
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurmounting the instability of atomically precise metal nanoclusters towards boosted photoredox organic transformation†
Yu-Bing
Li
and
Fang-Xing
Xiao
 *
*
College of Materials Science and Engineering, Fuzhou University, New Campus, 350108, China. E-mail: fxxiao@fzu.edu.cn
First published on 26th December 2024
Abstract
Atomically precise metal nanoclusters (NCs) have recently been recognized as an emerging sector of metal nanomaterials but suffer from light-induced poor stability, giving rise to the detrimental self-transformation into metal nanocrystals (NYs), losing the photosensitization effect and ultimately retarding their widespread applications in photoredox catalysis. Are metal NCs definitely superior to metal NYs in heterogeneous photocatalysis in terms of structural merits? To unlock this mystery, herein, we conceptually demonstrate how to rationally manipulate the instability of metal NCs to construct high-efficiency artificial photosystems and examine how the metal NYs self-transformed from metal NCs influence charge transfer in photoredox selective organic transformation. To our surprise, the results indicate that the Schottky-type electron-trapping ability of Au NYs surpasses the photosensitization effect of glutathione (GSH)-protected Au clusters [Au25(GSH)18 NCs] in mediating charge separation and enhancing photoactivities towards selective photoreduction of aromatic nitro compounds to amino derivatives and photocatalytic oxidation of aromatic alcohols to aldehydes under visible light irradiation. This work strategically provides new insights into the inherent instability of metal NCs utilized for photocatalysis and reinforces our fundamental understanding on metal NC-based artificial photosystems for solar energy conversion.
1. Introduction
Atomically precise metal nanoclusters (NCs) exhibit distinct physicochemical properties from those of their conventional metal nanocrystal (NY) counterparts such as unique atom-stacking mode, the quantum confinement effect, and enriched catalytically active sites.1–4 In particular, the characteristic discrete energy band structure of metal NCs makes them promising photosensitizers for solar energy conversion.1–4 Recent years have witnessed impressive progress on metal NC mediated photocatalysis wherein metal NCs serve as both photosensitizers and catalytic centers for stimulating multifarious photoredox catalytic reactions such as photocatalytic environmental remediation, selective organic transformations, hydrogen production, and CO2 reduction.5–9 However, developments of high-efficiency metal NC artificial photosystems are retarded by challenges including light or heat induced instability, ultra-short carrier lifetime, and difficulty in modulating the charge transport pathway. Addressing these issues is of paramount significance to advance the exploration of robust and efficient metal NC photocatalytic systems.Inspired by the self-transformation of metal NCs into metal NYs stemming from the intrinsic poor stability of metal NCs, we seek to figure out the charge transfer difference between these two metal nanomaterials of the same origin and unlock their impact on the photocatalytic mechanism. Metal NCs, by virtue of their discrete highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) energy band structure and favorable band gaps, have been evidenced to serve as efficient photosensitizers comparable to quantum dots, facilitating directional electron migration to the conduction band of semiconductors (e.g., TiO2), thus enhancing the charge separation efficiency.10–12 Conversely, metal NYs are characterized by distinct charge transfer mechanisms such as plasmon-induced hot electron injection and Schottky-type electron withdrawal.13–16 Despite the endeavors undertaken in the past few years, thus far, charge transfer correlation between atomically precise metal NCs and the corresponding metal NYs in photocatalysis has not yet been unlocked. Are metal NCs definitely superior to their metal NY counterparts in boosting the charge transport rate, accelerating charge transfer kinetics, and reinforcing the photosensitization effect considering the merits of the former? The intrinsic instability of metal NCs for self-transformation into metal NYs offers an applicable, convenient, and cherished opportunity to systematically compare the differences in their charge transport characteristics and photocatalytic mechanism.17–19
Herein, we conceptually report the thermal-induced self-transformation of metal NCs into metal NYs in composite photosystems and comprehensively evaluate the essential roles of metal NCs and metal NYs in stimulating the photocatalytic selective organic transformation. To these ends, one-dimensional (1D) CdS nanowires (NWs) are selected as the substrate which is modified by using an ultrathin mercaptoethylamine (MEA) layer to create a positively charged surface. Negatively charged atomically precise Au25(GSH)18 NCs are controllably deposited on the CdS NWs via electrostatic interaction under ambient conditions. These Au25(GSH)18 NCs are then self-converted into Au NYs through a simple annealing process, based on which interfacial charge transfer properties of the CdS/Au25(GSH)18 and CdS/Au heterostructures are rigorously and comprehensively compared, wherein two chemical states of Au in one integrated photosystem are accessed. We have also ascertained that the Schottky-type electron-withdrawing effect of Au NYs significantly surpasses the photosensitization effect of Au25(GSH)18 NCs in diverse photocatalytic reactions, including photocatalytic selective oxidation of aromatic alcohols to aldehydes, photoreduction of aromatic nitro compounds to amino derivatives, and photocatalytic hydrogen production under visible light irradiation. This study enhances our fundamental understanding on the charge transfer mechanisms over atomically precise metal NCs and metal NYs of the same origin towards solar energy conversion.
2. Experimental section
2.1 Preparation of CdS NWs20
Preparation of CdS NWs was referred to a previous study and detailed information is provided in the ESI.†2.2 Preparation of Au25(GSH)18 NCs21
Preparation of L-glutathione (GSH) capped Au25(GSH)18 NCs followed a previous study and detailed information is provided in the ESI.†2.3 Modification of the CdS NW surface
0.1 g CdS NWs was first dispersed in 100 mL of DI water by sonication for 10 min. Then, 9 mL of 1.0 mol L−1 2-mercaptoethylamine (MEA) was added under vigorous stirring (1000 rpm). The CdS NWs reacted with the modifier for 1 h at room temperature. Finally, the modified CdS@MEA NW samples were sufficiently rinsed with ethanol to wash away any remaining modifier moiety, and fully dried at 333 K in an oven.2.4 Preparation of the CdS/Au25(GSH)18 NC heterostructure
0.1 g modified CdS@MEA NWs was dispersed in 5 mL of Au25(GSH)18 NCs (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) the weight ratio of 3%) solution and stirred for 10 min. Then, different amounts of Au25(GSH)18 NCs were added to the modified CdS@MEA NW dispersion under vigorous stirring (1000 rpm). After mixing for 10 min, the mixture was centrifuged and washed with DI H2O and fully dried at 333 K in an oven to obtain the final CdS/Au25(GSH)18 composites.
the weight ratio of 3%) solution and stirred for 10 min. Then, different amounts of Au25(GSH)18 NCs were added to the modified CdS@MEA NW dispersion under vigorous stirring (1000 rpm). After mixing for 10 min, the mixture was centrifuged and washed with DI H2O and fully dried at 333 K in an oven to obtain the final CdS/Au25(GSH)18 composites.
