
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct aminolysis of methyl esters with ammonia in continuous flow through Bayesian optimization†
Bavo
Vandekerckhove
a,
Stefan
Desimpel
 a,
Bart
Ruttens
b,
Massimo
Bocus
c,
Wim
Temmerman
c,
Bert
Metten
b,
Veronique
Van Speybroeck
c,
Thomas S. A.
Heugebaert
a and
Christian V.
Stevens
*a
a,
Bart
Ruttens
b,
Massimo
Bocus
c,
Wim
Temmerman
c,
Bert
Metten
b,
Veronique
Van Speybroeck
c,
Thomas S. A.
Heugebaert
a and
Christian V.
Stevens
*a
aSynBioC Research Group, Department of Green Chemistry and Technology, Faculty of Bioscience Engineering, Ghent University, Coupure Links 653, 9000 Ghent, Belgium. E-mail: Chris.Stevens@UGent.be
bAjinomoto Omnichem, Cooppallaan 91, 9230 Wetteren, Belgium
cCenter for Molecular Modeling, Tech Lane Ghent Science Park, Campus Ardoyen, Ghent University, Technologiepark 46, 9052 Zwijnaarde, Belgium
First published on 13th May 2025
Abstract
Amides play a crucial role in the pharmaceutical, animal health and agrochemical industry. Despite the availability of various catalytic systems and coupling reagents, many methods suffered from long reaction times and poor atom economy. The direct synthesis of primary amides remained particularly challenging due to the limited availability of suitable nitrogen sources. In this study, continuous flow technology was explored as a process-intensification approach for the direct amidation of methyl esters to produce primary amides. Methanolic ammonia was employed as a nitrogen source to enhance process efficiency while circumventing the limitations of aqueous ammonia and the hazards of gaseous ammonia. Seventeen substrates were screened to assess their aminolysis reactivity under these conditions. As a proof of concept, methyl picolinate was selected for continuous flow optimization using Bayesian optimization. Therefore, a custom-designed high-pressure, high-temperature continuous flow reactor was utilized to achieve efficient, safe and scalable synthesis (200 °C, 50 bar).
Introduction
The amide functional group is crucial in the structure of biomolecules, including numerous clinically approved drugs. Amide bond formation is widespread in the production of pharmaceuticals, animal health products and agrochemicals, placing the various methods in a constant spotlight of chemical efficiency and environmental sustainability. The need for improved amide synthesis methods gained particular attention in 2007 when the American Chemical Society Green Chemistry Institute, in collaboration with global pharmaceutical companies, identified ‘amide formation avoiding poor atom economy reagents’ as the highest-priority research area.1 Furthermore, reports from three leading pharmaceutical companies revealed that amide bond formation was essential for 66% of drug candidates, underscoring its critical importance (Fig. 1).2 Despite the availability of various methods for amide bond formation, achieving efficient and environmentally friendly synthesis of primary amides remains a significant challenge. | ||
| Fig. 1 Examples of APIs containing a primary amide moiety. | ||
Extensive literature addresses the reactivity of carboxylic acids and esters, yet the direct synthesis of corresponding primary amides on a practical scale remains a persistent challenge. Various catalytic systems and coupling reagents have been investigated for converting carboxylic acids directly into amides, including DCC, DIC, EDC, HATU, HBTU and phosphonium salts. However, these typically complicate reaction work-up, substantially increase synthesis costs (including raw material expenses, recovery processes, regulatory compliance, and waste management), and lead to poor atom economy.3 An alternative approach involves the reaction of esters with ammonia or amines to produce the corresponding amides, with some catalysts being reported for the batch aminolysis of esters using ammonia in solution.4–6 Unfortunately, these methods often require harsh conditions, involve high costs, and are characterized by very long reaction times, often extending to several days. Given these limitations, the development of innovative and efficient approaches to amide synthesis is attracting growing interest.
The use of ammonia in organic synthesis poses several challenges, primarily stemming from its limited solubility in various solvents and the difficulties associated with handling its reactive and toxic gaseous form. In stirred tank reactors, gaseous ammonia often accumulates in the headspace above the solution, which reduces its reactivity and necessitates the use of excessive amounts to achieve desired outcomes. Commercially, ammonia is typically available either as an aqueous solution or dissolved in methanol. Although, aqueous solutions are the most sustainable, the hydrolysis caused by water is not compatible with many chemical processes and functional groups.
