
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the factors governing the ammonia oxidation reaction by a mononuclear ruthenium complex†
Guo
Chen‡
 ab,
Xiao-Lv
Ding‡
a,
Piao
He
a,
Tao
Cheng
a,
Yang
Chen
c,
Jian
Lin
c,
Xi
Zhang
a,
Shan
Zhao
a,
Na
Qiao
a and
Xiao-Yi
Yi
*a
ab,
Xiao-Lv
Ding‡
a,
Piao
He
a,
Tao
Cheng
a,
Yang
Chen
c,
Jian
Lin
c,
Xi
Zhang
a,
Shan
Zhao
a,
Na
Qiao
a and
Xiao-Yi
Yi
*a
aCollege of Chemistry and Chemical Engineering, Central South University, Changsha, Hunan 410083, P. R. China. E-mail: xyyi@csu.edu.cn
bSchool of Chemistry and Chemical Engineering, Southwest University, Chongqing, 400715, P. R. China
cCAS Key Laboratory of Science and Technology on Applied Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, P. R. China
First published on 19th March 2025
Abstract
Precise regulation of the active site of molecular catalysts is appealing because it could provide insights into the catalytic mechanism and possibly provide a new strategy for catalyst design. A ruthenium complex, [Ru(dppMe, COMe)(bipy)(Cl)] (CSU-3), containing –Me and –COMe substituted dipyridylpyrrole as a pincer ligand, was designed and synthesized. The CSU-3 complex featured a Cl− ligand at the axial position as the active site for ammonia oxidation (AO), and is structurally analogous to AO catalyst [Ru(trpy)(dmabpy)(NH3)][PF6]2 (1) bearing a terpyridine ligand, but different from AO catalyst [Ru(dpp)(bipy)(NH3)] (CSU-2) containing unsubstituted dipyridylpyrrole as a hemilabile ligand with the active site at an equatorial position. To gain insight into the role of active-site and ligand regulation in the AO reaction, the structure and electrochemical properties of CSU-3 and its catalytic performance and mechanism for the AO reaction were comparably studied. Complex CSU-3 has good selective catalytic performance for the oxidation of ammonia to hydrazine with a turnover frequency (TOF) of 258.8 h−1 and N2H4 formation selectivity of 84.7% at Eapp of 1.0 V. The DFT calculations reveal that N2H4 as a dominant product is generated via an ammonia nucleophilic attack of ruthenium(IV)-imide to form N2H4 followed by N2H4-by-NH3 substitution.
Introduction
Ammonia (NH3) as a carbon-free alternative fuel is considered as one of the most important hydrogen carriers;1–3 however, the classical heterogeneous catalytic cracking reaction for NH3-to-H2 conversion requires a precious metal catalyst and high temperature, and is therefore relatively high cost. Molecular catalytic ammonia splitting is one of the appealing alternative methodologies to produce H2.4,5 Ammonia oxidation (AO) is a critical half-reaction in NH3-to-H2 conversion, and due to the involvement of multiple electron and proton transfers it is kinetically demanding. Molecular catalysts based on transition metals for the oxidation of ammonia might offer many attractive attributes, and a myriad of spectroscopic, kinetic, thermodynamic, and electrochemical techniques have been used to gain detailed insights into the bond-breaking and bond-forming processes.6,7Numerous transition-metal complexes based on Ru,8–15 Fe,16–18 Cu,19 Ni,20 and Mn,21 with various ligands for catalytic AO have been developed,22–24 since the pioneering work using a [Ru(trpy)(dmabpy)(NH3)][PF6]2 (1, Scheme 1, trpy = 2,2′:6′,2′′-terpyridine, dmabpy = 4,4,-bis(dimethylamino)-2,2′-bipyridine) catalyst8 for AO was reported by Smith III, Hamann and co-workers. The electron rich –NMe2 group in 1 makes the catalyst decrease the onset potential of AO and triggers electrocatalytic oxidation of NH3 to generate N2via an ammonia nucleophilic attack (ANA) mechanism, with a TOFN2 of 0.7 h−1. The N2H4-ligated intermediates were determined through NMR spectra. As shown in Scheme 1, we recently reported a distinct example of a Ru-based catalyst,25a [Ru(K3-N,N′,N′′-dpp)(bipy)(dmso)][PF6] (CSU-1, bipy = 2,2′-bipyridine, Hdpp = 2,5-di(pyridin-2-yl)-1H-pyrrole) and [Ru(K2-N,N′-dpp)(bipy)(dmso)(NH3)][PF6] (CSU-2), which exhibits excellent electrocatalysis activity for the AO reaction to generate N2H4 with high selectivity (>99%) and high efficiency (TOFN2H4 > 100 h−1). The mechanism studies show that it benefits from the lower barrier in N2H4 formation involving a bimolecular coupling of RuII-aminyl or RuIII-iminyl intermediates, but is unfavorable for N2 formation via the ANA mechanism like in complex 1.