2.5 Self-transformation of metal NCs into metal NYs
The CdS/Au25(GSH)18 samples were calcined at different temperatures ranging from 200 to 500 °C for 1 h, including 200, 300, 400, and 500 °C, and the obtained samples were referred to as x-CdS/Au (x = 200, 300, 400 and 500 °C). The optimal sample calcined at 400 °C is denoted as CdS/Au.2.6 Characterization
Zeta potentials were probed by dynamic light scattering analysis (ZetasizerNano ZS-90). The crystal structure was determined by X-ray diffraction (XRD, Miniflex600, Rigaku Corporation, Japan) using Cu Kα as the radiation source at 40 kV and 15 mA. Field-emission scanning electron microscopy (FESEM, Supra55, Carl Zeiss, Germany) was used to probe the morphologies of the samples. Transmission electron microscopy (TEM), high-resolution (HR) TEM and energy dispersive X-ray spectroscopy (EDX) were conducted on a Tecnai G2 F20 transmission electron microscope with an accelerating voltage of 200 kV. UV-vis diffuse reflectance spectra (DRS, Cary50, Varian, America) were obtained using BaSO4 as the reflectance background ranging from 200 to 800 nm. Fourier transform infrared (FTIR) spectra were recorded on a TJ270-30A infrared spectrophotometer (Tianjin, China). X-ray photoelectron spectroscopy (XPS) spectra were recorded on a photoelectron spectrometer (Escalab 250, Thermo Scientific, America), where binding energies (B.E.) of the elements were calibrated at 284.60 eV. Photoluminescence (PL) spectra were measured on a Varian Cary Eclipse spectrometer. Raman measurements were carried out on a Raman spectrometer (DXR-2xi, Thermo Scientific, America) with scans conducted in an extended range from 0 to 3000 cm−1.2.7 Photocatalytic reduction performances22
2.8 Photocatalytic selective oxidation
Photocatalytic selective oxidation of a series of aromatic alcohols was performed as follows. Specifically, a mixture of alcohol (0.1 mmol) and 8 mg of the catalyst was dissolved in benzotrifluoride (BTF) (1.5 mL) solvent, which was saturated with pure molecular oxygen. BTF was selected as the solvent due to its inertness to oxidation and high solubility for molecular oxygen. The above mixture was transferred into a 10 mL Pyrex glass bottle filled with molecular oxygen at a pressure of 0.1 MPa and stirred for half an hour to blend the catalyst evenly in the solution. The suspension was then irradiated by using a 300 W Xe arc lamp (PLS-SXE300D, Beijing Perfect Light Co. Ltd, China) with a 420 nm cut-off filter. After the reaction, the mixture was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min to completely remove the catalyst. The remaining solution was analyzed with a gas chromatograph (Shimadzu GC2140).
000 rpm for 5 min to completely remove the catalyst. The remaining solution was analyzed with a gas chromatograph (Shimadzu GC2140).
2.9 Photoelectrochemical (PEC) measurements23
PEC measurements were carried out on an electrochemical workstation (CHI660E, CHI Shanghai, Inc.) with a conventional three-electrode system and 0.5 M Na2SO4 aqueous solution (pH = 6.69) was utilized as the electrolyte. The three-electrode system is composed of Pt foil (1 cm × 1 cm) which was used as the counter electrode and Ag/AgCl electrode as the reference electrode. Potentials of the electrode were calibrated against the reversible hydrogen electrode (RHE) based on the following formula:| ERHE = EAg/AgCl + 0.059 pH + E0Ag/AgCl (E0Ag/AgCl = 0.1976 V at 25 °C) | (1) |
The transient photocurrent response (i.e., I–t) was collected under chopped light irradiation (light on/off cycle: 30 s) at a fixed bias of 1.23 V vs. RHE. Electrochemical impedance spectra (EIS) were measured on an electrochemical workstation (IM6, Zahner Germany) with an amplitude of 10 mV in the frequency range from 105 to 0.1 Hz.
According to Mott–Schottky (M–S) results, the charge carrier density (ND) of the photoanodes can be calculated according to the following formula:
 | (2) |
As per OCVD results the average electron lifetime (τn) can be determined according to the formula below:
 | (3) |
3. Results and discussion
The fabrication process for CdS/Au25(GSH)18 and CdS/Au heterostructures is illustrated in Fig. 1a. Initially, MEA molecules are grafted onto the CdS NW surface to form a positively charged surface, thereby facilitating the electrostatic self-assembly between the positively charged CdS and negatively charged Au25(GSH)18 NCs (Fig. S1–S3†). Subsequently, the CdS/Au heterostructure is formed by the self-transformation of atomically precise Au25(GSH)18 NCs into conventional metallic Au NYs through annealing. This method is both convenient and logical for maintaining a consistent loading amount of the metal component in the nanocomposites.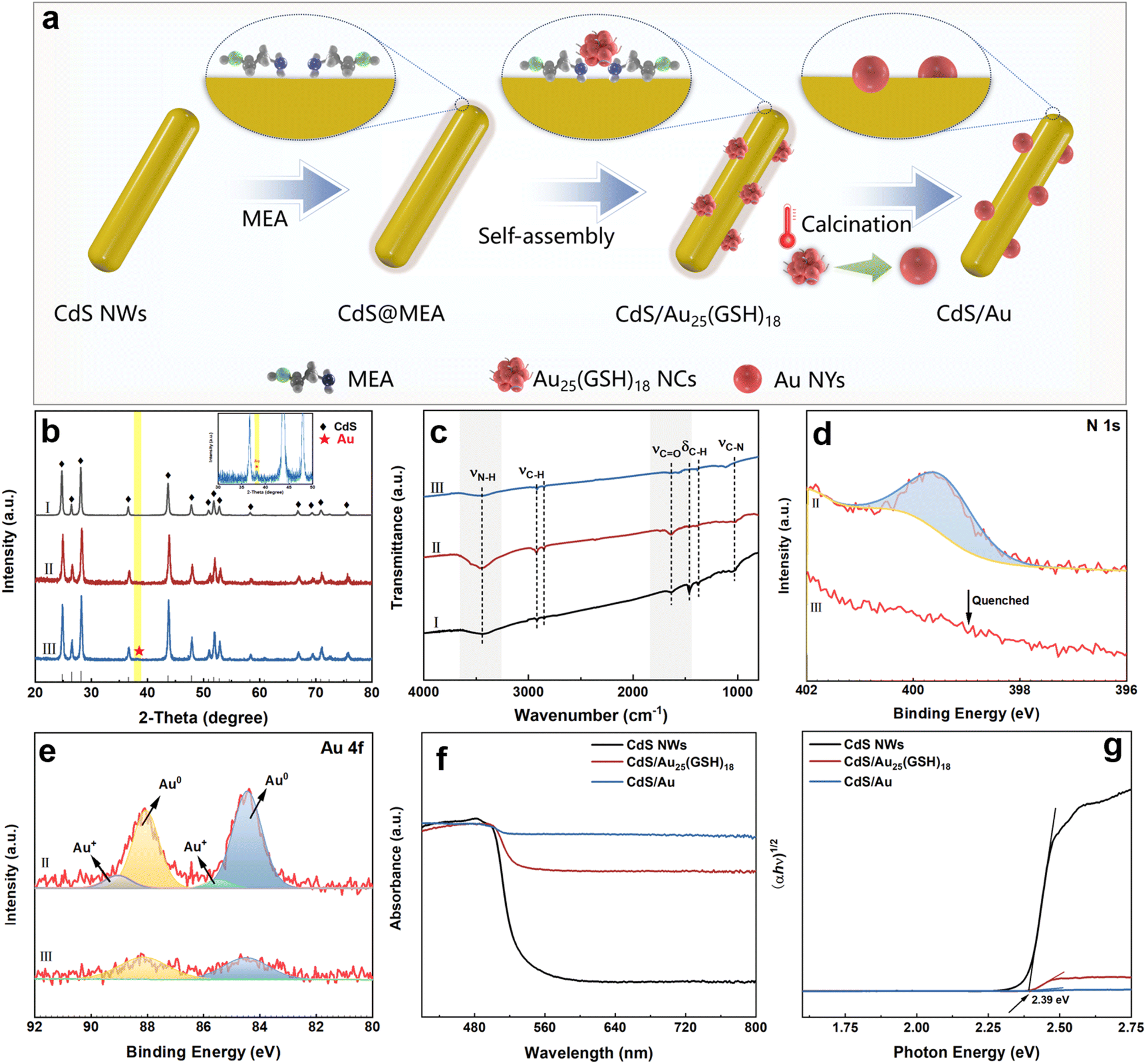 | ||
| Fig. 1 (a) Schematic illustration of the fabrication of CdS/Au25(GSH)18 and CdS/Au NY heterostructures. (b) XRD patterns of (I) CdS NWs, (II) CdS/Au25(GSH)18 and (III) CdS/Au; (c) FTIR spectra of (I) CdS NWs, (II) CdS/Au25(GSH)18 and (III) CdS/Au; (d) high-resolution N 1s and (e) Au 4f spectra of (II) CdS/Au25(GSH)18 and (III) CdS/Au; (f) DRS results and (g) transformed plots based on the Kubelka–Munk function vs. the energy of light for CdS NWs, CdS/Au25(GSH)18 and CdS/Au. | ||
As depicted in Fig. 1b, the X-ray diffraction (XRD) pattern of the CdS substrate accurately corresponds to the hexagonal phase of CdS (JCPDS no. 41-1049).25 It is observed that the XRD patterns of both CdS/Au25(GSH)18 and CdS/Au heterostructures are similar to that of raw CdS, which is mainly ascribed to the low loading amount of Au25 NCs and Au NYs. Notably, when Au NCs undergo self-transformation into Au NYs, a characteristic diffraction peak attributed to Au NYs appears at 38.3°, indicative of the formation of Au NYs after thermal treatment.22
