
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe VVBlue assay: a plate-readable, dye exclusion-based cell viability assay for the toxicological testing of chemicals†
Marianne Vitipon,
Esther Akingbagbohun and
Thierry Rabilloud *
*
Chemistry and Biology of Metals, Univ. Grenoble Alpes, CNRS UMR5249, CEA, IRIG-LCBM, F-38054 Grenoble, France. E-mail: thierry.rabilloud@cnrs.fr
First published on 29th May 2025
Abstract
A viability test for in vitro cultures, based on the intake of the textile dye alphazurine A by dead cells and its exclusion by viable cells, is described. This test uses the affinity of alphazurine A for proteins, so that the dye is retained in dead cells even after rinsing, while its anionic character prevents it from entering live cells. This feature makes this dye exclusion test amenable to a reading in a plate format. The alphazurine viability test provides an indicator of the absolute number of dead cells present in the culture well. To reach a cell viability index, a “dead cells” control (e.g. cells killed with ethanol) must be added. We also describe a double viability test, which first uses the alphazurine assay to provide the number of dead cells then a crystal violet assay to provide an index of the number of cells present in the plate. This double test provides a complete appraisal of the situation in the cell culture wells, and has been compared to other viability tests such as propidium iodide exclusion or tetrazolium reduction. Its performances to study the toxicity of substances such as pigments are also established, and allowed us to publish the first public toxicological data on the recently described Pigment Blue 86.
1 Introduction
There is more and more pressure on the chemical industry to market safe products for public consumers, and products with documented toxicology for professional customers. In some parts of the world, e.g. the European Union, it has become mandatory to document the toxicology of every product marketed over 1 ton per year. However, this assessment relies on regulatory tests that are both long and expensive. This poses a problem to industry, as the whole development of a new chemical may be canceled at the very end of the process by the toxicology assessment. There is thus a need for simple, fast and cheap toxicological tests to perform early toxicological tests during the development cycle of new products, to comply to the “fail early, fail cheap” principle that has long been adopted in the pharmaceutical industry.For this purpose, in vitro toxicological tests combine speed and relatively low costs, and are often amenable to automation, which allows many tests to be performed and to test several conditions simultaneously.
Within this frame on in vitro toxicology, the determination of cell viability is an operation performed routinely in all cell biology laboratories. While performed at a relatively low scale for routine cell culture maintenance, it becomes crucial in certain areas of biology such as toxicology, where the determination of cell viability is an essential foundation for all subsequent analyses.
Depending on the cellular parameter used to determine it, cell viability assays can be classified as direct or indirect assays. Direct assays rely on the contrast between the membrane permeability existing for dead cells compared to the much more tightly-regulated permeability observed on live cells. Thus, direct cell viability assays use either the permeability of dead cells to certain dyes or fluorescent probes added in the medium or the release of intracellular components in the medium.
Historically, the contrast between the impermeability of live cells to anionic dyes (and multicharged cationic dyes) and the permeability of dead cells to these dyes has been used to build cell viability assays, the forst dye used to this purpose being Trypan blue.1 When used in a classical microscopy format, dyes in addition to Trypan blue, such as eosin, erythrosin and safranin have been used and compared.2 However, these dyes have various properties, e.g. in their toxicity or speed of penetration into dead cells that limit their application for large scale experiments. Furthermore, they rely on human interpretation and are thus operator dependent, although automated solutions now exist when working with classical dyes such as Trypan blue. In a flow cytometry format, which is better suited for medium-large scale experiments and is less prone to operator dependency, propidium iodide is used as a fluorescent probe to discriminate between live and dead cells.3 However, this format is still a serial operation, requiring an expensive equipment and is prone to some artifacts, e.g. strongly negatively charged particles that bind propidium iodide can be misidentified as cells during gate analysis. Moreover, the definition of the gates to discriminate between live and dead cells is not necessarily obvious, as a fluorescence continuum is often observed between the two main clouds of points.
In addition, for these optical methods requiring cell observation or measurement of cellular fluorescence, the cells must ideally be transparent. Cells that are rendered opaque, e.g. by internalization of dark nanoparticles, cannot be tested by such methods.
In the other direction, i.e. the release of intracellular components into the extracellular medium, the only test that has gained popularity is based on the release of lactate dehydrogenase, i.e. a very abundant cytosolic enzyme that is relatively easy to assay.4 However, this assay still relies on the quantitative determination of an enzyme activity, i.e. a process that requires careful time and temperature control, and that can be biased if the extracellular medium contains substances interfering with the assay, which is sometimes encountered in toxicology experiments. This phenomenon has been described for metal ions, for example released by nanoparticles,5,6 but also for bacteria,7 and for nanoparticles that adsorb LDH.8
These limitations have stimulated the development of indirect assays that are substantially easier to automate, and thus less prone to inter-operator variations. Depending on the cellular parameter tested, several classes of indirect viability assays can be described.
The first and most popular is based on the cellular redox metabolism and the existence of the pool of reduced dinucleotides (NADH and NADPH). Due to their low redox potential, these reduced dinucleotides can convert either weakly colored tetrazolium salts into strongly colored formazans9,10 or convert non fluorescent molecules into fluorescent ones.11 Although extremely popular, these assays are not devoided of caveats. The first one is linked to their indirect nature. For example, a 50% reduction in formazan production in treated cells compared to controls does not tell whether half of the cells are dead and half of them have a completely normal metabolism, or whether all cells are alive but with a two-fold reduced metabolism. As an example, bromopyruvate, which is a metabolic poison, gives erratic results in tetrazolium and resazurin reduction assays.12 Other caveats encountered with such assays in toxicology experiments can be due to the intrinsic redox capacity of the substances to be tested,13 or to the superoxide production by the cells in response to the experimental conditions.14 This may lead to “viabilities” over 100% when such an interference or cell activation is encountered,13,14 which of course cannot happen. Further problems are encountered when the substance to be tested is a highly hydrophobic particle able to strongly adsorb the reduced molecule used as an indicator of cell metabolism, either formazans15 or resorufin.16 Furthermore, the resazurin reduction assay is prone to a specific artifact represented by the over-reduction of the desired fluorescent product resorufin, into the non-fluorescent product dihydroresorufin11
A second indirect cell viability assay relies on the proton pumping activity of lysosomes. As this phenomenon is ATP-dependent, it quickly stops after cell death. This phenomenon leads in turn to the ability of certain cationic dyes, such as neutral red, to be accumulated into lysosomes, in live cells only. This has led to the development of the neutral red uptake assay as an indirect cell viability assay.17 This assay is however also prone to biases, arising for example from light-absorbing compounds,18 from selective inhibition of lysosomal proton pumping in viable cells,19 or from cell activation. In the latter case, “viabilities” over 100% can be observed.20
Other indirect cell viability assay relies on cell number, which is not determined in an absolute way but rather via cellular macromolecules. Such assays include the crystal violet assay,21 which stains nuclei, and the sulforhodamine assay,22 which estimates the protein content. The rationale under these assays is that dead cells will not attach and/or detach from cell culture supports. In turn, these assays will apply only to adherent cells, which however represent the vast majority of cultured cells. Quite unfortunately, the underlying hypothesis of these assays on dead cells detachment has been shown to be incorrect in some cases.23
Due to these various limitations, rather than relying on indirect assay multiplexing,24 we sought to develop an assay that would rely on dye exclusion, but would not need either microscopy or flow cytometry, and would rather be readable in plate format. This would require a staining sequence similar to the one used for crystal violet or neutral red staining, i.e. staining with the dye, rinsing to eliminate the excess dye that would prevent reading in the plate reader, then dye elution to read the absorbance from the culture supports. When classical exclusion dyes or even sulforhodamine B2,22 were tested in such an approach, no signal was detectable after rinsing, probably due to the insufficient affinity of the dyes for intracellular components, thereby not withstanding the rinsing phase. Further substantiation of this hypothesis was obtained from a variation of the dye exclusion protocol using the fluorescence of erythrosin B in dead cells.25 This protocol does not use any rinsing step, but rather uses a very low probe concentration (5 μM) to limit the background fluorescence, thereby allowing sufficient contrast to identify the dead cells by photographic fluorescence light accumulation. As this method appeared as not suitable for medium-large scale experiments because of the need for a photographic process, we decided to test several dyes in order to find one that would not penetrate live cells, and have sufficient affinity for cellular components to allow for a stain-rinse-elution sequence.
