
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous-flow synthesis of 1,4,6,7-tetrahydro-5H-[1,2,3]triazolo[4,5-c]pyridines towards key intermediates of clinical candidates JNJ-54175446 and zanvipixant (JNJ-55308942) with antidepressant activity†
Zsolt Fülöp ab,
Péter Banab,
István Greinerb and
János Éles*b
ab,
Péter Banab,
István Greinerb and
János Éles*b
aDepartment of Organic Chemistry and Technology, Faculty of Chemical Technology and Biotechnology, Budapest University of Technology and Economics, Műegyetem rkp. 3., H-1111 Budapest, Hungary
bGedeon Richter Plc, P. O. Box 27, 1475 Budapest, Hungary. E-mail: j.eles@gedeonrichter.com
First published on 16th July 2025
Abstract
1,4,6,7-Tetrahydro-5H-[1,2,3]triazolo[4,5-c]pyridines are potent antagonists of the P2X7 receptor, a key regulator of neuroinflammation associated with depression. Two clinical candidates, JNJ-54175446 and JNJ-55308942 were developed based on this scaffold, together with a stereoselective synthesis involving a one-pot [3 + 2] cycloaddition/Cope-elimination cascade. Besides challenging regioselectivity, this route raises safety concerns, as both steps involve hazardous reagents. Herein, we report a two-step continuous-flow strategy for the synthesis of this valuable scaffold, which enables access to intermediates of JNJ-54175446 and JNJ-55308942 via temperature-controlled regioselectivity. Applying the optimized conditions, yields of 48% (58% regioselectivity) and 45% (91% regioselectivity) were achieved, respectively. The method ensures safe utilization of azide in the cycloaddition, and uses a safer oxidant for the elimination, offering a scalable and affordable alternative synthetic route.
Introduction
Depression affects 4.4% of the global population,1 yet over 75% of people with such mental disorders in low- and middle-income countries receive no treatment.2 The demand for affordable, innovative treatment is rising, especially after the COVID-19 pandemic, as up to 40% of recovered patients experienced depression, anxiety, or post-traumatic stress disorder.3 With high prevalence, increased mortality risk,4 and growing resistance to current drugs,5 new approaches are essential. One promising strategy is based on evidence linking mood disorders to neuroinflammation.6 Since the P2X7 receptor is involved in the release of the inflammatory cytokine IL-1β, which is a key mediator of neuroinflammation, developing P2X7 antagonists is being explored as a potential treatment for mood disorders.1,7,8Extensive medicinal chemistry research in this field9 led to Janssen's discovery of 1,4,6,7-tetrahydro-5H-[1,2,3]triazolo[4,5-c]pyridine as a valuable scaffold for potent, brain penetrant P2X7 antagonists.10 Derivatives having a methyl substituent on the tetrahydropyridine ring showed improved potency, among which the 4-methyl derivatives of the scaffold provided JNJ-54175446 ((R)-1), the first candidate to enter clinical studies.11 Meanwhile, its backup compound, containing the 6-methyl derivative of the same scaffold with an optimized acyl moiety, JNJ-55308942 ((S)-2) possessed improved solubility and in vitro profile.12 It was progressed into clinical development under the INN zanvipixant. Both compounds reached phase II clinical trials in indications including major depressive disorder and bipolar disorder.
The published synthesis of the first generation clinical candidate ((R)-1) constructed the 4-methyl substituted 1,4,6,7-tetrahydro-5H-[1,2,3]triazolo[4,5-c]pyridine scaffold via a racemic route involving multiple hydrogenations, and required chiral separation to obtain the product ((R)-1).11 The synthesis of the advanced clinical compound ((S)-2) was reevaluated to reduce the number of steps and avoid chiral separation. Therefore a stereoselective, one-pot reaction sequence was developed (Scheme 1), which forms the methylated bicyclic scaffold from the inexpensive chiral building block, tert-butyl (2S)-2-methyl-4-oxopiperidine-1-carboxylate ((S)-3).12 The first step of the sequence is a dipolar [3 + 2] cycloaddition between (S)-3 and 2-azido-5-fluoropyrimidine (4) in the presence of pyrrolidine, while the second step is an oxidative Cope elimination, leading to the key intermediate. The main issue is regioselectivity, as two regioisomers ((S)-5 and (S)-6) are generated. The researchers optimized this transformation thoroughly, to provide the desired 6-methyl isomer ((S)-5) as a major product, with a moderate isolated yield (65% for the mixture of regioisomers). Notably, the minor regioisomer could serve as a potential intermediate of the 4-methly clinical candidate ((R)-1), by using the opposite enantiomer of the piperidone starting material ((R)-3), but this synthetic path has not been explored.