 | ||
| Scheme 1 Molecular AO catalysts relevant to this work. | ||
The anionic dipyridylpyrrole dpp− ligand in CSU-1 and CSU-2 is structurally analogous with the neutral trpy ligand in 1; however, their ligated ruthenium complexes exhibit significant differences in their spatial configurations, selectivity, catalytic efficiency and even mechanism for AO. The factors governing ammonia oxidation seem very complicated and deserve further in-depth research. To gain an insight into the role of the active-site in catalysis for the oxidation of ammonia, herein we design a mononuclear ruthenium(II) complex [Ru(dppMe, COMe)(bipy)(Cl)] (CSU-3, HdppMe, COMe = 1-(4-methyl-2,5-di(pyridin-2-yl)-1H-pyrrol-3-yl)ethan-1-one, Scheme 1) and comparably study its electrocatalysis of the AO reaction in CH3CN media.
To avoid the hemilability of the dpp− ligand to form bidentate K2-N,N′-coordination modes like in CSU-2, we chose the dppMe, COMe− ligand in CSU-3. The steric effect of the substituted –Me and –COMe groups causes a smaller interior bond angle (av. 116.3°) between the pyrrole and pyridine in the free dppMe, COMe− ligand (Fig. S1–S4,† crystallographic data in Tables S1 and S2†) compared to the 121.4° in the free dpp− ligand.23 This leads to an increase in the binding strength of the Ru–N bond between the Ru and N of dppMe, COMe− in CSU-3, and causes dppMe, COMe− binding to the Ru center with a N^N^N coordination mode, like trpy in 1. Thus, the active site in CSU-3 was regulated to the axial position for a comparative study.
Results and discussion
Synthesis and characterization
Treatment of [Ru(dmso)4(Cl)2], HdppMe, COMe and bipy in the presence of Et3N under refluxing toluene gives a red solid, which is redissolved in MeOH and refluxed for 5 d to afford CSU-3 in 23% yield. Complex CSU-3 was fully characterized using NMR, elemental analysis, and infrared spectroscopy (Fig. S5–S8†). The ESI-MS results display a parent peak at m/z 569.0282 assigned to [M]+. Compared to the 1H NMR of CSU-1, the resonance signal of ligated dmso is absent in CSU-3. This result is consistent with what was observed in single crystal X-ray diffraction analysis. As shown in Fig. 1, CSU-3 displays a slightly distorted octahedral geometry around the ruthenium center. The Cl− ligand in CSU-3 coordinates with the ruthenium center at the axial position, unlike in CSU-1 with a dmso ligand at the axial position. Due to the steric effect of the substituted –Me and –COMe groups on the pyrrole unit, as mentioned in the introduction section, the dppMe, COMe− ligand in CSU-3 as an N^N^N pincer ligand strongly coordinates to the ruthenium center at the equatorial position. This is mirrored by the shorter bond