
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,2-Diaminocyclohexane-derived chiral tetradentate ligands for Mn(I)-catalyzed asymmetric hydrogenation of ketones†
Yi Su‡
 ab,
Dongzhi Zhu‡*ac,
Zhifeng Ma‡*cd,
Yizhou Wangc,
Zechen Wangb,
Zheng Wang*bc,
Yanping Mac and
Wen-Hua Sun*c
ab,
Dongzhi Zhu‡*ac,
Zhifeng Ma‡*cd,
Yizhou Wangc,
Zechen Wangb,
Zheng Wang*bc,
Yanping Mac and
Wen-Hua Sun*c
aGuangxi Key Laboratory of Advanced Structural Materials and Carbon Neutralization, School of Materials and Environment, Guangxi Minzu University, Nanning, 530105, China. E-mail: 20230021@gxmzu.edu.cn
bCollege of Science, Hebei Agricultural University, Baoding, 071001, China
cKey Laboratory of Engineering Plastics and Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: whsun@iccas.ac.cn; wangzheng@iccas.ac.cn
dSchool of Chemistry & Environment, Yunnan Key Laboratory of Chiral Functional Substance Research and Application, Yunnan Minzu University, Kunming, Yunnan 650504, China. E-mail: mazhifeng@ymu.edu.cn
First published on 4th July 2025
Abstract
A series of (R,R)-1,2-diaminocyclohexane-based chiral PNNP and SNNS tetradentate ligands were successfully employed as chiral chelating ligands for the asymmetric hydrogenation (AH) of substituted acetophenones (13 examples) with good activity and good enantioselectivity (up to 85% ee). In particular, two types of manganese(I) complexes (Mn1 and Mn2) with a “C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N” or “NH” group were isolated, and their comparative performance as catalysts revealed Mn1 as more effective in AH of ketones with a maximum enantiomeric excess (ee) value of 85%. DFT calculations revealed that the formation of the major S-type 1-phenylethanol by Mn1 was significantly influenced by steric repulsion between the substrate and ligand.
N” or “NH” group were isolated, and their comparative performance as catalysts revealed Mn1 as more effective in AH of ketones with a maximum enantiomeric excess (ee) value of 85%. DFT calculations revealed that the formation of the major S-type 1-phenylethanol by Mn1 was significantly influenced by steric repulsion between the substrate and ligand.
Introduction
Catalytic asymmetric reduction of ketones to chiral alcohols with H2 is of high academic and industrial interests owing to its widespread applications in many fields, such as in the production of drugs, pesticides, fragrances and other bioactive molecules.1 Notably, most of the currently available successful catalysts are based on noble metals such as Ru,2 Rh,3 and Ir4.1a,5 However, the earth-abundance of the above metals is limited, and their use causes environmental pollution and safety concerns, promoting the search for alternative catalysts based on earth-abundant, base metals (Scheme 1a).6 Consequently, AH catalysts based on manganese became particularly prominent owing to their high biocompatibility and earth-abundance.7 Furthermore, with the continuous advancements in chiral ligand design methodologies, the application of manganese(I)-based chiral organometallic complexes in AH catalysis has been greatly promoted,5a,b and it has been receiving sustained attention from academic and industrial circles.7 However, the activity and robustness of Mn-based catalytic systems generally fall short of those of noble metal-based catalytic systems.7a,8 | ||
| Scheme 1 (a) Various metal catalysts previously employed in the AH of ketones. (b) Selected tridentate-Mn(I) chiral catalysts for the AH of ketones; (c) Mn-based complexes studied in this work. | ||
In particular, Mn-based hydrogenation catalysis has become a subject of hot topic since 2016 when Beller's group first demonstrated its transformative potential in ketone hydrogenation.9 Since then, the groups of Clarke,10 Beller,11 Ding,12 Mezzetti,13 Zhong,14 Zhang,15 Morris,16 and Liu17 et al.18 have independently developed a series of Mn(I) complex catalysts (A–H, Scheme 1b), incorporating the chiral tridentate PNN or PNP ligands and successfully applying them in AH of ketones. Notably, the chiral 1,2-substituted ferrocene backbone was of particular interest. For example, Clarke et al. primarily described A bearing a ferrocene-incorporated chiral pincer ligand that could realize excellent turnover