In addition, this process should allow to develop a test that should be insensitive to optical interferences, and thus suitable for the toxicological testing of even the most difficult chemicals in in vitro toxicology, such as pigments, which are both particular in nature of optically opaque.
2 Material and methods
2.1. Materials
All dyes were purchased from Sigma at the highest purity available. Zinc oxide (<100 nm, catalog number # 721085) and silver (<100 nm, catalog number # 758329) nanoparticles dispersions were purchased from Sigma. Ethanol (96%) was purchased from RSE, acetic acid (analytical grade) from Sigma. Pigment Black 33 (mixed manganese iron oxide #47501) and Pigment Blue 86 (Yttrium manganese indium oxide #453208) were purchased from Kremer pigments, and Pigment Black 14 (manganese dioxide) was purchased from Zecchi (reference C0999). The pigments were suspended in gum arabic (100 mg mL−1, used as an anti-agglomeration reagent) that had been previously sterilized overnight at 80 °C. Then, the suspensions of pigments (100 mg mL−1) was sterilized overnight again before being was sonicated in a Vibra Cell VC 750 sonicator equipped with a Cup Horn probe (VWR, Fontenay-sous-Bois, France) with the following settings: time = 30 min – 1 second ON, 1 second OFF – Amplitude = 60%, corresponding to 90 W per pulse.Before adding to the cell culture, nanoparticles dispersions were diluted in sterile deionized water.
2.2. Cell culture
The mouse macrophage cell line J774A.1 and the alveolar epithelial cell line A549 were obtained from the European Cell Culture Collection (Salisbury, UK). The cells were cultured in DMEM medium + 10% fetal bovine serum (FBS). For routine culture, J774 cells were seeded on non-adherent flasks (e.g. suspension culture flasks from Greiner) at 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per mL and harvested 48 hours later, at 1000000 cells per mL. A549 cells were seeded on cell culture adherent flasks at a cell density of 10000 cells per cm2 and split by trypsinization every 3 days.
000 cells per mL and harvested 48 hours later, at 1000000 cells per mL. A549 cells were seeded on cell culture adherent flasks at a cell density of 10000 cells per cm2 and split by trypsinization every 3 days.
For treatment with particles, the following scheme was used: J774 cells were first seeded at 400000 cells per mL into 24-well adherent plates (FALCON®, Corning Incorporated) or 12-well non-adherent plates (CELLSTAR®, Greiner), depending on the viability test performed. They were exposed to zinc oxide or silver nanoparticles or silver nitrate or pigments on the following day and harvested after a further 24 h in culture. In some experiments, the cells were cultured in DMEM medium + 1% fetal bovine serum (FBS).
For A549 cells, they were first seeded at 100000 cells per cm2 into 24-well adherent plates (FALCON®, Corning Incorporated). They were exposed to silver nitrate on the following day and analyzed after a further 24 h in culture.
Cells were used at passage numbers from 5 to 15 post-reception from the repository.
2.3. Viability assays
:50) in phenol red-free DMEM and left incubate at 37 °C 5% CO2 for 20 min and 10–12 min, for J774A.1 and A549 respectively. Absorbances were measured at 440 nm with a plate reader (Infinite M200, Tecan, Durham, NC, USA).In other experiments, metabolic activity of cells was evaluated by using the MTT assay (Sigma Aldrich, #M5655). Briefly, after 24 hours exposure to chemicals, the culture medium was removed from the test conditions. The medium was replaced by DMEM without phenol red (PAN Biotech, #1190124), to which 10 μL of a 20 mg mL−1 MTT solution in absolute ethanol had been added. The cells were returned to the culture incubator for 1 hour. The culture medium was then removed and the precipitated formazan was eluted with a solution of 90% ethanol, 9% water and 1% acetic acid (all by volumes) for 45 min with agitation. Absorbances were measured at 570 nm with a plate reader.
When optical interference was expected, as in the case of pigments, two series of controls was implemented. These controls consisted of
(i) Plates devoid of any cells, but treated with the same concentrations of pigments used in the toxicological testing. These plates were then submitted to the VVBlue test and the obtained absorbances deduced from the ones obtained in the cells-containing plates.
(ii) Cells-containing plates, treated with the pigments but not with the dyes. The cells were just treated with the eluting solution and the absorbance of the eluate was determined at 590 and 637 nm, and substracted from the crystal violet and alphazurine absorbances, respectively.