distance of the ruthenium center and the terminal pyridine of the dppMe,COMe− ligand in CSU-3 with Ru1–N1 of 2.107(5) Å) and Ru1–N3 of 2.101(4) Å compared to that in CSU-1 (2.136(2) Å and 2.137(2) Å). Unlike in CSU-1, in complex CSU-3, the outer donor group is not readily decoordinated to supply a vacant site for coordination of the incoming NH3. The NMR and UV-vis monitoring experiments also confirm that CSU-3 is very stable in the presence of a coordinating solvent and even NH3 (Fig. S9†) due to the negative charge of the Cl− ligand, which makes it a poor leaving group. Thus, the corresponding NH3-ligated complex could not be obtained by direct Cl−-by-NH3 substitution of CSU-3.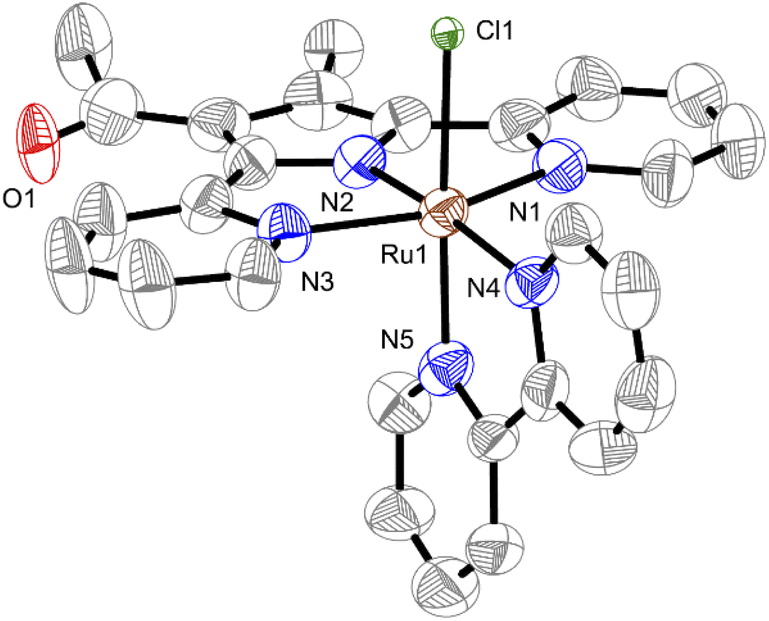 | ||
| Fig. 1 Solid-state structure of CSU-3. The hydrogen atoms are omitted for clarity. Bond distances (Å): Ru1–N1, 2.107(5); Ru1–N2, 1.907(4); Ru1–N3, 2.101(4); Ru1–N4, 2.033(5); Ru1–N5, 2.065(4); Ru1–Cl, 2.4297(14). | ||
Complex CSU-3 is treated with AgOTf (OTf− = trifluoromethylsulfonate) in CH3CN to remove the Cl− ligand, and then ammonia gas is bubbled into the filtrate solution to give NH3-ligated complex [Ru(dppMe, COMe)(bipy)(NH3)]OTf ([CSU-3-NH3]OTf) (Fig. S10–S13†). Its 1H NMR spectrum shows a newly added single broad peak at 2.47 ppm due to the incoming NH3 (Fig. S10†), which is consistent with what was observed in its ESI-MS spectrum with a parent peak at m/z 551.1135 for [CSU-3-NH3]+ (Fig. S12†) and elemental analysis.
Electrochemical and electrocatalytic performances
The electrochemical behavior of CSU-3 was studied using 0.1 M Bu4NPF6 in CH3CN as the electrolyte, glassy carbon as the working electrode, Pt wire as the counter electrode and Ag/AgCl in saturated KCl aqueous solution as the reference electrode. Unless otherwise specified, all potentials are converted into E1/2versus Cp2Fe+/0 in CH3CN by adding −0.43 V to the measured potential.As shown in Fig. 2a, the cyclic voltammogram (CV) of CSU-3 displays a reversible wave at −0.14 V followed by two irreversible waves at 0.98 and 1.21 V, which are assigned to RuIII/II, RuIV/III and the ligand oxidation, respectively. The redox potential of RuIII/II is significantly lower than that of 1 (RuIII/II 0.055 V vs. Cp2Fe+/0 in THF), CSU-1 (0.47 V vs. Cp2Fe+/0 in CH3CN) and CSU-2 (0.43 V vs. Cp2Fe+/0 in CH3CN). Obviously, the redox behavior of the metal center in CSU-3 is sensitive to the electron donor nature of the dppMe, COMe− ligand with a methyl substituent. Compared to CSU-1 and CSU-2 with a π-accepting dmso ligand at the axial position, the π-electron donating Cl− ligand at the axial position is also a possible reason for the significant negative-shift of redox potential in CSU-3.