Continuous flow technology has been extensively employed as a process intensification strategy to mitigate environmental challenges of batch processes and to develop green, scalable protocols.7–9 This study aims to investigate the use of continuous flow reactors as a process intensification technology for the amidation of methyl esters to produce primary amides. Flow chemistry presents key advantages, such as the safe handling of hazardous materials and the efficient utilization of gases.10 The objective is to drastically reduce reaction times from days to minutes while minimizing the amount of ammonia required, all without relying on coupling reagents, additives or catalysts.11
Recently, Bayesian optimization (BO) has emerged as a powerful tool for the optimization of process parameters in continuous flow chemistry, offering an efficient strategy to navigate complex, multidimensional parameter spaces with minimal experimental effort. By constructing a probabilistic model of the objective function, BO balances exploration and exploitation, enabling rapid identification of optimal conditions.12 Numerous applications have demonstrated its effectiveness in fine-tuning reaction parameters, leading to significant improvements in yield, selectivity and process robustness.13 Here, Bayesian optimization will be employed to determine the optimal reaction conditions for this chemistry.
Results and discussion
This research focuses on developing a green and environmentally friendly procedure for synthesizing primary amides. Amidation of unactivated esters is typically a slow process and the use of coupling reagents or catalysts is preferably avoided. The synthesis of primary amides presents a greater challenge due to the lower nucleophilicity of ammonia versus other amines. Protic solvents have a positive effect on addition reactions. To address this, methanolic ammonia is employed as the nitrogen source to optimize the process, avoiding aqueous ammonia, which is incompatible with water-sensitive substrates. In addition, previous experiments indicated hydrolysis conversions up to 40% when using aqueous ammonia, significantly complicating purification and workup. Gaseous ammonia cannot be dosed directly from the gas bottle at pressures since it liquifies at pressures >8 bar (25 °C). Liquid ammonia is difficult to pump with traditional gear, diaphragm, plunger and peristaltic pumps available in most laboratories and kilo labs. Therefore, the use of methanolic ammonia allows for reaction conditions that are not restricted to 8 bar, enabling significantly increased reaction rates by elevating the temperature.Scope
As previously discussed, many commercially available APIs contain a primary amide bond, often in combination with a heterocyclic structure (Fig. 1). Both a non-aromatic and aromatic proof-of-concept substrate were selected to explore and optimize the direct aminolysis of relevant heterocyclic compounds for API synthesis, respectively L-methyl prolinate hydrochloride 1a and methyl picolinate 1b (Fig. 2). Picolinamide 2b, known for its role in poly(ADP-ribose) synthetase inhibition (anticancer activity) and iron-induced renal damage reduction, has been previously synthesized via amidation.14 Pavlik et al. reported a 65% yield of picolinamide after a 12 hour reaction at room temperature using aqueous ammonia.15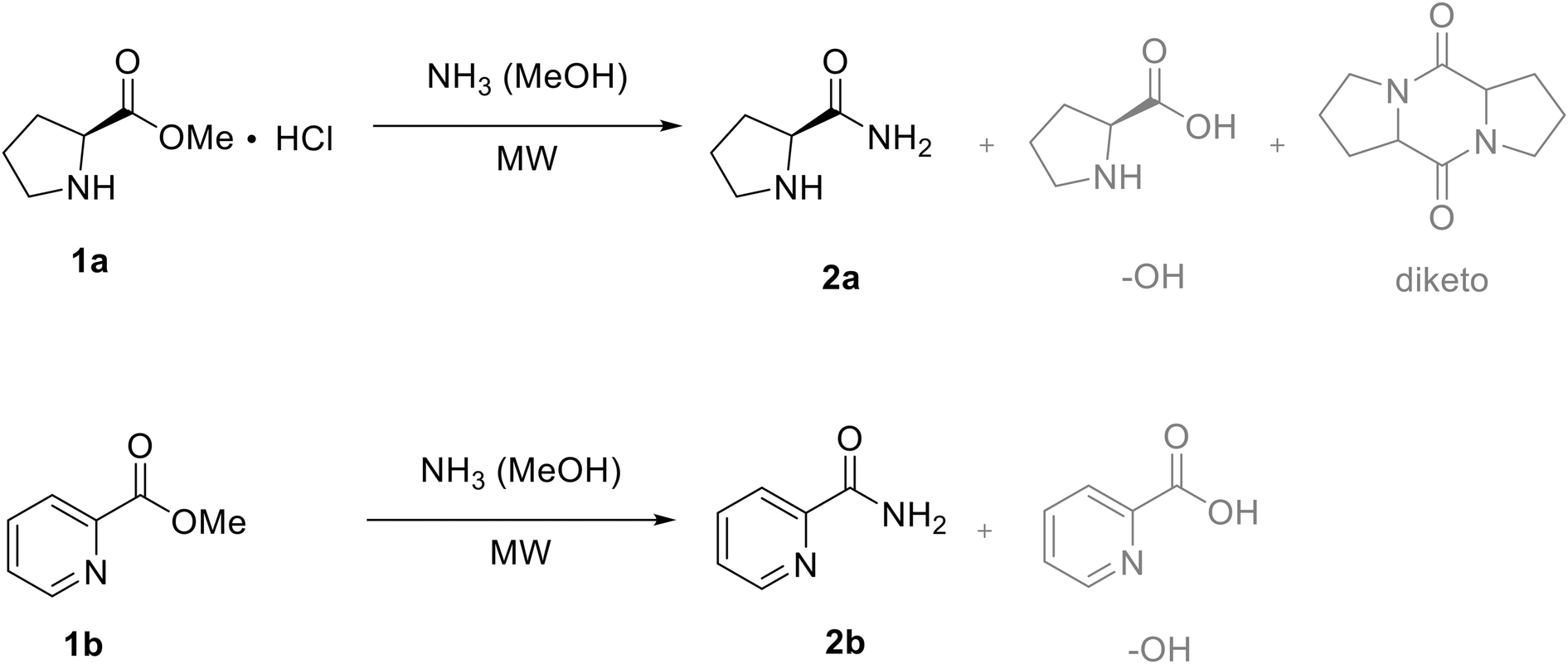 | ||
| Fig. 2 Direct amidation of methyl L-prolinate hydrochloride 1a (non-aromatic) and methyl picolinate 1b (aromatic) as non-aromatic and aromatic proof-of-concept substrates. | ||
Batch screening experiments