numbers (TONs up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and offered chiral alcohols (ee up to 97%).10a,b Clarke's group further modified A to H for attaining compatibility with cyclic ketones, offering the corresponding chiral cyclic alcohols with ee up to 98%.10c More recently, Hu et al. reported H for the Mn-catalyzed AH of heterobiaryl ketone N-oxides with an enantiomeric excess of up to 99%.18 Meanwhile, Zhong and co-workers developed a new chiral PNN catalyst (E) containing the “CN” or “NH” group for AH of simple ketones and unsymmetrical benzophenones, offering an outstanding activity (up to 13000 TON) and excellent enantioselectivities (>99% ee).14 Zhang et al. demonstrated that F catalyzed the hydrogenation of ketones with excellent enantioselectivities (up to 99% ee) and high activity (up to 2000 TON).15 We recently disclosed that strengthening the rigidity of the framework by adding the controllable “aliphatic cyclic” chiral amine motifs to a ferrocene moiety (G, Scheme 1b) can achieve the desired enantioinduction (up to 99% ee).17 Despite these great advances, several limitations, such as poor availability of the starting materials for catalysts and limited types of catalysts, pose great challenges in the field of the manganese-catalyzed asymmetric reduction of ketones. Furthermore, the current Mn-based catalytic systems are mostly based on chiral pincer ligands. In contrast, tetradentate manganese(I) complexes were rarely reported.19
000) and offered chiral alcohols (ee up to 97%).10a,b Clarke's group further modified A to H for attaining compatibility with cyclic ketones, offering the corresponding chiral cyclic alcohols with ee up to 98%.10c More recently, Hu et al. reported H for the Mn-catalyzed AH of heterobiaryl ketone N-oxides with an enantiomeric excess of up to 99%.18 Meanwhile, Zhong and co-workers developed a new chiral PNN catalyst (E) containing the “CN” or “NH” group for AH of simple ketones and unsymmetrical benzophenones, offering an outstanding activity (up to 13000 TON) and excellent enantioselectivities (>99% ee).14 Zhang et al. demonstrated that F catalyzed the hydrogenation of ketones with excellent enantioselectivities (up to 99% ee) and high activity (up to 2000 TON).15 We recently disclosed that strengthening the rigidity of the framework by adding the controllable “aliphatic cyclic” chiral amine motifs to a ferrocene moiety (G, Scheme 1b) can achieve the desired enantioinduction (up to 99% ee).17 Despite these great advances, several limitations, such as poor availability of the starting materials for catalysts and limited types of catalysts, pose great challenges in the field of the manganese-catalyzed asymmetric reduction of ketones. Furthermore, the current Mn-based catalytic systems are mostly based on chiral pincer ligands. In contrast, tetradentate manganese(I) complexes were rarely reported.19
Typically, ruthenium-,20 iron-,6b,21 nickel-22 and cobalt-6f,23 based tetradentate complex catalysts exhibit excellent performance in the AH and asymmetric transfer hydrogenation (ATH) of ketones into chiral alcohols. Compared with bidentate and tridentate ligands, tetradentate ligands exhibit versatile features and effectively regulate the electronic and steric properties, bond angles, and catalytic performance of the formed catalysts.24 In addition, tetradentate complex catalysts provide higher stability and unique chiral pocket towards carbonylation. Based on our long-term research interest in Mn-catalyzed (de)hydrogenation reactions,25 herein, we report a series of (R,R)-1,2-diaminocyclohexane-based chiral tetradentate ligands (L1–L4) and two types of PNNP manganese(I) complexes with a “CN” (Mn1) or “NH” (Mn2) group. Their comparative study on catalyst performance proved Mn1 to be more effective in AH of ketones. Moreover, DFT calculations were used to understand the influence of C–N types (CN (Mn1) vs. C–NH (Mn2)) and alkali-metal cations on the transition-state geometry of the hydrogen-transfer reaction. The distinct steric profiles of the CN (planar) and C–NH (tetrahedral) configurations were found to critically influence the chiral pocket geometry, explaining the observed enantioselectivity trends and guiding our ligand optimization strategy.