 | ||
| Fig. 2 (a) CV of CSU-3 and [CSU-3-NH3]OTf in MeCN solution; (b) CV of CSU-3 in MeCN in the presence of NH3 with various concentrations (0.01–0.05 mM); (c) CV of CSU-3 in MeCN in the presence of 0.10 M NH3; (d) CV of [CSU-3-NH3]OTf in MeCN in the presence of 0.10 M NH3. Conditions: [Ru]: = 1 mM, rate 0.1 V s−1, 0.1 M Bu4NPF6 as the supporting electrolyte, platinum wire as the working electrode, potential referenced to the Cp2Fe+/0 redox couple. | ||
For complex [CSU-3-NH3]OTf, the first reversible wave (0.09 V) and the second irreversible wave (0.97 V) were attributed to continued oxidation of the ruthenium center (RuII ⟶ RuIII ⟶ RuIV). The third irreversible wave at 1.34 V is due to ligand oxidation. Compared to CSU-3, the redox potential of the first reversible RuIII/II wave in [CSU-3-NH3]OTf is positively shifted. This suggests that the electron donating ability of NH3 is weaker than that of the negatively charged Cl− ligand. The latter acts as a π donor increasing the electron density of the ruthenium center.
The CV plot of CSU-3 in the presence of NH3 with various concentrations (0.01–0.05 M), as shown in Fig. 2b, illustrates that the RuIII/II couple remains unchanged, and a new oxidation wave (∼1.06 V) appears for the RuIV species. Subsequently, a strong catalytic current (icat) is observed (Fig. 2c), suggesting that the RuIV species triggers the oxidation of ammonia. Obviously, when the ruthenium center of CSU-3 is oxidized to the RuIV oxidation state, an EC process occurs to generate RuIV–NH3via Cl-by-NH3 substitution of the RuIV species from 2e− oxidation of CSU-3, which is also supported by the DFT calculations. According to the CV plot of [CSU-3-NH3]OTf (Fig. 2d), in addition to the RuIV species, the RuIII species from [CSU-3-NH3]OTf also triggers the oxidation of ammonia, which is very similar to what is observed in the CSU-1, CSU-2 and [Ru(K3-N,N′,N′′-dpp)(bpy)(L)]·PF6 (L = pyridine; 4-methylpyridine; pyrimidine; isoquinoline) catalyst systems and Ru(K3-N′,N′′-dpp)(trpy)(NH3)]·PF6.25b,c
As shown in Fig. S14 and S15,† complexes CSU-3 and [CSU-3-NH3]OTf exhibit satisfactory stability, corroborated by 100 consecutive cyclic voltammetry cycles, in which no new redox wave appears and the attenuation of the catalytic current is not significant. A post-catalysis and thoroughly rinsed carbon cloth working electrode displayed no catalytic activity (Fig. S16†). This indicates that the catalytic process of CSU-3 and [CSU-3-NH3]OTf is homogenous.
Controlled potential coulometry (CPC) experiments were conducted in a sealed Schlenk electrolytic cell with a 0.01 mM ruthenium catalyst solution containing 0.2 M (or 2.0 M) NH3 and 0.1 M [Bu4N][PF6] supporting electrolyte in anhydrous MeCN. The detection method for the possible products (H2, N2, N2H4, NO2− and NO3−) and blank experiments is described in the ESI (Fig. S17–S21 and Table S5).† The data of catalytic performance are listed in Table 1 and Fig. S22.†