Before initiating the ammonia-mediated amidation reaction in a continuous flow setup, a preliminary batch screening was conducted to adapt reaction conditions for flow compatibility. A microwave setup enabled exploration of temperatures exceeding methanol's boiling point (64.7 °C), significantly reducing reaction times from several hours to minutes (Table 1). However, during the aminolysis of methyl L-prolinate hydrochloride 1a, notable side reactions such as methyl ester hydrolysis to the amino acid and dimerization to the diketopiperazine were observed (Fig. 2, Table 1). Additionally, NMR analysis proved challenging due to peak shifts caused by variations in sample acidity. Unlike methyl L-prolinate hydrochloride, the aminolysis of methyl picolinate 1b proceeded smoothly without interference from these side reactions, and NMR analysis was significantly more straightforward and reliable (Table 1). Although the obtained conditions were still suboptimal, they are considered adequate as batch optimization is not the primary objective of this study. Efforts to reduce the ammonia equivalents in this batch setup were not entirely successful. However, it was anticipated that flow technology can provide advantages related to this aspect by eliminating the headspace.| Entry | NH3 eq.a | Time (min) | Temp. (°C) | Conversion 1ab (%) | 2a/–OH/diketob (%) | Conversion 1bb (%) | 2b/–OHb (%) |
|---|---|---|---|---|---|---|---|
| a A 7 N solution of NH3 in MeOH is used. b Conversion and NMR yield calculated relative to internal standard (dimethyl sulfone). c Reagent concentration remained constant. | |||||||
| 1 | 12.0 | 30 | 50 | 42 | 26/6/0 | 85 | 56/0 |
| 2 | 12.0 | 30 | 80 | 75 | 43/10/1 | 85 | 75/0 |
| 3 | 12.0 | 30 | 100 | 83 | 64/3/0 | 89 | 82/0 |
| 4 | 12.0 | 15 | 110 | 76 | 62/4/0 | 96 | 86/0 |
| 5 | 12.0 | 15 | 120 | 82 | 61/4/0 | 62 | 51/0 |
| 6 | 12.0 | 10 | 120 | 75 | 22/3/14 | 50 | 48/0 |
| 7 | 12.0 | 15 | 130 | 83 | 70/7/0 | 50 | 45/0 |
| 8 | 12.0 | 10 | 130 | 69 | 53/8/0 | 54 | 51/0 |
| 9c | 9.0 | 10 | 130 | 70 | 40/5/0 | 33 | 26/0 |
| 10c | 6.0 | 10 | 130 | 59 | 22/5/1 | 33 | 28/0 |
| 11c | 3.0 | 10 | 130 | 41 | 14/13/3 | 22 | 21/0 |
As mentioned before, the formation of both a hydrolysis byproduct and diketopiperazine was observed during the amidation of methyl L-prolinate hydrochloride (Fig. 2). To improve the selectivity of the aminolysis reaction towards the desired product, controlling the reagent concentration to suppress dimerization side reactions, along with careful optimization of reaction time and temperature, could further enhance amide formation. Especially, minimizing the presence of water is crucial to reduce hydrolysis. Karl–Fisher analysis of the 7 N ammonia solution in methanol confirmed that the solvent was ultra-dry, with a water content of 2.4 ppm. A reaction mixture was then prepared using methyl L-prolinate hydrochloride and the ammonia solution, stirred for 1 hour, and its water content was measured. This resulted in 1808 ppm (0.1808%, considered very wet) and these levels could interfere with moisture-sensitive reactions or promote unwanted side reactions. This corresponds with a water content of 0.18 equivalents relative to the methyl L-prolinate hydrochloride, making hydrolysis levels of up to 13% quite likely. The same procedure for methyl picolinate 1b resulted in a water content of 466 ppm (0.0466% or 0.045 eq.), which is acceptable for most standard reactions. The extent of hydrolysis observed during the aminolysis of methyl L-prolinate hydrochloride can likely be attributed to its hygroscopic nature. Therefore, various drying methods were evaluated to reduce possible hydrolysis (Table 2). Performing the reaction on a larger scale (×3) did not significantly reduce the formation of the hydrolysis side product, thus excluding glassware adsorbed water. The addition of trimethyl orthoformate (TMO) as an organic drying agent resulted in the formation of additional side products, while oven-dried methyl L-prolinate hydrochloride exhibited some degradation. As expected, using ammonia solution dried on molecular sieves did not effectively reduce hydrolysis to any notable extent either.