2.4. Microscopy visualization experiments
After the adequate treatments, the culture medium was replaced by phenol red-free DMEM for imaging. Images were taken before and after the alphazurine treatment, including the PBS washes, but before any elution step. The cells were visualized under an optical microscope (Motic AE21) set at ×10 magnification. The images were captured with an Axiocam 105 color camera and processed uniformly using ImageJ to standardize contrast and saturation levels.3 Results
3.1. Dye selection
In order to select suitable dyes, we performed preliminary experiments in a simplified format. Cells were grown in 6 well plates, and in one half of the wells the cells were killed with a lethal dose of silver nanoparticles. All wells were then treated with 0.2 mg mL−1 dye dissolved in culture medium for 30 minutes. The dye solution was then removed, the wells rinsed twice with PBS, and the dye was eluted with an acidic ethanol solution (1% acetic acid, 50% ethanol). The absorbance of the wells corresponding to live and dead cells was then compared. The following dyes were tested through this method: erythrosin (C.I. 45430), crystal violet (C.I. 42555), methyl green (C.I. 42585), brilliant green (C.I. 42040), safranin (C.I. 50240), methylene green (C.I.52020), ponceau S (C.I. 27195), light green SF (C.I. 42095), acid violet 17 (C.I. 42650), erioglaucine (C.I. 42090), alphazurine A (C.I. 42080), Coomassie blue G250 (C.I. 42655), copper phthalocyanine tetrasulfonate, lissamine green (C.I. 44090), thioflavin S (C.I. 49010), thiazole yellow (C.I. 19540), Nile blue (C.I. 51180) and naphthol yellow (C.I. 10316).Besides classical exclusion dyes such as erythrosin and safranin, other dyes were selected on the basis of their use as wool (protein) dyes (light green SF, acid violet 17, erioglaucine, alphazurine A, Coomassie blue G250 thioflavin S, thiazole yellow and lissamine green), as nucleic acid dyes (crystal violet, methyl green, brilliant green) or as protein stains in blotting (Ponceau S, copper phthalocyanine tetrasulfonate).
Of all those, only the textile dyes acid violet 17 and alphazurine A offered an interesting absorbance difference between live cells and dead cells, as shown in Fig. 1. Coomassie blue did stain dead cells well but appeared too hydrophobic, as its excess was not easily removed from the culture wells. Acid violet 17 also gave a rather high absorbance in the wells with live cells. Hence, we finally focused on the alphazurine A dye.
 | ||
| Fig. 1 Microscopy viewing of live and dead cells after treatment with alphazurine A J774A.1 cells were seeded at a density of 500000 cells per mL in 24-well adherent plates containing DMEM supplemented with 1% horse serum and 1% streptomycin–penicillin. Cells were either kept untreated (panel A), killed with 50% ethanol for 5 minutes (panel B) or treated with 10 or 20 μg mL−1 silver nitrate for 24 hours (panels C and D). The black arrows in the panels show the dead cells occurring in the various conditions. | ||
3.2. Assay development
We first optimized the rinsing and elution phases. For rinsing, a buffer saline solution (i.e..with an ionic strength equivalent to 150 mM sodium chloride) was tested. Alternatively, a low ionic strength solution containing 10 mM Hepes pH 7.5, 1 mM MgCl2 and 300 mM sucrose was also tested. The rinsing efficiency was visually evaluated by the retention of the green color in the wells containing 100% dead cells wells, while the green color should have been removed in the wells containing live cells. Since both solutions performed equally well, the saline solution was selected for its better shelf life.For elution, we tested either an acidic solution (1% acetic acid, 50% ethanol) or a basic solution (20 mM Tris base in 50% ethanol), as basic solutions have been used to elute triphenylmethane dyes from stained polyacrylamide gels bands.26,27 However, basic elution proved inappropriate, as evaluation by microscopy showed that it removed the cell layer from the plates, which prevented the construction of a robust assay.
We then investigated the linearity of the assay. For this purpose, a defined number of cells were seeded in 24-well plates, let to adhere for 1 hour, and then killed by the addition of 50% ethanol. After rehydration of the dead cell layers in DMEM, the cells were stained with alphazurine, rinsed and the dye eluted with the acid ethanol solution. The absorbance at 637 nm was recorded, and the results are shown in Fig. 2A. The results show that this calibration curve is linear in the 0–05 OD range, corresponding to cell numbers up to 1 million cells per well in a 24-wells plate, and that the method indeed indicated the number of dead cells present in the wells. However, it could not be excluded that in some experiments dead cells may detach, or that depending on the experimental conditions, the adherence of cells may vary. To normalize the number of dead cells (alphazurine-positive) by the total number of cells present in each well, we used an assay that is based on cell numbers as a second step, i.e. the crystal violet assay, as described in 28. When applied to the wells with defined cells numbers after the alphazurine staining and elution, the results, described in Fig. 2B, showed that the crystal violet signal correlated well with the cell numbers seeded in the plates, with a correlation coefficient of 0.96.
 | ||
| Fig. 2 Relationship between J774A.1 cell numbers inoculated into 24- well plates and absorbance after staining (A) staining with alphazurine A in DMEM (B) staining with crystal violet. The absorbance was measured at 637 nm for alphazurine A and 590 nm for crystal violet. Bars indicate SD of readings from triplicate experiments. | ||
When several experiments were performed, a variability in the absorbance values was observed, as shown in Fig. 3.
 | ||
| Fig. 3 Inter-experiment variability of the relationship between J774A.1 cell numbers inoculated into 24-well plates and absorbance after staining with alphazurine A in DMEM. Three independent experiments were conducted. In each experiment, a linear correlation was observed, with a R2 coefficient of 0.9899 for the grey dots, 0.9951 for the yellow dots, and 0.995 for the blue dots. | ||
Consequently, the utilization of a single numerical equation that would convert the OD signals directly into cell numbers is impossible. However, within the confines of a single experiment, linearity is upheld with a high R2 coefficient as shown in Fig. 3. Thus, the incorporation of additional control points in each experiment is crucial to use the linearity correlation observed for these two dyes.
For the alphazurine stain, a “cells only” control is imperative to ascertain the initial viability of the cells and to relate to the theoretical cell count in the wells. Additionally, a “cells only 100% dead” control is essential. These two controls enable the establishment, within the same experimentation, of the 0% dead cells and 100% dead cells points found in the standard ranges, facilitating the subsequent application of linearity correlation. Another control condition needed is the “empty well”, which have undergone the same coloration steps as the others conditions as outlined in Table 1, to be used as a blank.
Once all the absorbance have been obtained, they are processed as follows to deduce cell viability.
The absorbances resulting from these stains were normalized against the blank to eliminate plastic and solvent absorbance. Subsequently, for crystal violet, a normalized absorbance ratio was calculated between the absorbance of each condition and the average absorbance of the control conditions, where the cell count remains unchanged, serving as the reference. This index gave the proportion of cells still present in the plate at the end of the assay.
The alphazurine data were then used. The background-corrected absorbance were used to calculate the absorbance due to dead cells in every well. The average absorbance of the ethanol-killed cells and the average absorbance of the control cells were used to determine the absorbance linked to 0% viability and 100% viability, respectively, leading to the calculation of the absorbance linked to dead cells in each well. The subtraction of this value to the crystal violet value led to the proportion of live cells. These calculations are detailed in ESI Document 1.†
3.3. Cell viability assessments comparisons: VVBlue vs. propidium iodide and WST1 assays
To ensure the robustness of our assay, we conducted a comparative analysis of J774A.1 cell viability using the VVBlue assay against a well-established cell viability assay employing propidium iodide (PI). Cells were seeded at a density of 400000 cells per mL in a 24-well adherent plate for VVBlue assay, and, in a 12-well non-adherent plate for PI assay. Cells were exposed to increasing concentrations of particles, either zinc oxide (ZnO) or silver nanoparticles (AgNP), on the following day and harvested after 24 h of exposure to particles.
For the PI cell viability assay, cells were pre-stained with propidium iodide at 1 μg mL−1 in PBS for 5 min at RT, washed with PBS before analysis using flow cytometry. The cell viability under different conditions was normalized to the control group composed of cells without particles. As for the VVBlue assay, the samples were processed as explained previously in Table 1. The obtained results are illustrated in Fig. 4.
 | ||
| Fig. 4 Comparison of the propidium iodide and VVBlue assays (A) cell viability after 24 hours of exposure to zinc oxide nanoparticles Blue curve: viability measured by the propidium iodide assay (cells cultured on non-adherent plates). Green curve: viability measured by the VVBlue assay (cells cultured on adherent plates). Experiments were conducted in quadruplicate and represented as mean ± standard deviation (B) cell viability after 24 hours of exposure to silver nanoparticles. Blue curve: viability measured by the propidium iodide assay (cells cultured on non-adherent plates). Green curve: viability measured by the VVBlue assay (cells cultured on adherent plates). Experiments were conducted in triplicate and represented as mean ± standard deviation. | ||
Remarkably, the results displayed in Fig. 4 showed similarities between the viability obtained from these two assays. Specifically, upon exposure to zinc oxide, a notable decline in cell viability was evident, particularly above the concentration threshold of 8 μg mL−1. Similarly, when exposed to silver nanoparticles, a consistent trend in cell viability was observed in both assays. These findings highlight the robustness and reliability of the VVBlue assay, allowing to obtain similar results as the PI assay. However, it should be noted as well that the curves do not superimpose. This may be due to the fact that the cells tested by the VVBlue protocol do not sustain any disturbance in their analysis, while cells tested by the PI protocol must be dislodged from their culture support prior to analysis.
In order to test if an interference could occur by adsorption of the dyes on the nanoparticles tested (zinc oxide or silver), we performed additional experiments that rules out this hypotheses. These experiments are described in ESI Document 2.†
Another popular viability assay is represented by the metabolic assays, e.g. the tetrazolium/formazan assays. We thus performed a comparison of the VVBlue assay with the WST1 reduction assay.29 To this purpose, we used J774A.1 cells exposed to silver nitrate as the toxicant. Cells were seeded at a density of 400000 cells per mL in a 24-well adherent plate for both assays. Cells were exposed to increasing concentrations of silver nitrate on the following day and assayed after 24 h of exposure to the toxicant. The results, displayed on Fig. 5a, showed a difference in the LD50 obtained by the two methods. The LD50 determined by the WST1 method is around 37 μg mL−1, while the LD50 determined by the VVBlue assay was around 160 μg mL−1. Such a phenomenon has already been described in the literature (e.g. in ref. 30) and will be discussed later.
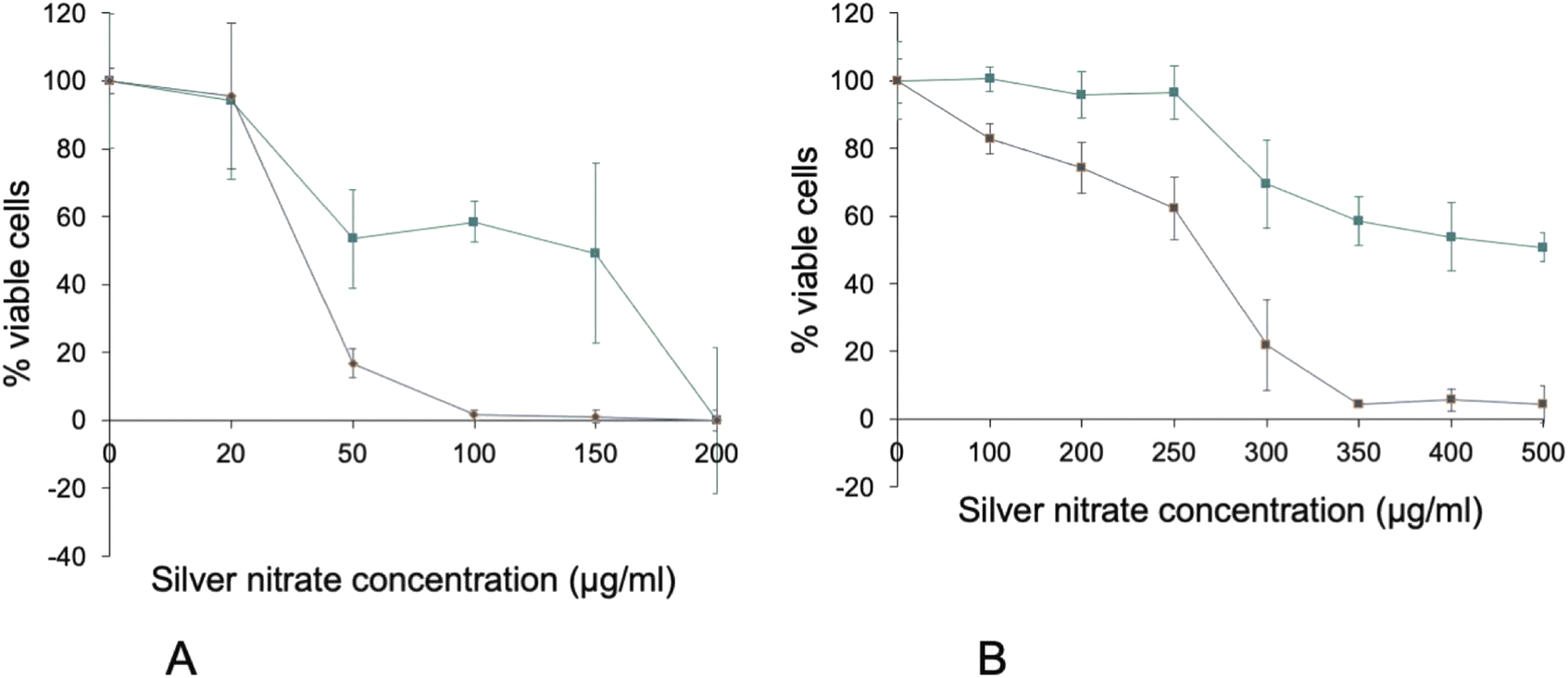 | ||
| Fig. 5 Comparison of the WST1 and VVBlue assays cell viability curves after 24 hours of exposure to silver nitrate. Green: VVBlue assay. Blue: WST1 assay. Experiments conducted in quadruplicates and expressed as mean ± standard deviation. Panel A: J774A.1 murine macrophage cell line. Panel B: A549 human alveolar epithelial cell line. | ||
In order to test our assay on a different cell type, we repeated the WST1 vs. VVBlue assay comparison on a different cell type, i.e. the A549 epithelial cells, still using silver nitrate as a toxicant. The results, displayed on Fig. 5b, showed once again an important difference in the viabilities determined by the two assays. This figure also showed the interest of the combined alphazurine A/crystal violet assay. While the alphazurine step showed a relatively low number of dead cells, this low number was explained in part by the fact that some of the dead cells detached, a phenomenon taken into account by the crystal violet step. The combination allowed a much more precise determination of the real numbers of viable cells remaining at the end of the assay, and thus of the toxicity.