| Entry | Cat. | c NH3 (mol L−1) | E app (V) | TOFH2 (h−1) nH2 (μmol) | TOFN2H4 (h−1) nN2H4 (μmol) | TOFN2 (h−1) nN2(μmol) | Q (C) | FEN2H4c(%) | S N2H4 (%) |
|---|---|---|---|---|---|---|---|---|---|
| a [Cat.] = 0.01 mM; electrolysis time, 1 h; Eappvs. Cp2Fe+/0; carbon cloth (1 cm2) as the working electrode; molar ratio of N2, N2H4, and H2 determined by taking the average of two tests for the electrolyte in CPC experiments, and the generation of these compounds in the control CPC experiment (Table S5) is subtracted. The maximum relative errors of 1.5%, 2.2% and 3.5% for production of H2, N2H4 and N2. b Charge passed in CPC experiments in 1 h. c FEN2H4 = nN2H4/Q × 100%. d S N2H4 = nN2H4/(nN2H4 + nN2H4) × 100%. | |||||||||
| 1 | CSU-3 | 0.2 | 0.2 | Trace | Trace | Trace | — | — | |
| 2 | CSU-3 | 2.0 | 0.2 | Trace | Trace | Trace | — | — | |
| 3 | CSU-3 | 0.2 | 1.0 | 141.4 | 140.3 | 1.0 | 25.3 | 85.6 | 99.3 |
| 113.1 | 112.2 | 0.8 | |||||||
| 4 | CSU-3 | 2.0 | 1.0 | 276.0 | 258.5 | 2.9 | 47.1 | 84.7 | 98.9 |
| 220.8 | 206.8 | 2.3 | |||||||
| 5 | [CSU-3-NH3]OTf | 0.2 | 0.2 | 5.0 | 4.8 | Trace | 0.8 | 87.9 | 100 |
| 4.0 | 3.8 | ||||||||
| 6 | [CSU-3-NH3]OTf | 2.0 | 0.2 | 19.5 | 19.1 | Trace | 3.3 | 89.5 | 100 |
| 15.6 | 15.3 | ||||||||
| 7 | [CSU-3-NH3]OTf | 0.2 | 1.0 | 169.3 | 165.3 | 1.8 | 28.5 | 89.5 | 98.9 |
| 135.4 | 132.2 | 1.4 | |||||||
| 8 | [CSU-3-NH3]OTf | 2.0 | 1.0 | 366.9 | 350.5 | 3.8 | 60.7 | 89.1 | 98.9 |
| 293.5 | 280.4 | 3.0 | |||||||
For complex CSU-3, the applied potential (Eapp) is fixed at 0.2 V (entry 1, 2) to only generate RuIII species, and as expected, in CV studies, no oxidation products of ammonia are detected. When holding the Eapp at 1.0 V for a low concentration ammonia solution (0.2 M) for 1 h, the gas products of H2 (113.1 μmol, 141.4 equiv. based on Ru) and N2 (0.8 μmol, 1 equiv. based on Ru) in the headspace and N2N4 (112.2 μmol, 140.3 equiv. based on Ru) in the electrolyte solution are determined (entry 3). When the concentration of ammonia is increased to 2.0 M, the catalytic efficiency of CSU-3 is approximately doubled. In addition, the selectivity of N2H4 formation and Faraday efficiency (FE) is almost unchanged with changes in ammonia concentration, maintaining a level of 85.6 and 84.7%, respectively. This indicates that NH3 is possibly involved in the N2H4 formation step via an ammonia nucleophilic attack mechanism and/or N2H4 release via N2H4-by-NH3 substitution.
In the [CSU-3-NH3]OTf catalyst system, only N2H4 as an anodic product is generated at low electrolytic potential (0.2 V) to only generate RuIII species as the intermediate (entry 5, 6), which is very similar to the results for CSU-1, CSU-2 and [Ru(K3-N,N′,N′′-dpp)(bpy)(L)]·PF6.25 This suggests that a bimolecular coupling mechanism of ruthenium amide is possibly involved. Holding the Eapp at 1.0 V (RuIV species generated at this potential), the catalytic efficiency is greatly enhanced (entry 7, 8). TOFN2H4, FEN2H4, and SN2H4 reach 350.5 s−1, 87.9% and 98.9%, respectively.