Fourier transform infrared (FTIR) spectra were examined to confirm the attachment of Au25(GSH)18 NCs or Au NYs on the CdS NWs. As shown in Fig. 1c, all the samples exhibit two broad peaks at 1038 cm−1 and 3439 cm−1, corresponding to the stretching vibration modes of the C–N and N–H functional groups from the CdS NW precursor (Table S1†),25 respectively. In the FTIR spectrum of the CdS/Au25(GSH)18 heterostructure (Fig. 1cII), compared with that of raw CdS NWs (Fig. 1cI), the intensity of these peaks increases gradually due to the overlapping stretching vibration modes of the C–N and N–H bonds from MEA,21 indicating successful MEA integration in the heterostructures.21 Additionally, the FTIR spectrum of CdS/Au25(GSH)18 (Fig. 1cII) shows apparent peaks at 2920 and 1634 cm−1 (Table S1†), corresponding to the C–H and CO functional groups from the GSH ligands.23 The results strongly confirm the successful deposition of Au25(GSH)18 in the CdS/Au25(GSH)18 heterostructure. Furthermore, after calcining the CdS/Au25(GSH)18 heterostructure at 400 °C, the intensity of C–N and N–H bonds of MEA on the CdS NW surface, as well as the C–H and CO functional groups of the GSH ligand are concurrently reduced (Fig. 1cIII).26 This indicates the removal of MEA and GSH ligands and successful transformation of Au25(GSH)18 NCs into Au NYs, resulting in close interfacial contact between CdS NWs and Au NYs.
Survey X-ray photoelectron spectra (Fig. S4†) of all the samples show the expected elements including Cd, S and Au. Fig. S5aI† illustrates the high-resolution Cd 3d spectrum of CdS NWs, which displays the peaks with binding energies (BEs) of 404.4 eV (Cd 3d5/2) and 411.1 eV (Cd 3d3/2), signifying the presence of Cd in the +2 valence state.27 Furthermore, the high-resolution S 2p spectrum of CdS NWs (Fig. S5bI†), which features two peaks at 160.82 eV (S 2p3/2) and 161.96 eV (S 2p1/2), is indicative of S in the −2 valence state.28 Compared with the CdS NW substrate, high-resolution spectra of Cd 3d and S 2p of CdS/Au25(GSH)18 and CdS/Au heterostructures reveal distinct red shifts in the binding energies, which suggests the presence of interfacial and electronic interaction between Au NCs/NYs and the CdS substrate. The high-resolution N 1s signal (Fig. 1d) confirms that the key component, MEA, used in electrostatic self-assembly, was successfully functionalized on the surface of CdS NWs. After the thermal-induced self-conversion, the disappearance of the N 1s signal indicates the removal of MEA for forming a close-contact interface between CdS NWs and Au NYs. TG results also suggest the rapid elimination of MEA upon calcination at 400 °C (Fig. S6†). As depicted in Fig. 1e, the high-resolution Au 4f spectrum of CdS/Au25(GSH)18 exhibits two double peaks corresponding to Au+ (85.5 eV, Au 4f7/2 and 89.0 eV, Au 4f5/2) and metallic Au0 species (84.4 eV, Au 4f7/2 and 88.1 eV, Au 4f5/2)29–31 (Table S2†), respectively, confirming the deposition of Au25(GSH)18 NCs on the CdS NWs.32 The observed shift in the Au 4f peak positions could be attributed to the electronic interactions between Au25(GSH)18 NCs and the CdS surface, which modify the electronic environment of gold atoms. Following calcination at 400 °C to form Au NYs, the Au+ species disappears, leaving only the Au0 species observable on the CdS surface, which agrees with the high-resolution N 1s spectrum and FTIR results.
As shown in Fig. S7,† Raman spectra of CdS NWs and CdS/Au25(GSH)18 exhibit two vibration peaks at 301 and 603 cm−1, corresponding to the longitudinal optical modes (1 LO and 2 LO) of hexagonal CdS,33 respectively. No peaks attributed to Au25(GSH)18 NCs are observed. Furthermore, after the successful self-assembly of Au25(GSH)18 NCs, these peaks are significantly blue-shifted, indicating the strong electronic interaction.
UV-vis diffuse reflectance spectroscopy (DRS) results of CdS NWs, CdS/Au25(GSH)18, and CdS/Au heterostructures imply that deposition of Au25(GSH)18 NCs and Au NYs enhances the light-harvesting ability of CdS NWs in the visible light region (Fig. 1f). As shown in Fig. 1g, band gaps of CdS NWs, CdS/Au25(GSH)18, and CdS/Au heterostructures are approximately 2.40, 2.39, and 2.39 eV, respectively. The results indicate that although deposition of Au25(GSH)18 NCs and Au NYs significantly enhances the light absorption of CdS NWs, while the absorption band edge of nanocomposites remains unaffected.
As shown in Fig. S8 and Table S3,† all the samples exhibit type-IV isotherms.34 The specific surface areas of CdS, CdS/Au25(GSH)18, and CdS/Au heterostructures are measured to be 19.55, 18.60, and 16.32 m2 g−1, respectively. The slight reduction in the specific surface area of the CdS/Au heterostructure compared with that of CdS NWs and CdS/Au25(GSH)18 suggests that Au NY deposition does not significantly impact the specific surface area of CdS NWs.
As shown in Fig. 2a, the field emission scanning electron microscopy (FESEM) image of CdS NWs exhibit a typical 1D morphology. Compared with zero-dimensional (0D) and two-dimensional (2D) structures, 1D CdS NWs enable faster electron transport along the nanowire direction, thereby more efficiently reducing electron–hole recombination.35 After functionalizing CdS NWs with Au25(GSH)18 NCs, SEM (Fig. 2b) and transmission electron microscopy (TEM, Fig. 2d) images of the CdS/Au25(GSH)18 heterostructure indicate that it retains a morphology similar to that of pristine CdS NWs, owing to the ultra-small size of the Au25(GSH)18 NCs. The high-resolution TEM (HRTEM) image of the CdS/Au25(GSH)18 heterostructure (Fig. 2d) shows no lattice spacing of Au25(GSH)18 NCs, attributed to its amorphous properties. Upon annealing, which enables the transformation of Au25(GSH)18 NCs into Au NYs, the morphology of the CdS/Au25(GSH)18 heterostructure remains largely unchanged, although the surface becomes rougher (Fig. 2c). Notably, small, evenly distributed dots on the CdS NW surface correspond to the Au NYs. Consistent with the SEM image, the TEM image of the CdS/Au heterostructure (Fig. 2e) confirms the uniform distribution of Au NYs on the CdS NW surface. Consistently, the HRTEM image of the CdS/Au heterostructure (Fig. 2f) shows the lattice spacings of 0.34 nm and 0.225 nm, corresponding to the (002) and (111) crystal planes of CdS and Au NYs, respectively.22,36 Additionally, elemental mapping results (EDS) further verify the deposition of Au NYs on the CdS NWs. As shown in Fig. 2g–k, uniform distribution patterns of Cd, S, and Au elements confirm the successful Au NC deposition. This uniform dispersion prevents agglomeration and enhances the utilization of active sites.
 | ||
| Fig. 2 Panoramic FESEM images of (a) CdS NWs, with a high-magnification image in the inset; (b and c) high-magnification FESEM images of CdS/Au25(GSH)18 and CdS/Au heterostructures; TEM images of (d) CdS/Au25(GSH)18 with the corresponding HRTEM image in the inset; (e) low and (f) HRTEM images of the CdS/Au heterostructure with a low magnification TEM image in the inset; (g) FESEM image of CdS/Au with (h–k) elemental mapping results. | ||
Photocatalytic activities of pristine CdS, CdS/Au25(GSH)18, and x-CdS/Au (x = 200, 300, 400 and 500 °C) heterostructures were evaluated through the photoreduction of 4-nitroaniline (4-NA) to 4-phenylenediamine (4-PDA) in aqueous solution under visible light irradiation (λ > 420 nm) with ammonium formate as a quencher for photogenerated holes and N2 purging under ambient conditions.37 Control experiments without light or a catalyst exhibit negligible photoactivities (Fig. S9†), confirming the photocatalytic nature of the reaction. Fig. 3a shows that photoactivity of the CdS/Au25(GSH)18 heterostructure remains largely unchanged compared with that of pristine CdS NWs, and both of them show low photoactivities, indicating that Au25(GSH)18 NC deposition on the CdS substrate is insufficient to stimulate the photoreduction of nitro compounds. Notably, x-CdS/Au heterostructures, when calcined at different temperature, demonstrate significantly superior photoactivities compared with both blank CdS and the CdS/Au25(GSH)18 heterostructure in the reduction of 4-NA under visible light. This enhancement underscores the crucial role of Au NYs in boosting the photoactivity. As shown in Fig. 3a, photoactivity of x-CdS/Au heterostructures progressively increases on increasing the calcination temperature. This enhancement is chiefly attributed to the progressive removal of GSH ligands from the surface of Au25(GSH)18 NCs and self-transformation of Au25(GSH)18 NCs into Au NYs on increasing the annealing temperature. This facilitates the formation of Schottky junctions, thereby significantly enhancing the photoactivity of the CdS/Au heterostructure. The calcination temperature profoundly influences the photocatalytic activity, with optimal CdS/Au achieving a 100% conversion rate within 10 min, and it is about 20 times larger than that of blank CdS and the CdS/Au25(GSH)18 heterostructure. These findings reveal that the Au NYs transformed from Au25(GSH)18 NCs play an indispensable role in enhancing the photoreduction performance of the x-CdS/Au heterostructures for efficiently capturing electrons photoexcited from the CdS NWs, while excessively high temperature (>400 °C) diminishes the photoactivity probably due to Au NY aggregation, obstructing the active sites. Consequently, the CdS/Au and CdS/Au25(GSH)18 heterostructure was identified as the best sample for the subsequent systematic comparison. As exhibited in Fig. 3b–j, apart from 4-NA, similar results were also observed in the photoreduction of other aromatic nitro compounds over CdS, CdS/Au25(GSH)18 and CdS/Au heterostructures including 3-nitroaniline (3-NA), 2-nitroaniline (2-NA), 4-nitrophenol (4-NP), 3-nitrophenol (3-NP), 2-nitrophenol (2-NP), 4-nitrotoluene (4-NT), 4-nitroanisole, 1-chloro-4-nitrobenzene, and 1-bromo-4-nitrobenzene. The results demonstrate that the CdS/Au heterostructure consistently exhibits superior photoactivity compared with both CdS and CdS/Au25(GSH)18 counterparts under identical conditions, with the latter two showing similar photoactivities. Fig. S10† shows that addition of K2S2O8 as an electron scavenger significantly reduces the photoactivity of the CdS/Au heterostructure, indicating that the photoreduction reaction is driven by electrons. Fig. S11† shows a distinct peak from 400 to 550 nm in the action spectrum of the CdS/Au heterostructure under different monochromatic light irradiation, underscoring the dominant role of band-gap-photoexcitation of the CdS NW matrix. Besides the photocatalytic performance, stability of the catalyst is also crucial for practical application. The recyclability of the CdS/Au heterostructure in the photoreduction of 4-NA was evaluated through six consecutive cyclic reactions under identical conditions. Fig. S10b† demonstrates that the CdS/Au heterostructure retains about 95% of its initial photoactivity, exhibiting negligible deactivation. This stability is further confirmed by FTIR and XPS analysis (Fig. S12–S14†), which show no significant changes in the crystal structure, elemental chemical state, and morphology of CdS/Au after repeated cycles, verifying its excellent photostability.
 | ||
| Fig. 3 (a) Photoactivities of blank CdS NWs, CdS/Au25(GSH)18 and x-CdS/Au (x = 200, 300, 400 and 500 °C) heterostructures toward the photoreduction of 4-NA with the addition of ammonium formate as a hole scavenger and N2 bubbling under ambient conditions and visible light irradiation (λ > 420 nm). Photocatalytic reduction of aromatic nitro compounds including (b) 3-NA, (c) 2-NA, (d) 4-NP, (e) 3-NP, (f) 2-NP, (g) 4-NT, (h) 4-nitroanisole, (i) 1-chloro-4-nitrobenzene and (j) 1-bromo-4-nitrobenzene over the samples under the same experimental conditions. (k) Photoreduction reaction model under the current experimental conditions. | ||
Photocatalytic hydrogen evolution performances of pristine CdS, CdS/Au25(GSH)18 and CdS/Au heterostructures were also evaluated. As displayed in Fig. S15,† the CdS/Au25(GSH)18 heterostructure shows an enhanced H2 evolution rate of 2.3 mmol g−1 h−1, approximately 9 times larger than that of blank CdS NWs (0.25 mmol g−1 h−1) under 2 h of visible light irradiation, which results from the advantageous photosensitization effect of Au25(GSH)18 NCs. In contrast, the CdS/Au heterostructure demonstrates an even more impressive H2 evolution rate of 5.2 mmol g−1 h−1, about 21 and 2 times larger than those of pure CdS NWs and the CdS/Au25(GSH)18 heterostructure, respectively. Noteworthily, the CdS/Au heterostructure consistently exhibits substantially higher photocatalytic H2 generation performance compared with its CdS/Au25(GSH)18 counterpart, strongly highlighting the superiority of metallic Au NYs over atomically precise Au25(GSH)18 NCs in enhancing the solar-to-hydrogen conversion efficiency.