Results and discussion
Based on the available literature,20,26,27 1,2-diamino-cyclohexane-based chiral tetradentate ligands (L1–L4) were readily prepared via the condensation reaction between (R,R)-1,2-diaminocyclohexane and commercially available aldehydes or carboxylic acids, respectively (see ESI†). Subsequently, the four tetradentate ligands (L1–L4, see ESI†) containing chiral 1,2-diaminocyclohexane motif were explored for the Mn-catalyzed asymmetric hydrogenation of acetophenone (a1) in ethanol solution (Scheme 2). The catalyst was prepared by treating Mn(CO)5Br with 1.1 molar equivalents of the respective ligand in dry EtOH under reflux (80 °C, 4 h). The substrate and K2CO3 (as base) were dissolved in ethanol, followed by the addition of the EtOH solution to the in situ formed complex. In these AH tests, 2 mol% [Mn] with respect to acetophenone (a1) was employed, while 20 mol% K2CO3 was used as the base (Tables S1–S3, see ESI†). Notably, among these four in situ generated complexes, only the combination of MnBr(CO)5/L1 displayed higher activity (86% yield) for AH of a1, and the enantiomeric excess (ee) of b1 regarding the chiral ligand (Scheme 2) dropped in the order of L2 (70%) > L1 (65%) > L3 (45%) > L4 (17%); this result aligned well with the previous finding reported by the groups of Gao and Li.19a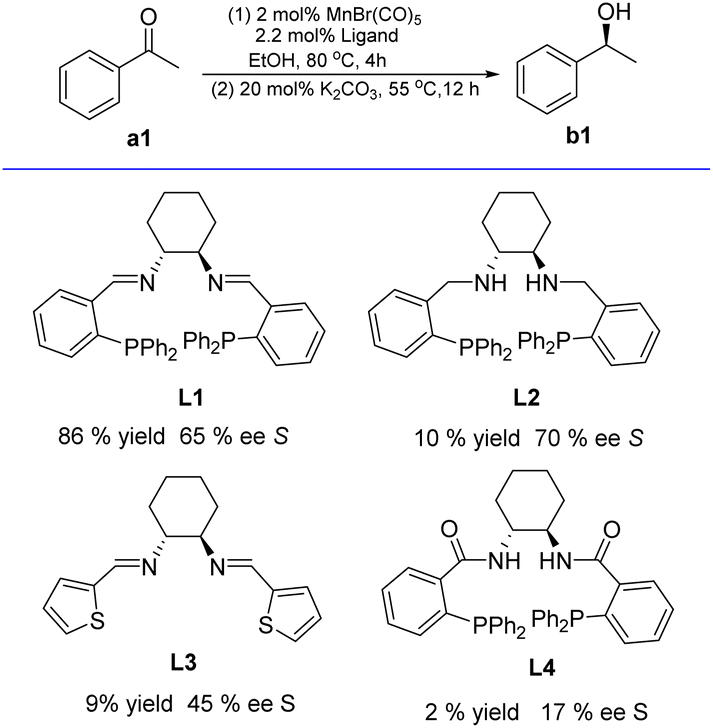 | ||
| Scheme 2 Ligand screening. Conditions: (1) Mn(CO)5Br (2.0 mol%), L1–L4 (2.2 mol%), 5 mL EtOH, 80 °C, 4 h; (2) a1 (0.25 mmol), K2CO3 (20 mol%), 50 bar H2, 55 °C, 12 h. Yields (%) and enantioselectivities (% ee) were determined using GC and chiral-phase GC, respectively, using a CP-Chirasil-Dex CB column. | ||
After identifying L1 as the best ligand in the above tests, we proceeded to check the veracity of the complex structure and the catalytic process. Initial efforts focused on isolating the complex generated from the coordination reaction of Mn(CO)5Br with L1 or L2. The P2N2-Mn1 complex was efficiently prepared (84% yield) in toluene at 90 °C for 10 h, whereas P2N2-Mn2 required reaction with dry toluene, refluxing for 12 h (Scheme 3a and see ESI†). Both P2N2-Mn1 and P2N2-Mn2 were characterized using 1H/13C/31P NMR, elemental analysis, IR spectroscopy and crystal X-ray analysis. Notably, two strong peaks were displayed in the IR spectra at 1852 and 1950 cm−1, which were characteristic of CO ligands coordinated to manganese in each complex. Additionally, Mn1 exhibited a prominent peak at 1612 cm−1, matching the expected CN stretching frequency. In contrast, Mn2's IR spectrum displayed strong absorption bands around 1622 cm−1 and 3233 cm−1, which corresponded to the NH group.
 | ||
| Scheme 3 Synthesis and reactivity of Mn(I) complexes Mn1 and Mn2. | ||
Meanwhile, their 31P NMR spectra (recorded in DMSO-d6) offered key structural insights, with the phosphine signal for Mn1 appearing at 63.45 ppm and at 61.93 ppm for Mn2 (cf. δ −13.68 ppm for L1 and δ −15.92 ppm for L2). Furthermore, single crystals of Mn1 were successfully obtained for X-ray diffraction analysis, confirming its molecular structure. Crystallographic data revealed that the cationic unit of Mn1 (Scheme 3b and Table S4,† CCDC: 2408177) adopted a distorted octahedral geometry in the P3121 space group, in which the two imine moieties and carbonyl groups were coordinated in a cis-fashion, while the two phosphine groups were bound in a trans-fashion to each other. Structural parameters, including bond lengths and angles for Mn1, are summarized in Table S5 (see ESI†).