Mechanism
The full mechanism of the AO reaction catalysed by CSU-3 is proposed by theoretical calculations. As shown in Fig. 3a, CSU-3 is firstly oxidized to [RuIII–Cl]+ (ΔG = −8.3 kcal mol−1). The corresponding calculated RuIII/II redox potential is −0.36 V. Direct Cl-by-NH3 substitution of [RuIII–Cl]+ and CSU-3 to generate [RuIII–NH3]2+ and [RuII–NH3]+, respectively, is unfavourable due to the high energy barrier (Fig. S25†). For example, two possible substitution pathways, namely the concerted associative pathway (Ia) and dissociative pathway (D), are considered. The energy barriers of the Ia pathway (ΔG‡ = 26.2 kcal mol−1) and D pathway (ΔG‡ = 24.7 kcal mol−1) are high enough to hinder direct Cl-by-NH3 substitution of [RuIII–Cl]+, which is in agreement with what is observed in CV studies and synthetic experiments. A subsequent oxidation of [RuIII–Cl]+ generates [RuIV–Cl]2+ (ΔG = 22.5 kcal mol−1, Ecal. = 0.98 V). The subsequent Cl-by-NH3 substitution of [RuIV–Cl]2+ to produce [RuIV–NH3]3+ is an endergonic step with ΔG of = 12.3 kcal mol−1). Subsequently, deprotonation of [RuIV–NH3]3+ affords ruthenium(IV)-imido complex [RuIV–NH2]2+, which is a key intermediate for N2H4 formation. | ||
| Fig. 3 The possible mechanism of AO catalyzed by CSU-3. (a) Precatalyst formation, (b) N–N formation, and (c) N2H4 release. The free energy changes (ΔG) are presented in the individual reaction steps in kcal mol−1, with the calculated potentials in parentheses versus Cp2Fe+/0 in CH3CN. | ||
Complex [RuIV–NH2]2+ is nucleophilically attacked by NH3 to produce terminal hydrazinium-ligated [RuII–NH2NH3]2+, only overcoming an energy barrier of 1.7 kcal mol−1, followed by an energetically favourable deprotonation process to generate terminal hydrazine-ligated RuII-intermediate [RuII–NH2NH2]+ (ΔG = −17.4 kcal mol−1). Obviously, an N–N bond is readily formed via ammonia nucleophilic attack of [RuIV–NH2]2+ of CSU-3 (ΔG‡ = 1.7 kcal mol−1), unlike 1 and CSU-2via ammonia nucleophilic attack of RuIV-imide with higher barriers (ΔG‡ = 24.1 and 7.7 kcal mol−1, respectively). The single-site molecular catalytic pathway of CSU-3 is confirmed by the linear relationship between the catalytic current and concentration of ammonia and catalyst (Fig. S23 and S24†). Furthermore, the pathway of generating hydrazine-bridged bimetallic [RuIII–μ–N2H4–RuIII]4+via bimolecular N–N coupling [RuIV–NH2]2+ (grey line in Fig. 3b) is excluded due to the high energetic barrier (ΔG‡ = 10.8 kcal mol−1).
N2H4/N2 selectivity is usually based on the hydrazine-ligated RuII-intermediate, which could oxidize the ruthenium centre leading to hydrazine oxidation to generate N2,8,17,19a,20a but also could cause N2H4-by-NH3 substitution to produce N2H4. As shown in Fig. 3c, N2H4 release through N2H4-by-NH3 substitution to generate [RuII–NH3]+via the Ia and D pathways was considered. Compared to the Ia mechanism with two transition states with large energetic barriers (ΔG‡ = 21.6 and 30.4 kcal mol−1), the release of N2H4via the D pathway is more favourable due to the lower energetic barrier of 17.8 kcal mol−1, which is also lower than that in the CSU-2 catalytic AO system (ΔG‡ = 23.4 kcal mol−1).
Orbital interaction and electrostatic force between Ru and hydrazine in [RuII–NH2NH2]+ play a key role in the stabilization of the binding of the dative ligand. The energy of the lowest unoccupied molecular orbital of [RuII]+ from CSU-3 shows a higher value of −0.078 au compared to [Ru(trpy)(dmabpy)]+ from 1 (−0.093 au), indicating the relatively weaker Ru–N2H4 bond in [RuII–NH2NH2]+, which is more labile (Fig. S26†). Meanwhile, natural population analysis shows that the partial charge at the ruthenium centre of [RuII–NH2NH2]+ from CSU-3 is more positive than that from 1 (Table S6†), indicating that the influence of the electrostatic interaction is not as large as that of orbital interaction because N2H4 binds less strongly to the complex, where the partial charge at the ruthenium is larger. After N2H4-by-NH3 substitution, the formed [RuII–NH3]+ is continuously oxidized to [RuIII–NH3]2+, and the catalytic cycle restarts. Obviously, except for the Cl-by-NH3 substitution in the precatalyst formation step, N2H4-by-NH3 substitution (or N2H4 release) is the rate-determining step for the catalytic oxidation of ammonia to hydrazine. According to the literature,7 one-electron metal-based oxidation of [RuII–NH2NH2]+ to [RuIII–NH2NH2]2+ in the complex 1 catalytic system is calculated to be the most endergonic step (31.7 kcal mol−1) in the AO reaction. This electron transfer step seems to be a key ingredient in NH3 conversion into N2. Hence, we believe that this very thermodynamically demanding step is the possible reason that N2 generation is unfavourable in the CSU-3 catalytic system.