| Entry | NH3 eq.a | Time (min) | Temp. (°C) | Attempt | Conversion 1ac (%) | 2a/–OH/diketoc (%) |
|---|---|---|---|---|---|---|
| a A 7 N solution of NH3 in MeOH is used. b 450 mg scale instead of 150 mg. c Conversion and NMR yield calculated relative to internal standard (dimethyl sulfone). | ||||||
| 1 | 12.0 | 10 | 130 | Original | 69 | 53/8/0 |
| 2b | 12.0 | 10 | 130 | Scale ×3 | 86 | 65/6/1 |
| 3 | 12.0 | 10 | 130 | Oven dried | 59 | 32/14/2 |
| 4 | 12.0 | 10 | 130 | TMO (0.5 eq.) | 95 | 14/17/2 |
| 5 | 12.0 | 10 | 130 | MS dried solvent | 72 | 51/10/1 |
To further explore the scope of methyl ester aminolysis in the formation of primary amides, several alternative substrates with a particular focus on heterocyclic structures, were evaluated at suboptimal conditions (130 °C, 10 minutes, Fig. 3). Eight additional substrates demonstrated moderate to excellent reactivity in forming the primary amide. Notably, pyrimidine 2g and pyrazine 2h exhibited remarkably high NMR yields. While the aminolysis of these compounds has been previously reported in literature, the reactions typically required prolonged times ranging from 1 to 3 days.16,17
 | ||
| Fig. 3 NMR yield for the ammonia-mediated aminolysis of different substrates (amethyl ester hydrochlorides are used). | ||
Some substrates exhibited yields that could not be readily rationalized based on chemical intuition. To study the lack of reactivity observed for the other substrates, additional first-principles simulations were conducted.18,19 The problem was simplified by considering the 0 K electronic energy difference between the most stable conformers that could be identified for reactants and products. Simulations were performed both in the gas phase and with implicit methanol solvation. We found that the reaction energy, while straightforward, is a relatively good reactivity descriptor for the amidation reaction. The results, presented in Fig. 4, indicate an inverse correlation with the experimental yield. A more negative reaction energy indicates a greater thermodynamic driving force, suggesting that the reaction should be more favorable. This suggest that relative thermodynamic stability plays an important role in driving the reaction. The inclusion of implicit solvation effects and temperature corrections via reaction free energies did not significantly alter these trends. It must be noted that these calculations provide thermodynamic insights only and do not account for reaction kinetics (see ESI† for details). It is expected that, at high temperatures and in the presence of a protic solvent, specific intermolecular interactions and the hydrogen bonding patterns will play a major role during the reaction from a kinetic point of view. However to perform such extensive kinetic analysis from theoretical point of view, it would be necessary to model the reaction in explicit solvation and with molecular dynamics simulations, which is beyond the scope of the current study.
 | ||
| Fig. 4 Experimental yield of the different substrates (a–q) in function of 0 K electronic reaction energy for gas-phase (left) and implicit solvation (right) (red: high yielding substrates; grey: moderate yielding substrates; blue: low yielding substrates). | ||
We hypothesize that an important structural parameter influencing the reaction yield is the ability of the amide to form an intramolecular hydrogen bond with an available electron donor. This could involve a nitrogen atom with a lone pair positioned ortho to the amide group. Such interactions may contribute to the inverse correlation observed between reaction energies and experimental yields. From a kinetic point of view, it could also facilitate favorable interactions between ammonia and the carbonyl group, potentially accelerating the reaction rate (analogue to coupling reagents e.g. HATU; kinetic study beyond scope of current work). The optimized geometries from the conformational search (Fig. 5) confirm that the observed stabilization of the amide is relatively well explained by the formation of such intramolecular hydrogen bond between the amide NH2 group and an electron-rich atom in the ortho position. Among the studied compounds, only two, 2e and 2m (highlighted in orange in Fig. 5), exhibit this intramolecular hydrogen bond while still having an experimental yield of zero. In the case of 2e, the only available hydrogen bond acceptor is the nitrogen atom of the pyrazole linker. However, its proximity to a second nitrogen atom reduces its electron density, making it a weaker hydrogen bond acceptor. For 2m, additional factors due to its relatively large size may contribute to the observed lack of yield, for example the steric repulsion of the isopropyl and benzyl groups.