 | ||
| Scheme 1 Synthetic approaches towards P2X7 receptor antagonists JNJ-54175446 and JNJ-55308942, and their safety issues. | ||
Despite its brevity and versatility, this reaction sequence poses considerable safety concerns, as both steps use hazardous reagents, such as the explosive azide13 (4) in a heated reaction, and the shock sensitive meta-chloroperoxybenzoic acid (m-CPBA) as the oxidant.14,15 Flow chemistry16–18 is well-suited for one-pot transformations avoiding intermediate work-up procedures and thus minimizing human intervention, which is an important safety consideration when working with explosive and shock sensitive reagents.19–23 The use of microreactors further improves safety by limiting the amount of hazardous material present in the reaction at any given time.24 In addition, continuous-flow protocols enable precise regulation of parameters such as temperature and residence time, ensuring a controlled environment for high degrees of selectivity. Considering these aspects, herein we have developed a safe, two-step telescoped continuous-flow procedure for the synthesis of the 1,4,6,7-tetrahydro-5H-[1,2,3]triazolo[4,5-c]pyridine scaffold. We aimed to investigate the possibility of tuning the reaction parameters to afford both key intermediates towards Janssen's clinical candidates, while ensuring affordability by employing readily available and low-cost starting materials.
Results and discussion
To translate the one-pot reaction sequence to a continuous-flow method, we designed a telescoped system consisting of two modules, one for the cycloaddition and an other for the subsequent oxidation. We began by optimizing the first transformation, followed by coupling the oxidation to the first module's output, and the parameters of the second synthetic step were optimized separately. In the first step, the commercially available (S)-enantiomer of the oxopiperidine derivative ((S)-3) was used as the substrate to form two regioisomers of the pyrrolidine adduct of the bicyclic scaffold ((S)-7 and (S)-8, Scheme 2), differing in the position of the methyl group attached to the tetrahydropyridine ring. The absolute configuration of the methyl group is determined by the oxopiperidine starting material, in this case (S)-configuration is needed for JNJ-55308942 ((S)-2). | ||
| Scheme 2 Continuous-flow experimental set-up for the synthesis of (S)-7 and (S)-8 through cycloaddition (BPR: back pressure regulator). | ||
Preliminary experiments revealed that toluene (which was the optimal solvent for the batch procedure) was not suitable for flow experiments, since solid particles precipitated as the reaction progressed (even at room temperature), which could lead to clogging of the flow reactor. This precipitation was isolated by filtration and NMR analysis identified its main component as the ring opening product (9) formed in the reaction between 2-azido-5-fluoropyrimidine (4) and pyrrolidine (Scheme 3). Solubility studies (Fig. S1†) showed that the solvent mixture of toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH 1:1 could solubilize this side-product, without detrimental effects on the reaction in batch experiments. Furthermore, to prevent such decomposition in the feed solution, pyrrolidine was kept separately from the azide (4) before entering the flow reactor. On the other hand, mixing of (S)-3 and pyrrolidine in a single feed solution was viable. The formation of the presumed dienophile enamine could be observed in this mixture, but pre-mixing didn't have a significant effect on the reaction.
:MeOH 1:1 could solubilize this side-product, without detrimental effects on the reaction in batch experiments. Furthermore, to prevent such decomposition in the feed solution, pyrrolidine was kept separately from the azide (4) before entering the flow reactor. On the other hand, mixing of (S)-3 and pyrrolidine in a single feed solution was viable. The formation of the presumed dienophile enamine could be observed in this mixture, but pre-mixing didn't have a significant effect on the reaction.
 | ||
| Scheme 3 Presumed side-reaction in the cycloaddition step. | ||
Based on their respective solubilities, 4 (0.25 mol L−1) was dissolved in toluene, and the mixture of (S)-3 (0.25 mol L−1) and pyrrolidine (0.5 mol L−1) was dissolved in methanol. The reagent solutions were filled in two separate injector loops and were pumped at the same flow rate to ensure the ideal reaction medium after mixing in the reactor. The pumps continuously transferred toluene:MeOH 1:1 solvent mixture on both channels. The reagent streams were mixed in a T-piece before entering the small-volume heated reactor constructed from PTFE tubing. After the back-pressure regulator (set to 17 bar), the reaction mixture was collected and subjected to analysis (Scheme 2).