Conclusions
In summary, a mononuclear ruthenium complex CSU-3 and its selective catalysis for ammonia oxidation is reported. The dppMe, COMe− as a pincer ligand coordinates to the ruthenium center, and the Cl− ligand occupies the axial position. The redox potential of RuIII/II in CSU-3 is negatively shifted to about 0.14 V and 0.61 V due to the electron donor nature of the dipyridylpyrrole ligand and the Cl− axially coordinated ligand, compared to structurally analogous complex 1 and CSU-1, respectively. Complex CSU-3 could selectively catalyze the oxidation of ammonia to generate N2H4 as a dominant product via ammonia nucleophilic attack of the ruthenium(IV) imide forming a N–N bond, followed by an N2H4-by-NH3 substitution, which is significantly distinguished from the structurally analogous 1 producing N2via an ANA mechanism and is also different from CSU-1 and CSU-2 bearing similar hemilabile dipyridylpyrrolide ligands, which more efficiently give the N2H4 product via a bimolecular coupling mechanism of the ruthenium(III)-iminyl radical. DFT calculation indicates that N2H4 release is the rate-determining step for NH3-to-N2H4 conversion catalysed by CSU-3. The weak orbital interaction between the HOMO of N2H4 and the LUMO of D (the LUMO is the orbital that the dissociated N2H4 binds to) in the CSU-3 catalyst system may be the main reason why N2H4 is more easily released than in the 1 catalyst system.Data availability
All data included in this paper are available upon request by contact with the corresponding author.Author contributions
X.-Y. Yi designed research; G. Chen, X.-L. Ding, P. He, and T. Cheng. performed research; G. Chen., X.-L. Ding, Y. Chen, J. Lin, X. Zhang, S. Zhao and N. Qiao analyzed data; G. Chen and X.-Y. Yi wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate Ms Haolin Zou (School of Chemistry and Chemical Engineering, Southwest University, China) for FTIR tests. This work was supported by the National Natural Science Foundation of China (22471291), the Foundation of Central South University Research Programme of Advanced Interdisciplinary (2023QYJC019) and the Southwest University Talent Introduction Program (SWU-KR24050).References
- D. T. Tran, T. H. Nguyen, H. Jeong, P. K. L. Tran, D. Malhotra, K. U. Jeong, N. H. Kim and J. H. Lee, Nano Energy, 2022, 94, 106929 CrossRef CAS.
- L. Jiang and X. Fu, Engineering, 2021, 7, 1688–1691 CrossRef.
- O. Elishav, B. Mosevitzky Lis, E. M. Miller, D. J. Arent, A. Valera-Medina, A. Grinberg Dana, G. E. Shter and G. S. Grader, Chem. Rev., 2020, 120, 5352–5436 CrossRef CAS.
- H. Y. Liu, H. M. C. Lant, C. C. Cody, J. Jelusic, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2023, 13, 4675–4682 CrossRef CAS.
- P. L. Dunn, B. J. Cook, S. I. Johnson, A. M. Appel and R. M. Bullock, J. Am. Chem. Soc., 2020, 142, 17845–17858 CrossRef CAS PubMed.
- M. Barona, S. I. Johnson, M. Mbea, R. M. Bullock and S. Raugei, Top. Catal., 2022, 65, 341–353 CrossRef CAS.
- A. Najafian and T. R Cundari, J. Phys. Chem. A, 2019, 123, 7973–7982 CrossRef CAS PubMed.
- F. Habibzadeh, S. L. Miller, T. W. Hamann and M. R. Smith III, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2849–2853 CrossRef CAS.
- K. Nakajima, H. Toda, K. Sakata and Y. Nishibayashi, Nat. Chem., 2019, 11, 702–709 CrossRef CAS.
- P. L. Dunn, S. I. Johnson, W. Kaminsky and R. M. Bullock, J. Am. Chem. Soc., 2020, 142, 3361–3365 CrossRef CAS.
- J. Holub, N. Vereshchuk, F. J. Sanchez-Baygual, M. Gil-Sepulcre, J. Benet-Buchholz and A. Llobet, Inorg. Chem., 2021, 60, 13929–13940 CrossRef CAS.