3.4. Performance of the VVBlue under severe optical situations
The VVBlue assay is also performing well under severe situations where other dye-exclusion assays become impracticable. A typical case is the determination of the toxicity of pigment particles, which are opaque, highly colored, and often poorly toxic. These features rendered conventional tests, such as PI or Trypan blue, unsuitable for accurate toxicity assessment. In this precise experimental setup, cells were initially seeded at 500000 cells per mL in DMEM supplemented with 1% horse serum for 24 hours, and subsequently exposed to Pigment Black 33 for an additional 24 hours period. The VVBlue assay was employed for viability determination, facilitating the discrimination between live and dead cells, even exposed to elevated concentration of particles, as shown in Fig. 6. Notably, unexposed live cells (live cells only) exhibited a distinct refractivity and appeared uncolored (Fig. 6A), whereas unexposed dead cells lost their refringence and retained a blue alphazurine staining (Fig. 6B). Interestingly, the numerous black particles did not impede the accurate distinction of dead cells within the exposed cells conditions (Fig. 6C and D).
 | ||
| Fig. 6 Viability assessment of J774A.1 cells using VVBlue assay in the presence of colored particle dispersions. Optical microscopy images of live (A and C) or ethanol-killed (B and D) macrophages exposed (C and D) to 1000 μg mL−1 Pigment Black 33. Images were taken with an optical microscope (Motic AE21) at a magnification of 40×. The images were captured with an Axiocam 105 color camera and processed uniformly using ImageJ to standardize contrast and saturation levels. | ||
This robustness of the VVBlue assay allowed us to determine the toxicity curve of the two manganese-based pigments Pbk14 and PBk33. The results, displayed on Fig. 7, showed that PBk33 was not toxic at concentrations up to 1 mg mL−1, while PBk14 became toxic at concentrations as low as 100 μg mL−1.
 | ||
| Fig. 7 Toxicity curves of two manganese-based pigments (A) cell viability after 24 hours of exposure to PBk33 (mixed manganese iron oxide). The abscissae are the concentration of pigment added to the cultures in μg mL−1 (B) cell viability after 24 hours of exposure to PBk14 (manganese dioxide). The abscissae are the concentration of pigment added to the cultures in μg mL−1 experiments conducted in triplicates and expressed as mean ± standard deviation. | ||
Thus, the VVBlue assay was able to work under conditions where no other dye exclusion method could work, while avoiding the use of indirect assays and keeping cell integrity as the cell viability parameter.
Finally, we wondered whether the VVBlue assay would still work with blue or green pigments, which may interfere with the reading of the alphazurine absorbance. We thus tested the toxicity of the recently discovered Pigment Blue 86,31 which is authorized for commercialization under a low volume exemption.32 As the pigment deposited at the bottom of the cell culture wells and formed an opaque blue layer, that stayed at the bottom of the wells, the eluates from the alphazurine and crystal violet staining steps were transferred to a clean reading plate before absorbance reading, as described in the protocol, removing any interference fro the pigments. Control experiments with the pigment alone without cells showed negligible absorbance at 637 nm, comparable to the one of control live cells untreated with the pigment but treated with alphazurine. In these experiments, we compared the viability data obtained via the indirect MTT assay9 and the ones obtained via the VVBlue assay. The results, displayed on Fig. 8, showed once again a discrepancy between the results obtained via the MTT metabolic assay and the membrane permeability VVBlue assay. The MTT assay led to a letal dose 20% of 75 μg mL−1 and a letal dose 50% of 185 μg mL−1, while the VVBlue assay led to a letal dose 20% of 150 μg mL−1 and a letal dose 50% of 600 μg mL−1. Furthermore, the different shapes of the survival curves were consistent with the mechanisms underlying the two assays. The metabolic assay showed a gradual decrease of the metabolism with increasing pigment concentrations, while the cell permeability assay showed an extended survival followed by a rapid drop in cell viability with increasing pigment concentrations.
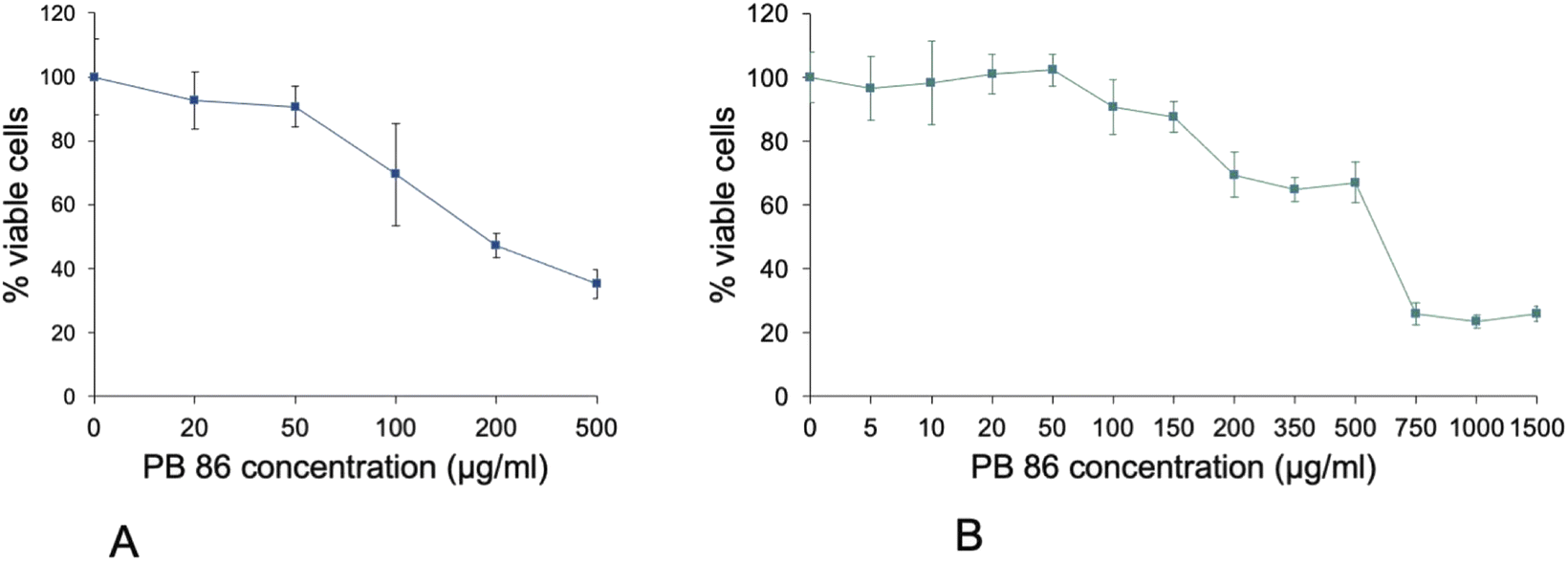 | ||
| Fig. 8 Comparison of the MTT and VVBlue assays on J774A.1 cells exposed to Pigment Blue 86. Cell viability curves after 48 hours of exposure to Pigment Blue 86. The abscissae are the concentration of pigment added to the cultures in μg mL−1. (A) Data obtained with the MTT assay. (B) Data obtained with the VVBlue assay. Experiments conducted in quadruplicates and expressed as mean ± standard deviation. | ||
4 Discussion
In viability assays, it is necessary to interpolate numerical values from the experimental points, as the desired parameters (e.g. LD20, LD50) may not be directly reached from the actual data points. To this purpose, regression analysis and most often linear regression analysis is most commonly used.In regression analysis, the R2 coefficient of determination serves as a statistical measure of how well the regression predictions approximate the actual data points. A close-to-1 R2 indicates that the model predicts the biological data accurately, while a slightly lower R2, within the range of 0.8 < R2 < 0.91, suggest a close approximation to real data points.
Prior studies on the correlation between cell numbers and absorbance of bound dyes have revealed linear relationships between these two variables.33 It has been ascertained that crystal violet staining may exhibit a plateau phenomenon at high cells numbers. In this study, distinctions between the two employed dyes have been observed. In contrast to the alphazurine staining, crystal violet displays a curvilinear regression pattern. Upon splitting the crystal violet dot chart into two segments, distinct linear trends emerge, each characterized by different correlation coefficients. The first segment, showing the absorbance of CV over the range from 0 to 105 cells, reveal a correlation coefficient of 0.9981. The second segment, spanning from 2 × 105 to 106 cells, reveal a lower correlation coefficient of 0.9694. Despite the slightly diminished R2 value in the latter linear phase, it attests to a certain correlation between absorbance and cell numbers within range.
When developing a new assay, it must be standardized against established reference assays to ensure that its reliability and accuracy, and thus its robustness. To this purpose, we evaluated our new method against a validated assay of a similar type (i.e. dye exclusion), namely the propidium iodide (PI) viability assay. Within the current dye exclusion assays, propidium iodide does not rely on human eye to determine live from dead cells, opposite to more ancient assays such as the Trypan blue assay, only requiring a few gating steps that are propagated throughout the series of samples to be analyzed. It was thus considered as more robust than assays relying on human eye. The VVBlue assay demonstrated a good correlation with the propidium iodide method, although always indicating higher LD20s. This observation can be attributed to the fact that cell detachment of the cells from the plates was required in the PI method. Indeed this process can induce artificial cell death, especially for very adherent cells such as macrophages that can get damaged upon the harsh conditions required to detach them from the culture plates, and are of course even more fragile when their viability starts to be compromised.
When comparing the results of the VVBlue assay with those obtained with a classical metabolic test (WST1), important differences in the toxicity curves were observed. The same type of discrepancy between metabolic tests and leakage-based ones has been described in the literature,30 with different formats (MTT and LDH leakage in30 compared to WST1 and alphazurine A here). Such apparent discrepancies shall not come as a surprise, and are just linked to the indirect nature of the metabolic tests. To give a stylistic example, a halved signal in a metabolic test may indicate either that all the cells are still viable nut with a 50% reduced metabolism, or that half of the cells are dead while the other half exhibit a full metabolic activity. Conversely, if cells become more metabolically active to fight a stress that is not lethal, viabilities above 100% can be obtained, as previously described in the literature.13,14 Thus, indirect test such as metabolic test or neutral red uptake19 should be taken with caution in their interpretation.
It can also be noted from Fig. 4 that different cell lines can show different responses to the same toxicant, in this case silver ion. Silver ion has been shown to be quite toxic toward J774A.1 cells, where it induces strong oxidative stress and metabolic poisoning.34 Thus, the rate of poisoning and thus the toxicity may depend on the metabolic status of the cells. Indeed, we observed that the J774A.1 cells consumed their culture medium much faster than the confluent A549 cells that we used, indicating a higher glucose consumption. Macrophages are known to utilize extensively glycolysis, e.g. for phagocytosis.35 As silver ion is known to inhibit key glycolytic enzymes such as glyceraldehyde phosphate dehydrogenase,36 triose phosphate isomerase,37 phosphoglycerate dehydrogenase38 or lactate dehydrogenase,39 this may explain the higher sensitivity of macrophages to silver ion.
However, it must be noted, for example on the Pigment Blue 86 case, that the data obtained by the metabolic assay and the cell permeability assay fit well with what is known about the toxic mechanisms shown by metals. Indeed, pigments gradually dissolved when internalized by cells, and release free metal ions, which then bind to some proteins active sites and block their functions.40 This applies to metabolic enzymes and leads to a decrease in metabolism that is detected by the tetrazolium-based assays (MTT or WST). However, cells are able to survive with a hampered metabolism, up to the point were they cannot compensate any longer and cannot maintain the integrity of their membrane, leading to cell death detection by the permeability assay.
Our experiments also provide the first public data for the toxicological analysis of Pigment Blue 86, a new metal-based pigment31 that has received limited market authorization.32 This pigment has shown limited toxicity in our assays, which is worth noting and supports its current authorization.
Overall, the VVBlue assay represents, to our knowledge, the first exclusion dye assay that is plate-readable. This statement is based on the fact that from the beginning up to the end of the assay, only pipetting and centrifuging (indeed optional) steps are required, and they are easily automatizable. Classical dye exclusion dye methods (e.g. Trypan blue) require either manual reading under a microscope, which is tedious and makes the assay prone to inter-operator variability, or an expensive and dedicated camera type setup. The PI assay requires a flow cytometer, and operator-defined delimitation of the threshold values for live and dead cells, which may be questionable too. At the expense of additional internal controls, namely “live cells” and “dead cells”, the VVBlue assay provides an operator independent reading of the cell viability, ideally using a versatile plate reader instrument. Alternatively, the absorbance of each well can be read individually in a spectrophotometer, although this will severely limit the throughput of the assay.
Furthermore, this assay can operate under conditions in which many of the other cell viability assays become impracticable, as demonstrated here for pigment particles. Of course, further control conditions needed to prevent data misinterpretation caused by dye-particle adsorption issues, but this can be easily implemented, as shown in the tests displayed in Fig. 8.
It must be kept in mind that the VVBlue assay reads the number of dead cells in the well, based on the accessibility of cellular proteins to the alphazurine dye. Thus, different cell death mechanisms (e.g. necrosis vs. apoptosis) may lead to different sensitivities, as the dye requires a compromised membrane to access the inside of the dead cells or apoptotic bodies. However, this feature is shared by all dye exclusion assays, including the trypan blue and propidium iodide assays.
Depending on whether the centrifugation step can keep all the dead cells in the well or not, or if the assay is conducted under conditions where the cells proliferate, the second crystal violet step, which provides the total number of cells still attached to the plate, may be useful or not. In any case, the double reading proposed here covers all situations, and may help distinguishing cytostatic effects from cytotoxic ones.
In conclusion, the VVBlue assay offers the advantages of dye exclusion assays under a plate-readable format. When the adequate controls are added to standardize the assay, it offers a reliable of the cell viability, even under severe optical conditions. However, it also shows some downsides. First of all, because of its mechanism, the alphazurine assay must be coupled with a cell enumeration-based assay, crystal violet assay in our case. This means in turn that the overall assay consists of two successive individual assays, which renders the global assay slow, somewhat cumbersome, and increases its variability. Indeed, our experiments showed that the standard deviation of the coupled alphazurine-crystal violet assay is 10% on average, while the average standard deviation of more standard assays (propidium iodide, WST1) is lesser than 5%. Although we have not yet found conditions where the assay becomes ineffective, it can be predicted that toxicants that are able to release a blue or green color in the elution medium will strongly interfere with the alphazurine assay. However, these interference can probably be dealt with by designing the correct controls (e.g. plates with the colored toxicant alone) to determine the level of interference. Interference with proteins, i.e. binding alphazurine, shall not be a problem, as the assay takes place in culture medium containing 10% serum, i.e. 4 mg mL−1 of protein.
Last but not least, as every assay based on numbers and operating in the plate format (such as metabolic assays, neutral red assay etc.), and opposite to assays operating on cell suspensions, (such as propidium iodide or trypan blue assays), which operate directly as proportions, the VVBlue assay will be sensitive to cell proliferation, which may increase cell numbers in some conditions and thus bias the toxicity results. The signal is also dependent on cell number, which requires rather dense cultures to start with. Nevertheless, in the 24 well plate format used here, the assay could easily detect a 10% increase in mortality, as shown by our data on zinc oxide.
Because of its intrinsic drawbacks (e.g. slow speed, rather high standard deviation), the VVBlue assay is not anticipated to supersede more classical and established assays, and has not been designed in this perspective. It has been designed as a plate-readable dye exclusion assay, which does not require dislodging cells from the culture support, and oppositely works only on attached cells (which represent the vast majority of cell types). It has also been designed to operate under adverse optical conditions where other assays fail to operate properly, while keeping the direct operation of dye exclusion or LDH release assays, as opposed to the indirect nature of metabolic assays. Moreover, its possible automation and direct mechanism may widen its application to other classes of chemicals than particles and pigments.
Data availability
DOI for the data. doi:10.6019/S-BSST1506.40 Link for the reviewers prior to release.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work used the flow cytometry facility hosted at IRIG, which received funding from GRAL, a program from the Chemistry Biology Health (CBH) Graduate School of University Grenoble Alpes (ANR-17-EURE-0003). This work on pigments was funded by the Agence Nationale de la Recherche (Tattooink grant, ANR-21-CE34-0025).References
- A. M. Pappenheimer, Experimental studies upon lymphocytes : I. The reactions of lymphocytes under various experimental conditions, J. Exp. Med., 1917, 25, 633–650, DOI:10.1084/jem.25.5.633.
- D. K. Yip and N. Auersperg, The dye-exclusion test for cell viability: persistence of differential staining following fixation, In Vitro, 1972, 7, 323–329, DOI:10.1007/BF02661722.
- D. B. Jacobs and C. Pipho, Use of propidium iodide staining and flow cytometry to measure anti-mediated cytotoxicity: resolution of complement-sensitive and resistant target cells, J. Immunol. Methods, 1983, 62, 101–108, DOI:10.1016/0022-1759(83)90115-1.
- T. Decker and M. L. Lohmann-Matthes, A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity, J. Immunol. Methods, 1988, 115, 61–69, DOI:10.1016/0022-1759(88)90310-9.
- X. Han, R. Gelein, N. Corson, P. Wade-Mercer, J. Jiang and P. Biswas, et al., Validation of an LDH assay for assessing nanoparticle toxicity, Toxicology, 2011, 287, 99–104, DOI:10.1016/j.tox.2011.06.011.
- L. Liang, M. Cui, M. Zhang, P. Zheng, Z. Deng and S. Gao, et al., Nanoparticles' interference in the evaluation of in vitro toxicity of silver nanoparticles, RSC Adv., 2015, 5, 67327–67334, 10.1039/C5RA05863E.
- S. Van Den Bossche, E. Vandeplassche, L. Ostyn, T. Coenye and A. Crabbé, Bacterial Interference With Lactate Dehydrogenase Assay Leads to an Underestimation of Cytotoxicity, Front. Cell. Infect. Microbiol., 2020, 10, 494, DOI:10.3389/fcimb.2020.00494.
- A. L. Holder, R. Goth-Goldstein, D. Lucas and C. P. Koshland, Particle-Induced Artifacts in the MTT and LDH Viability Assays, Chem. Res. Toxicol., 2012, 25, 1885–1892, DOI:10.1021/tx3001708.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63, DOI:10.1016/0022-1759(83)90303-4.
- D. A. Scudiero, R. H. Shoemaker, K. D. Paull, A. Monks, S. Tierney and T. H. Nofziger, et al., Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines, Cancer Res., 1988, 48, 4827–4833 CAS.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity, Eur. J. Biochem., 2000, 267, 5421–5426, DOI:10.1046/j.1432-1327.2000.01606.x.
- A. Van Tonder, A. M. Joubert and A. D. Cromarty, Limitations of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay when compared to three commonly used cell enumeration assays, BMC Res. Notes, 2015, 8, 47, DOI:10.1186/s13104-015-1000-8.
- P. Wang, S. M. Henning and D. Heber, Limitations of MTT and MTS-based assays for measurement of antiproliferative activity of green tea polyphenols, PLoS One, 2010, 5, e10202, DOI:10.1371/journal.pone.0010202.
- S. Wang, H. Yu and J. K. Wickliffe, Limitation of the MTT and XTT assays for measuring cell viability due to superoxide formation induced by nano-scale TiO2, Toxicol. In Vitro, 2011, 25, 2147–2151, DOI:10.1016/j.tiv.2011.07.007.
- A. Kroll, M. H. Pillukat, D. Hahn and J. Schnekenburger, Interference of engineered nanoparticles with in vitro toxicity assays, Arch. Toxicol., 2012, 86, 1123–1136, DOI:10.1007/s00204-012-0837-z.