Apart from the photoactivities toward reduction of aromatic nitro compounds and photocatalytic hydrogen evolution, photocatalytic selective oxidation of aromatic alcohols to their corresponding aldehydes under visible light was also carried out to gain deeper understanding on the roles of Au NCs and Au NYs in modulating interfacial charge transfer. Blank experiments in the absence of a catalyst or light irradiation suggest that no conversion of alcohols occurred (Fig. S16†), strongly confirming that the reaction is driven by a photocatalytic process. As shown in Fig. 4a–i, photooxidation activity of the CdS/Au25(GSH)18 heterostructure is significantly enhanced compared with that of CdS NWs, owing to the photosensitization effect of Au25(GSH)18 NCs. Notably, the CdS/Au heterostructure exhibits superior photoactivity in the selective oxidation of aromatic alcohols to aldehydes, achieving both high conversion and selectivity (75% to 100%). In contrast, blank CdS NWs display very inferior photoactivity, likely due to its ultra-fast charge recombination rate. Calcination enables the self-conversion of Au25(GSH)18 NCs to Au NYs with the same metal loading and removes the coverage of the GSH ligand, increases the interface contact, and forms a Schottky junction with strong electron-withdrawing ability between CdS NWs and Au NYs, thereby enhancing the conversion rate and selectivity of aromatic alcohol photoreduction. Consequently, the calcined CdS/Au heterostructure demonstrates optimal photoactivity for photocatalytic selective oxidation of aromatic alcohols, far exceeding that of blank CdS NWs and the CdS/Au25(GSH)18 heterostructure. Interestingly, the CdS/Au25(GSH)18 heterostructure shows different performances for enhancing the photocatalytic selective oxidation of aromatic alcohols and reduction of aromatic nitro compounds. In other words, the photosensitization effect of Au25(GSH)18 NCs in the CdS/Au25(GSH)18 heterostructure can enhance the photooxidation performance but does not foster nitro compound reduction. Upon the formation of Au NYs, electron-withdrawing ability of Schottky junctions is greatly stimulated, enhancing charge separation. Therefore, transforming Au25(GSH)18 NCs into Au NYs results in a significantly better Schottky junction effect compared with the photosensitization effect.
 | ||
| Fig. 4 Photoactivities of (I) blank CdS NWs, (II) CdS/Au25(GSH)18 and (III) CdS/Au toward the selective oxidation of aromatic alcohols including (a) benzyl alcohol, (b) p-methylbenzyl alcohol, (c) p-methoxybenzyl alcohol, (d) p-fluorobenzyl alcohol, (e) p-chlorobenzyl alcohol, (f) p-nitrobenzyl alcohol, (g) cinnamyl alcohol, and (h) 3-methyl-2-buten-1-ol, to their corresponding aldehydes under visible light irradiation (λ > 420 nm) for 4 h. (i) Photooxidation reaction model under the current experimental conditions. | ||
The dominant active species responsible for the photocatalytic selective oxidation performances of CdS/Au heterostructures were determined by quenching experiments through adding different scavengers. Fig. S17† shows the photoactivities of the CdS/Au heterostructure with the addition of benzoquinone (BQ), tert-butyl alcohol (TBA), triethanolamine (TEOA) and silver nitrate (AgNO3), which are used as scavengers for trapping superoxide radicals  hydroxyl radicals (OH·), holes (h+) and electrons (e−), respectively.38 It is evident that photooxidative activity of CdS/Au was substantially inhibited upon the addition of these four scavengers, indicating that OH·, h+, and e− are all the key active species in the photocatalytic selective oxidation of alcohols. Among these, TEOA demonstrates the most pronounced inhibitory effect, suggesting that h+ is the dominant active species for photooxidative performance. The contribution of the active species follows the order: h+ > ·OH > e− >
hydroxyl radicals (OH·), holes (h+) and electrons (e−), respectively.38 It is evident that photooxidative activity of CdS/Au was substantially inhibited upon the addition of these four scavengers, indicating that OH·, h+, and e− are all the key active species in the photocatalytic selective oxidation of alcohols. Among these, TEOA demonstrates the most pronounced inhibitory effect, suggesting that h+ is the dominant active species for photooxidative performance. The contribution of the active species follows the order: h+ > ·OH > e− >  Notably,
Notably,  radicals primarily originate from the trapping of photogenerated electrons by oxygen dissolved in the reaction system.39
radicals primarily originate from the trapping of photogenerated electrons by oxygen dissolved in the reaction system.39
Photoelectrochemical (PEC) measurements were performed under visible light irradiation to evaluate the interfacial charge separation efficiency.40 As shown in Fig. 5a, the CdS NW substrate demonstrates a relatively low photocurrent density of ca. 5 μA cm−2, which is lower than that of the CdS/Au25(GSH)18 heterostructure. The improved photocurrent of the CdS/Au25(GSH)18 heterostructure relative to that of pristine CdS verifies the photosensitization effect of Au25(GSH)18 NCs. Note that the CdS/Au heterostructure shows considerably enhanced photocurrent in comparison with those of the CdS/Au25(GSH)18 heterostructure and pristine CdS, implying the superiority of Au NYs to Au25(GSH)18 NCs in boosting charge separation. The photocurrent follows the order of CdS/Au > CdS/Au25(GSH)18 > CdS NWs. Furthermore, as shown in Fig. 5b, EIS Nyquist plots of the CdS/Au heterostructure reveal smaller semicircular arc radii and lower charge transfer resistance (Rct) compared with that of the CdS/Au25(GSH)18 heterostructure and pristine CdS, which indicates that the Schottky junction formed by Au NYs and CdS NWs significantly improves the interfacial charge separation efficiency compared with that of interface formed between Au25(GSH)18 NCs and CdS. Open-circuit voltage decay (OCVD) (Fig. 5c) and the corresponding electron lifetime analysis (Fig. 5d) were further utilized to probe the interfacial charge transfer kinetics of the samples. As shown in Fig. 5c, the CdS/Au heterostructure exhibits the largest open-circuit voltage (Voc) relative to that of CdS/Au25(GSH)18 and blank CdS, which discloses that the CdS/Au heterostructure demonstrates optimal charge separation efficiency, consistent with the photocurrent results (Fig. 5a). According to the OCVD result after turning off the light irradiation, the average photoelectrons lifetime (τn) is evaluated. Consistent with the photovoltage result, as shown in Fig. 5d, CdS/Au demonstrates a more prolonged electron lifetime relative to that of the CdS/Au25(GSH)18 heterostructure and pristine CdS. To determine the carrier density, Mott–Schottky (M–S) analysis was conducted (Fig. 5e). The CdS/Au heterostructure exhibits the largest carrier density of 15.85 × 1019 cm−3 (Fig. 5f), markedly surpassing those of the CdS/Au25(GSH)18 heterostructure (8.16 × 1019 cm−3) and raw CdS (3.56 × 1019 cm−3). These findings, along with the results from photocurrent, EIS, OCVD, and electron lifetime measurements, validate the enhanced optimal photoactivity of the CdS/Au heterostructure. Consequently, PEC results confirm that the substantial improvement in the photoactivity of the CdS/Au heterostructure is primarily due to the significantly enhanced charge separation afforded by the Schottky-type interface. Furthermore, compared with CdS NWs and the CdS/Au25(GSH)18 heterostructure, the CdS/Au heterostructure consistently shows the lowest photoluminescence (PL) intensity in comparison with that of pristine CdS and the CdS/Au25(GSH)18 heterostructure (Fig. S18†), indicating that charge carrier recombination over the CdS/Au heterostructure has been effectively suppressed. The synergistic effect of these components in the CdS/Au heterostructure results in efficient charge separation efficiency, leading to superior photocatalytic and PEC performances.
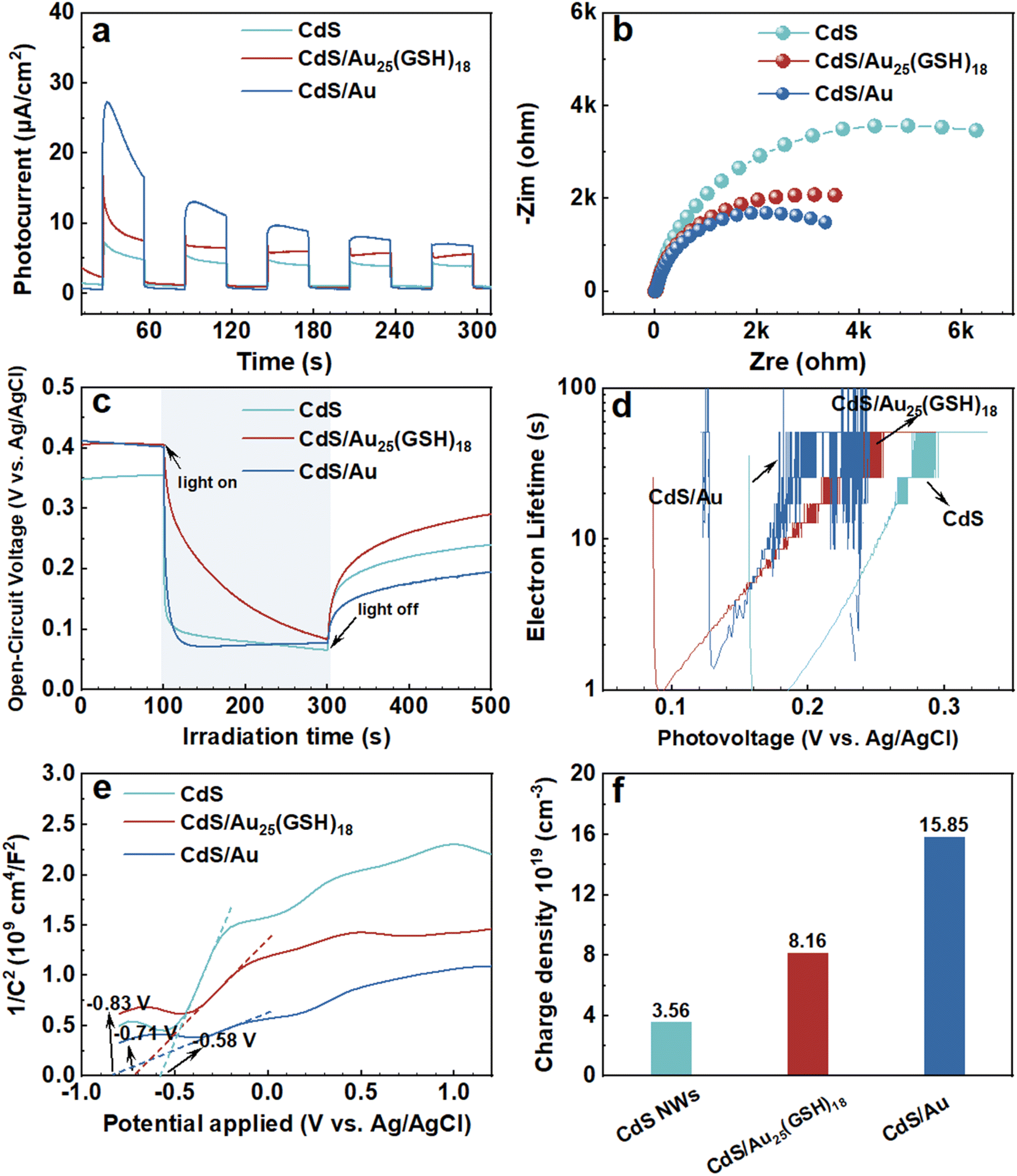 | ||
| Fig. 5 PEC water splitting performances of CdS NWs, CdS/Au25(GSH)18 and CdS/Au heterostructures in Na2SO4 aqueous solution (pH = 6.69) under visible light irradiation (λ > 420 nm) including (a) transient photocurrent responses (bias: 0.61 V vs. RHE), and (b) EIS results, (c) OCVD & (d) electron lifetime, and (e) Mott–Schottky plots & (f) charge carrier density (ND). | ||
Based on the above analysis, photocatalytic mechanisms of CdS/Au25(GSH)18 and CdS/Au heterostructures are schematically illustrated in Scheme 1. CV results of Au25(GSH)18 NCs, as shown in Fig. S19,† reveal a LUMO energy level of −0.76 V vs. NHE, derived from the band gap (2.66 eV) and HOMO energy level (+1.90 V vs. NHE) (Fig. S3†).41 Under visible light irradiation, Au25(GSH)18 NCs act as a photosensitizer, and are rapidly photoexcited to generate electron–hole pairs due to their favorable band gap. On the other hand, the Mott–Schottky analysis (Fig. S20†) indicates that the flat band potential of CdS NWs is −0.45 V vs. NHE. In n-type semiconductors, the conduction band edge typically aligns with the flat band potential. Drawing from the DRS data, the valence band position of CdS NWs is inferred to be +1.95 V vs. NHE (Eg = VVB − VCB).42 This suitable energy level alignment and close interfacial contact between CdS NWs and Au25(GSH)18 NCs allow electrons to be transferred from the LUMO level of Au25(GSH)18 NCs to the conduction band (CB) of CdS NWs passing through the interfacial ultra-thin MEA interim layer. At the same time, holes are transferred from the valence band (VB) of CdS to the HOMO level of the Au25(GSH)18 NCs. For the photocatalytic reduction of aromatic nitro compounds over the CdS/Au25(GSH)18 heterostructure, photoelectrons generated by Au25(GSH)18 NCs alone cannot efficiently drive the reduction of nitroaromatic compounds, which require multi-electron participation. However, these electrons can effectively reduce water to produce hydrogen. Simultaneously, holes are transferred from the VB of CdS NWs to the HOMO level of Au25(GSH)18 NCs, enabling efficient photocatalytic selective oxidation of aromatic alcohols to aldehydes. Consequently, charge separation over Au25(GSH)18 NCs is significantly enhanced through proper energy level alignment, leading to substantial improvements in both photocatalytic hydrogen production and the selective oxidation of aromatic alcohols over the CdS/Au25(GSH)18 heterostructure.
 | ||
| Scheme 1 Schematic illustration depicting the photoredox mechanisms of CdS/Au25(GSH)18 and CdS/Au heterostructures. | ||
For the CdS/Au photosystem, Au NYs self-transformed from Au25(GSH)18 NCs serve as electron-withdrawing mediators to trap the electrons from the CB of CdS, by which charge separation over CdS was considerably enhanced. Owing to the Schottky junction-driven charge flow initiated by the Au NYs, electron transport from the CB of CdS NWs to the Au NYs occurs spontaneously and efficiently, and then electrons migrate to the active sites to trigger the photoreduction reaction. Based on our experimental results, we speculate that the electrons captured by the Au NYs further boost the photoreduction of nitro compounds and improve the photocatalytic hydrogen production efficiency, surpassing those of the CdS/Au25(GSH)18 heterostructure. Alternatively, the Schottky-type electron-capturing effect of Au NYs also significantly contributes to the photocatalytic selective oxidation of aromatic alcohols to their corresponding aldehydes. That is, the Schottky junction formed in CdS/Au significantly enhances the separation efficiency of photogenerated charge carriers. In terms of the photocatalytic selective oxidation of aromatic alcohols, the electrons captured by Au NYs combine with dissolved O2 to generate a substantial amount of superoxide radicals  within the reaction system,43 which act as the main active species, enabling the direct oxidation of aromatic alcohols to aldehydes. Simultaneously, holes (h+) exhibit strong oxidizing properties and thus directly oxidize H2O or hydroxide ions (OH−) adsorbed on the surface of the photocatalyst to generate hydroxyl radicals (OH·).44,45 These holes along with the hydroxyl radicals directly oxidize the alcohols, thereby enhancing the overall photocatalytic oxidation performances.
within the reaction system,43 which act as the main active species, enabling the direct oxidation of aromatic alcohols to aldehydes. Simultaneously, holes (h+) exhibit strong oxidizing properties and thus directly oxidize H2O or hydroxide ions (OH−) adsorbed on the surface of the photocatalyst to generate hydroxyl radicals (OH·).44,45 These holes along with the hydroxyl radicals directly oxidize the alcohols, thereby enhancing the overall photocatalytic oxidation performances.
4. Conclusions
In summary, this study provides a comprehensive investigation into the thermally induced self-transformation of atomically precise Au25(GSH)18 NCs into Au NYs within composite photosystems and their distinct roles in photocatalytic redox. Our results reveal that while Au25(GSH)18 NCs exhibit notable photosensitization effects due to their favorable energy-level alignment, the Schottky-type electron-withdrawing ability of the resulting Au NYs significantly surpasses this effect, leading to enhanced charge separation and superior photocatalytic performance. The transformation from metal NCs into NYs not only provides a strategic pathway for optimizing photocatalytic systems but also offers new insights into the inherent instability of metal NCs as a functional advantage rather than a limitation. This study enriches our fundamental understanding of the charge transfer mechanisms in metal NC-based artificial photosystems, paving the way for the development of more efficient photocatalytic materials for solar energy conversion.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Yu-Bing Li performed the experiments, analyzed all data, and drafted the manuscript. Fang-Xing Xiao guided this work and corrected the manuscript. All the authors contributed to a critical discussion on the data and the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support by the award Program for Minjiang Scholar Professorship is greatly acknowledged. Financial support from the National Natural Science Foundation of China (No. 21703038 and 22072025) is also acknowledged.References
- F.-X. Xiao, Z. Zeng and B. Liu, Bridging the Gap: Electron Relay and Plasmonic Sensitization of Metal Nanocrystals for Metal Clusters, J. Am. Chem. Soc., 2015, 137, 10735–10744 CrossRef CAS PubMed.
- T. Oshima, D. Lu, O. Ishitani and K. Maeda, Intercalation of highly dispersed metal nanoclusters into a layered metal oxide for photocatalytic overall water splitting, Angew. Chem., Int. Ed., 2015, 127, 2736–2740 CrossRef.
- Q.-L. Mo, B.-J. Liu and F.-X. Xiao, Solar-powered photocatalysis and photoelectrocatalysis over atomically precise metal nanoclusters, J. Phys. Chem. C, 2021, 125, 22421–22428 CrossRef CAS.
- Y. Liu, Y. Wang and N. Pinna, Atomically Precise Metal Nanoclusters for Photocatalytic Water Splitting, ACS Mater. Lett., 2024, 6, 2995–3006 CrossRef CAS.
- X.-Y. Fu, Z.-Q. Wei, S. Xu, X. Lin, S. Hou and F.-X. Xiao, Maneuvering intrinsic instability of metal nanoclusters for boosted solar-powered hydrogen production, J. Phys. Chem. Lett., 2020, 11, 9138–9143 CrossRef CAS PubMed.
- H. Liang, B.-J. Liu, B. Tang, S.-C. Zhu, S. Li, X.-Z. Ge, J.-L. Li, J.-R. Zhu and F.-X. Xiao, Atomically precise metal nanocluster-mediated photocatalysis, ACS Catal., 2022, 12, 4216–4226 CrossRef CAS.
- X. Cui, J. Wang, B. Liu, S. Ling, R. Long and Y. Xiong, Turning Au nanoclusters catalytically active for visible-light-driven CO2 reduction through bridging ligands, J. Am. Chem. Soc., 2018, 140, 16514–16520 CrossRef CAS PubMed.
- Y.-S. Chen and P. V. Kamat, Glutathione-capped gold nanoclusters as photosensitizers. Visible light-induced hydrogen generation in neutral water, J. Am. Chem. Soc., 2014, 136, 6075–6082 CrossRef CAS PubMed.
- Y. Sun, X. Cai, W. Hu, X. Liu and Y. Zhu, Electrocatalytic and photocatalytic applications of atomically precise gold-based nanoclusters, Sci. China:Chem., 2021, 64, 1065–1075 CrossRef CAS.
- R. Khan, M. H. Naveen, M. A. Abbas, J. Lee, H. Kim and J. H. Bang, Photoelectrochemistry of Au nanocluster-sensitized TiO2: intricacy arising from the light-induced transformation of nanoclusters into nanoparticles, ACS Energy Lett., 2020, 6, 24–32 CrossRef.
- Y. Liu, E. Wierzbicka, A. Springer, N. Pinna and Y. Wang, Influence of the Electronic Properties of the Ligand on the Photoelectrochemical Behavior of Au25 Nanocluster-Sensitized TiO2 Photoanode, J. Phys. Chem. C, 2022, 126, 1778–1784 CrossRef CAS.
- S. Li, S. Fu, Y. Gong, N. Wu, L. Wang and L. Wang, Design of Support Effect in the Catalytic Application of Ligand-Protect Gold Clusters, Chin. J. Chem., 2024, 42, 73–86 CrossRef CAS.
- Q.-L. Mo, X. Lin, Z.-Q. Wei, X.-C. Dai, S. Hou, T. Li and F.-X. Xiao, All-in-one: branched macromolecule-protected metal nanocrystals as integrated charge separation/motion centers for enhanced photocatalytic selective organic transformations, J. Mater. Chem. A, 2020, 8, 16392–16404 RSC.
- M. Gharib, A. Kornowski, H. Noei, W. J. Parak and I. Chakraborty, Protein-Protected Porous Bimetallic AgPt Nanoparticles with pH-Switchable Peroxidase/Catalase-Mimicking Activity, ACS Mater. Lett., 2019, 1, 310–319 CrossRef CAS.
- J. Huang, W. Niu, C. Li, C. Tan, P. Yin, H. Cheng, Z. Hu, N. Yang, Q. He, G.-H. Nam and H. Zhang, In Situ Probing of Crystal-Phase-Dependent Photocatalytic Activities of Au Nanostructures by Surface-Enhanced Raman Spectroscopy, ACS Mater. Lett., 2020, 2, 409–414 CrossRef CAS.
- I. V. Lightcap, S. Murphy, T. Schumer and P. V. Kamat, Electron Hopping Through Single-to-Few-Layer Graphene Oxide Films. Side-Selective Photocatalytic Deposition of Metal Nanoparticles, J. Phys. Chem. Lett., 2012, 3, 1453–1458 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Atomically Precise Colloidal Metal Nanoclusters and Nanoparticles: Fundamentals and Opportunities, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- M.-H. Huang, Y.-B. Li, T. Li, X.-C. Dai, S. Hou, Y. He, G. Xiao and F.-X. Xiao, Self-transformation of ultra-small gold nanoclusters to gold nanocrystals toward boosted photoreduction catalysis, Chem. Commun., 2019, 55, 10591–10594 RSC.
- S. Liu, W. Xu, Y. Niu, B. Zhang, L. Zheng, W. Liu, L. Li and J. Wang, Ultrastable Au nanoparticles on titania through an encapsulation strategy under oxidative atmosphere, Nat. Commun., 2019, 10, 5790 CrossRef CAS.
- Y.-B. Li, T. Li, X.-C. Dai, M.-H. Huang, Y. He, G. Xiao and F.-X. Xiao, Cascade charge transfer mediated by in situ interface modulation toward solar hydrogen production, J. Mater. Chem. A, 2019, 7, 8938–8951 RSC.
- X. Yan, X.-Y. Fu and F.-X. Xiao, Filling the Gap: Atomically Precise Metal Nanoclusters-Induced Z-Scheme Photosystem toward Robust and Stable Solar Hydrogen Generation, Adv. Funct. Mater., 2023, 33, 2303737 CrossRef CAS.
- T. Li, M.-H. Huang, Y.-B. Li, X.-C. Dai, Y. He, G. Xiao and F.-X. Xiao, General self-assembly of metal/metal chalcogenide heterostructures initiated by a surface linker: modulating tunable charge flow toward versatile photoredox catalysis, J. Mater. Chem. A, 2019, 7, 21182–21194 RSC.