- M. J. Trenerry, C. M. Wallen, T. R. Brown, S. V. Park and J. F. Berry, Nat. Chem., 2021, 13, 1221–1227 CrossRef CAS.
- S. I. Jacob, A. Chakraborty, A. Chamas, R. Bock, L. Sepunaru and G. Ménard, ACS Energy Lett., 2023, 8, 3760–3766 CrossRef CAS.
- A. M. Beiler, A. Denisiuk, J. Holub, F. J. Sánchez-Baygua, M. Gil-Sepulcre, M. Z. Ertem, D. Moonshiram, A. Piccioni and A. Llobet, ACS Energy Lett., 2023, 8, 172–178 CrossRef CAS.
- S. Feng, J. Chen, R. Wang, H. Li, J. Xie, Z. Guo, T.-C. Lau and Y. Liu, J. Am. Chem. Soc., 2024, 146, 21490–21495 CrossRef PubMed.
- (a) M. D. Zott, P. Garrido-Barros and J. C. Peters, ACS Catal., 2019, 9, 10101–10108 CAS; (b) M. D. Zott and J. C. Peters, J. Am. Chem. Soc., 2021, 143, 7612–7616 Search PubMed; (c) M. D. Zott and J. C. Peters, ACS Catal., 2023, 13, 14052–14057 CAS.
- Y. Li, J.-Y. Chen, Q. Miao, X. Yu, L. Feng, R.-Z. Liao, S. Ye, C.-H. Tung and W. A. Wang, J. Am. Chem. Soc., 2022, 144, 4365–4375 Search PubMed.
- L. Liu, S. I. Johnson, A. M. Appel and R. M. Bullock, Angew. Chem., Int. Ed., 2024, 63, e202402635 Search PubMed.
- (a) M. E. Ahmed, M. Raghibi Boroujeni, P. Ghosh, C. Greene, S. Kundu, J. A. Bertke and T. H. Warren, J. Am. Chem. Soc., 2022, 144, 21136–21145 Search PubMed; (b) H. Y. Liu, H. M. C. Lant, J. L. Troiano, G. Hu, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2022, 144, 8449–8453 CAS.
- (a) D. N. Stephens, R. K. Szilagyi, P. N. Roehling, N. Arulsamy and M. T. Mock, Angew. Chem., Int. Ed., 2023, 62, e202213462 CAS; (b) N. X. Gu, P. H. Oyala and J. C. Peters, Angew. Chem., Int. Ed., 2021, 60, 4009–4013 CAS.
- H. Toda, K. Kuroki, R. Kanega, S. Kuriyama, K. Nakajima, Y. Himeda, K. Sakata and Y. Nishibayashi, ChemPlusChem, 2021, 86, 1511–1516 CAS.
- P. L. Dunn, B. J. Cook, S. I. Johnson, A. M. Appel and R. M. Bullock, J. Am. Chem. Soc., 2020, 142, 17845–17858 CAS.
- J. Li, F. Zhang, H. Xiong, Y. Cai and B. Zhang, Sci. China: Chem., 2024, 67, 3976–3993 Search PubMed.
- D. N. Stephens and M. T. Mock, Eur. J. Inorg. Chem., 2024, 27, e202400039 CAS.
- (a) G. Chen, P. He, C. Liu, X.-F. Mo, J.-J. Wei, Z.-W. Chen, T. Cheng, L.-Z. Fu and X. Y. Yi, Nat. Catal., 2023, 6, 949–958 CrossRef CAS; (b) C. Zhou, X. Zhang, S. Zhao, S.-D. Zhong, X.-L. Ding, S.-P. Yang, F. Pan, P. He and X.-Y. Yi, ACS Catal., 2025, 15, 3535–3545 CrossRef CAS; (c) S. Zhao, X. Zhang, G. Chen, T. Cheng, X.-L. Ding, S.-D. Zhong, S.-P. Yang, P. He and X.-Y. Yi, Inorg. Chem., 2024, 63, 23150–23157 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Complex synthesis and structure characterization, electrochemical property measurement methods, ammonia, hydrazine, hydrogen and nitrogen detection methods, and supplementary figures and tables. CCDC 2330568 and 2329730. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02360a |
| ‡ Guo Chen and Xiao-Lv Ding contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |