
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast and scalable continuous flow synthesis of butenolides and coumarins†
Lucas Coral
Ferreira
a,
Renan
de Souza Galaverna
 ab,
Tom
McBride
b,
Rodrigo
Costa e Silva
a,
Duncan L.
Browne
*c and
Julio Cezar
Pastre
*a
ab,
Tom
McBride
b,
Rodrigo
Costa e Silva
a,
Duncan L.
Browne
*c and
Julio Cezar
Pastre
*a
aInstitute of Chemistry, State University of Campinas – UNICAMP, PO Box 6154, 13083970, Campinas, SP, Brazil. E-mail: jpastre@unicamp.br
bSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK
cSchool of Pharmacy, University College London, London WC1N 1AX, UK. E-mail: duncan.browne@ucl.ac.uk
First published on 10th February 2025
Abstract
Herein, we present a versatile and efficient continuous flow protocol for the synthesis of structurally diverse butenolides and coumarins through the in situ generation of acylketenes via the retro hetero-Diels–Alder reaction of dioxinones with α-hydroxy-ketones and salicylaldehydes, respectively. This protocol enabled the synthesis of 5 examples of butenolides with yields ranging from 30 to 91% and 16 examples of coumarins with yields ranging from 30 to 99%. The versatility and practicality of the protocol were demonstrated by the gram-scale synthesis of a biologically relevant γ-spiro butenolide core, as well as the production of benzo-coumarins. Modern medicinal chemistry demands both scalability and the ability to synthesize structurally diverse compounds in a single synthetic platform, and our methodology effectively addresses these challenges in a fast and safer manner.
Introduction
Advances in drug development have significantly improved both life expectancy and quality of life for individuals, while also profoundly impacting society on a global scale.1 However, the drug discovery process is an intricate, time-consuming, and costly endeavour, often spanning 12 to 15 years and requiring an investment of up to $1 billion.2 This multifaceted journey encompasses various stages of development, critical to achieving a clinically effective therapeutic outcome.3 Throughout this rigorous process, molecules undergo exhaustive physicochemical, pharmacological, and biological testing and validation, necessitating the evaluation of hundreds of millions of potentially bioactive compounds.4,5 To meet this demand, it is crucial to develop a unique synthetic platform capable of enabling fast, safe, and scalable production of structurally diverse organic molecules (Fig. 1A).6–12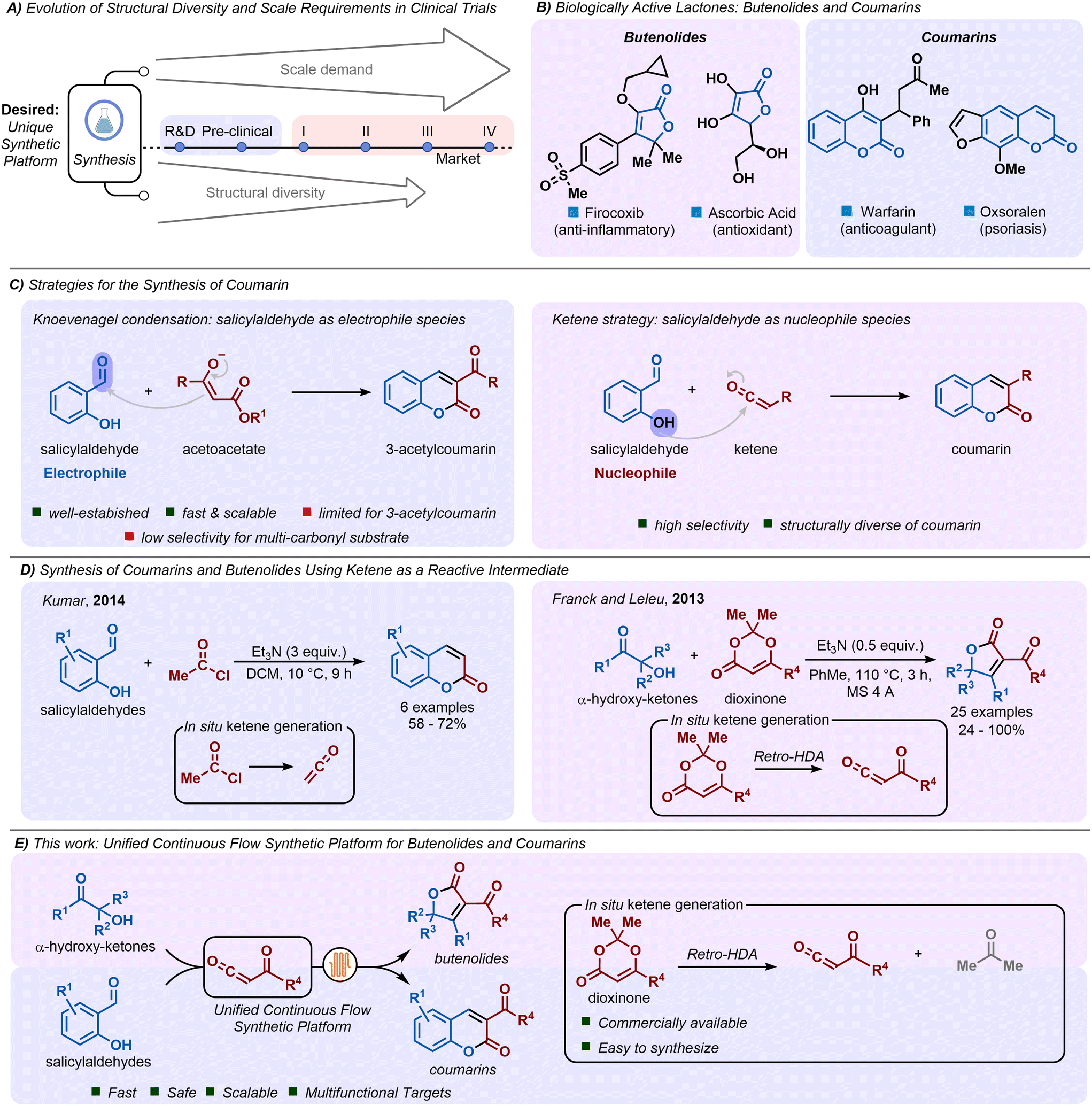 | ||
| Fig. 1 A) Evolution of the synthetic requirements during the drug development process. B) Drugs from butenolides and coumarins derivatives. C) Previous synthetic platform for synthesis of both butenolide and coumarins. D) Synthesis of coumarins and butenolides using acylketene as a reactive intermediate. E) This work: synthesis of butenolides and coumarins using a unified continuous flow synthetic platform. | ||
Among the many classes of compounds explored for their therapeutic potential, lactones stand out as a diverse group widely distributed in nature, characterized by their structural variability, which gives rise to different subclasses, such as sesquiterpene lactones, coumarins, and butenolides.13 This structural diversity underpins their extensive range of biological activities.14–17 Butenolides and coumarins, in particular, are notable for their broad spectrum of effects, including anti-inflammatory, anti-cancer, antiviral, anticoagulant, antioxidant, and anti-psoriatic properties, among others.18–21 Numerous drugs have been developed from these core structures, including firocoxib and ascorbic acid from the butenolide class,21 as well as warfarin and oxsoralen from the coumarin class (Fig. 1B).19,20
A wide range of methodologies has been established for the synthesis of both butenolides and coumarins, including the Perkin reaction, Knoevenagel condensation, Pechmann condensation, Wittig reaction, and Baylis–Hillman reaction.22 Among these, the Knoevenagel condensation has been widely employed for the preparation of 3-acetylcoumarins, with salicylaldehyde acting as the electrophilic component (Fig. 1C). Nevertheless, this method requires selective generation of nucleophilic enolate species, a challenge that becomes even more pronounced when working with multi-carbonyl compounds.
Alternatively, the ketene chemistry has also been yielded in the synthesis of coumarins. Ketenes, carbonyl compounds containing the CO group connected by a double bond to a carbon atom,23 are known for their high reactivity and remarkable versatility, facilitating a wide array of chemical transformations.24–29 In ketene-based strategies, salicylaldehyde acts as the nucleophile, reacting selectively with the electrophilic ketene species (Fig. 1C). In 2014, Kumar and co-workers, for instance, described the synthesis of 6 coumarin derivatives via the reaction of salicylaldehydes with ketenes, which were generated in situ by the HCl elimination of acetyl chloride in the presence of triethylamine, achieving yields of up to 72% (Fig. 1D).30
The ketene strategy enables the synthesis of significant scaffolds through various nucleophilic species. In 2013, Franck, Leleu, and co-workers demonstrated the synthesis of 25 butenolide examples, with yields ranging from 24 to 100%. Their approach involved the use of α-hydroxy-ketones, triethylamine, and dioxinone as a ketene precursor at 110 °C by 3 hours in the presence of molecular sieves. In this particular case, the reactive species was an acylketene, generated in situ thermically through a retro hetero-Diels–Alder reaction of the dioxinone (Fig. 1D).31
Acylketene chemistry has been explored in our group in several ways. In 2019, using 2,2,6-trimethyl-4H-1,3-dioxin-4-one (TMD) as a precursor, we demonstrated the synthesis of dioxinones, β-dicarbonyls and 1,3-oxazine-2,4-diones under flow conditions. One example of a butenolide was also demonstrated in 45% yield.32 In 2022, a flow platform was again applied for the generation of acylketene through extrusion of nitrogen gas. In this example, a diazo dimedone was used as a precursor.33
This breakthrough laid the foundation for the unified continuous flow synthetic platform we describe herein, enabling on demand production of both butenolides and coumarins (Fig. 1E). By identifying acylketenes as reactive and versatile intermediates, we capitalized on the precise control, enhanced safety, and scalability inherent to continuous flow synthesis.34–39 This approach not only ensures greater reproducibility but also facilitates the efficient handling of reactive intermediates like acylketenes, making it an ideal strategy for a unified synthetic platform.40 Additionally, continuous flow technology allows for safe operation at temperatures exceeding solvent boiling points, a challenging feat in batch setups.41 The use of a back pressure regulator (BPR) maintains proper system pressurization, boosting safety and accelerating reaction kinetics for more efficient and controlled outcomes.42,43
Results and discussion
To start our study, we selected 2-hydroxy-2-methyl-1-phenylpropan-1-one (1) and 2,2,6-trimethyl-4H-1,3-dioxin-4-one (TMD, 2), in the presence of triethylamine, as model substrates and reagents to evaluate the synthesis of the corresponding 2(5H)-furanone 3 under continuous flow conditions (mechanism details are shown in the ESI†). The reaction conditions were optimized, with representative examples detailed in Table 1, and a comprehensive evaluation provided in the ESI.† The optimal conditions involved a mixture of α-hydroxy-ketone 1a and 2 equivalents of TMD (2), along with 2 equivalents of triethylamine in toluene (0.1 M), at 130 °C for a 20 minute residence time (τ). Under these conditions, the target furanone 3 was obtained with a 76% yield as determined by 1H NMR analysis, and 71% yield as an isolated product (Table 1, entry 1). Comparable yields were achieved under batch conditions, albeit requiring a significantly longer reaction time of 3 hours.|
|
||
|---|---|---|
| Entry | Changes | Yielda (%) |
| A solution of 1a (0.25 mmol), 2a (2 equiv.), and Et3N (2 equiv.) in PhMe (0.1 M) was pumped at 0.7 mL min−1 through a 14 mL HPFA tubing (ID 0.79 mm) and heated at 130 °C, unless otherwise specified.a Yield was determined by 1H NMR analysis using 1,3,5-trimethoxylbenzene as an internal standard.b Reaction performed at 110 °C.c Reaction performed at 0.5 M. | ||
| 1 | None | 76 (71) |
| 2 | THF instead of PhMeb,c | 5 |
| 3 | 110 °C instead of 130 °Cc | 13 |
| 4 | 1 equiv. of 2a instead of 2 equiv.c | 25 |
| 5 | 0.5 M of 1 instead of 0.1 M | 42 |
| 6 | DBU instead of Et3N | 65 |
| 7 | 1 equiv. of Et3N instead of 2 equiv. | 54 |
| 8 | Residence time of 10 min instead of 20 min | 0 |

Using alternative solvents such as THF drastically reduced the yield to 5% (Table 1, entry 2). Lowering the temperature to 110 °C resulted in a marked decrease in the yield of compound 3, down to 13% (Table 1, entry 3). According to our previous work,32 the retro hetero-Diels–Alder reaction of TMD 2, which generates the reactive acylketene intermediate, is significantly less efficient at lower temperatures.
Ketenes are known to be prone to dimerization in solution,32 a side reaction that can be mitigated by using an excess of the ketene source and maintaining dilute solutions. This was evident in our experiments, where reducing the amount of TMD 2 or increasing the reaction concentration led to decreased yields of 25% (Table 1, entry 4) and 42% (Table 1, entry 5), respectively. Moreover, the use of other organic bases, such as DBU, did not improve the yield of 3 (Table 1, entry 6). Lowering the amount of Et3N also reduced the yield, underscoring the necessity of using 2 equivalents of this reagent (Table 1, entry 7). Reducing the residence time to 10 minutes completely inhibited the formation of the target product 3 (Table 1, entry 8). Further details, including solvent screening, temperature testing, TMD equivalents, concentration of 1, organic base screening, and base equivalents, can be found in the ESI.†
With the optimized continuous flow conditions established, we proceeded to explore the scope of the transformation. TMD (2) was reacted with various hydroxyketones in the presence of Et3N, successfully yielding a range of butenolides for all tested aryl ketone derivatives, with yields ranging from 30 to 91% (Fig. 2). Notably, phenyl ketones bearing electron-withdrawing groups, such as CF3, were particularly effective, with the corresponding butenolide obtained in 91% yield 3c, likely due to the increased electrophilic nature imparted by this substituent on the carbonyl group of the hydroxyketone. In contrast, a lower yield of 51% was observed with 4-methoxy-phenylketone 3b. Despite this success, the methodology did not yield any product for the alkyl ketone derivative 3f, suggesting a limitation of this approach.
 | ||
| Fig. 2 Scope of butenolides. Reaction conditions: a solution of 1 (0.25 mmol), 2 (2 equiv.), and Et3N (2 equiv.) in PhMe (0.1 M) was pumped at 0.7 mL min−1 through a 14 mL HPFA tubing (ID 0.79 mm) and heated at 130 °C. Isolated yield by chromatography column. aFed-batch procedure. bPurified by crystallization. NR = no reaction. | ||
Ketones containing a tertiary α-hydroxyl group provided the corresponding butenolides in yields ranging from 51 to 91% (3a–3c, 3g). On the other hand, secondary alcohols resulted in a more complex mixture (3d). Primary alcohols demonstrated lower stability under flow conditions, thus butenolide 3e was prepared under fed-batch conditions in 30% isolated yield. In this case, the reaction mixture was collected in a flask containing base to promote the final cyclization step. Butenolides derived from primary hydroxy-ketones are, indeed, valuable intermediates for synthesizing other biologically relevant compounds, such as cadiolides.31,44,45
Notably, this continuous flow protocol effectively afforded a spirocyclic Δα,β butanolide, also known as γ-spiro butenolide in 60% yield (3j). The product was crystallized directly from the crude reaction mixture and readily isolated for characterization by X-ray diffraction, which confirmed its structure. These γ-spiro butenolides are crucial structural motifs in various biologically active compounds and serve as versatile building blocks for synthesizing diverse families of bioactive natural products, including alkaloids, terpenes, steroids, and macrolides.46–48 The synthesis of this relevant compound was successfully scaled up to gram-scale example, yielding 1.25 g of the compound 3g in 46% isolated yield without the need for chromatography. This outcome underscores the capability of the platform to not only produce structurally diverse compounds but also to scale production as needed.
The versatility of this protocol was extended to the synthesis of coumarins (mechanism details are shown in the ESI†). As previously demonstrated by Kumar and co-workers, coumarins can be obtained by reacting salicylaldehyde with ketenes.30 Gratifyingly, our continuous flow protocol enabled the reaction of TMD (2) with salicylaldehyde, and in just 20 minute residence time afforded the 3-acetylcoumarin (Fig. 3, 5a) in quantitative yield. Due to solubility issues for some coumarins, the solvent was changed to ethyl acetate and the temperature increased to 150 °C. Under these conditions, the protocol demonstrated broad compatibility, yielding the target products across a range of functional groups, including alkyl, nitro, methoxy, bromide and chloride substituents, with yields ranging from 28 to 99%.
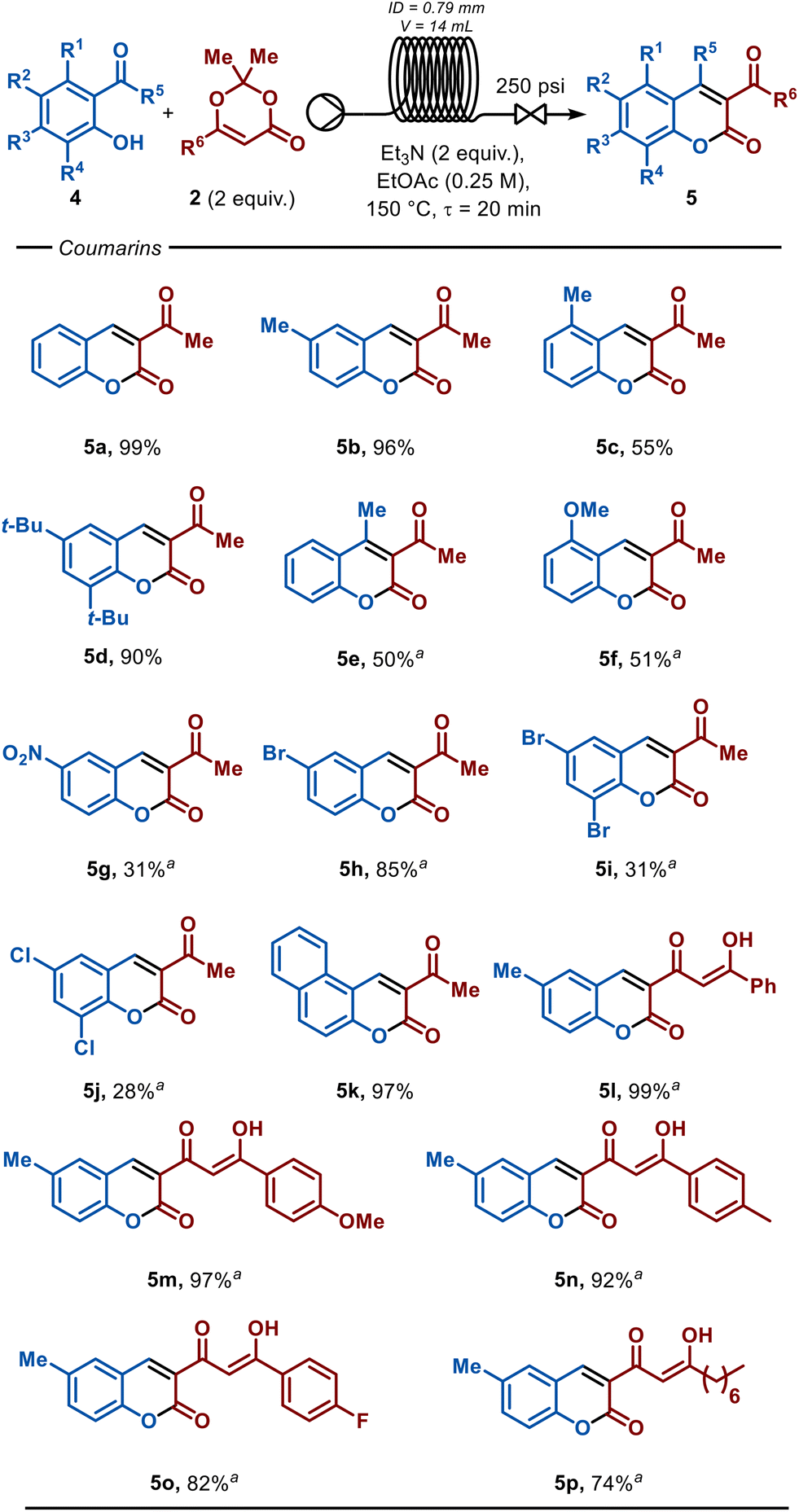 | ||
| Fig. 3 Scope of coumarins. Reaction conditions: a solution of 4 (0.50 mmol), 2 (2 equiv.), and Et3N (2 equiv.) in EtOAc (0.25 M) was pumped at 0.7 mL min−1 through a 14 mL HPFA tubing (ID 0.79 mm) and heated at 130 °C. Isolated yield by chromatography column. aTHF used as the solvent due to poor solubility in EtOAc. NR = no reaction. | ||
The effectiveness of the reaction was strongly influenced by the electronic nature of the substituents and their position relative to the hydroxy and aldehyde moieties of the salicylaldehyde starting material. Electron-donating groups, such as methyl (5b) and tert-butyl (5d), when positioned at the 6-position, afforded the corresponding 3-acetyl coumarins in high yields of 96% and 90%, respectively, likely due to the increased nucleophilicity imparted by these groups on the hydroxyl moiety. In contrast, salicylaldehyde substituted at the same position with an electron-withdrawing group, such as NO2, proved less effective, yielding the target product in only 31% (5g). However, electron-donating groups, such as methyl (5c) and methoxy (5f), substituted at the 5-position of the coumarin, resulted in moderate yields of 55 and 51%, respectively, likely due to the reduced electrophilicity of the carbonyl group in the salicylaldehyde.
Halogenated salicylaldehydes effectively yield the corresponding 3-acetylcoumarins. For instance, 6-bromo coumarin (5h) was obtained in a high yield of 85%. However, the disubstituted 6,8-dibromo-(5i) and 6,8-dichloro-coumarins (5j) were obtained in only 31 and 28% yields, respectively. This reduction in yield is likely attributed to the α-electron-withdrawing effect of the 8-halo substituents, which may diminish the nucleophilic nature of the hydroxyl moiety. Furthermore, it is noteworthy that coumarin 5e was obtained in 50% yield through the annulation of the ketone moiety of o-hydroxy-acetophenone, rather than the aldehyde, as seen with salicylaldehyde derivatives. This observation suggests potential for expanding annulations to other ortho-substituted phenol functional groups, thereby broadening the scope of this transformation.
The structural diversity of this transformation was also evaluated using other o-aldehyde and dioxinone derivatives. Gratifyingly, the reaction between 2 and 2-hydroxy-naphthaldehyde afforded 3-acetyl benzo-coumarin (5k) in an excellent 97% yield, demonstrating the efficiency of the method with extended aromatic systems. Additionally, the use of various 6-benzoyl-dioxinone derivatives afforded the corresponding coumarins 5l–5p in yields ranging from 74 to 99%.
Conclusions
Our continuous flow protocol enabled the synthesis of butenolides in yields comparable to those achieved under batch conditions, but with a significantly reduced residence time of only 20 minutes, thereby enhancing productivity. This notable reduction in reaction time is attributed to improved heat transfer, mixing and the safe operation at temperatures exceeding the boiling point of solvent, facilitated by the flow system. Similarly, the flow protocol proved highly effective for synthesizing 16 structurally diverse coumarins within the same short residence time. Gratifyingly, it demonstrated compatibility with multi-carbonyl substrates, a challenging aspect for the Knoevenagel condensation strategy.In summary, we established a unified continuous flow platform that has proven to be a powerful tool for the efficient synthesis of structurally diverse butenolides and coumarins, offering both scalability and practicality. The gram-scale synthesis and crystallographic confirmation of a γ-spiro butenolide, further underscore the robustness and reliability of this methodology. Additionally, the platform's application to the synthesis of coumarins from salicylaldehydes and o-hydroxy-phenylacetyl groups, achieving quantitative yields, highlights its potential for broadening synthetic scope. By streamlining the synthesis of diverse structures, this continuous flow approach addresses the demands of modern synthetic and medicinal chemistry, providing a scalable, efficient, and safe solution for complex chemical transformations.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 3g has been deposited at the CCDC under 2380970 and can be obtained from DOI: https://doi.org/10.5517/ccdc.csd.cc2kxlhl.Author contributions
LCF, RSG and TM contributed to data curation, formal analysis, investigation, and validation. RSG also contributed to conceptualization. RCS, DLB, and JCP contributed to investigation and writing. JCP and DLB also contributed to conceptualization, resources, project administration, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the São Paulo Research Foundation – FAPESP (RCS 2023/07466-7; JCP, 2021/06661-5), the Brazilian National Council for Scientific and Technological Development – CNPq (JCP, 308540/2021-2), and the Coordination for the Improvement of Higher Education Personnel – CAPES (Finance Code 001; LCF 88887.910613/2023-00).Notes and references
- H. Beck, M. Härter, B. Haß, C. Schmeck and L. Baerfacker, Drug Discovery Today, 2022, 27, 1560–1574 CrossRef CAS PubMed.
- J. Hughes, S. Rees, S. Kalindjian and K. Philpott, Br. J. Pharmacol., 2011, 162, 1239–1249 CrossRef CAS PubMed.
- J. L. Dahlin and M. A. Walters, Future Med. Chem., 2014, 6, 1265–1290 CrossRef CAS PubMed.
- A. Sertkaya, T. Beleche, A. Jessup and B. D. Sommers, JAMA Netw. Open, 2024, 7, e2415445 CrossRef PubMed.
- G. A. Van Norman, J. Am. Coll. Cardiol. Basic Trans. Science, 2016, 1, 170–179 Search PubMed.
- D. L. Hughes, Org. Process Res. Dev., 2018, 22, 13–20 CrossRef CAS.
- L. F. Peña, P. González-Andrés, L. G. Parte, R. Escribano, J. Guerra, A. Barbero and E. López, Mar. Drugs, 2023, 21, 402 CrossRef PubMed.
- B. T. Ramanjaneyulu, N. K. Vishwakarma, S. Vidyacharan, P. R. Adiyala and D.-P. Kim, Bull. Korean Chem. Soc., 2018, 39, 757–772 CrossRef CAS.
- F. C. Braga, F. L. N. da Silva, T. de O. Ramos, J. G. H. Rosa, É. de A. Araujo, N. F. C. Junior, E. P. Wendler, A. Beatriz, R. O. M. A. de Souza, T. J. Brocksom and K. T. de Oliveira, Chem. – Asian J., 2024, 19, e202400689 CrossRef CAS PubMed.
- G. M. Martins, F. C. Braga, P. P. de Castro, T. J. Brocksom and K. T. de Oliveira, Chem. Commun., 2024, 60, 3226–3239 RSC.
- D. V. Silva-Brenes, S. Agrawal, V. López-Mejías, J. Duconge, C. P. Vlaar, J.-C. M. Monbaliu and T. Stelzer, React. Chem. Eng., 2024, 9, 2728–2739 RSC.
- A. Bonner and M. Baumann, Org. Biomol. Chem., 2024, 22, 8323–8327 RSC.
- S. K. Sartori, M. A. N. Diaz and G. Diaz-Muñoz, Tetrahedron, 2021, 84, 132001 CrossRef CAS.
- P. Dhyani, P. Sati, E. Sharma, D. C. Attri, A. Bahukhandi, B. Tynybekov, A. Szopa, J. Sharifi-Rad, D. Calina, H. A. R. Suleria and W. C. Cho, Cancer Cell Int., 2022, 22, 305 CrossRef CAS PubMed.
- H. Ma, K. Wang, B. Wang, Z. Wang, Y. Liu and Q. Wang, J. Agric. Food Chem., 2024, 72, 4658–4668 CrossRef CAS PubMed.
- H. Mou, J. Shi, J. Chen and D. Hu, Pestic. Biochem. Physiol., 2021, 178, 104913 CrossRef CAS PubMed.
- D. V. Silva-Brenes, S. K. Reyes-Vargas, J. Duconge, C. Vlaar, T. Stelzer and J.-C. M. Monbaliu, Org. Process Res. Dev., 2024, 28, 1704–1712 CrossRef CAS.
- M. Mazur and D. Masłowiec, Antibiotics, 2022, 11, 1327 CrossRef CAS PubMed.
- L. A. Sposato and O. Fustinoni, in Handbook of Clinical Neurology, Elsevier B.V., 1st edn, 2014, vol. 121, pp. 1635–1671 Search PubMed.
- E. K. Akkol, Y. Genç, B. Karpuz, E. Sobarzo-Sánchez and R. Capasso, Cancers, 2020, 12, 1959 CrossRef CAS PubMed.
- A. Husain, S. A. Khan, F. Iram, M. A. Iqbal and M. Asif, Eur. J. Med. Chem., 2019, 171, 66–92 CrossRef CAS PubMed.
- M. Lončarić, D. Gašo-Sokač, S. Jokić and M. Molnar, Biomolecules, 2020, 10, 151 CrossRef PubMed.
- S. C. Moldoveanu, in Pyrolysis of Organic Molecules, Elsevier, 2019, pp. 391–418 Search PubMed.
- K. P. Reber, S. D. Tilley and E. J. Sorensen, Chem. Soc. Rev., 2009, 38, 3022 RSC.
- C. N. Eid and J. P. Konopelski, Tetrahedron, 1991, 47, 975–992 CrossRef CAS.
- O. Ishibashi, Y. Meguro, S. Kuwahara and M. Enomoto, Eur. J. Org. Chem., 2024, 27, e202301006 CrossRef CAS.
- H. Lu, K. L. Handore, T. E. Wood, G. K. Shimokura, A. D. Schimmer and R. A. Batey, Org. Lett., 2023, 25, 7502–7506 CrossRef CAS PubMed.
- R. M. O'Mahony, D. Lynch, H. L. D. Hayes, E. Ní Thuama, P. Donnellan, R. C. Jones, B. Glennon, S. G. Collins and A. R. Maguire, Eur. J. Org. Chem., 2017, 2017, 6533–6539 CrossRef.
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed.
- S. Chandrasekhar and H. V. Kumar, Synth. Commun., 2015, 45, 232–235 CrossRef CAS.
- P. A. Peixoto, A. Boulangé, S. Leleu and X. Franck, Eur. J. Org. Chem., 2013, 2013, 3316–3327 CrossRef CAS.
- R. Galaverna, T. McBride, J. C. Pastre and D. L. Browne, React. Chem. Eng., 2019, 4, 1559–1564 RSC.
- H. R. Smallman, G. A. Brancaglion, J. C. Pastre and D. L. Browne, J. Org. Chem., 2022, 87, 12297–12305 CrossRef CAS PubMed.
- J. Sanjosé-Orduna, R. C. Silva, F. Raymenants, B. Reus, J. Thaens, K. T. de Oliveira and T. Noël, Chem. Sci., 2022, 13, 12527–12532 RSC.
- J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849 RSC.
- N. Kockmann, P. Thenée, C. Fleischer-Trebes, G. Laudadio and T. Noël, React. Chem. Eng., 2017, 2, 258–280 RSC.
- M. C. F. C. B. Damião, H. M. Marçon and J. C. Pastre, React. Chem. Eng., 2020, 5, 865–872 RSC.
- A. Bonner, P. Naik, R. Crawford and M. Baumann, Curr. Opin. Green Sustainable Chem., 2024, 47, 100907 CrossRef CAS.
- A. I. Alfano, J. García-Lacuna, O. M. Griffiths, S. V. Ley and M. Baumann, Chem. Sci., 2024, 15, 4618–4630 RSC.
- A. Bonner, P. Naik, R. Crawford and M. Baumann, Curr. Opin. Green Sustainable Chem., 2024, 47, 100907 CrossRef CAS.
- P. Bianchi and J.-C. M. Monbaliu, Acc. Chem. Res., 2024, 57, 2207 CrossRef CAS PubMed.
- M. C. F. C. B. Damião, R. Galaverna, A. P. Kozikowski, J. Eubanks and J. C. Pastre, React. Chem. Eng., 2017, 2, 896–907 RSC.
- J. M. De Souza, R. Galaverna, A. A. N. De Souza, T. J. Brocksom, J. C. Pastre, R. O. M. A. De Souza and K. T. De Oliveira, An. Acad. Bras. Cienc., 2018, 90, 1131–1174 CrossRef CAS PubMed.
- A. Boulangé, J. Parraga, A. Galán, N. Cabedo, S. Leleu, M. J. Sanz, D. Cortes and X. Franck, Bioorg. Med. Chem., 2015, 23, 3618–3628 CrossRef PubMed.
- C. J. Smith, R. L. Hettich, J. Jompa, A. Tahir, M. V. Buchanan and C. M. Ireland, J. Org. Chem., 1998, 63, 4147–4150 CrossRef CAS.
- S. Mandal and B. Thirupathi, Org. Biomol. Chem., 2020, 18, 5287–5314 RSC.
- Y. Li, T. Zhang, H. Ma, L. Xu, Q. Zhang, L. He, J. Jiang, Z. Zhang, Z. Zhao and M. Wang, J. Agric. Food Chem., 2023, 71, 6249–6267 CrossRef CAS PubMed.
- V. F. Batista, D. C. G. A. Pinto and A. M. S. Silva, Expert Opin. Drug Discovery, 2022, 17, 603–618 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For general procedure, experimental setup, optimization reactions, and NMR data. See DOI: https://doi.org/10.1039/d4re00567h |
| This journal is © The Royal Society of Chemistry 2025 |