
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulticomponent Hosomi–Sakurai reaction on chiral, bio-based, alcohols†
Chiara Lambruschini *a,
Asunción Barberob,
Alberto Cherubina,
Lisa Monia,
Lorenzo Palioa,
Renata Rivaa and
Luca Banfi*a
*a,
Asunción Barberob,
Alberto Cherubina,
Lisa Monia,
Lorenzo Palioa,
Renata Rivaa and
Luca Banfi*a
aDepartment of Chemistry and Industrial Chemistry, University of Genova, via Dodecaneso 31, 16146 Genova, Italy. E-mail: chiara.lambruschini@unige.it; luca.banfi@unige.it
bDepartment of Organic Chemistry, University of Valladolid, Paseo de Belen, 7, Valladolid, 47011, Spain
First published on 29th May 2025
Abstract
The multicomponent Hosomi–Sakurai reaction, which gives homoallyl ethers from aldehydes, trimethylsilyl ethers and allyltrimethylsilane, was thoroughly studied using a bio-based aldehyde (protected 5-(hydroxymethyl)tetrahydrofuran-2-carboxyaldehyde) along with trimethylsilyl ethers derived from a variety of (mainly bio-based) alcohols. For comparison, a reactive aromatic aldehyde was also employed. These studies helped to identify the optimal conditions for each aldehyde and provided insight into the scope and limitations of this relatively underexplored multicomponent reaction, one of the few that can incorporate alcohols as diversity inputs.
Introduction
Multicomponent reactions (MCRs) are a powerful tool for the diversity-oriented preparation of complex organic molecules from easily available starting materials, with high atom- and step-economy.1–3 These features make MCRs also very useful in green chemistry, fulfilling several of its principles.4 The sustainability of this approach can be further enhanced when some or all of the components are bio-based, that is derived from renewable natural sources.5So far, the exploitation of bio-based starting materials has been mainly focused on the production of low-cost commodities and materials, such as biopolymers.6
However, it is of great interest to expand their use also in the preparation of bio-based fine chemicals (e.g. Active Pharmaceutical Ingredients) with high added value. The diversity-oriented synthesis of complex molecules from biomass is predestined to become an important tool in relieving dependence from oil.
In this context, in recent years we have often investigated the use of bio-based reagents in the Passerini,7–10 Ugi,11–14 and Ugi-Joullié15 MCRs. In all these efforts, the bio-based components were carbonyl compounds, amines, carboxylic acids or isocyanides, but never alcohols. Although alcohols are probably the most abundant among low-cost natural substances, multicomponent reactions that exploit these building blocks are very rare, unless they are in situ converted into carbonyl compounds.16–19
Recently, we have pointed our attention to the use, in multicomponent reactions, of aldehydes 3 and 4, which can be derived, in high enantiomeric excess, from 5-hydroxymethylfurfural (5-HMF). 5-HMF is in turn one of the twelve most interesting bio-based compounds accessible by dehydration processes of lignocellulosic biomass20,21 and it can be further converted to the meso diol 1 by catalytic hydrogenation.22,23 In 2020, we have reported the successful bio-catalytic desymmetrization of 1 to give aldehydes 3 and 4 (Scheme 1), which have been employed in Zn(II)-mediated diastereoselective Passerini reactions.9
 | ||
| Scheme 1 Previously reported conversion of diol 1 into desymmetrized aldehydes 3 and 4. | ||
In order to extend the potential applications of 3 and 4, and given our continuous interest in MCRs, as well as in their stereochemical aspects,24–26 we were interested to explore other types of multicomponent reactions, especially by combining 3 and 4 with bio-based alcohols.
One of the very few examples of MCRs using alcohols27 is represented by the so called “multicomponent Hosomi–Sakurai” reaction,28–34 where a carbonyl compound 5, an alcohol (or its trimethylsilyl ether 6) are combined with an allyl silane 7 in the presence of a Lewis or protic acid, to afford a homoallyl ether 8, with the generation of a new stereogenic centre (unless a symmetric ketone or formaldehyde are used) (Scheme 2).
 | ||
| Scheme 2 The Hosomi–Sakurai multicomponent reaction using trimethylsilyl ethers. TMS = trimethylsilyl. | ||
Homoallylic ethers are important building blocks recurrent in natural products.35 When a silyl ether of the alcohol is used, this reaction is also called “silyl modified Sakurai reaction”.29 The control of the stereochemical outcome has been studied using chiral silyl ethers28,36–38 and chiral crotyl silanes.39–42 To the best of our knowledge, there are very few examples employing chiral aliphatic aldehydes.33,34 Thus, we decided to study the scope of this MCR on our aldehyde 3 or its analogues bearing different protecting groups. However, we encountered some experimental issues that prompted us to carry out a more systematic study of this reaction. This involved using both achiral and chiral alcohols, including bio-based ones, as well as a less problematic aromatic aldehyde for comparison. Eventually, our efforts have allowed to establish the best experimental conditions for this reaction and to evaluate its scope and limitations. We report here our results, hoping that they can be useful for all those who want to exploit this underexplored MCRs in both target-oriented or diversity-oriented synthesis.
Results and discussion
We preliminarily investigated the Hosomi–Sakurai multicomponent reaction by subjecting enantiopure aldehyde 3 to the classical conditions reported by Markó et al.,34 using, as partners, cyclohexyloxytrimethyl silane (CyOTMS) 13b, and allyltrimethyl silane (Scheme 3). The preparation of 13b and of the other TMS ethers used at a later time to test the scope of the reaction (Scheme 5) can be easily performed from the corresponding alcohols using stoichiometric (TMS)2NH in the presence of catalytic iodine.43 At the end of the reaction, TMS ethers are obtained by quick filtration through a silica plug and evaporation to dryness. They are pure enough to be used for the MCR, and can be stored at −20 °C for months.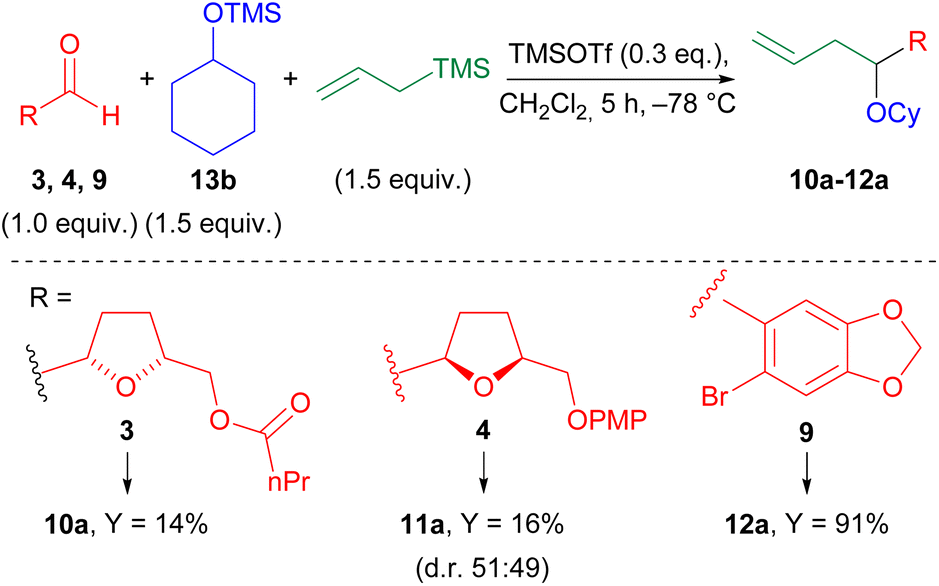 | ||
| Scheme 3 Preliminary results. TMSOTf (trimethylsilyl trifluoromethanesulfonate) was used as freshly prepared 0.3 M solution in CH2Cl2; equiv. = equivalent; Cy = cyclohexyl; Y = yield; d.r. = diastereomeric ratio. | ||
In the Hosomi–Sakurai multicomponent reaction, we used CH2Cl2 as the solvent and a freshly prepared solution of TMSOTf in CH2Cl2 as the catalyst. As shown in Scheme 3, the desired product 10a was obtained in low yield. Hypothesizing an incompatibility of the ester group with TMSOTf, we shifted to aldehyde 4, characterized by a more stable protecting group. This aldehyde is obtained in high yield and as a single enantiomer from desymmetrized alcohol 2 as previously reported by us.9 Again, only a low yield of product 11a was obtained. Furthermore, the d.r. (HPLC-UV) was found to be quite low (near to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Scheme 4 shows the putative mechanism of this multicomponent reaction.‡ The possible side products are the bicomponent product II and the acetal VI. Compound II was formed in significant amounts when the TMS ether of the alcohol was used in stoichiometric amounts, but can be suppressed using 1.5 or 2.0 equiv. of it. On the other hand, we were able to isolate variable amounts of the acetal VI. However, its formation seems to be reversible, as demonstrated by its conversion into multicomponent adduct VII, when treated again with TMSOTf and allyl trimethylsilane.
 | ||
| Scheme 4 Putative mechanism of multicomponent Hosomi–Sakurai reaction. | ||
Also Markó et al.34 have reported low yields under the same conditions using α-alkoxyaldehydes. These authors suggested that the low yield could be due to side reactions occurring during work-up and that the product could be converted into hemiacetal VIII because of the presence of triflic acid. Although we could detect the acetal VI, the hemiacetal VIII was never observed, and it is likely to be unstable. Finally, from our evidence, the formation of product VII seems to be irreversible under the reaction conditions.§.
Reasoning that these disappointing results could stem from the particular nature of our aldehyde, we tested 6-bromopiperonal 9, and the expected product 12a was eventually obtained in 91% yield in a short reaction time. Aldehyde 9 is non-enolizable, does not contain potentially reactive group, and is also activated towards this reaction, since the dioxolane group can stabilize the intermediate oxocarbenium ion.
This last data pointed out that the conditions that are efficient for “easy” aldehydes, such as 9, may be not well suited for more complex, aliphatic and not activated aldehydes. We tried to decrease the amount of catalyst to 0.1 equiv. but the reactions became too slow with 4, and even with 9 it was far from completion after 24 h.
We then used 9 to explore potential substitutes for TMSOTf, aiming to identify a less aggressive Lewis acid that could also work effectively on “difficult” aldehydes like 4 (Table 1). None of them were effective in promoting the reaction, and only using stoichiometric Bi(OTf)3 we observed tiny traces of the product (entry 3). It is worth noting that Bi(OTf)3 and iodine left the aldehyde untouched (entries 2, 3 and 7). On the other hand, stronger Lewis acids led to consumption of the aldehyde, but affording other unidentified products (entries 4–6).
|
|||
|---|---|---|---|
| Entry | Catalyst (equiv.) | Yieldb of 12a (%) | Unreacted aldehydeb (%) |
| a 6-Bromopiperonal 9 (0.30 mmol, 1 equiv.), CyOTMS (1.5 equiv.), Allyl-TMS (1.5 equiv.), catalyst (equiv. indicated in the Table), CH2Cl2 (1.5 mL), −78 °C, 5 h.b Calculated by 1H NMR of the crude using 3,4,5-trimethyoxybenzyl alcohol as internal standard (see ESI).c Freshly prepared 0.3 M solution in CH2Cl2.d Isolated yield. | |||
| 1 | TMSOTf (0.3)c | d91% | — |
| 2 | Bi(OTf)3 (0.3) | 0% | 93% |
| 3 | Bi(OTf)3 (1.1) | 2% | 90% |
| 4 | BF3·Et2O (1.1) | 0% | 66% |
| 5 | SnCl4 (1.1) | 0% | 37% |
| 6 | TiCl4 (1.1) | 0% | 26% |
| 7 | I2 (0.1) | 0% | 93% |

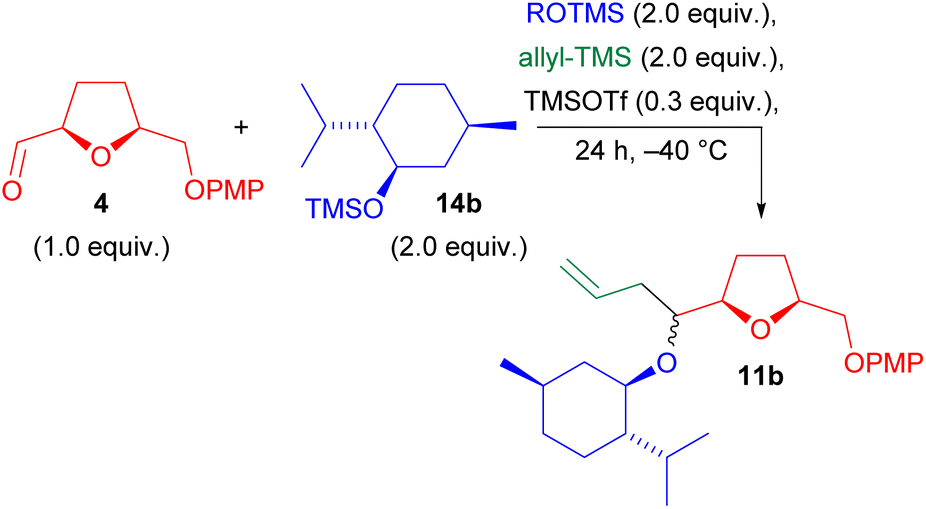
Thus, we returned to using TMSOTf and aldehyde 4, focusing on optimizing the reaction conditions. This time, we decided to use the TMS ether of (−)-menthol, a cheap bio-based substrate (Table 2). First of all, we noticed that, contrary to aldehyde 9, the reaction was too slow at −78 °C, and substantial amount of the acetal VI (Scheme 4) were isolated. Thus, we decided to work at −40 °C. At this temperature, the multicomponent adduct 11b was formed in slightly better, but still unsatisfactory, yield, although with a good d.r. (entry 1). A possible explanation of this low yield was attributed to the presence of water. We indeed noted that aldehyde 4 has a strong tendency to stay in its hydrated form thanks to a 5-membered intramolecular hydrogen bond involving the tetrahydrofuran oxygen. Water may be detrimental by destroying the catalyst, at the same time producing triflic acid, which in turn may cause opening of the tetrahydrofuran ring. Actually, during the first set of experiments, we always observed that the recovery of the crude product was much lower than the theoretical, likely because opening of the ring would afford highly polar adducts, which are lost in the aqueous phase during extraction.
|
|||||
|---|---|---|---|---|---|
| Entry | Aldehyde pre-treatment | Solvent | Additive | Yield b | d.rc |
| a Scale: 0.24 mmol of 4. Concentration: 0.2 M. TMSOTf was used as a freshly prepared 0.3 M solution in CH2Cl2.b Determined by HPLC-UV using 1,2-dimethoxybenzene as internal standard (see ESI).c The diastereomeric ratio was determined by HPLC.d The aldehyde was used directly after chromatography.e After the chromatography the aldehyde was taken up in the desired reaction solvent, and kept overnight over freshly activated 3 Å MS (rods).f After the chromatography the aldehyde was kept overnight in a desiccator over P2O5 under vacuum.g Unreacted aldehyde was detected by TLC.h Isolated yield on 0.50 mmol was 65%.i 55 mg of powdered 3 Å MS were added to the reaction mixture. | |||||
| 1 | None | CH2Cl2 | None | 20% | 81:19 |
| 2 | Noned | Toluene | None | 20% | 92:8 |
| 3 | 3 Å MSe | CH2Cl2 | None | 67% | 81:19 |
| 4 | 3 Å MSe | Toluene | None | 58% | 92:8 |
| 5 | P2O5f | Toluene | None | —g | — |
| 6 | 3 Å MSe | Pentane/toluene 85:15 |
None | 54% | 92:8 |
| 7 | 3 Å MSe | THF | None | 20% | 84:16 |
| 8 | 3 Å MSe | Toluene | CaCO3 (0.3 equiv.) | h71% | 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :8 :8 |
| 9 | 3 Å MSe | Toluene | CaCO3 (1.0 equiv.) | 57% | 92:8 |
| 10 | 3 Å MSe | Toluenei | 3 Å MSi | 42% | 92:8 |
In order to overcome these problems, we adopted two strategies: (a) pre-treat the aldehyde with dessicants, to force its dehydration; (b) add a non-soluble base, such as CaCO3, able to buffer the system without suppressing catalyst activity. These two changes turned out to be beneficial for the yield. On the other hand, the change of solvent from CH2Cl2 to toluene was beneficial from the point of view of stereoselectivity (entry 3 vs. entry 4). Further decrease of polarity of the solvent (entry 6) was not useful.
The best way to dehydrate aldehyde 4 makes use of molecular sieves, whereas drying in dessiccator on P2O5 was ineffective (entry 5). Addition of molecular sieves in the reaction mixture was deleterious (entry 10): it is better to use them to dry the aldehyde, but not to have them left during the reaction. A breakthrough result was obtained using CaCO3 as insoluble buffering agent (entries 8 and 9) and 0.3 equiv. seemed the optimal amount.
With this optimization, the yield was increased to 71%, with an excellent diastereoselective ratio (92:8) (entry 8).
Having identified the optimal conditions for 4 (Table 2, entry 8) and 9 (Table 1, entry 1), we moved to the reaction scope (Scheme 5) testing primary (15b), secondary (13b–14b and 16b–21b) and tertiary (22b–24b) silyl ethers. As can be seen, most of the secondary alcohols were chosen due to their natural origin. All chiral alcohols of Scheme 5 employed were enantiopure.

Apart from allyloxytrimethylsilane 15b, trimethylsilyl ethers are not commercially available, but they can be easily prepared from the corresponding alcohol as above described.43
Some of the alcohols were commercially available: cyclohexanol 13a, (−)-menthol 14a, (+)-menthol ent-14a, (+)-fencyl alcohol 17a, (−)-isopinocampheol 18a, (−)-isopulegol 19a, and tertiary alcohols 22a–24a. The others were prepared as reported in Scheme 6.
 | ||
| Scheme 6 Synthesis of non-commercial alcohols. | ||
The racemic alcohol rac-16a was obtained in one step from phenylacetaldehyde44 and resolved by biocatalysis using Amano PS lipase.45 The non-reactive enantiomer 16a46 was isolated with excellent yield and e.e., whereas the acylated (S)-25 was hydrolysed under basic conditions affording the other enantiomer ent-16a in excellent yield and e.e.47
Although the diastereomeric secondary alcohols 20a–21a were previously synthesized by Seebach by addition of diethylzinc under asymmetric catalysis,48 we found more convenient a biocatalytic approach. The addition of EtMgBr to commercially available (−)-myrtenal 25 proceeds in high yield, but with poor substrate induced diastereoselectivity and a 57:47 mixture of inseparable epimers was detected by 1H NMR.
We exploited again the use of biocatalysis to selectively convert one epimer into the acetate, which can be easily separated from the unreacted epimeric alcohol by chromatography. We ran a screening of lipases (see ESI†) and Amano AK lipase emerged as the best in terms of selectivity and reaction rate, giving selectively alcohol 20a. Finally, the acetate group of (R)-26 was removed under basic hydrolysis affording the corresponding alcohol 21a in excellent yield. The configuration of epimeric 20a and 21a was assessed by Mosher's esters analysis (see ESI†) and the results are in agreement with the Kazlauskas's rule for lipase preference in acylation of secondary alcohols.49
The results of multicomponent Hosomi–Sakurai reaction with the TMS ethers shown in Scheme 5 are reported in Table 3 for chiral aldehyde 4 and in Table 4 for achiral 6-bromopiperonal 9. Ethers 12a and 12c were obtained as racemic mixtures. All the remaining ethers 11 or 12, derived from chiral enantiopure substrates were obtained as pure enantiomers.¶
|
|||||
|---|---|---|---|---|---|
| Entry | TMS ether | Prod | Special conditions | Yieldb | Syn/anti ratioc |
| a Standard condition of entry 8 of Table 2 were followed, unless otherwise noted.b Isolated yield.c The diastereomeric ratio was determined by HPLC or 1H NMR (see ESI for details).d Anti isomer was faster running in TLC (PE/Et2O).e Syn isomer was faster running in TLC (PE/Et2O).f The acetal VI was the main product (79:21 ratio acetal VI/11d).g Prins adduct 30 was formed (see text).h A dimeric ether was formed (see text).i No reaction. |
|||||
| 1 | 13b | 11a | None | 70% | 65:36d |
| 2 | 14b | 11b | None | 65% | 92:8e |
| 3 | ent-14b | 11c | None | 70% | 32:68d |
| 4 | 15b | 11d | None | f<21% | n.d |
| 5 | 15b | 11d | 0 °C | 55% | 48:52d |
| 6 | 16b | 11e | None | 66% | 57:43d |
| 7 | ent-16b | 11f | None | 57% | 27:73d |
| 8 | 17b | 11g | None | 66% | 90:10e |
| 9 | 19b | — | None | —g | n.d |
| 10 | 20b | — | None | —h | n.d |
| 11 | 21b | — | None | —h | n.d |
| 12 | 22b | — | None | —i | n.d |
| 13 | 24b | — | None | —i | n.d |

|
|||||
|---|---|---|---|---|---|
| Entry | TMS ether | Product | Time | Yieldb | D.rc |
| a Standard condition of entry 1 of Table 1 were followed, unless otherwise noted.b Isolated yield.c The diastereomeric ratio was determined by HPLC (see ESI). Relative configuration not determined.d Prins products were formed (see text). | |||||
| 1 | 13b | 12a | 5 h | 91% | — |
| 2 | 14b | 12b | 4 h | 87% | 5545 |
| 3 | 15b | 12c | 2 h | 82% | — |
| 4 | ent-16b | 12d | 1 h | 89% | 82:18 |
| 5 | 17b | 12e | 1, 5h | 60% | 52:48 |
| 6 | 18b | 12f | 6 h | 49% | 52:48 |
| 7 | 19b | — | 4 h | -d | — |
| 8 | 23b | — | 6 h | No reaction | — |

Allyloxytrimethylsilane 15b, a primary silyl ether, afforded the expected product in excellent yield by reaction with 9 (Table 4, entry 3). On the other hand, starting from 4, and using standard conditions (−40 °C), the 1H NMR of the crude product showed a 79:21 ratio of acetal VI/11d. However, when we submitted again the crude product to the same reaction conditions (catalyst, allyl trimethylsilane, −40 °C), a progress of the reaction was observed (45:55 acetal VI/11d). This confirms that the acetal is a possible intermediate of the reaction. We postulated that in the case of primary groups, the acetal has a lower tendency to give the key intermediate oxocarbenium ion V, due to a lower benefit following the steric release. In this scenario, a higher temperature might be beneficial; in fact, the desired products were obtained in 55% yield when the reaction was carried out at 0 °C (entry 5). The diastereomeric ratio was near to 1:1. However, the epimers can be easily separated by chromatography, as in all the other examples of Table 3, and this was crucial for the assessment of the relative stereochemistry (see below).
Secondary silyl ethers proved to be the best partner in the Hosomi–Sakurai multicomponent reaction and a good variety of bio-based silyl ethers were employed.
As expected, the reactions starting from 6-bromopiperonal 9 were cleaner and the yields were higher than those from aldehyde 4. This is due to the high reactivity of 9, which allowed the reactions to be performed at −78 °C in few hours, whereas 4 required a higher temperature (−40 °C) and longer reaction times. Anyway, even TMS ethers of bulky secondary alcohols were able to afford in satisfactory yields adducts 11 or 12 starting from both aldehydes.
With the aim to explore post MCR cyclizations based on ring closing metathesis (RCM), we were particularly interested in TMS ethers containing double bonds, especially those of natural origin, such as 19b–21b. However, the outcomes were somehow surprising. While allyl ethers 16b and ent-16b led to the expected products, starting from allyl ethers 20b and 21b, we could only isolate, either for 4 or 9, a diastereomeric mixture of ethers 27 (Scheme 7), together with unreacted aldehyde.
 | ||
| Scheme 7 Unexpected products of some multicomponent Hosomi–Sakurai reactions. | ||
Ethers 27 may derive from the formation of the carbocation 28. The different behaviour of 20b–21b compared to 16b, may be due to a subtle difference in stability of carbocation 28 compared to 29. Indeed, carbocation 28 has two resonance forms that are both secondary, whereas one of the forms of 29 is primary (Scheme 7).
Also the failure to obtain the expected products using tertiary TMS ethers 22b–24b may be ascribed to the formation of stable carbocations, which can lead to oligomerization–polymerization reactions, leaving the aldehydes unreacted.
In the literature, there is only one example of multicomponent Hosomi–Sakurai reaction employing a tertiary TMS ether, namely 24b,34 but, in our hands, we were not able to detect any multicomponent product with it. Also by removing the double bond, and using 22b or 23b, no product was obtained.
We can conclude that the reaction is possible unless the TMS ether is able to form a relatively stable carbocation, either tertiary or allylic and doubly secondary, such as 28.
On the other hand, when using the TMS ether 19b, obtained from (−)-isopulegol, aldehyde 4 was consumed, although not affording the expected product. In this case, after the formation of the oxocarbenium intermediate, an intramolecular Prins cyclization occurred affording compound 30, that was isolated in 41% yield, along with minor amounts of its stereoisomers/regioisomers (Scheme 7).50 The relative configuration of this interesting product, being (R) at the new stereogenic center, was assumed because the hydrogen bound to it seems to be axial from the coupling constants. Also with 9 a similar behaviour was observed, but the mixture was in this case more complex and we were not able to isolate a single isomer.
As far as it concerns diastereoselectivity, with achiral alcohols aldehyde 4 reacts giving typically low to moderate induction, in favour of the syn isomer. Interestingly, in our previous work concerning Passerini reaction with the same aldehyde, the anti isomers were instead favoured.9
With chiral TMS ethers, matched-mismatched cases were observed. It is well known that in “double asymmetric synthesis”, where two enantiopure substrates interact, the two diastereofacial selectivities may either oppose each other (“mismatched case”) or act in concert (“matched case”).51,52 In particular, with (–)-menthol 14b, a matched case allowed to reach the highest d.r. of 92:8 for the syn product. With (+)-menthol ent-14b (mismatched case), the anti isomer was preferred, indicating that the main control was put forth by 14b. With 16b and ent-16b, a matched–mismatched combination was again in action, but this time the matched combination favoured the anti isomer of 11g. With (+)-fenchyl alcohol 17b, probably we are in presence of another matched case, although, due to unavailability of the (–) enantiomer, we could not demonstrate that.
With 6-bromopiperonal 9 a good diastereoselectivity was achieved only with 16b (or ent-16b, which gave identical d.r. ratios), whereas with other chiral TMS ethers, the asymmetric induction was low, even with (–)-menthol 14a and (+)-fenchyl alcohol 17a, which seemed quite selective with aldehyde 4.
As a first preliminary example of post-MCR transformation, we carried out a cyclization through Ring Closing Metathesis on the anti stereoisomer of 11f to give 31is shown in Scheme 8, which involves cyclization of the anti stereoisomer of 11f to give 31.
 | ||
| Scheme 8 Ring closing metathesis and transformations made for the determination of relative configuration. | ||
The relative configuration of compounds 11f, was assessed through NMR studies on dihydropyran 31. In the case of 11d, the relative configuration was demonstrated by an independent synthesis of the syn isomer from 32, in turn obtained in high yield (79%) and d.r. (99:1) by allylation of aldehyde 4 with allyltributyltin at −78 °C under the catalysis of MgBr2 (Scheme 8) to afford bicomponent product 32, whose configuration was demonstrated by Mosher ester analysis (see ESI†). Moreover, although 32 is a new compound, the analogue with another protecting group (benzyl) has been previously obtained by the same allylation reaction and the configuration established by us is in agreement with the one proposed in that work.53,54
For 11a–c, and 11g, we used NMR analogies with the spectra of 11d and 11f (see ESI†).
On the contrary, the relative configuration of ethers 12b, 12d, 12e, 12f, derived from 6-bromopiperonal 9, was not determined.
Conclusions
In conclusion, we have carried out a comprehensive study on the scope and limitations of the multicomponent Hosomi–Sakurai reaction using TMS ethers of various alcohols, with a particular emphasis on those of terpenic origin. For this study, we employed two complementary aldehydes: one is aromatic and highly activated for this reaction, making it less critical, while the other, derived from biomass, is more critical due to being acid-labile and highly prone to hydration.Nevertheless, we have successfully developed conditions well suited for both. As far as it concerns the alcohol counterpart, reactions works very well with TMS ethers of secondary alcohols, even the bulky ones, unless they are able to form relatively stable carbocations. TMS ethers of tertiary alcohols are not suited for this MCR. TMS ethers of primary alcohols may be used, but at higher temperatures, being inclined to form acetals too. Thus we have mainly focussed on secondary alcohols. The use of these conditions for other bio-based TMS ethers (including also carbohydrate-derived ones), in combination with various (biobased or not) aldehydes is in progress.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: LB, RR, CL, AB. Funding acquisition: LB, RR. Resources: CL, LM. Data analysis: CL, LB, RR, LM. Investigation: AC, LP, CL. Supervision: CL, LM. Writing of draft: CL, LB. Writing: editing and review: RR, LM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support under the National Recovery and Resilience Plan (NRRP), Mission 4, Component 2, Investment 1.1, Call for tender no. 104 published on 2.2.2022 by the Italian Ministry of University and Research (MUR), funded by the European Union – NextGenerationEU– Project Title SUSTCARB – CUP D53D23010360006, – Grant Assignment Decree No. 1064 adopted on july 18, 2023, by the Italian Ministry of Ministry of University and Research (MUR). We thank Federica Minuto for her collaboration in this work and Valeria Rocca for HPLC analyses. We thank Amano for kind gift of its enzymes.Notes and references
- A. Dömling, W. Wang and K. Wang, Chemistry and Biology Of Multicomponent Reactions, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed.
- L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, Synthesis of seven-membered nitrogen heterocycles through the Ugi multicomponent reaction, Chem. Heterocycl. Compd., 2017, 53, 382–408 Search PubMed.
- L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, The 100 facets of the Passerini reaction, Chem. Sci., 2021, 12, 15445–15472 RSC.
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Multicomponent reactions: advanced tools for sustainable organic synthesis, Green Chem., 2014, 16, 2958–2975 RSC.
- L. Banfi, C. Lambruschini, L. Moni and R. Riva, in RSC Green Chemistry, ed. R. Ballini, Royal Society of Chemistry, 2019, vol. 2019-January, pp. 115–140 Search PubMed.
- Z. Wang, M. S. Ganewatta and C. Tang, Sustainable polymers from biomass: Bridging chemistry with materials and processing, Prog. Polym. Sci., 2020, 101, 101197 CrossRef CAS.
- L. Moni, L. Banfi, A. Basso, E. Martino and R. Riva, Diastereoselective Passerini Reaction of Biobased Chiral Aldehydes: Divergent Synthesis of Various Polyfunctionalized Heterocycles, Org. Lett., 2016, 18, 1638–1641 CrossRef CAS PubMed.
- G. Vitali Forconesi, L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, Synthesis of Polyoxygenated Heterocycles by Diastereoselective Functionalization of a Bio-Based Chiral Aldehyde Exploiting the Passerini Reaction, Molecules, 2020, 25, 3227 Search PubMed.
- L. Moni, L. Banfi, D. Cartagenova, A. Cavalli, C. Lambruschini, E. Martino, R. V. A. Orru, E. Ruijter, J. M. Saya, J. Sgrignani and R. Riva, Zinc(ii)-mediated diastereoselective Passerini reactions of biocatalytically desymmetrised renewable inputs, Org. Chem. Front., 2020, 7, 380–398 RSC.
- G. Vitali Forconesi, A. Basso, L. Banfi, D. Gugliotta, C. Lambruschini, M. Nola, R. Riva, V. Rocca and L. Moni, Total Synthesis of 4-epi-Bengamide E, Molecules, 2024, 29, 1715 CrossRef PubMed.
- C. Lambruschini, D. Galante, L. Moni, F. Ferraro, G. Gancia, R. Riva, A. Traverso, L. Banfi and C. D'Arrigo, Multicomponent, fragment-based synthesis of polyphenol-containing peptidomimetics and their inhibiting activity on beta-amyloid oligomerization, Org. Biomol. Chem., 2017, 15, 9331–9351 RSC.
- D. Galante, L. Banfi, G. Baruzzo, A. Basso, C. D'Arrigo, D. Lunaccio, L. Moni, R. Riva and C. Lambruschini, Multicomponent Synthesis of Polyphenols and Their In Vitro Evaluation as Potential β-Amyloid Aggregation Inhibitors, Molecules, 2019, 24, 2636 Search PubMed.
- C. Lambruschini, A. Basso, L. Moni, A. Pinna, R. Riva and L. Banfi, Bicyclic Heterocycles from Levulinic Acid through a Fast and Operationally Simple Diversity-Oriented Multicomponent Approach, Eur. J. Org Chem., 2018, 2018, 5445–5455 CrossRef CAS.
- C. Lambruschini, I. Demori, Z. El Rashed, L. Rovegno, E. Canessa, K. Cortese, E. Grasselli and L. Moni, Synthesis, Photoisomerization, Antioxidant Activity, and Lipid-Lowering Effect of Ferulic Acid and Feruloyl Amides, Molecules, 2021, 26, 89 CrossRef CAS PubMed.
- V. Cerulli, L. Banfi, A. Basso, V. Rocca and R. Riva, Diversity oriented and chemoenzymatic synthesis of densely functionalized pyrrolidines through a highly diastereoselective Ugi multicomponent reaction, Org. Biomol. Chem., 2012, 10, 1255–1274 RSC.
- B. Paul, M. Maji, K. Chakrabarti and S. Kundu, Tandem transformations and multicomponent reactions utilizing alcohols following dehydrogenation strategy, Org. Biomol. Chem., 2020, 18, 2193–2214 RSC.
- T. Ngouansavanh and J. Zhu, Alcohols in Isonitrile-Based Multicomponent Reaction: Passerini Reaction of Alcohols in the Presence of O-Iodoxybenzoic Acid, Angew. Chem., Int. Ed., 2006, 45, 3495–3497 CrossRef CAS PubMed.
- F. Drouet, G. Masson and J. Zhu, Ugi Four-Component Reaction of Alcohols: Stoichiometric and Catalytic Oxidation/MCR Sequences, Org. Lett., 2013, 15, 2854–2857 CrossRef CAS PubMed.
- L. Moni, L. Banfi, R. Riva and A. Basso, External-Oxidant-Based Multicomponent Reactions, Synthesis, 2016, 48, 4050–4059 CrossRef CAS.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Hydroxymethylfurfural, a versatile platform chemical made from renewable resources, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- G. V. López and W. Porcal, Renewable carbon resource from biomass: building molecular architectures from furanic platforms, Pure Appl. Chem., 2024, 96, 1271–1277 Search PubMed.
- Y. Nakagawa, M. Tamura and K. Tomishige, Supported Metal Catalysts for Total Hydrogenation of Furfural and 5-Hydroxymethylfurfural, J. Jpn. Petrol. Inst., 2017, 60, 1–9 CrossRef CAS.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Caprolactam from Renewable Resources: Catalytic Conversion of 5-Hydroxymethylfurfural into Caprolactone, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 Search PubMed.
- C. Lambruschini, L. Moni and L. Banfi, Diastereoselectivity in Passerini Reactions of Chiral Aldehydes and in Ugi Reactions of Chiral Cyclic Imines, Eur. J. Org Chem., 2020, 2020, 3766–3778 Search PubMed.
- S. Caputo, L. Banfi, A. Basso, A. Galatini, L. Moni, R. Riva and C. Lambruschini, Diversity-Oriented Synthesis of Various Enantiopure Heterocycles by Coupling Organocatalysis with Multicomponent Reactions, Eur. J. Org Chem., 2017, 2017, 6619–6628 CrossRef CAS.
- A. Pinna, A. Basso, C. Lambruschini, L. Moni, R. Riva, V. Rocca and L. Banfi, Stereodivergent access to all four stereoisomers of chiral tetrahydrobenzo[f][1,4]oxazepines, through highly diastereoselective multicomponent Ugi–Joullié reaction, RSC Adv., 2020, 10, 965–972 Search PubMed.
- A. G. Németh, G. M. Keserű and P. Ábrányi-Balogh, A novel three-component reaction between isocyanides, alcohols or thiols and elemental sulfur: a mild, catalyst-free approach towards O-thiocarbamates and dithiocarbamates, Beilstein J. Org. Chem., 2019, 15, 1523–1533 CrossRef PubMed.
- A. Mekhalfia and I. E. Markó, The silyl modified sakurai (SMS) reaction. An efficient and versatile one-pot synthesis of homoallylic ethers, Tetrahedron Lett., 1991, 32, 4779–4782 CrossRef CAS.
- T. Jacques, I. E. Markó and J. í. Pospíšil, in Multicomponent Reactions, ed. J. Zhu and H. Bienaymé, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005, ch. 13, pp. 398–452 Search PubMed.
- D. Kampen, A. Ladépêche, G. Claßen and B. List, Brønsted Acid-Catalyzed Three-Component Hosomi–Sakurai Reactions, Adv. Synth. Catal., 2008, 350, 962–966 Search PubMed.
- H. Sakurai, K. Sasaki, J. Hayashi and A. Hosomi, Chemistry of organosilicon compounds. 191. Acetalization of carbonyl compounds with alkoxysilanes catalyzed by iodotrimethylsilane. One-pot allylation reactions of carbonyl compounds to homoallyl ethers using an allylsilane, J. Org. Chem., 1984, 49, 2808–2809 CrossRef CAS.
- T. Watahiki, Y. Akabane, S. Mori and T. Oriyama, Iron(III) Chloride-Catalyzed Convenient One-Pot Synthesis of Homoallyl Benzyl Ethers Starting from Aldehydes, Org. Lett., 2003, 5, 3045–3048 CrossRef CAS PubMed.
- J. Pospíšil and I. E. Markó, Total Synthesis of Jerangolid D, J. Am. Chem. Soc., 2007, 129, 3516–3517 Search PubMed.
- J. Pospisil, T. Kumamoto and I. E. Marko, Highly diastereoselective silyl-modified sakurai multicomponent reaction, Angew. Chem., Int. Ed., 2006, 45, 3357–3360 Search PubMed.
- M. Yus, J. C. Gonzalez-Gomez and F. Foubelo, Diastereoselective allylation of carbonyl compounds and imines: application to the synthesis of natural products, Chem. Rev., 2013, 113, 5595–5698 CrossRef CAS PubMed.
- M. Teruaki, O. Masahiro and M. Norikazu, An Efficient and Convenient Method for the Asymmetric Allylation of Aldehydes with Allyltrimethylsilane Catalyzed by Diphenylboryl Triflate, Chem. Lett., 1987, 16, 1121–1124 CrossRef.
- L. F. Tietze, K. Schiemann and C. Wegner, Enantioselective Synthesis of Tertiary Homoallylic Alcohols via Diastereoselective Addition of Allylsilanes to Ketones, J. Am. Chem. Soc., 1995, 117, 5851–5852 Search PubMed.
- J. Cossrow and S. D. Rychnovsky, Optically pure alpha-(trimethylsilyl)benzyl alcohol: a practical chiral auxiliary for oxocarbenium ion reactions, Org. Lett., 2002, 4, 147–150 Search PubMed.
- J. S. Panek, M. Yang and F. Xu, Diastereoselective additions of chiral (E)-crotylsilanes to in situ generated oxonium ions: a direct asymmetric synthesis of functionalized homoallylic ethers, J. Org. Chem., 1992, 57, 5790–5792 CrossRef CAS.
- J. Wu, Y. Chen and J. S. Panek, Vinylogous Aldol Products from Chiral Crotylsilanes Obtained by Enantioselective Rh(II) and Cu(I) Carbenoid Si–H Insertion, Org. Lett., 2010, 12, 2112–2115 CrossRef CAS PubMed.
- D. Kim, J. S. Lee, L. Lozano, S. B. Kong and H. Han, Enantioenriched Bifunctional Crotylsilanes for the Asymmetric Synthesis of Orthogonally Protected 2-Methyl-1,3-diols, Org. Lett., 2013, 15, 5142–5145 CrossRef CAS PubMed.
- Y. Chu, Q. Pu, Z. Tang, L. Gao and Z. Song, Enantioselective synthesis of crotyl geminal bis(silane) and its usage for asymmetric Sakurai allylation of acetals, Tetrahedron, 2017, 73, 3707–3713 Search PubMed.
- B. Karimi and B. Golshani, Mild and Highly Efficient Method for the Silylation of Alcohols Using Hexamethyldisilazane Catalyzed by Iodine under Nearly Neutral Reaction Conditions, J. Org. Chem., 2000, 65, 7228–7230 Search PubMed.
- A. Ramdular and K. A. Woerpel, Using Neighboring-Group Participation for Acyclic Stereocontrol in Diastereoselective Substitution Reactions of Acetals, Org. Lett., 2020, 22, 4113–4117 CrossRef CAS PubMed.
- A. Chojnacka, R. Obara and C. Wawrzeńczyk, Kinetic resolution of racemic secondary aliphatic allylic alcohols in lipase-catalyzed transesterification, Tetrahedron:Asymmetry, 2007, 18, 101–107 CrossRef CAS.
- S. Purushotham Reddy, B. Chinnababu, V. Shekhar, D. Kumar Reddy, G. V. Bhanuprakash, L. R. Velatoor, J. Venkateswara Rao and Y. Venkateswarlu, Stereoselective synthesis of alpinoid-C and its analogues and study of their cytotoxic activity against cancer cell lines, Bioorg. Med. Chem. Lett., 2012, 22, 4182–4184 CrossRef CAS PubMed.
- Q. Gao, Y. Li, L. Chen, L.-J. Xie, X. Shao, Z. Ke and S. Xu, Enantioselective α-C(sp3)–H Borylation of Masked Primary Alcohols Enabled by Iridium Catalysis, J. Am. Chem. Soc., 2025, 147, 88–95 Search PubMed.
- D. Seebach, A. K. Beck, B. Schmidt and Y. M. Wang, Enantio- and diastereoselective titanium-TADDOLate catalyzed addition of diethyl and bis(3-buten-1-yl) zinc to aldehydes a full account with preparative details, Tetrahedron, 1994, 50, 4363–4384 Search PubMed.
- R. J. Kazlauskas, A. N. Weissfloch, A. T. Rappaport and L. A. Cuccia, A rule to predict which enantiomer of a secondary alcohol reacts faster in reactions catalyzed by cholesterol esterase, lipase from Pseudomonas cepacia, and lipase from Candida rugosa, J. Org. Chem., 1991, 56, 2656–2665 Search PubMed.
- M. N. Timofeeva, V. N. Panchenko, A. Gil, S. V. Zakusin, V. V. Krupskaya, K. P. Volcho and M. A. Vicente, Effect of structure and acidity of acid modified clay materials on synthesis of octahydro-2H-chromen-4-ol from vanillin and isopulegol, Catal. Commun., 2015, 69, 234–238 CrossRef CAS.
- S. Masamune, W. Choy, J. S. Petersen and L. R. Sita, Double Asymmetric Synthesis and a New Strategy for Stereochemical Control in Organic Synthesis, Angew. Chem., Int. Ed., 1985, 24, 1–30 Search PubMed.
- W. R. Roush, L. K. Hoong, M. A. J. Palmer, J. A. Straub and A. D. Palkowitz, Asymmetric synthesis using tartrate ester modified allylboronates. 2. Single and double asymmetric reactions with alkoxy-substituted aldehydes, J. Org. Chem., 1990, 55, 4117–4126 Search PubMed.
- G. Keum, S. B. Kang, Y. Kim and E. Lee, Stereoselection in Radical Cyclization of β-Alkoxyvinyl Sulfoxides: Synthesis of Tetrahydrofuranyl Allyl Carbinols, Org. Lett., 2004, 6, 1895–1897 Search PubMed.
- G. Keum, C. H. Hwang, S. B. Kang, Y. Kim and E. Lee, Stereoselective Syntheses of Rolliniastatin 1, Rollimembrin, and Membranacin, J. Am. Chem. Soc., 2005, 127, 10396–10399 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures, optimizations, characterization data, NMR spectra, details of HPLC analyses, and discussion on the assessment of relative configuration. See DOI: https://doi.org/10.1039/d5ra03165f |
| ‡ Although we were not able to detect hexamethyldisiloxane, which is highly volatile and probably hydrolysed during work-up, its formation can be inferred from the stoichiometry of the reaction. The product has one oxygen less than the three reagents, the same number of hydrogens, and two TMS groups less. Since the reaction is carried out under dry conditions (no water present, unless in traces) and TMS-OTf is catalytic, hexamethyldisiloxane is the only possible stoichiometric product containing the missing oxygen and the two TMS groups. |
| § Markó et al. reported that the yield could be improved by a basic non aqueous work-up (pyridine34 or EtN(iPr)2) (ref. 33). However, in our hands, these basic work-up conditions were ininfluential, whereas addition from the beginning of those bases suppressed completely the activity of the catalyst. |
| ¶ Loss of enantiopurity under reaction conditions is very unlikely and was not observed by HPLC with chiral stationary phase (see ESI†). In the case of aldehyde 4, which has two stereogenic centers, no epimerization was observed in all cases. Racemization would imply simultaneous inversion of both stereocenters, which is impossible. Furthermore, in reaction of 4 with chiral TMS ethers (e.g. 14b), more than 2 diastereomers would have been observed, and this was not the case. Racemization of chiral TMS ethers is also highly unlikely, and again, in reactions with aldehyde 4, this would have led to more than two diastereomers. |
| This journal is © The Royal Society of Chemistry 2025 |