 | ||
| Fig. 5 Optimized structures of the most stable conformers for the various amide products of the different substrates (a–q). The colored rectangles highlight the correlation between the presence of an intramolecular hydrogen bond with the amide and an observed yield greater than 0. | ||
Continuous flow experiments
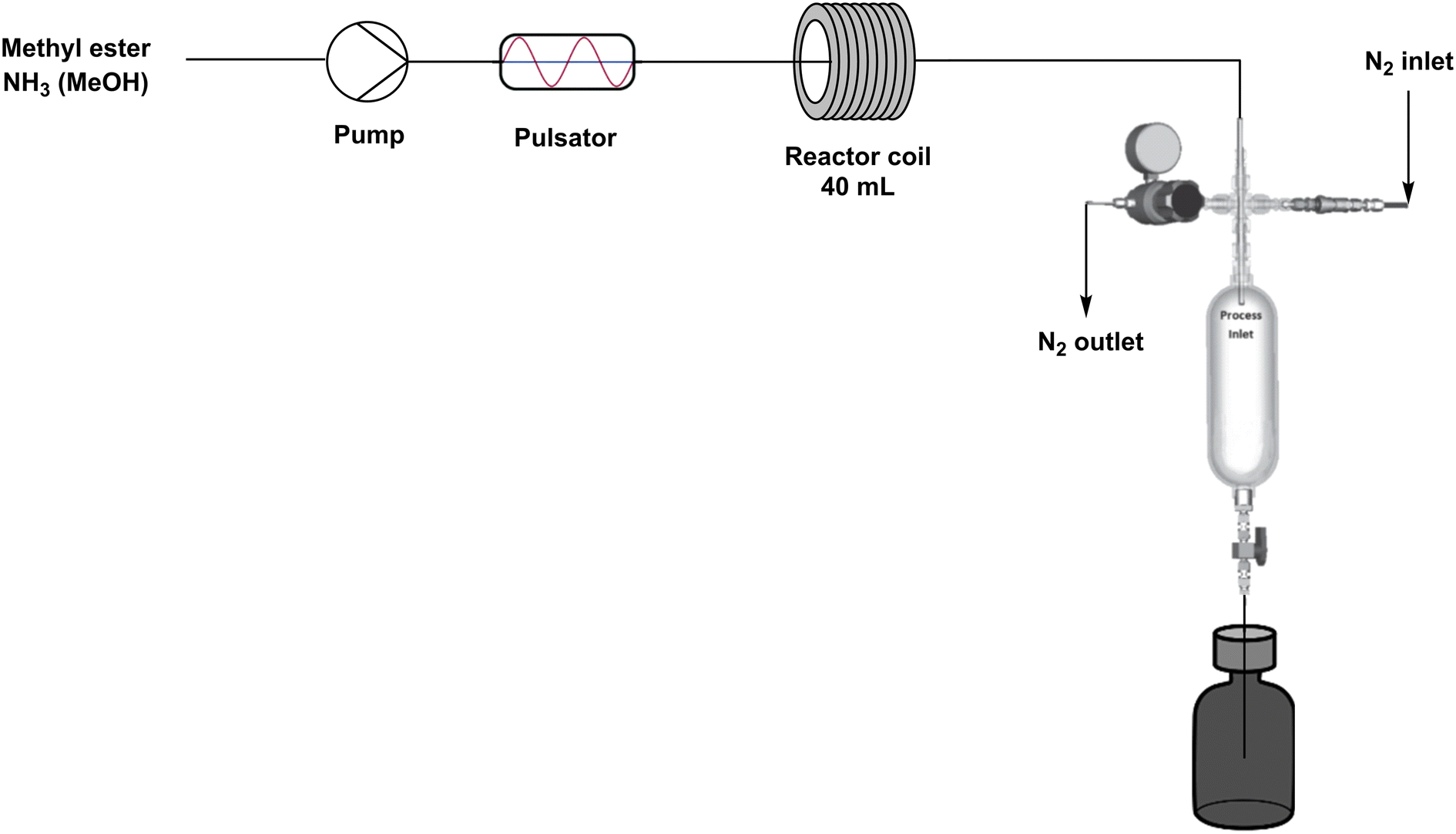 | ||
| Fig. 6 Schematic overview of continuous flow setup (mini CØPE reactor). *This system was constructed as a down-scaled model of the CØPE reactor of Ajinomoto Omnichem, Belgium. | ||
| Entry | NH3 eq. | Time (min) | Temp. (°C) | Conversion 1b (%) | NMR yield 2b (%) |
|---|---|---|---|---|---|
| 1 | 8.9 | 5.5 | 159 | 54 | 26 |
| 2 | 8.0 | 0.7 | 182 | 39 | 13 |
| 3 | 5.7 | 9.8 | 197 | 53 | 27 |
| 4 | 4.2 | 4.9 | 136 | 22 | 12 |
| 5 | 10.2 | 7.9 | 146 | 62 | 41 |
| 6 | 2.0 | 2.2 | 165 | 20 | 4 |
| 7 | 12.0 | 10.0 | 188 | 80 | 59 |
| 8 | 20.0 | 20.0 | 188 | 89 | 58 |
| 9 | 14.7 | 15.1 | 199 | 85 | 53 |
| 10 | 20.0 | 9.7 | 197 | 75 | 47 |
| 11 | 13.3 | 10.9 | 194 | 79 | 57 |
| 12 | 11.6 | 20.0 | 194 | 92 | 53 |
| 13 | 17.7 | 30.0 | 200 | 97 | 69 |
| 14 | 20.0 | 30.0 | 139 | 98 | 70 |
Due to its ease of analysis and reliability compared to methyl L-prolinate, methyl picolinate 1b was selected for optimizing the flow process using Bayesian optimization. The primary objective was to simultaneously maximize both the conversion and the NMR yield of the target product, picolinamide. While these objectives are obviously correlated, particularly in the absence of competing reactions or degradation pathways, both objectives are used to identify high-yielding conditions. Although minimizing the equivalents of ammonia was considered as a potential objective, it was not incorporated into the optimization strategy. Chemical intuition suggests that higher ammonia equivalents would be favored due to ammonia's relatively unreactive nature. It was therefore anticipated that explicitly minimizing ammonia equivalents within the Bayesian optimization framework would lead to significantly suboptimal yields (Fig. 7).
 | ||
| Fig. 7 The optimization campaign for the aminolysis of methyl picolinate is illustrated through a progression plot. The blue line and points represent the obtained conversion/yield as a function of the experiment number, while the green curve tracks the maximum yield achieved up to a given experiment. Below the plots, the conditions that resulted in the highest yield are highlighted, along with the defined parameter boundaries. | ||
Initially, the parameter boundaries were set as follows: NH3 equivalents (1–12 eq.), reaction time (0.5–10 min) and reaction temperature (130–200 °C) (entries 1–6). However, the algorithm's first suggestion after the initialization set (entry 7) already reached the maximum for two out of three parameters. To expand the optimization space, the boundaries for reaction time and ammonia equivalents were expanded to 20 minutes and 20 equivalents, respectively, demonstrating the flexibility of this approach. When boundaries are extended during optimization, the algorithm exhibits high uncertainty within the newly expanded process window. As a result, the next experiment (entry 8) reached the new maximum limits to explore this region. Afterwards, conversion gradually increased over subsequent iterations. Since entry 