Structural comparison between Mn1 and a reported achiral cyclic manganese(I) complex Mn3,19b exhibited difference in their structures with respect to bond distances (Table 1). Mn1 contained two unsaturated CN bonds. The N1–C19 and N2–C28 bond distances of 1.238(9) Å and 1.242(9) Å, respectively, were typical of CN distances. As for the case of the complex Mn3 reported by Fang et al.,19b both C–N bonds demonstrated lengths of typical saturated covalent connections. Mn1 has Mn–N bonds of 2.081(6) Å and 2.078(6) Å, while the reported complex Mn3 exhibited longer Mn–N bonds of 2.161(7) Å and 2.143(6) Å. Mn1 also exhibited shorter Mn–P bonds than Mn3 [2.308(2) Å and 2.322(2) Å vs. 2.293(2) Å and 2.269(2) Å]. Mn1 displayed greater steric encumbrance in its cyclic arrangement than Mn3 owing to its rigid CN connectivity. Accordingly, Mn3 displayed higher activity for the hydrogenation of ketones.19b Currently, few examples exist for these carefully constructed chiral manganese(I) cyclic complexes with P2N2 donor sets. When applied to acetophenone (a1) hydrogenation (Scheme 3b), Mn1 showed remarkable catalytic activity (98% conversion) and stereoselectivity (85% ee), significantly outperforming Mn2 (6% yield, 42% ee). This difference underscores the critical role of imine functionality in the cyclohexanediamine-derived ligand framework for effective metal–ligand cooperative catalysis.14a
| Complex | N–C (Å) | Mn–N (Å) | Mn–P (Å) |
|---|---|---|---|
| Mn1 | N1–C19 1.238(9) | Mn1–N1 2.081(6) | Mn1–P1 2.293(2) |
| N2–C28 1.242(9) | Mn1–N2 2.078(6) | Mn1–P2 2.269(2) | |
| Mn3,19b | N1–C01V 1.50(1) | Mn1–N1 2.161(7) | Mn1–P1 2.308(2) |
| N2–C016 1.483(9) | Mn1–N2 2.143(6) | Mn1–P2 2.322(2) |
With the best manganese complex catalyst in hand, we decided to explore Mn1 as a catalyst for the benchmark AH transformation of acetophenone (a1) to S-1-phenylethanol (b1). Initial optimization attempts revealed that Mn1 (2 mol%, S/C = 50) combined with t-BuOK (20 mol%) at 55 °C produced b1 in near-quantitative yield (99%), with 65% ee (entry 1, Table S1, see ESI†). With the aim to establish the most compatible base, the AH of a1 was then screened for various bases, including t-BuOK, t-BuONa, t-BuOLi, CH3ONa, K3PO4, KOH, NaOH, LiOH·H2O, K2CO3, and Cs2CO3 in EtOH at 55 °C (Table S1, see ESI†). As shown in Fig. 1, both the yields and enantioselectivity were strongly dependent on the selected alkali metal cation. Among them, K+ offered the greatest promoting effect, viz., K2CO3 proved as a standout example (ee ≥ 85%) owing to its moderate alkalinity and better compatibility between the size of the potassium ion and ligand's cavity.
 | ||
| Fig. 1 Screening of bases. Conditions: 0.25 mmol a1, 0.05 mmol base (20 mol%), 5 μmol Mn1 (2 mol%), 50 bar H2, 5 mL EtOH, 55 °C, 12 h; yields (%) and enantioselectivities (% ee) were determined using GC and chiral-phase GC, respectively, using a CP-Chirasil-Dex CB column. | ||
Considering the importance of temperature on catalytic activity and enantioselectivity, the AH of a1 was systematically investigated at temperatures ranging from 35 °C to 85 °C, with K2CO3 employed consistently as the base (Table S3, see ESI†). Notably, excellent conversions to b1 in the range of 97% to 99% were observed across the temperature range from 55 °C to 85 °C. However, a lower temperature (45 °C) was found to be unfavorable for the transformations, with a distinct loss in conversion observed at 45 °C, which can attributed to the fact that low temperature could not overcome the energy barrier. Furthermore, enantioselectivity with a distinct loss in selectivity was observed at 75 °C.
With the standard reaction conditions (2 mol% Mn1, 20 mol% K2CO3, 55 °C, EtOH) in hand, we further explored the capacity of this Mn-based catalytic system for the AH of ketones (Table 2). Various acetophenones and their derivatives (a2–a15) were explored for the AH of ketones. In general, substituted acetophenones containing electron-withdrawing substituents (Cl and Br: a3–a4, a7–a8 and a11–a12) displayed higher activities and diminished optical purities than their analogues containing electron-donating methyl groups (Me: a5, a9 and a13). Notably, the pattern of substituents on the phenyl ring exhibited a significant influence on the conversion of the product, and steric hindrance on the ortho-position (b2–b5) led to a decrease in the yield. In terms of steric effects, enhanced steric hindrance on the aromatic ring (e.g. 2,6-disubstituted b14 and b15) led to trace product conversions. Furthermore, the results showed that enantio-selectivities decreased in the sequence of meta (58–79% ee) > para (65–72% ee) > ortho (27–68% ee). Compared with the reported manganese complex catalysts (A, C, D, E, F and H), the current system based on Mn1 displayed lower activities and lower enantioselectivities for the substituted acetophenones.