During the optimization process, temperature was varied between −15 °C and 150 °C (Fig. 1), with a residence time of 5 minutes. According to the literature, the reaction favored the formation of the 6-methyl adduct (S)-7 at room temperature with a selectivity of 60:40%, which improved at higher temperatures, up to 88:12% at 100 °C, interpreted by Janssen's researchers as the result of thermodynamic control.12 In our flow experiments, we achieved a similar level of selectivity towards (S)-7 at 130 °C, above which a slight decrease was observed, due to decomposition. Notably, under milder conditions, our results deviated from the reported trend: we observed a reversal in regioselectivity under rapid flow conditions, with the 4-methyl bicycle (S)-8 becoming the major product. This observation suggests that the kinetic control can be exploited under flow conditions. The optimal selectivity of 37:63% was reached at 60 °C, which couldn't be improved further by lowering the temperature. These findings suggests that our flow system allows the temperature-dependent control of regioselectivity, enabling the preferential formation of either regioisomer. Specifically, 130 °C and 60 °C were identified as the optimal conditions for the formation of (S)-7 and (S)-8, respectively.
 | ||
| Fig. 1 Optimization of selectivity for the cycloaddition reaction from (S)-3. Reaction conditions: 0.25 mol L−1 (S)-3 concentration, 1.5 mL reactor volume, 17 bar, 5 min residence time, 1 equivalent of azide (4), 2 equivalent of pyrrolidine. | ||
However, using these two distinct sets of parameters, the isolated regioisomeric mixtures (which contained (S)-7 and (S)-8 in the expected ratios) revealed low yields (ca. 20%) in both cases, making further improvements necessary. In order to accurately quantify the decrease of the starting material and the formation of the products, 2-methoxynaphthalene was used as an internal standard (detailed in the ESI†). During the subsequent optimization, the molar amount of the azide (4) was found to be a significant factor (Table S2†). These experiments were conducted in batch at 60 °C, due to the risk of precipitation of the side-product (9), which was expected to form in more concentrated solutions. Using two equivalents of 4 led to a marked increase in conversion, with minimal oxopiperidine ((S)-3) remaining. Higher amounts of 4 resulted in marginal improvements, but the observed precipitation rendered these latter conditions unfeasible for flow chemistry. Applying two equivalents of 4 in the continuous-flow system (with other parameters unaltered), the mixture of regioisomeric adducts ((S)-7 and (S)-8) was isolated with 50% and 46% yields at 60 °C and 130 °C, respectively. Importantly, the previously achieved regioselectivities were preserved.
Despite the nearly complete consumption of the oxopiperidine ((S)-3), the yields remained moderate. This is explained by the presence of unreacted enamine intermediate derived from pyrrolidine and (S)-3 in the reaction mixtures, due to the short reaction time. Still, this was designated as the endpoint of the optimization, prioritizing a rapid flow method over achieving total conversion, which could possibly require additional hours, and could compromise regioselectivity, especially for the hypothesized kinetically controlled product.
In the second step, oxidation of the regioisomer mixture ((S)-7 and (S)-8) gives the isomers of the targeted bicyclic scaffold ((S)-5 and (S)-6). Preliminary batch experiments showed that the shock sensitive m-CPBA can be replaced by magnesium bis(monoperoxyphthalate) hexahydrate (MMPP), a stable alternative oxidant. MMPP exhibits comparable chemical properties to m-CPBA, but it is safer to handle and commercially available at lower cost.25 Furthermore, its water solubility facilitates simplified work-up procedures. However, the application of MMPP is limited by its low solubility in non-polar solvents. To address this, the oxidation was conducted in a biphasic medium, consisting of the organic solvent mixture used in the first step, and basic solution of the oxidant, which accommodated the solubility requirements of both the substrates and the oxidant and enabled effective flow conditions.26
Batch experiments using MMPP showed total conversion overnight, and no solid particles were observed in the biphasic mixture. For the flow experiments, the previously optimized flow system was complemented by introducing the basic aqueous solution of MMPP (0.12 mol L−1), transferred by a pump connected directly to the output of the first cycloaddition step. The resulting biphasic reaction mixture was then directed to a reactor, kept at ambient temperature, as the increased safety provided by MMPP and flow processing eliminates the need for cooling. The two-reactor system was pressurized using a back-pressure regulator (set to 17 bar), after which samples were collected, and the organic phase was subjected to analysis (Scheme 4).
 | ||
| Scheme 4 Continuous-flow synthesis of (S)-5 and (S)-6 through cycloaddition followed by oxidation (M: mol L−1). | ||
The parameters and output flow rate of the first step were kept unchanged, optimization of the second step focused on the volume of the reactor (which determined residence time), molar ratio of MMPP and mixing (Fig. 2). Given the use of excess pyrrolidine in the first step, it was anticipated that an excess of MMPP would be required as residual pyrrolidine could rapidly quench part of the oxidant.15 Using a T-shaped mixer to introduce the oxidant with 1 mL reactor volume, conversion improved up until 3 equivalents of MMPP (50 s residence time), but further increase had no effects. Application of a 4 mL reactor (residence time 2.7–6.7 min, depending on the MMPP equivalent) resulted in significantly higher conversion, but the reaction was still incomplete.
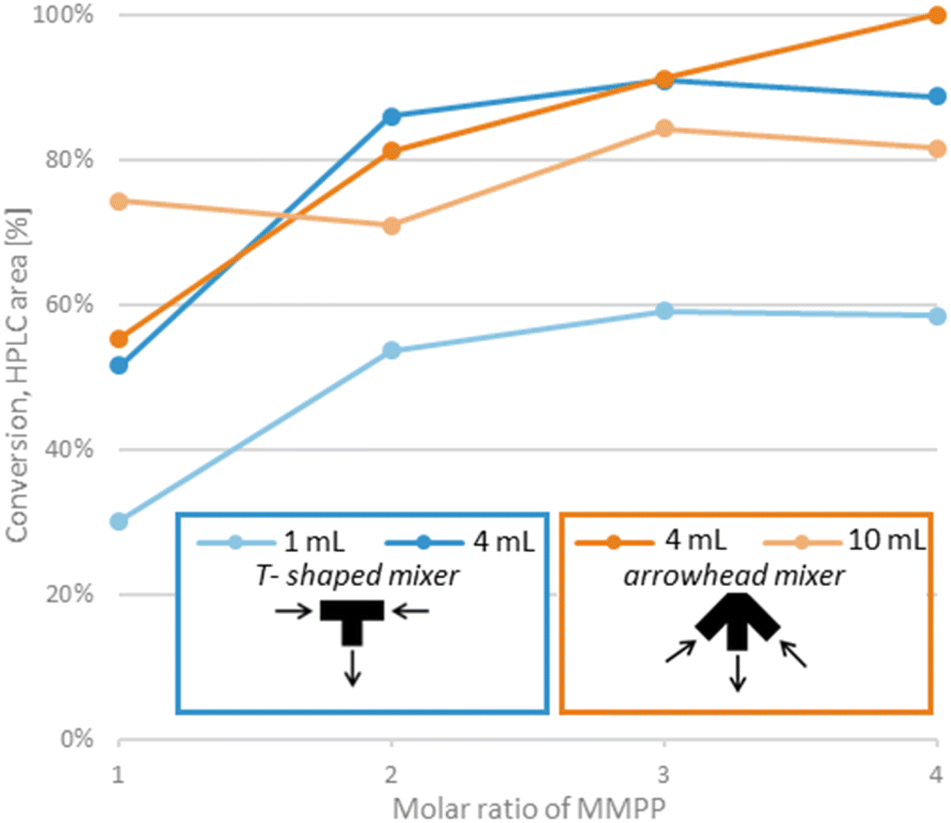 | ||
| Fig. 2 Optimization of reaction parameters for the oxidation reaction. Reaction conditions: ambient temperature, 17 bar. Parameters of the first step were kept unchanged. Residence time varies depending on the MMPP equivalents, the corresponding ranges are described in the text. | ||
The effect of mixing was presumed to be an important factor in the biphasic reaction mixture. Therefore, the T-shaped mixer was replaced by a static mixer (containing a polymer frit) with an arrowhead channel configuration, which provides enhanced mixing.27 At lower reagent ratios, the difference seemed negligible, but it still improved with 4 equivalents of MMPP, which gave full conversion (2.7 min residence time). Further increase in reactor size (10 mL, residence time 6.7–16.7 min, depending on the MMPP equivalent) proved to be unnecessary, as the conversion deteriorated.
By combining the optimized oxidation conditions with the two distinct sets of optimized parameters for the cycloaddition step, two telescoped continuous-flow methods were developed for the synthesis of the methylated 1,4,6,7-tetrahydro-5H-[1,2,3]triazolo[4,5-c]pyridine scaffolds (Scheme 5). Starting from (R)-3 and conducting the cycloaddition step at 60 °C, the regioisomer mixture containing the corresponding major product ((R)-6) was obtained with an overall yield of 48% (361 mg h−1 throughput) after purification, with a modest regioselectivity of 58% in favor of the 4-methyl scaffold ((R)-6), the intermediate of the first generation clinical candidate P2X7 antagonist JNJ-54175446 ((R)-1). On the other hand, when the opposite enantiomer of the starting material ((S)-3) was used as starting material and the cycloaddition was performed at 130 °C, the corresponding major product ((S)-5) was obtained with an overall yield of 45% (338 mg h−1 throughput) after purification as a mixture of regioisomers. In this case, a remarkable regioselectivity of 91% was achieved in favor of (S)-5, the intermediate of the advanced P2X7 antagonist clinical candidate zanvipixant (JNJ-55308942, (S)-2). Despite the rapid residence time, the yields of the flow synthesis were comparable to the literature. Regioisomer ratios were determined by a supercritical fluid chromatography (SFC) method using a chiral stationary phase, which was primarily developed for the separation of regioisomers, but also resolved the enantiomers. No signs of racemization were shown in each case, despite the high temperature flow conditions.
 | ||
| Scheme 5 Continuous-flow strategies for the synthesis of (R)-1 and (S)-2. | ||
For both mixtures, preparative separation of the regioisomers were carried out by SFC to obtain analytical samples of each of the four bicyclic products ((R)-5, (S)-5, (R)-6 and (S)-6; detailed in the ESI†). The intermediates obtained as major products ((R)-6 and (S)-5) are suitable for accessing both clinical candidates ((R)-1 and (S)-2, respectively) through already established and straightforward N-deprotection followed by N-acylation protocols.11,12
Conclusions
We have designed a two-step continuous-flow system for synthesizing the 1,4,6,7-tetrahydro-5H-[1,2,3]triazolo[4,5-c]pyridine scaffold. In the first cycloaddition step, two sets of optimal conditions were identified, revealing a temperature-dependent regioselective control. Specifically, the 4-methyl adduct (S)-8 was preferentially formed at 60 °C, while the 6-methyl isomer (S)-7 was predominant at 130 °C. In the subsequent oxidation, the shock sensitive and potentially explosive m-CPBA was replaced with MMPP, and conditions were optimized for total conversion at ambient temperature. Telescoping the two steps enables a compact, safe and scalable approach to this scaffold, including both JNJ-intermediates (48% yield with 58% regioselectivity for (R)-6 and 45% yield with 91% regioselectivity for (S)-5), controlled by temperature and the absolute configuration of the starting material. We believe, that our flow method offers a more convenient and affordable access to this important scaffold, including clinical candidates JNJ-54175446 and JNJ-55308942. In case of the former, this work may provide a novel, alternative synthetic route.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed in the frame of the FIEK_16-1-2016-0007 project, implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the FIEK_16 funding scheme. The work was carried out under the framework of the Important Project of Common European Interest (IPCEI) Med4Cure project. The authors are grateful to Anita Prechl and Bonifác Komáromi for their assistance with SFC analysis and separation, János Kóti for HRMS analysis and Márton Weber for NMR analysis. Z. F. thanks the Gedeon Richter Talentum Foundation for financial support.Notes and references
- J. M. Deussing and E. Arzt, Trends Mol. Med., 2018, 24, 736–747 CrossRef CAS PubMed.
- S. Evans-Lacko, S. Aguilar-Gaxiola, A. Al-Hamzawi, J. Alonso, C. Benjet, R. Bruffaerts, W. T. Chiu, S. Florescu, G. de Girolamo, O. Gureje, J. M. Haro, Y. He, C. Hu, E. G. Karam, N. Kawakami, S. Lee, C. Lund, V. Kovess-Masfety, D. Levinson, F. Navarro-Mateu, B. E. Pennell, N. A. Sampson, K. M. Scott, H. Tachimori, M. ten Have, M. C. Viana, D. R. Williams, B. J. Wojtyniak, Z. Zarkov, R. C. Kessler, S. Chatterji and G. Thornicroft, Psychol. Med., 2018, 48, 1560–1571 CrossRef CAS.
- N. Seighali, A. Abdollahi, A. Shafiee, M. J. Amini, M. M. Teymouri Athar, O. Safari, P. Faghfouri, A. Eskandari, O. Rostaii, A. H. Salehi, H. Soltani, M. Hosseini, F. S. Abhari, M. R. Maghsoudi, B. Jahanbakhshi and M. Bakhtiyari, BMC Psychiatry, 2024, 24, 105 CrossRef PubMed.
- P. Cuijpers and F. Smit, J. Affective Disord., 2002, 72, 227–236 CrossRef PubMed.
- A. M. Alqahtani, C. Kumarappan, V. Kumar, R. Srinivasan and V. Krishnaraju, Eur. Rev. Med. Pharmacol. Sci., 2020, 24, 7784–7795 CAS.
- K. A. Jones and C. Thomsen, Mol. Cell. Neurosci., 2013, 53, 52–62 CrossRef CAS PubMed.
- A. Bhattacharya, Front. Pharmacol, 2018, 9, 30 CrossRef PubMed.
- W. Qi, X. Jin and W. Guan, Biochem. Pharmacol., 2024, 219, 115959 CrossRef CAS PubMed.
- J. C. Rech, A. Bhattacharya, M. A. Letavic and B. M. Savall, Bioorg. Med. Chem. Lett., 2016, 26, 3838–3845 CrossRef CAS.
- B. M. Savall, D. Wu, M. De Angelis, N. I. Carruthers, H. Ao, Q. Wang, B. Lord, A. Bhattacharya and M. A. Letavic, ACS Med. Chem. Lett., 2015, 6, 671–676 CrossRef CAS PubMed.
- M. A. Letavic, B. M. Savall, B. D. Allison, L. Aluisio, J. I. Andres, M. De Angelis, H. Ao, D. A. Beauchamp, P. Bonaventure, S. Bryant, N. I. Carruthers, M. Ceusters, K. J. Coe, C. A. Dvorak, I. C. Fraser, C. F. Gelin, T. Koudriakova, J. Liang, B. Lord, T. W. Lovenberg, M. A. Otieno, F. Schoetens, D. M. Swanson, Q. Wang, A. D. Wickenden and A. Bhattacharya, J. Med. Chem., 2017, 60, 4559–4572 CrossRef CAS PubMed.
- C. C. Chrovian, A. Soyode-Johnson, A. A. Peterson, C. F. Gelin, X. Deng, C. A. Dvorak, N. I. Carruthers, B. Lord, I. Fraser, L. Aluisio, K. J. Coe, B. Scott, T. Koudriakova, F. Schoetens, K. Sepassi, D. J. Gallacher, A. Bhattacharya and M. A. Letavic, J. Med. Chem., 2018, 61, 207–223 CrossRef CAS PubMed.
- D. S. Treitler and S. Leung, J. Org. Chem., 2022, 87, 11293–11295 CrossRef CAS PubMed.
- R. N. McDonald, R. N. Steppel and J. E. Dorsey, Org. Synth., 1970, 50, 15 CrossRef CAS.
- H. Hussain, A. Al-Harrasi, I. R. Green, I. Ahmed, G. Abbas and N. U. Rehman, RSC Adv., 2014, 4, 12882–12917 RSC.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- F. M. Akwi and P. Watts, Chem. Commun., 2018, 54, 13894–13928 RSC.
- Z. Fülöp, P. Szemesi, P. Bana, J. Éles and I. Greiner, React. Chem. Eng., 2020, 5, 1527–1555 RSC.
- B. Gutmann, J. Roduit, D. Roberge and C. O. Kappe, Angew. Chem., Int. Ed., 2010, 49, 7101–7105 CrossRef CAS PubMed.
- P. B. Palde and T. F. Jamison, Angew. Chem., Int. Ed., 2011, 50, 3525–3528 CrossRef CAS PubMed.
- F. Carpentier, F.-X. Felpin, F. Zammattio and E. Le Grognec, Org. Process Res. Dev., 2020, 24, 752–761 CrossRef CAS.
- P. Szemesi, P. Bana, Z. Szakács, I. Greiner and J. Éles, Curr. Org. Chem., 2023, 26, 2223–2229 CrossRef.
- Z. Fülöp, P. Bana, M. Temesvári, J. Barabás, Z. Kazsu, Z. Béni, I. Greiner and J. Éles, Org. Process Res. Dev., 2024, 28, 3685–3690 CrossRef.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- P. Brougham, M. S. Cooper, D. A. Cummerson, H. Heaney and N. Thompson, Synthesis, 1987, 1987, 1015–1017 CrossRef.
- N. Weeranoppanant, React. Chem. Eng., 2019, 4, 235–243 RSC.
- S. Schwolow, J. Hollmann, B. Schenkel and T. Röder, Org. Process Res. Dev., 2012, 16, 1513–1522 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra03780h |
| This journal is © The Royal Society of Chemistry 2025 |