- D. Breznan, D. Das, C. MacKinnon-Roy, B. Simard, P. Kumarathasan and R. Vincent, Non-specific interaction of carbon nanotubes with the resazurin assay reagent: Impact on in vitro assessment of nanoparticle cytotoxicity, Toxicol. in Vitro, 2015, 29, 142–147, DOI:10.1016/j.tiv.2014.09.009.
- E. Borenfreund and J. A. Puerner, Toxicity determined in vitro by morphological alterations and neutral red absorption, Toxicol. Lett., 1985, 24, 119–124, DOI:10.1016/0378-4274(85)90046-3.
- R. Guadagnini, K. B. Halamoda, L. Walker, G. Pojana, Z. Magdolenova and D. Bilanicova, et al., Toxicity screenings of nanomaterials: challenges due to interference with assay processes and components of classic in vitro tests, Nanotoxicology, 2015, 9, 13–24, DOI:10.3109/17435390.2013.829590.
- S. Triboulet, C. Aude-Garcia, M. Carriere, H. Diemer, F. Proamer and A. Habert, et al., Molecular responses of mouse macrophages to copper and copper oxide nanoparticles inferred from proteomic analyses, Mol. Cell. Proteomics, 2013, 12, 3108–3122 CrossRef CAS PubMed.
- S. Triboulet, C. Aude-Garcia, L. Armand, A. Gerdil, H. Diemer and F. Proamer, et al., Analysis of cellular responses of macrophages to zinc ions and zinc oxide nanoparticles: a combined targeted and proteomic approach, Nanoscale, 2014, 6, 6102–6114 RSC.
- K. Saotome, H. Morita and M. Umeda, Cytotoxicity test with simplified crystal violet staining method using microtitre plates and its application to injection drugs, Toxicol. In Vitro, 1989, 3, 317–321, DOI:10.1016/0887-2333(89)90039-8.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon and D. Vistica, et al., New colorimetric cytotoxicity assay for anticancer-drug screening, J. Natl. Cancer Inst., 1990, 82, 1107–1112, DOI:10.1093/jnci/82.13.1107.
- G. R. Pullen, P. J. Chalmers, A. P. Nind and R. C. Nairn, Criteria of cell killing in vitro, J. Immunol. Methods, 1981, 43, 87–93, DOI:10.1016/0022-1759(81)90039-9.
- K. Chiba, K. Kawakami and K. Tohyama, Simultaneous evaluation of cell viability by neutral red, MTT and crystal violet staining assays of the same cells, Toxicol. In Vitro, 1998, 12, 251–258, DOI:10.1016/s0887-2333(97)00107-0.
- A. W. Krause, W. W. Carley and W. W. Webb, Fluorescent erythrosin B is preferable to trypan blue as a vital exclusion dye for mammalian cells in monolayer culture, J. Histochem. Cytochem., 1984, 32, 1084–1090, DOI:10.1177/32.10.6090533.
- C. Fenner, R. R. Traut, D. T. Mason and J. Wikman-Coffelt, Quantification of Coomassie Blue stained proteins in polyacrylamide gels based on analyses of eluted dye, Anal. Biochem., 1975, 63, 595–602, DOI:10.1016/0003-2697(75)90386-3.
- L. B. Gilmore and G. E. Hook, Quantitation of specific proteins in polyacrylamide gels by the elution of Fast Green FCF, J. Biochem. Biophys. Methods, 1981, 5, 57–66, DOI:10.1016/0165-022x(81)90033-6.
- B. Dalzon, A. Torres, J. Devcic, D. Fenel, J.-A. Sergent and T. Rabilloud, A Low-Serum Culture System for Prolonged in Vitro Toxicology Experiments on a Macrophage System, Front. Toxicol., 2021, 3, 780778, DOI:10.3389/ftox.2021.780778.
- M. Ishiyama, M. Shiga, K. Sasamoto, M. Mizoguchi and P. He, A New Sulfonated Tetrazolium Salt That Produces a Highly Water-Soluble Formazan Dye, Chem. Pharm. Bull., 1993, 41, 1118–1122, DOI:10.1248/cpb.41.1118.
- W. Cybulski, L. Radko and W. Rzeski, Cytotoxicity of monensin, narasin and salinomycin and their interaction with silybin in HepG2, LMH and L6 cell cultures, Toxicol. in Vitro, 2015, 29, 337–344, DOI:10.1016/j.tiv.2014.11.013.
- A. E. Smith, H. Mizoguchi, K. Delaney, N. A. Spaldin, A. W. Sleight and M. A. Subramanian, Mn3+ in Trigonal Bipyramidal Coordination: A New Blue Chromophore, J. Am. Chem. Soc., 2009, 131, 17084–17086, DOI:10.1021/ja9080666.
- Shepherd Color's YInMn Blue gains full EPA approval, Addit. Polym., 2020, 2020, 7, 4, DOI:10.1016/S0306-3747(20)30081-6.
- M. A. Scragg and L. R. Ferreira, Evaluation of different staining procedures for the quantification of fibroblasts cultured in 96-well plates, Anal. Biochem., 1991, 198, 80–85, DOI:10.1016/0003-2697(91)90509-r.
- M. Gumińska, W. Ptak and M. Zembala, Macrophage metabolism during phagocytosis and digestion of normal and IgG antibody-coated sheep erythrocytes, Enzyme, 1975, 19, 24–37, DOI:10.1159/000458969.
- H. Wang, M. Wang, X. Yang, X. Xu, Q. Hao and A. Yan, et al., Antimicrobial silver targets glyceraldehyde-3-phosphate dehydrogenase in glycolysis of E. coli, Chem. Sci., 2019, 10, 7193–7199, 10.1039/c9sc02032b.
- R. H. Davis, P. Delaney and C. S. Furfine, Chemical modification of sulfhydryl residues of rabbit triosephosphate isomerase, Arch. Biochem. Biophys., 1973, 159, 11–24, DOI:10.1016/0003-9861(73)90424-4.
- J. Rosenbloom, The mechanism of end product inhibition of serine biosynthesis. 3. Physical and chemical properties of phosphoglycerate dehydrogenase, J. Biol. Chem., 1968, 243, 2099–2107 CrossRef CAS.
- M. P. Menon and C. E. Wright, A radiotracer probe to study metal interaction with human lactate dehydrogenase isoenzymes, J. Protein Chem., 1989, 8, 757–766, DOI:10.1007/BF01024900.
- P. Chen, J. Bornhorst and M. Aschner, Manganese metabolism in humans, Front. Biosci.-Landmark, 2018, 23, 1655–1679, DOI:10.2741/4665.
- U. Sarkans, M. Gostev, A. Athar, E. Behrangi, O. Melnichuk and A. Ali, et al., The BioStudies database—one stop shop for all data supporting a life sciences study, Nucleic Acids Res., 2018, 46, D1266–D1270, DOI:10.1093/nar/gkx965.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08606f |
| This journal is © The Royal Society of Chemistry 2025 |