| a Conditions: 0.25 mmol ketone (a1–a15), 0.05 mmol K2CO3 (20 mol%), 5 μmol Mn1 (2 mol%), 50 bar H2, 5 mL EtOH, 55 °C, 12 h. Yields (%) and enantioselectivities (% ee) were determined using GC and chiral-phase GC, respectively. |
|---|
 |
Based on the bifunctional concept,6f,7b,22c and our early mechanistic studies,17 DFT calculations using the PCM B3LYP-D3//B3LYP-D3 method were performed to investigate the influence of the types of C–N bonds (imine CN in Mn1 vs. amine C–NH in Mn2) and potassium cations on the transition-state geometry during hydrogen transfer (see Tables S6–S8†). Results revealed that the free energy barriers for hydrogen atom transfer (HAT) in the generation of (S)-type chiral product with Mn1 and Mn2 catalyst were lower than those for (R)-type chiral product, with a ΔΔG of 1.4 and 0.7 kcal mol−1, respectively (Fig. 2a and b). The distortion energy (ΔΔ Edist ∼4.5 kcal mol; Fig. 2c) played a key role in controlling the enantioselectivity (%V_Burs = 76.7 and 75.3 for TSRMn1 and TSSMn1, respectively). In contrast, the noncovalent interaction (π–π stacking) played a key role in controlling the enantioselectivity of TSRMn2 and TSSMn2 (ΔΔ Eint = ∼6.0 kcal mol; Fig. 2d). In comparison to the (S)-type chiral product generated with the Mn2 catalyst, the formation of the (S)-product catalyzed by Mn1 was favored through the transition state TSSMn1. The observed free-energy difference (ΔΔG = ∼3.4 kcal mol−1) primarily stemmed from the steric interactions between the ketone substrate (a1) and catalyst (ΔΔEdist ∼10.9 kcal mol; Fig. 3). In general, the DFT results indicated that the generation of the major S-type product by Mn1 with good ee values was mainly influenced by the steric repulsion between the substrate and ligand. Our DFT results were consistent with the experimental observations.
 | ||
| Fig. 2 Bond distances (in Å) and corrected Gibbs free energies (in kcal mol−1) of transition states (TSs) calculated using the PCM B3LYP-D3/SDD+6-311+G**//B3LYP-D3/SDD+6-31G* method in an ethanol solution: (a) TS involving K+ ion derived from Mn1; (b) TS derived from Mn2; (c) distortion–interaction energy analysis of the transition states, showing the distortion energy (ΔΔEdist) and interaction energy (ΔΔEint) between acetone and the remaining Mn-L1 parts, the steric map with the computed buried volumes (in %); and (d) noncovalent interaction (NCI) plots (red: strong repulsion; green: weak attraction; blue: strong attraction). | ||
 | ||
| Fig. 3 Relative corrected free energies (in kcal mol−1) for TSSMn1 and TSSMn2 calculated using the PCM B3LYP-D3/BS2//B3LYP-D3/BS1 method in an ethanol solution. | ||
Conclusions
In conclusion, we have successfully synthesized a series of chiral tetradentate ligands and two new coordination P2N2 manganese(I) complexes with “CN” (Mn1) or “NH” (Mn2) groups and demonstrated their successful applications in the AH of aryl and alkyl ketones. Applying Mn1, featuring a (R,R)-1,2-diaminocyclohexane scaffold, high activity and good stereocontrol (up to 85% ee) were achieved across diverse acetophenone derivatives to produce enantio-enriched 1-phenylethanols. DFT calculations revealed that the origin of the distinctive anti stereoselectivity in the catalysis was mainly owing to the steric repulsion between the substrate and ligand in the favored transition state. Further investigations on the reaction mechanism are currently ongoing in our laboratory.