- Z.-Q. Wei, S. Hou, X. Lin, S. Xu, X.-C. Dai, Y.-H. Li, J.-Y. Li, F.-X. Xiao and Y.-J. Xu, Unexpected Boosted Solar Water Oxidation by Nonconjugated Polymer-Mediated Tandem Charge Transfer, J. Am. Chem. Soc., 2020, 142, 21899–21912 CrossRef CAS PubMed.
- Y.-B. Li, T. Li, X.-C. Dai, M.-H. Huang, S. Hou, X.-Y. Fu, Z.-Q. Wei, Y. He, G. Xiao and F.-X. Xiao, Precise Tuning of Coordination Positions for Transition-Metal Ions via Layer-by-Layer Assembly To Enhance Solar Hydrogen Production, ACS Appl. Mater. Interfaces, 2020, 12, 4373–4384 CrossRef CAS PubMed.
- K. Wang, X.-Z. Ge, Q.-L. Mo, X. Yan, Y. Xiao, G. Wu, S.-R. Xu, J.-L. Li, Z.-X. Chen and F.-X. Xiao, Steering bi-directional charge transfer via non-conjugated insulating polymer, J. Catal., 2022, 416, 92–102 CrossRef CAS.
- Z.-Q. Wei and F.-X. Xiao, Photosensitization Efficiency Modulation of Atomically Precise Silver Nanoclusters for Photoelectrocatalysis, Inorg. Chem., 2023, 62, 6138–6146 CrossRef CAS.
- Q. Chen, X.-Z. Ge, L. Yu and F.-X. Xiao, Atomically Precise Metal Nanocluster Photosystem: Electron Relay Boosts Photocatalytic Organic Transformation, Inorg. Chem., 2023, 62, 19358–19365 CrossRef CAS PubMed.
- X.-Z. Ge, K. Wang, Q.-L. Mo, Y. Xiao, J.-L. Li, G. Wu, S.-R. Xu and F.-X. Xiao, Non-conjugated polymer ligand: stimulating charge transfer towards photocatalytic selective organic transformation, Catal. Sci. Technol., 2023, 13, 479–489 RSC.
- S. Yang, S. Chen, L. Xiong, C. Liu, H. Yu, S. Wang, N. L. Rosi, Y. Pei and M. Zhu, Total structure determination of Au16(S-Adm)12 and Cd1Au14(StBu)12 and implications for the structure of Au15(SR)13, J. Am. Chem. Soc., 2018, 140, 10988–10994 CrossRef CAS PubMed.
- S. Yang, J. Chai, Y. Song, J. Fan, T. Chen, S. Wang, H. Yu, X. Li and M. Zhu, In situ two-phase ligand exchange: a new method for the synthesis of alloy nanoclusters with precise atomic structures, J. Am. Chem. Soc., 2017, 139, 5668–5671 CrossRef CAS PubMed.
- Y. Tan, G. Sun, T. Jiang, D. Liu, Q. Li, S. Yang, J. Chai, S. Gao, H. Yu and M. Zhu, Symmetry Breaking Enhancing the Activity of Electrocatalytic CO2 Reduction on an Icosahedron-Kernel Cluster by Cu Atoms Regulation, Angew. Chem., Int. Ed., 2024, 63, e202317471 CrossRef CAS PubMed.
- Q.-L. Mo, X.-C. Dai, Y. Xiao and F.-X. Xiao, Atomically precise Au25(GSH)18 nanoclusters versus plasmonic Au nanocrystals: evaluating charge impetus in solar water oxidation, Chin. Chem. Lett., 2023, 34, 107901 CrossRef CAS.
- Q.-L. Mo, X.-Y. Fu, K. Wang, X.-Z. Ge, S. Hou, B.-J. Liu and F.-X. Xiao, Precise interface modulation cascade enables unidirectional charge transport, J. Catal., 2022, 410, 31–41 CrossRef CAS.
- H.-J. Lin, S. Xu, X.-Y. Fu, Z.-Q. Wei, M.-H. Huang, X. Lin, Y. He, G. Xiao and F.-X. Xiao, Layer-by-Layer Self-Assembly of Metal/Metal Oxide Superstructures: Self-Etching Enables Boosted Photoredox Catalysis, Inorg. Chem., 2020, 59, 4129–4139 CrossRef CAS PubMed.
- T. Li, Y.-B. Li, X.-C. Dai, M.-H. Huang, Y. He, G. Xiao and F.-X. Xiao, Ligand-Triggered Tunable Charge Transfer toward Multifarious Photoreduction Catalysis, J. Phys. Chem. C, 2019, 123, 4701–4714 CrossRef CAS.
- Z. Xu, W. Yue, C. Li, L. Wang, Y. Xu, Z. Ye and J. Zhang, Rational synthesis of Au–CdS composite photocatalysts for broad-spectrum photocatalytic hydrogen evolution, ACS Nano, 2023, 17, 11655–11664 CrossRef CAS PubMed.
- S.-C. Zhu, S. Li, B. Tang, H. Liang, B.-J. Liu, G. Xiao and F.-X. Xiao, MXene-motivated accelerated charge transfer over TMCs quantum dots for solar-powered photoreduction catalysis, J. Catal., 2021, 404, 56–66 CrossRef CAS.
- M.-H. Huang, X.-C. Dai, T. Li, Y.-B. Li, Y. He, G. Xiao and F.-X. Xiao, Stimulating Charge Transfer Over Quantum Dots via Ligand-Triggered Layer-by-Layer Assembly toward Multifarious Photoredox Organic Transformation, J. Phys. Chem. C, 2019, 123, 9721–9734 CrossRef CAS.
- F.-X. Xiao, J. Miao and B. Liu, Layer-by-Layer Self-Assembly of CdS Quantum Dots/Graphene Nanosheets Hybrid Films for Photoelectrochemical and Photocatalytic Applications, J. Am. Chem. Soc., 2014, 136, 1559–1569 CrossRef CAS PubMed.
- Z.-Q. Wei, S. Hou, S.-C. Zhu, Y. Xiao, G. Wu and F.-X. Xiao, Polymer-Mediated Electron Tunneling Towards Solar Water Oxidation, Adv. Funct. Mater., 2022, 32, 2106338 CrossRef CAS.
- Y.-B. Li and F.-X. Xiao, Tuning the photosensitization efficiency of atomically precise metal nanoclusters by super-efficient and exquisite interface modulation, J. Mater. Chem. A, 2023, 11, 589–599 RSC.
- Z.-Y. Li, Y.-H. Chen, J.-R. Zhu, Q. Chen, S.-J. Lu and F.-X. Xiao, Self-Transformation of Atomically Precise Alloy Nanoclusters to Plasmonic Alloy Nanocrystals: Evaluating Photosensitization in Solar Water Oxidation, Inorg. Chem., 2023, 62, 16965–16973 CrossRef CAS PubMed.
- M. Li, P. Wang, Z. Ji, Z. Zhou, Y. Xia, Y. Li and S. Zhan, Efficient photocatalytic oxygen activation by oxygen-vacancy-rich CeO2-based heterojunctions: synergistic effect of photoexcited electrons transfer and oxygen chemisorption, Appl. Catal., B, 2021, 289, 120020 CrossRef CAS.
- S. Xu, M.-H. Huang, T. Li, Z.-Q. Wei, X. Lin, X.-C. Dai, S. Hou, X.-Y. Fu and F.-X. Xiao, Modulating charge migration in photoredox organic transformation via exquisite interface engineering, J. Mater. Chem. A, 2020, 8, 8360–8375 RSC.
- Y. Wang, X. Li, S. Liu, Y. Liu, T. Kong, H. Zhang, X. Duan, C. Chen and S. Wang, Roles of catalyst structure and gas surface reaction in the generation of hydroxyl radicals for photocatalytic oxidation, ACS Catal., 2022, 12, 2770–2780 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06256f |
| This journal is © The Royal Society of Chemistry 2025 |