12 achieved near-full conversion but once again approached the parameter boundaries, the process window was expanded one final time, extending the reaction time to a maximum of 30 minutes. It is important to note that when ammonia equivalents exceed 12, the concentration of methyl picolinate had to be decreased due to the limitation of using a commercially available 7 N methanolic ammonia solution. Finally, nearly complete conversion was achieved using 20 equivalents of ammonia, a reaction time of 30 minutes and a temperature of 139 °C. Further application of the algorithm did not lead to any improvements. Thus, the optimal conditions were established after 8 additional experiments, next to the initial 6 screening experiments (Fig. 7). It is estimated that the algorithm would have achieved convergence faster if more suitable boundaries had been chosen from the start (based on technical feasibilities of equipment). Initially, it was hypothesized that eliminating headspace and using continuous flow could enhance the reactivity of dissolved ammonia gas, potentially reducing the required excess of reagent. However, Bayesian Optimization revealed that achieving desirable yields still necessitates a high excess of ammonia, even within a continuous flow setup. In addition, NMR analysis shows no byproduct formation, suggesting that the difference between conversion and yield may be attributed to unknown volatile byproducts.
 | ||
| Fig. 8 Conversion, NMR and isolated yield for the ammonia-mediated aminolysis of different substrates in continuous flow (amethyl ester hydrochlorides are used). | ||
After optimization of the reaction parameters within the continuous flow setup, these substrates showed improved conversion rates and NMR yields compared to the non-optimal batch conditions (except 2i). Notably, no precipitation was observed for substrates utilizing hydrochloride salts as starting materials during the reaction or sample collection (2a and 2i). Final purification of all substrates was achieved via recrystallization (see ESI† for details).
| Productivity (flow) = 0.00034 mol mL−1 × 1.333 mL min−1 × 60% yield × 122.1 g mol−1 = 0.033 g min−1 = 1.99 g h−1 | (1) |
In alignment with the principles of green chemistry, the presented protocol operates without the use of additives, catalysts, or stoichiometric amounts of coupling reagents, thereby minimizing waste generation and simplifying purification processes. Reaction times could be shortened from several hours down to 30 minutes, although high equivalents of ammonia are still necessary. Furthermore, scale-up can be readily achieved through continuous production compared to previously reported batch protocols, while simultaneously mitigating the hazards associated with the handling of ammonia gas at larger scales. Particular emphasis was set on developing a broadly applicable setup and protocol, leading to the decision to employ methanolic ammonia exclusively (acknowledged as green solvent). This decision also accounts for potential challenges related to hydrolysis or the presence of water-sensitive functional groups in other substrates (beyond those investigated in this study). Potential solubility limitations of substrates in methanol (because of this decision) could still be effectively managed due to the solid-handling capabilities of the system.
Conclusions
This study demonstrates the potential of continuous flow technology as a process-intensification approach for the direct amidation of methyl esters using methanolic ammonia to produce primary amides. Following an initial batch screening, the reactivity of seventeen API-relevant substrates toward direct amidation was evaluated. Conformational simulations were conducted to provide a deeper understanding of the observed reactivity trends. Bayesian optimization was successfully applied to optimize the amidation of methyl picolinate using a custom-built high-pressure, high-temperature continuous flow setup capable of handling potential precipitation (200 °C, 50 bar). However, it was determined that achieving desirable yields still required a high excess of ammonia. Finally, the optimized setup was employed for the amidation of substrates that exhibited moderate reactivity toward ammonia, further validating the effectiveness of continuous flow processing for these challenging transformations (even without optimization for each substrate).Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Bavo Vandekerckhove – designed the project, conducted all wet lab experiments, analysis, calculations, setup, writing. Stefan Desimpel – support Bayesian optimization. Bart Ruttens – supported the project, edited the manuscript. Massimo Bocus – support molecular modeling and calculations. Wim Temmerman – support molecular modeling and calculations. Bert Metten – designed and supervised the project, edited the manuscript. Veronique Van Speybroeck – support molecular modeling and calculations. Thomas S. A. Heugebaert – designed and supervised the project, edited the manuscript. Christian V. Stevens – designed and supervised the project, edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are indebted to VLAIO – Belgium (Baekeland HBC.2021.0198) for financial support. M. B. whishes to thank the Research Foundation Flanders (FWO) for a junior postdoctoral fellowship (grant no. 1269725N). The computational resources (Stevin Supercomputer Infrastructure) and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by Ghent University, FWO and the Flemish Government – department EWI. VVS acknowledges the financial support from the Research Board of Ghent University (BOF).References
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Key green chemistry research areas—a perspective from pharmaceutical manufacturers, Green Chem., 2007, 9, 411–420, 10.1039/B703488C.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Analysis of the reactions used for the preparation of drug candidate molecules, Org. Biomol. Chem., 2006, 4, 2337–2347, 10.1039/B602413K.
- R. C. Dabhi, U. P. Patel, V. B. Rathod, S. N. Shah and J. J. Maru, Process optimizatio of acid-amine coupling: a catalytic approach, Current, Chem. Lett., 2022, 12, 133–140, DOI:10.5267/j.ccl.2022.8.010.
- R. L. Betts and L. P. Hammett, A Kinetic Study of the Ammonolysis of Phenylacetic Esters In Methanol Solution 1, J. Am. Chem. Soc., 1937, 59(8), 1568–1572, DOI:10.1021/ja01287a043.
- A. Basha, M. Lipton and S. M. Weinreb, A Mild, General Method for Conversion of Esters to Amides, Tetrahedron Lett., 1977, 18(48), 4171–4172, DOI:10.1016/S0040-4039(01)83457-2.
- M. F. Lipton, A. Basha and S. M. Weinreb, Conversion of Esters to Amides with Dimethylaluminum Amides: N,N -Dimethylcyclohexanecarboxamide, Inorg. Synth., 2003, 49, DOI:10.1002/0471264180.os059.08.
- M. D. Edwards, M. T. Pratley, C. M. Gordon, R. I. Teixeira, H. Ali, I. Mahmood, R. Lester, A. Love, J. G. H. Hermens, T. Freese, B. L. Feringa, M. Poliakoff and M. W. George, Process intensification of the continuous synthesis of bio-derived monomers for sustainable coatings using a taylor vortex flow rector, Org. Process Res. Dev., 2024, 28(5), 1917–1928, DOI:10.1021/acs.oprd.3c00462.
- M. Andresini, M. Colella, R. Savina Dibenedetto, E. Graziano, G. Romanazzi, A. Aramini, L. Degennaro and R. Luisi, Sustainable continuous flow synthesis of β-aminocarbonyls via acid-catalyzed hydration of N-Boc-2-azetines, React. Chem. Eng., 2023, 8, 3203–3209, 10.1039/D3RE00342F.
- A. Chaudhuri, W. F. C. de Groot, J. H. A. Schuurmans, S. D. A. Zondag, A. Bianchi, K. P. L. Kuijpers, R. Broersma, A. Delparish, M. Dorbec, J. van der Schaaf and T. Noël, Scaling up gas-liquid photo-oxidations in flow using rotor-stator spinning disc reactors and a high-intensity light source, Org. Process Res. Dev., 2025, 29(2), 460–471, DOI:10.1021/acs.oprd.4c00458.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Taming hazardous chemistry by continuous flow, Chem. Soc. Rev., 2016, 45, 4892–4928, 10.1039/C5CS00902B.
- T. Ginman, J. Viklund, J. Malmström, J. Blid, R. Emond, R. Forsblom, A. Johansson, A. Kers, F. Lake, F. Sehgelmeble, K. J. Sterky, M. Bergh, A. Lindgren, P. Johansson, F. Jeppsson, J. Fälting, Y. Gravenfors and F. Rahm, Core Refinement toward Permeable β-Secretase (BACE-1) Inhibitors with Low hERG Activity, J. Med. Chem., 2013, 56(11), 4181–4205, DOI:10.1021/jm3011349.
- B. Shahriari, K. Swersky, Z. Wang, R. P. Adams and N. de Freitas, Taking the human out of the loop: A review of Bayesian optimization, Proc. IEEE, 2016, 104(1), 148–175, DOI:10.1109/JPROC.2015.2494218.
- F. L. Wagner, P. Sagmeister, T. G. Tampone, V. Manee, D. Yerkozhanov, F. G. Buono, J. D. Williams and C. O. Kappe, Self-optimizing flow reactions for sustainability: an experimental Bayesian optimization study, ACS Sustainable Chem. Eng., 2024, 12(26), 10002–10010, DOI:10.1021/acssuschemeng.4c03253.
- A. O. L. Evora, R. A. E. Castro, T. M. R. Maria, M. T. S. Rosado, M. R. Silva, J. Canotilho and M. E. Eusébio, Resolved structures of two picolinamide polymorphs. Investigation of the dimorphic system behaviour under conditions relevant to co-crystal synthesis, CrystEngComm, 2012, 14, 8649–8657, 10.1039/C2CE26244D.
- J. W. Pavlik and S. Laohhasurayotin, Synthesis and spectroscopic properties of some dideuterated cyanopyridines, J. Heterocyclic Chem., 2009, 42, 73–76, DOI:10.1002/jhet.5570420110.
- P. Norcott, P. J. Rayner, G. G. R. Green and S. B. Duckett, Achieving high 1H nuclear hyperpolarization levels with long lifetimes in a range of tuberculosis drug scaffolds, Chem. – Eur. J., 2017, 23, 16990–16997, DOI:10.1002/chem.201703278.
- R. A. Scism, D. F. Stec and B. O. Bachmann, Synthesis of nucleotide analogues by a promiscuous phosphoribosyltransferase, Org. Lett., 2007, 9(21), 4179–4182, DOI:10.1021/ol7016802.
- P. Pracht, S. Grimme, C. Bannwarth, F. Bohle, S. Ehlert, G. Feldmann, J. Gorges, M. Müller, T. Neudecker, C. Plett, S. Spicher, P. Steinbach, P. A. Wesołowski and F. Zeller, CREST—A program for the exploration of low-energy molecular chemical space, J. Chem. Phys., 2024, 160(11), 114110, DOI:10.1063/5.0197592.
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions, J. Chem. Theory Comput., 2019, 15(3), 1652–1671, DOI:10.1021/acs.jctc.8b01176.
- B. Vandekerckhove, L. Van Coillie, B. Mette, T. S. A. Heugebaert and C. V. Stevens, Development of a solid-compatible continuous flow reactor for the paraformaldehyde slurry mediated α-hydroxymethylation of methyl vinyl ketone, React. Chem. Eng., 2024, 9, 2240, 10.1039/d4re00220b.
- J. Claes, A. Vancleef, M. Segers, B. Brabants, M. E. Leblebici, S. Kuhn, L. Moens and L. C. J. Thomassen, Synthesis of amines: from a microwave batch reactor to a continuous milliflow reactor with heterogeneous feed and product, Chem. Eng. Process., 2023, 182, 0255–2701, DOI:10.1016/j.cep.2022.109252.
- S. Desimpel, J. Dijkmans, K. P. L. Kuijpers, M. Dorbec, K. M. Van Geem and C. V. Stevens, Efficient multi-objective Bayesian optimization of gas-liquid photochemical reactions using an automated flow platform, Chem. Eng. J., 2024, 501, 157685, DOI:10.1016/j.cej.2024.157685.
Footnote |
| † Electronic supplementary information (ESI) available: A detailed description of the experimental work is provided in the ESI. See DOI: https://doi.org/10.1039/d5re00163c |
| This journal is © The Royal Society of Chemistry 2025 |