Data availability
The data that support the findings of this study are available in the ESI† of this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. Z. would like to acknowledge the financial support from the Nature Science Foundation of Guangxi Province (2024JJB120135). Z. W. acknowledge the support from the Hebei provincial central guiding local science and Technology Development Fund project (236Z1402G). Z. W. would like to acknowledge the financial support from the Hebei Graduate Innovation Funding Project (CXZZSS20250041). Z. M and Z. W. would like to acknowledge the financial support from the Yunnan Key Laboratory of Chiral Functional Substance Research and Application (202402AN360010; 2024SXFKO5).References
- (a) V. Ratovelomanana-Vidal and P. Phansavath, Asymmetric hydrogenation and transfer hydrogenation, Wiley-VCH, 2021 CrossRef; (b) P.-C. Yan, G.-L. Zhu, J.-H. Xie, X.-D. Zhang, Q.-L. Zhou, Y.-Q. Li, W.-H. Shen and D.-Q. Che, Org. Process Res. Dev., 2013, 17, 307–312 CrossRef CAS; (c) G.-L. Zhu, X.-D. Zhang, L.-J. Yang, J.-H. Xie, D.-Q. Che, Q.-L. Zhou, P.-C. Yan and Y.-Q. Li, Org. Process Res. Dev., 2015, 20, 81–85 CrossRef; (d) S. Duan, B. Li, R. W. Dugger, B. Conway, R. Kumar, C. Martinez, T. Makowski, R. Pearson, M. Olivier and R. Colon-Cruz, Org. Process Res. Dev., 2017, 21, 1340–1348 CrossRef CAS.
- (a) M. Kitamura, T. Ohkuma, S. Inoue, N. Sayo, H. Kumobayashi, S. Akutagawa, T. Ohta, H. Takaya and R. Noyori, J. Am. Chem. Soc., 2002, 110, 629–631 CrossRef; (b) R. N. A. T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 Search PubMed; (c) K. Matsumura, N. Arai, K. Hori, T. Saito, N. Sayo and T. Ohkuma, J. Am. Chem. Soc., 2011, 133, 10696–10699 CrossRef CAS; (d) F. Naud, F. Spindler, C. J. Rueggeberg, A. T. Schmidt and H. U. Blaser, Org. Process Res. Dev., 2007, 11, 519–523 CrossRef CAS; (e) Y. Li, K. Ding and C. A. Sandoval, Org. Lett., 2009, 11, 907–910 CrossRef CAS; (f) T. Ohkuma and N. Arai, Chem. Rec., 2016, 16, 2797–2815 Search PubMed.
- (a) H. Nie, G. Zhou, Q. Wang, W. Chen and S. Zhang, Tetrahedron: Asymmetry, 2013, 24, 1567–1571 CrossRef CAS; (b) D.-H. Bao, H.-L. Wu, C.-L. Liu, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2015, 54, 8791–8794 CrossRef CAS; (c) F.-H. Zhang, F.-J. Zhang, M.-L. Li, J.-H. Xie and Q.-L. Zhou, Nat. Catal., 2020, 3, 621–627 CrossRef CAS; (d) S. F. Zhu and Q. L. Zhou, Acc. Chem. Res., 2017, 50, 988–1001 CrossRef CAS; (e) F. Yang, J. H. Xie and Q. L. Zhou, Acc. Chem. Res., 2023, 56, 332–349 Search PubMed; (f) K. Chang, L. Yang, Y. Liu, J. Cao, L. Zuo, Q. Liu, X. Zhang, C. Yin and H. Zhou, Org. Lett., 2024, 26, 9841–9846 CrossRef CAS.
- (a) H. Yang, N. Huo, P. Yang, H. Pei, H. Lv and X. Zhang, Org. Lett., 2015, 17, 4144–4147 Search PubMed; (b) F. Bruning, H. Nagae, D. Kach, K. Mashima and A. Togni, Chem.–Eur. J., 2019, 25, 10818–10822 Search PubMed; (c) H. Sun, L. Xu, S. Ruan, V. Ratovelomanana-Vidal, G. Q. Chen and X. Zhang, Org. Lett., 2024, 26, 10008–10012 CrossRef CAS.
- (a) H. Wang, J. Wen and X. Zhang, Chem. Rev., 2021, 121, 7530–7567 CrossRef CAS; (b) D. H. Liang, C. J. Hou, Q. Li, H. Qin, L. Li and X. P. Hu, Adv. Synth. Catal., 2024, 366, 2165–2185 CrossRef CAS.
- (a) T. Zell and R. Langer, ChemCatChem, 2018, 10, 1930–1940 CrossRef CAS; (b) Y.-Y. Li, S.-L. Yu, W.-Y. Shen and J.-X. Gao, Acc. Chem. Res., 2015, 48, 2587–2598 CrossRef CAS; (c) G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC; (d) F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS; (e) P. Wang, Z. L. He, Z. F. Xia, J. Wei and X. Q. Dong, Chin. J. Chem., 2024, 42, 3135–3156 CrossRef CAS; (f) Z. Wang, M. Li and W. Zuo, J. Am. Chem. Soc., 2024, 146, 26416–26426 CrossRef CAS.
- (a) D. Fu, Z. Wang, Q. Liu, S. Prettyman, G. A. Solan and W.-H. Sun, ChemCatChem, 2024, e202301567 CrossRef CAS; (b) K. Das, S. Waiba, A. Jana and B. Maji, Chem. Soc. Rev., 2022, 51, 4386–4464 RSC.
