromaticity in an Antiaromatic Cyclophane, Nat. Commun., 2019, 10(1), 1–7, DOI:10.1038/s41467-019-11467-4.
- K. Hanida, J. Kim, N. Fukui, Y. Tsutsui, S. Seki, D. Kim and H. Shinokubo, Antiaromatic 1,5-Diaza-s-Indacenes, Angew. Chem., Int. Ed., 2021, 60(38), 20765–20770, DOI:10.1002/ANIE.202109003.
- H. Kawashima, S. Ukai, R. Nozawa, N. Fukui, G. Fitzsimmons, T. Kowalczyk, H. Fliegl and H. Shinokubo, Determinant Factors of Three-Dimensional Aromaticity in Antiaromatic Cyclophanes, J. Am. Chem. Soc., 2021, 143(28), 10676–10685, DOI:10.1021/JACS.1C04348.
- N. A. Jenek, A. Helbig, S. M. Boyt, M. Kaur, H. J. Sanderson, S. B. Reeksting, G. Kociok-Köhn, H. Helten and U. Hintermair, Understanding and Tuning the Electronic Structure of Pentalenides, Chem. Sci., 2024, 15(32), 12765–12779, 10.1039/D3SC04622B.
- A. Stanger, Nucleus-Independent Chemical Shifts (NICS): Distance Dependence and Revised Criteria for Aromaticity and Antiaromaticity, J. Org. Chem., 2006, 71(3), 883–893, DOI:10.1021/JO051746.
- A. Stanger, G. Monaco and R. Zanasi, NICS-XY-Scan Predictions of Local, Semi-Global, and Global Ring Currents in Annulated Pentalene and s-Indacene Cores Compared to First-Principles Current Density Maps, ChemPhysChem, 2020, 21(1), 65–82, DOI:10.1002/CPHC.201900952.
- N. D. Charistos, A. G. Papadopoulos, T. A. Nikopoulos, A. Muñoz-Castro and M. P. Sigalas, Canonical Orbital Contributions to the Magnetic Fields Induced by Global and Local Diatropic and Paratropic Ring Currents, J. Comput. Chem., 2017, 38(30), 2594–2604, DOI:10.1002/JCC.24917.
- M. H. Garner, J. T. Blaskovits and C. Corminboeuf, Double-Bond Delocalization in Non-Alternant Hydrocarbons Induces Inverted Singlet–Triplet Gaps, Chem. Sci., 2023, 14(38), 10458–10466, 10.1039/D3SC03409G.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, additional analytical data (NMR, XRD). CCDC 2335451. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06439a |
| This journal is © The Royal Society of Chemistry 2025 |