- (a) C. Liu, R. van Putten, P. O. Kulyaev, G. A. Filonenko and E. A. Pidko, J. Catal., 2018, 363, 136–143 CrossRef CAS; (b) W. Yang, T. Y. Kalavalapalli, A. M. Krieger, T. A. Khvorost, I. Y. Chernyshov, M. Weber, E. A. Uslamin, E. A. Pidko and G. A. Filonenko, J. Am. Chem. Soc., 2022, 144, 8129–8137 CrossRef CAS; (c) W. Yang, E. Nieuwlands, I. Y. Chernyshov, G. A. Filonenko and E. A. Pidko, ChemCatChem, 2025, 17, e202401237 CrossRef CAS.
- S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS.
- (a) M. B. Widegren, G. J. Harkness, A. M. Z. Slawin, D. B. Cordes and M. L. Clarke, Angew. Chem., Int. Ed., 2017, 56, 5825–5828 CrossRef CAS; (b) M. B. Widegren and M. L. Clarke, Catal. Sci. Technol., 2019, 9, 6047–6058 RSC; (c) C. L. Oates, A. S. Goodfellow, M. Buhl and M. L. Clarke, Angew. Chem., Int. Ed., 2023, 62, e202212479 CrossRef CAS.
- M. Garbe, K. Junge, S. Walker, Z. Wei, H. Jiao, A. Spannenberg, S. Bachmann, M. Scalone and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 11237–11241 CrossRef CAS.
- (a) L. Zhang, Y. Tang, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2019, 58, 4973–4977 CrossRef CAS; (b) L. Zhang, Z. Wang, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2020, 59, 15565–15569 CrossRef CAS.
- A. Passera and A. Mezzetti, Adv. Synth. Catal., 2019, 361, 4691–4706 CrossRef CAS.
- (a) F. Ling, H. Hou, J. Chen, S. Nian, X. Yi, Z. Wang, D. Song and W. Zhong, Org. Lett., 2019, 21, 3937–3941 CrossRef CAS; (b) F. Ling, J. Chen, S. Nian, H. Hou, X. Yi, F. Wu, M. Xu and W. Zhong, Synlett, 2020, 31, 285–289 CrossRef CAS; (c) Z. Wang, X. Zhao, A. Huang, Z. Yang, Y. Cheng, J. Chen, F. Ling and W. Zhong, Tetrahedron Lett., 2021, 82, 153389 CrossRef CAS; (d) J. He, W. Mao, J. Lin, Y. Wu, L. Chen, P. Yang, D. Song, P. Zhu, W. Zhong and F. Ling, Org. Chem. Front., 2023, 10, 3321–3327 RSC.
- L. Zeng, H. Yang, M. Zhao, J. Wen, J. H. R. Tucker and X. Zhang, ACS Catal., 2020, 10, 13794–13799 CrossRef CAS.
- C. S. G. Seo, B. T. H. Tsui, M. V. Gradiski, S. A. M. Smith and R. H. Morris, Catal. Sci. Technol., 2021, 1, 3153–3163 RSC.
- (a) J. Yang, L. Yao, Z. Wang, Z. Zuo, S. Liu, P. Gao, M. Han, Q. Liu, G. A. Solan and W.-H. Sun, J. Catal., 2023, 418, 40–50 CrossRef CAS; (b) Y. Su, Z. Ma, J. Wang, L. Li, X. Yan, N. Ma, Q. Liu, G. A. Solan and Z. Wang, J. Org. Chem., 2024, 89, 12318–12325 CrossRef CAS; (c) Z. Wang, S. Zhang, Z. Ma, L. Li, X. Yan, Q. Cao, Y. Su, N. Ma and Z. Wang, Mol. Catal., 2024, 564, 114274 CrossRef CAS; (d) S. Zhang, Z. Ma, Y. Li, Y. Su, N. Ma, X. Guo, L. Li, Q. Liu and Z. Wang, J. Catal., 2024, 437, 115682 CrossRef CAS.
- Y.-B. Wan and X.-P. Hu, ACS Catal., 2024, 14, 17633–17641 CrossRef CAS.
- (a) G.-Y. Zhang, S.-H. Ruan, Y.-Y. Li and J.-X. Gao, Chin. Chem. Lett., 2021, 32, 1415–1418 CrossRef CAS; (b) Y. Zhang, B. Li, T. Wang, N. Duan, J. Zheng, H. Li, F. Zhang and X. Fang, Dalton Trans., 2024, 53, 16475–16479 RSC.
- (a) J.-X. Gao, T. Ikariya and R. Noyori, Organometallics, 1996, 15, 1087–1089 CrossRef CAS; (b) H. Zhang, C. B. Yang, Y. Y. Li, Z. R. Donga, J. X. Gao, H. Nakamura, K. Murata and T. Ikariya, Chem. Commun., 2003, 1, 142–143 RSC.
- (a) W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1080–1083 CrossRef CAS; (b) Y. Li, S. Yu, X. Wu, J. Xiao, W. Shen, Z. Dong and J. Gao, J. Am. Chem. Soc., 2014, 136, 4031–4039 CrossRef CAS; (c) R. Bigler, R. Huber and A. Mezzetti, Angew. Chem., Int. Ed., 2015, 54, 5171–5174 CrossRef CAS; (d) K. Z. Demmans, C. S. G. Seo, A. J. Lough and R. H. Morris, Chem. Sci., 2017, 8, 6531–6541 RSC; (e) Q. Xue, R. Wu, D. Wang, M. Zhu and W. Zuo, Organometallics, 2020, 40, 134–147 CrossRef; (f) R. T. Endean, L. Rasu and S. H. Bergens, ACS Catal., 2019, 9, 6111–6117 CrossRef CAS.
- (a) Z. R. Dong, Y. Y. Li, S. L. Yu, G. S. Sun and J. X. Gao, Chin. Chem. Lett., 2012, 23, 533 CrossRef CAS; (b) Z. Wang, S.-L. Yu, Z.-B. Wei, D.-L. An, Y.-Y. Li and J.-X. Gao, J. Organomet. Chem., 2019, 898, 120882 CrossRef; (c) H. Chen, Z. Wang, M. Li and W. Zuo, ACS Catal., 2023, 13, 4261 CrossRef CAS.
- (a) M. Li, Z. Wang, H. Chen, Q. Huang and W. Zuo, Chem, 2024, 10, 250–264 CrossRef CAS; (b) S.-H. Ruan, Z.-W. Fan, W.-J. Zhang, H. Xu, D.-L. An, Z.-B. Wei, R.-M. Yuan, J.-X. Gao and Y.-Y. Li, J. Catal., 2023, 418, 100–109 CrossRef CAS.
- (a) W. Li, J.-H. Xie, M.-L. Yuan and Q.-L. Zhou, Green Chem., 2014, 16, 4081–4085 RSC; (b) X. Tan, Y. Wang, Y. Liu, F. Wang, L. Shi, K. H. Lee, Z. Lin, H. Lv and X. Zhang, Org. Lett., 2015, 17, 454–457 CrossRef CAS; (c) U. Sharma, N. Kumar, P. K. Verma, V. Kumar and B. Singh, Green Chem., 2012, 14, 2289–2293 RSC; (d) P. K. Verma, U. Sharma, N. Kumar, M. Bala, V. Kumar and B. Singh, Catal. Lett., 2012, 142, 907–913 CrossRef CAS; (e) P. K. Verma and S. D. Sawant, Coord. Chem. Rev., 2022, 450, 214239 CrossRef CAS.
- (a) Z. Wang, Q. Lin, N. Ma, S. Liu, M. Han, X. Yan, Q. Liu, G. A. Solan and W.-H. Sun, Catal. Sci. Technol., 2021, 11, 8026–8036 RSC; (b) Z. Wang, N. Ma, X. Lu, M. Liu, T. Liu, Q. Liu, G. A. Solan and W.-H. Sun, Dalton Trans., 2023, 52, 10574–10583 RSC; (c) D. Fu, Z. Wang, M. Liu, S. Liu, Y. Wang, C. Wei, Y. Ma, Q. Liu, G. A. Solan and W.-H. Sun, J. Catal., 2024, 436, 115601 CrossRef CAS; (d) Z. Wang, X. Lu, Z. Li, L. Li, Z. Ma, N. Ma, X. Yan, X. Liu, P. Han and Q. Liu, J. Catal., 2024, 430, 115337 CrossRef CAS.
- X.-Q. Zhang, Y.-Y. Li, Z.-R. Dong, W.-Y. Shen, Z.-B. Cheng and J.-X. Gao, J. Mol. Catal. A:Chem., 2009, 307, 149–153 CrossRef CAS.
- (a) M. Cettolin, P. Puylaert, L. Pignataro, S. Hinze, C. Gennari and J. G. de Vries, ChemCatChem, 2017, 9, 3125–3130 CrossRef CAS; (b) Y. Zhang, E. Chong, J. A. H. White, S. Radomkit, Y. Xu, S. C. Kosnik and J. C. Lorenz, Synlett, 2022, 33, 1287–1289 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, spectra (NMR, FT-IR), Fig. S1–S27, Chart S1, Tables S1–S8 and X-ray crystallographic data in CIF for CCDC 2408177 (Mn1). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra03062e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |