
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct conversion of methane to value-added hydrocarbons using alkali metal-promoted cobalt catalysts†
Sarannuch Sringamab,
Punyanut Thansiriphata,
Thongthai Witoon ab,
Waleeporn Donphaia,
Metta Chareonpanichab,
Chularat Wattanakitc,
Hiesang Sohnd,
Nevzat Yigite,
Günther Rupprechtere and
Anusorn Seubsai*ab
ab,
Waleeporn Donphaia,
Metta Chareonpanichab,
Chularat Wattanakitc,
Hiesang Sohnd,
Nevzat Yigite,
Günther Rupprechtere and
Anusorn Seubsai*ab
aDepartment of Chemical Engineering, Faculty of Engineering, Kasetsart University, Bangkok 10900, Thailand. E-mail: fengasn@ku.ac.th
bCenter of Excellence on Petrochemical and Materials Technology, Kasetsart University, Bangkok 10900, Thailand
cSchool of Energy Science and Engineering, Vidyasirimedhi Institute of Science and Technology, Rayong 21210, Thailand
dDepartment of Chemical Engineering, Kwangwoon University, Seoul, 01897, South Korea
eInstitute of Materials Chemistry, Technische Universität Wien, Vienna 1060, Austria
First published on 7th July 2025
Abstract
The oxidative coupling of methane (OCM) is a promising pathway for directly converting methane into higher hydrocarbons (C2+). This research investigated the influence of alkali metal promoters (Li, Na, K, or Rb) on Co/Al2O3 catalysts prepared based on incipient wetness impregnation for the OCM reaction. The catalyst investigations demonstrated that the catalysts promoted with K and Rb had superior performance, with the 4.6K–Co/Al2O3 catalyst achieving a maximum C2+ yield of 8.1%, C2+ selectivity of 24.0%, and CH4 conversion of 32.1% at 640 °C. Catalyst characterization, based on XRD, HR-TEM, BET, XPS, CO2-TPD, and H2-TPR analyses, revealed the structural and physicochemical properties responsible for the enhanced catalytic activity. Specifically, K and Rb promoters increased surface basicity and enhanced the electron density of active sites, thereby promoting selective methane activation. In-situ DRIFTS and mechanistic studies highlighted the role of reactive oxygen species in promoting C2+ hydrocarbon formation. These results should position K–Co/Al2O3 as a promising catalyst for OCM and provide valuable guidance for designing more efficient catalytic systems for methane utilization.
1. Introduction
Methane (CH4) and carbon dioxide (CO2) are the most potent greenhouse gases driving global warming and climate change.1 Methane, in particular, has a global warming potential approximately 25 times greater than CO2 over a 100 year period, making it a critical target for emission reduction and alternative utilization strategies.2 Despite its environmental impact, methane is also a valuable raw material for producing more complex and economically essential compounds. Efficient conversion of methane into higher-value chemicals can provide a dual benefit of mitigating climate impact and creating valuable products.3Methane conversion can proceed through two primary pathways: indirect and direct.4 Indirect routes involve a two-step process, where methane is first reformed (via steam reforming, dry reforming, or partial oxidation) to produce syngas, a mixture of hydrogen (H2) and carbon monoxide (CO). Then, these syngas can be transformed into valuable chemicals, such as olefins and fuels, through processes such as Fischer–Tropsch synthesis. However, the indirect pathway is energy-intensive and requires multiple stages, driving interest toward more efficient direct conversion approaches.5
Direct methane conversion aims to simplify the process by producing valuable chemicals in a single step. Such methods include partial oxidation to formaldehyde and methanol or converting methane to higher hydrocarbons (C2+), such as ethylene (C2H4), ethane (C2H6), propylene (C3H6), propane (C3H8), and butanes (C4H10), via oxidative coupling of methane (OCM)6 or non-oxidative coupling of methane (NOCM). Although the NOCM process offers a promising route to convert methane without oxygen, it involves considerable thermodynamic challenges and requires high energy input, limiting its industrial viability.7,8 Consequently, OCM has attracted substantial attention as a feasible pathway for directly converting methane to C2+. In the OCM process, methane reacts with molecular oxygen at high temperatures (above 700 °C) to produce these valuable compounds and byproducts, including water, hydrogen, carbon monoxide, and carbon dioxide.9
Early research on OCM explored a range of catalysts, including pure oxides of rare earth, alkaline earth, and transition metals.10 However, the focus has shifted towards more sophisticated catalyst formulations to enhance methane conversion and selectivity toward higher hydrocarbons. Among the most extensively studied catalysts is Na2WO4–Mn/SiO2,11,12 which, despite its promising activity, has not been commercialized due to low C2+ yield and selectivity, coupled with issues of catalyst deactivation during prolonged operation.13,14 Challenges, such as sintering, phase changes, and coking, continue to limit the catalyst's industrial applicability, emphasizing the need for innovations that enhance performance and long-term stability.6
In 2023, we introduced a novel hybrid catalyst system for the direct conversion of CH4 to C2+, combining 15 wt% Ni supported on Al2O3 (15Ni/Al2O3) and 20 wt% Co supported on Al2O3 doped with 4.6 wt% K (4.6K–20Co/Al2O3). Operating at relatively low temperatures (490 °C), this catalyst demonstrated impressive results, achieving C2+ yields of 3.6–4.3%, with selectivity ranging from 7.9% to 15.8% and CH4 conversion rates between 27.2% and 46.3%.15,16 When compared to the individual catalysts under identical conditions, the hybrid catalyst outperformed both, showcasing the synergistic effect of combining Ni and K-promoted Co catalysts. Notably, the 4.6K–20Co/Al2O3 catalyst produced exceptionally high catalytic activity for methane conversion to C2+, whereas the unpromoted 20Co/Al2O3 catalyst was essentially inactive, yielding 0% C2+ products. This highlighted the crucial role of K as a promoter in facilitating the direct activation of CH4—a finding that warranted further investigation.
The selection of appropriate promoters is crucial for addressing the limitations of traditional OCM catalysts. When integrated into the catalyst, alkali metals function as modifiers that enhance the surface basicity.17 This modulation of catalytic properties can redirect reaction pathways, promoting the formation of higher hydrocarbons (C2+).18 However, comprehensive studies that have systematically compared the effects of various alkali metals on cobalt-based catalysts have not been explored. In refining catalyst designs, it is crucial to understand how different alkali metal promoters influence the catalyst structure, activity, and selectivity.
Given that K belongs to the alkali metals group, which also includes lithium (Li), sodium (Na), rubidium (Rb), and cesium (Cs), it raises the intriguing possibility that other alkali metals may produce similar effects when used as promoters. Therefore, in this study, we explored the influence of various viable alkali metals (Li, Na, K, and Rb) on the performance of 20Co/Al2O3 catalysts in the OCM reaction. We systematically investigated how these promoters impacted catalytic activity, product selectivity, and CH4 conversion. In addition, we examined the effect of metal loading on optimizing the OCM process. Various advanced characterization techniques were applied to understand the relationships between the physical and chemical properties of the catalysts and their performance, offering insights into the design of more efficient and stable catalysts for direct methane conversion.
2. Experimental
2.1 Catalyst preparation
All the catalysts were prepared using the incipient wetness impregnation method. The Co/Al2O3 catalyst was promoted with different weights of four alkali metals: Li, Na, K, and Rb. Several metal nitrates were used as precursors, consisting of LiNO3 (99.99%, Sigma-Aldrich), NaNO3 (99.5%, Alfa Aesar), RbNO3 (99%, Alfa Aesar), KNO3 (99%, Alfa Aesar), and Co (NO3)2·6H2O (99%, Alfa Aesar). The support used for all catalysts was γ-Al2O3 (denoted as Al2O3, with a surface area of 75.32 m2 g−1, 99.97%, Alfa Aesar). Each metal precursor was dissolved in deionized water as a stock solution in the first step. Then, each solution was dropped onto Al2O3. Each mixture was stirred for 1 hour at room temperature before heating and continuously stirring at 90 °C until dry. Each dried sample was ground and calcined at 400 °C for 1 hour at a heating rate of 10 °C min−1. The weight percentage of Co in all catalysts was fixed at 20, while the weight percentage of each promoter was in the range 0.1–10.0, with the balance comprising the weight percentage of the Al2O3 support. For example, one catalyst was denoted as 4.6K–Co/Al2O3, representing 4.6wt% K, 20wt% Co, and 75.4wt% Al2O3. Thus, there were five catalyst groups studied: Li–Co/Al2O3, Na–Co/Al2O3, K–Co/Al2O3, Rb–Co/Al2O3, and Co/Al2O3.2.2 Catalyst characterization
The crystalline structure of each sample was identified using X-ray diffractometry (XRD; Rigaku Smart Lab XE, 9 kW), using Cu-Kα radiation at 40 kV and 100 mA, a step size of 0.01°, a scan speed of 3° min−1, and a 2θ range of 10–80°.The morphology of the samples was observed using high-resolution transmission electron microscopy (HR-TEM; JEM-ARM200F) and a high-angle annular dark-field (HAADF) scanning transmission electron microscopy (TEM) and energy dispersive X-ray spectrometry (EDS) (JEM-ARM200F). The operating voltage for the TEM was 200 kV. Before analysis, each sample was prepared by dispersing it in an ethanol solution for 30 min and dropping it onto a copper TEM grid. Then, it was dried in a chamber filled with nitrogen at room temperature.
The surface area, pore volume, and pore size of each catalyst was determined using a nitrogen-physisorption analyzer (3Flex Physisorption Micrometrics). Before measurement, each sample was degassed overnight at 200 °C to remove moisture and other adsorbed molecules. The Brunauer–Emmett–Teller model was used to calculate the surface area, while the Barrett–Joyner–Halenda model was used to calculate the pore size distribution.
The binding energy of Co in each catalyst was analyzed using X-ray photoelectron spectroscopy (XPS; Kratos Model Axis ultra DLD), with a monochromator (Al Kα) as the X-ray source and beam current of 10 mA, with a voltage of 15 kV. The spectra of Co 2p were collected at a pass energy of 40 eV in steps of 0.1 eV. All spectra were calibrated using the C1s signal of the carbon support material at 284.6 eV.
The surface basicity of the catalyst was analyzed using temperature-programmed desorption of carbon dioxide (CO2-TPD) using an AutoChem II 2920 (Micromeritics). Each sample (200 mg) was contained in a quartz U-tube and heated to 400 °C under a flow of helium (He) gas for 30 min and cooled to 200 °C. Subsequently, a flow of 10% CO2 in He gas was applied for 60 min and then purged with He gas until the baseline was stable. Next, it was heated again from 200 to 800 °C (heating rate of 10 °C min−1), and the CO2 desorbed was detected using a thermal conductivity detector (TCD).
The reducibility of the catalysts was analyzed using temperature-programmed reduction of hydrogen (H2-TPR) using an AutoChem II 2920 (Micromeritics). Each sample (200 mg) was contained in a quartz U-tube and heated to 150 °C under a flow of argon (Ar) gas for 30 min and cooled to 50 °C. When the baseline was stable, a flow of 10% H2 in Ar gas was applied, and the temperature was increased to 1000 °C (heating rate of 5 °C min−1). The quantity of H2 consumption was detected using a TCD.
Thermogravimetry/differential thermal analysis (TG/DTA, PerkinElmer TGA 8000) was performed under atmospheric pressure. Prior to analysis, the samples were dried at 80 °C overnight to eliminate residual moisture. TG/DTA measurements were conducted over a temperature range of 30–800 °C, using a heating rate of 5 °C min−1 and N2 flow rate of 50 mL min−1.
A single-beam infrared spectrometer (Bruker VERTEX 70v FT-IR) coupled with a wide-band mercury-cadmium-telluride detector and a liquid-nitrogen-cooled system was used to perform in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). The catalyst (20 mg) was placed inside a stainless-steel flow cell oven with a CaF2 window. The catalyst was pretreated at 400 °C under Ar gas with a flow rate of 40 mL min−1 for 1 hour, followed by cooling to room temperature. Subsequently, the gas was converted to a CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2:N2 mixture gas with a 2:1:4 ratio and a total flow rate of 50 mL min−1, and the temperature was increased to 490 °C at a heating rate of 10 °C min−1, with the spectrum being recorded at 1 min intervals for 30 min. Each spectrum was collected based on 128 scans at a resolution of 4 cm−1 over a spectral range of 900–4000 cm−1.
:O2:N2 mixture gas with a 2:1:4 ratio and a total flow rate of 50 mL min−1, and the temperature was increased to 490 °C at a heating rate of 10 °C min−1, with the spectrum being recorded at 1 min intervals for 30 min. Each spectrum was collected based on 128 scans at a resolution of 4 cm−1 over a spectral range of 900–4000 cm−1.
2.3 Catalyst activity testing
Each catalyst (40 mg) was packed between quartz wool in a quartz tube with a diameter of 0.5 cm in a plug flow reactor. The reactant gases, consisting of CH4 (99.999%, Labgaz) and O2 (99.999%, Linde), with a CH4:O2 ratio of 2 and a total flow rate of 40 mL min−1, were fed to the quartz tube at atmospheric pressure and a reaction temperature of 440−740 °C. The feed gases were controlled using mass flow controllers (Aalborg GFC17). The effluent gases were analyzed using an online gas chromatograph (GC-14A; Shimadzu) equipped with a flame ionization detector to evaluate C2H4, C2H6, C3H6, C3H8, C4H8, and C4H10 and a TCD was used to assess CO, CO2, and CH4. A standard calibration curve of five calibration points was established for each gas, with an R-squared value exceeding 0.995. This enabled accurate quantification of the mole of effluent gas. The activity of each catalyst was expressed as %CH4 conversion, %C2+ selectivity, and %C2+ yield, as shown in eqn (1)–(3), respectively.
 | (1) |
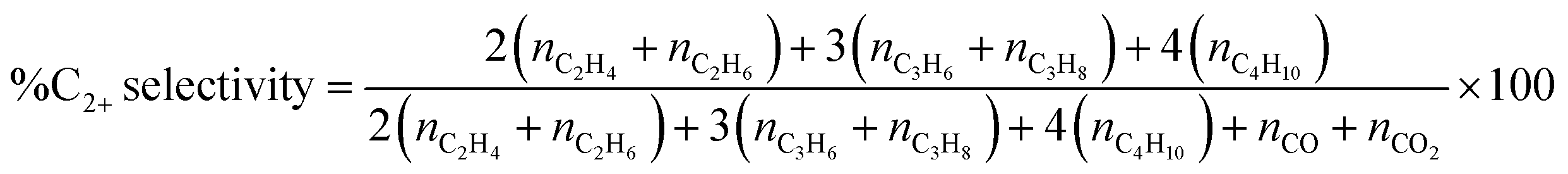 | (2) |
 | (3) |
Each catalyst testing study was conducted at least three times, and the results were repeatable within 10%. The data were presented as average values except for the catalyst stability test. Furthermore, the catalytic performance data contained less than 5% carbon balance errors.
3. Results and discussion
3.1 Performance of catalysts
The catalytic performance levels of the 20 wt% Co/Al2O3 catalysts, both unpromoted and promoted with 4.6 wt% of alkali metals (Li, Na, K, and Rb), were evaluated in a plug flow reactor under reaction conditions of 490 °C and atmospheric pressure. As depicted in Fig. 1, incorporating the different alkali promoters led to considerable variations in catalytic activity, allowing the catalysts to be categorized into two distinct groups based on their performance. Group I comprised 4.6K–Co/Al2O3 and 4.6Rb–Co/Al2O3, which had superior catalytic activity, while Group II included 4.6Li–Co/Al2O3, 4.6Na–Co/Al2O3, and the unpromoted Co/Al2O3, all of which had comparatively lower performance. | ||
| Fig. 1 Catalytic performance of Co/Al2O3 catalysts with different promoters for OCM reaction. Reaction conditions: CH4:O2 ratio = 2:1, catalyst weight = 40 mg, total feed flow rate = 40 mL min−1, reactor temperature = 490 °C. | ||
The catalysts in Group I achieved C2+ hydrocarbon yields in the range 3.4–6.5%, with C2+ selectivity in the range 15.2–22.3% and CH4 conversion rates in the range 22.8–29.5%. These results highlighted the enhanced catalytic behavior when K or Rb was used as a promoter. In contrast, the Group II catalysts produced negligible C2+ yields (0%) and minimal C2+ selectivity (0–0.1%), with CH4 conversion rates limited to 0.7–1.7%. This stark difference from Group 1 underscored the effectiveness of the K and Rb promoters in enhancing the activity of Co/Al2O3 catalysts compared to Li, Na, or no promoter at all.
Based on the results, it was clear that the 4.6K–Co/Al2O3 and 4.6Rb–Co/Al2O3 catalysts outperformed their counterparts, making them the most promising candidates for further investigation. Consequently, these two catalysts were selected for detailed characterization, as described in Section 3.2, to elucidate the reasons behind their superior catalytic behavior. For comparative purposes, the unpromoted Co/Al2O3 catalyst was also characterized to provide a comprehensive analysis of the effects of alkali promotion.
3.2 Catalyst characteristics
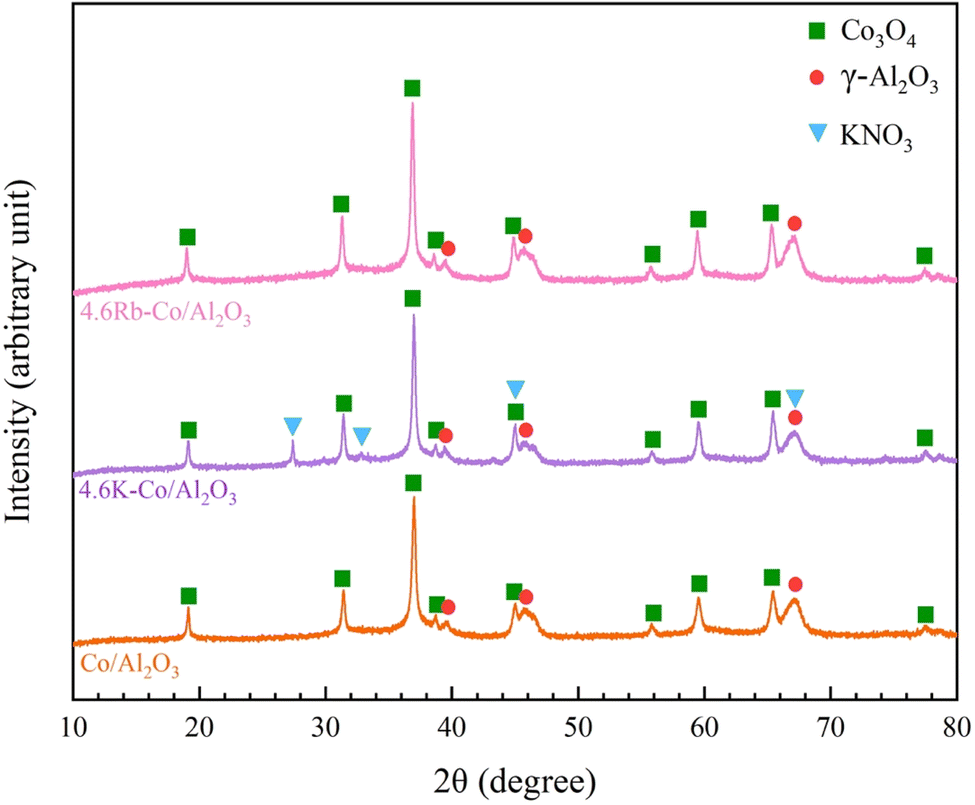 | ||
| Fig. 2 XRD patterns of Co/Al2O3, 4.6K–Co/Al2O3, and 4.6Rb–Co/Al2O3 catalysts. | ||
 | ||
| Fig. 3 HR–TEM images of (a and b) Co/Al2O3, (c and d) 4.6K–Co/Al2O3, and (e and f) 4.6Rb–Co/Al2O3. | ||
In Fig. 4, the HAADF images with EDS elemental mapping reveal a robust elemental distribution of Co, Al, O, K, and Rb. These elements were distributed across all catalysts. Notably, while the crystalline phase of Rb species was undetected in XRD, the HAADF-EDS images distinctly indicate the dispersion of Rb. The even elemental distribution—particularly of the oxygen species—is advantageous for catalytic efficiency in OCM processes.22
 | ||
| Fig. 4 Images of HAADF with EDS elemental mapping of (a) Co/Al2O3, (b) 4.6K–Co/Al2O3, and (c) 4.6Rb–Co/Al2O3. | ||
| Catalyst | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore diameter (nm) |
|---|---|---|---|
| Co/Al2O3 | 1.21, 59.18 | 0.0001, 0.34 | 3.71, 47.90 |
| 4.6K–Co/Al2O3 | 27.80 | 0.27 | 40.46 |
| 4.6Rb–Co/Al2O3 | 4.90, 41.50 | 0.002,0.32 | 3.49, 44.89 |
 | ||
| Fig. 5 Pore size distribution of Co/Al2O3, 4.6K–Co/Al2O3, and 4.6Rb–Co/Al2O3 catalysts. | ||
 | ||
| Fig. 6 N2 adsorption–desorption isotherms of Co/Al2O3, 4.6K–Co/Al2O3, and 4.6Rb–Co/Al2O3 catalysts. | ||
 | ||
| Fig. 7 XPS spectra of Co/Al2O3, 4.6K–Co/Al2O3, and 4.6Rb–Co/Al2O3 catalysts. | ||
For the 4.6K–Co/Al2O3 and 4.6Rb–Co/Al2O3 catalysts, there were shifts to lower binding energy values for both Co 2p3/2 and Co 2p1/2 compared to the catalyst without the dopant. The binding energy of each catalyst is summarized in Table S2.† These shifts to lower binding energies in the XPS spectra suggested an increase in electron density around the active sites, which promoted selective methane activation while reducing the likelihood of complete oxidation to CO and CO2.27 Consequently, the 4.6K–Co/Al2O3 catalyst showed better selectivity for C2 hydrocarbons than the 4.6Rb–Co/Al2O3 catalyst.
 | ||
| Fig. 8 CO2-TPD profiles of Co/Al2O3, 4.6K–Co/Al2O3, and 4.6Rb–Co/Al2O3 catalysts. | ||
| Catalyst | Basicity amount (μmol g−1) | |
|---|---|---|
| Moderate basic sites (200–540 °C) | Strong basic sites (540–800 °C) | |
| Co/Al2O3 | 0.83 | 0.63 |
| 4.6K–Co/Al2O3 | 1.37 | 1.22 |
| 4.6Rb–Co/Al2O3 | 0.98 | 0.72 |
 | ||
| Fig. 9 H2-TPR profiles of Co/Al2O3, 4.6K–Co/Al2O3, and 4.6Rb–Co/Al2O3 catalysts. | ||
3.3 Optimal promoter weight percentage
According to Section 3.1, the Co/Al2O3 catalyst doped with K and Rb had high activity for the OCM reaction. This section describes the testing of different loadings (0.1, 0.5, 1.0, 2.0, 4.6, 6.0, 8.0, and 10.0 wt%) of these two promoters on the catalyst for the OCM reaction at atmospheric pressure and the reaction temperature of 490 °C. The activity results are shown in Fig. 10. The C2+ formation could be seen for the catalysts doped with K (0–0.5wt%) and Rb (0–2.0wt%). Then, the levels of catalytic performance (C2+ yield, C2+ selectivity, and CH4 conversion) increased with an increasing percentage of promoters because the promoters formed active sites essential for methane activation and the ensuing coupling processes.33 The highest performance percentages were 6.5% C2+ yields, 22.3% C2+ selectivity, and 29.5% CH4 conversion for the 4.6K–Co/Al2O3 catalyst and 5.7% C2+ yield, 21.7% C2+ selectivity, and 26.3% CH4 conversion for the 8Rb–Co/Al2O3 catalyst. However, excessive promoter loading resulted in aggregation or inadequate dispersion of active sites, reducing the effective surface area available for the reaction and potentially decreasing catalytic activity.34 These testing results indicated that the most effective catalyst was the 4.6K–Co/Al2O3 catalyst. | ||
| Fig. 10 Catalytic performance of (a) K–Co/Al2O3 catalyst and (b) Rb–Co/Al2O3 with varying weight percentages of promoter for OCM reaction. Reaction conditions: CH4:O2 ratio = 2:1, catalyst weight = 40 mg, total feed flow rate = 40 mL min−1, reactor temperature = 490 °C. | ||
3.4 Optimal reaction temperature
To ensure a fair comparison based on the alkali metal content, the two catalysts were reformulated to possess equivalent molar amounts of potassium and rubidium. This adjustment resulted in catalysts with revised weight loadings: 4.6K–Co/Al2O3 (i.e., 2.05 molar of K on Co/Al2O3) and 10Rb–Co/Al2O3 (i.e., 2.05 molar of Rb on Co/Al2O3). The impact of reaction temperature on catalytic performance was examined for both 4.6K–Co/Al2O3 and 10Rb–Co/Al2O3 catalysts over a temperature range of 440–740 °C, as shown in Fig. 11. The two catalysts exhibited similar performance. At low reaction temperatures (440 °C), CH4 conversion was poor due to insufficient thermal energy to activate CH4 molecules and promote coupling reactions, resulting in a low C2+ yield and selectivity. Then, the catalytic performance progressively increased with temperature up to the optimum reaction temperature, which was 640 °C for the 4.6K–Co/Al2O3 catalyst and 690 °C for the 10Rb–Co/Al2O3 catalyst, after which a decline was observed. Notably, 4.6K–Co/Al2O3 consistently demonstrated higher catalytic activity than 10Rb–Co/Al2O3. At 640 °C, the optimal performance of 4.6K–Co/Al2O3 achieved a C2+ yield of 8.1% with 24.0% C2+ selectivity and 32.1% CH4 conversion, while the optimal performance of 10Rb–Co/Al2O3 occurred at 690 °C, resulting in a C2+ yield of 7.4% with 21.9% C2+ selectivity and 27.8% CH4 conversion. Above the optimum temperature, both catalysts exhibited decreased catalytic performance, likely due to the increased formation of CO and CO2 through the combustion of CH4 and C2+ hydrocarbons. In summary, K-doped Co/Al2O3 exhibited superior performance in C2+ hydrocarbon formation compared to the Rb-doped counterpart at the same molar loading. | ||
| Fig. 11 Catalytic performance of the 4.6K–Co/Al2O3 and 10Rb–Co/Al2O3 catalyst with varying reaction temperatures for OCM reaction. Reaction conditions: CH4:O2 ratio = 2:1, catalyst weight = 40 mg, total feed flow rate = 40 mL min−1, reactor temperature = 440–740 °C. | ||
3.5 Catalytic stability of the 4.6K–Co/Al2O3 catalyst for the OCM reaction
The long-term stability of the 4.6K–Co/Al2O3 catalyst was assessed under continuous operation at 640 °C over a 24 h period, as illustrated in Fig. 12. At the beginning of the time-on-stream testing, the C2+ yield was 8.1%, with C2+ selectivity of 24.0% and a corresponding CH4 conversion of 32.1%. As the reaction proceeded, a gradual change in performance was observed, followed by a relatively steady. After 24 h of testing, the catalyst maintained a C2+ yield of 8.2%, C2+ selectivity of 23.6%, and CH4 conversion of 33.3%. According to these findings, the 4.6K–Co/Al2O3 catalyst shows excellent durability under reaction conditions, maintaining most of its initial activity over time. | ||
| Fig. 12 The time-on-stream performance of the 4.6K–Co/Al2O3 catalyst over 24 h. Reaction conditions: CH4:O2 ratio = 2:1, catalyst weight = 40 mg, total feed flow rate = 40 mL min−1, reactor temperature = 640 °C. | ||
The XRD analysis of the spent 4.6K–Co/Al2O3 catalyst, as shown in Fig. S2.† Peaks corresponding to Co3O4 and KNO3 disappeared, while new reflections attributed to the CoAl2O4 spinel phase emerged prominently. This phase transformation likely occurred due to strong interactions between Co species and the Al2O3 support under high reaction temperature conditions (640 °C) and long operation time. The analysis shown in Fig. S3† was conducted to investigate potential coke formation on the catalyst after 24 h of use. A minor signal below 100 °C is likely due to moisture evaporation. Typically, coke formation is detected by TG–DTA analysis between 200 and 600 °C.35 However, in this case, no such signal was observed across that range, indicating that coke did not accumulate on the catalyst surface. Although XRD patterns revealed the presence of the CoAl2O4 phase in the spent catalyst, this had minimal influence on its behavior. Overall, the TG–DTA results confirmed the absence of coke, demonstrating that the catalyst maintained excellent performance under the tested conditions.
3.6 In situ DRIFTS analysis of 4.6K–Co/Al2O3 catalyst
In the OCM reaction, the electrophilic oxygen species, including the peroxide (O22−) and superoxide (O2−) anions, play a critical role in enhancing CH4 conversion and promoting C2 selectivity during the OCM reaction.36 The in situ DRIFTS analysis of the 4.6K–Co/Al2O3 catalyst, presented in Fig. 13, revealed a peak at 1014 cm−1, attributed to surface O2− species.30 The peaks at 1307 and 3012 cm−1 correspond to the presence of CH4 in the gas phase, while the peak at 1356 cm−1 is associated with bidentate carbonate species (CO32−).37 Notably, no new surface carbonate species were detected after 30 min of reaction feed exposure. This suggested that the surface O2− species was regenerated by the O2 present in the reaction feed. Additionally, a peak at 967 cm−1 was observed, signifying the formation of C2H4 on the catalyst surface under reaction conditions.38 Furthermore, the catalyst had a peak at 2390 cm−1, characteristic of adsorbed CO2, and a peak at 1756 cm−1, attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency, indicating CO formation during the OCM reaction.37 A peak detected at 3630 cm−1 was assigned to the formation of O–H bonds. This suggests that a hydrogen atom from CH4 was chemisorbed onto reactive oxygen sites on the catalyst surface through the formation of O–H bonds.39 The results indicated that methane activation occurred through interaction with the active oxygen species on the 4.6K–Co/Al2O3 catalyst. This interpretation was consistent with other studies that identified oxygen as the active site for methane activation in gas–solid phase reactions.40
O stretching frequency, indicating CO formation during the OCM reaction.37 A peak detected at 3630 cm−1 was assigned to the formation of O–H bonds. This suggests that a hydrogen atom from CH4 was chemisorbed onto reactive oxygen sites on the catalyst surface through the formation of O–H bonds.39 The results indicated that methane activation occurred through interaction with the active oxygen species on the 4.6K–Co/Al2O3 catalyst. This interpretation was consistent with other studies that identified oxygen as the active site for methane activation in gas–solid phase reactions.40
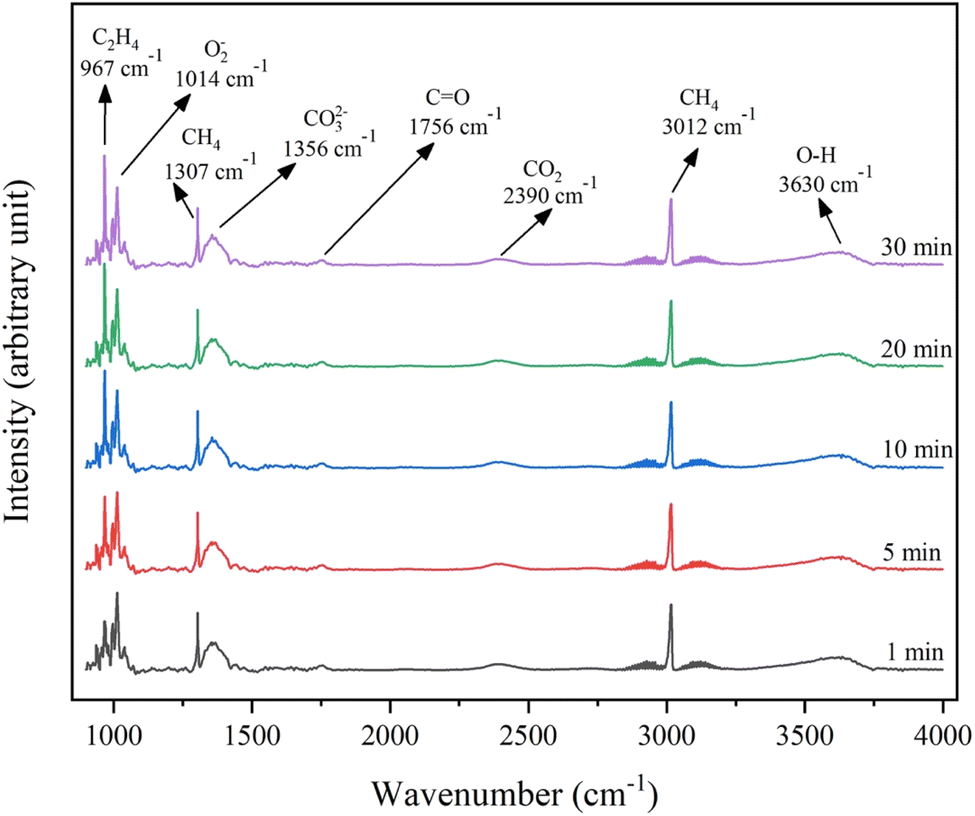 | ||
| Fig. 13 In-situ DRIFTS spectra of the 4.6K–Co/Al2O3 catalyst. | ||
3.7 Proposed mechanism of the 4.6K–Co/Al2O3 catalyst for the OCM reaction
The analysis of the in situ DRIFTS results in Fig. 13, combined with insights from other studies on catalysts used in the OCM reaction, provided essential information for understanding the catalytic mechanism of the 4.6K–Co/Al2O3 catalyst. Initially, molecular O2 dissociated on the catalyst surface, producing O2− species, which appeared at 1014 cm−1 in Fig. 13. It is also noted that the O2− band's constant intensity indicated that the consumption and regeneration of O2− proceed at a sufficiently rapid rate to achieve equilibrium at the reaction temperature.36 Then, the O2− species extracted hydrogen from CH4, forming methyl radicals (˙CH3) and surface hydroxyl groups (—OH),39 which was confirmed by the in situ DRIFTS peak at 3630 cm−1. The methyl radicals combined in the gas phase to form C2H6, which can subsequently produce C2H4, via dehydrogenation processes.41 The synthesis of C2H4 on the catalyst surface was indicated by the peak at 967 cm−1, with the concentration increasing with reaction time. The adsorbed –OH species may desorb from the surface as either ˙OH or ˙H radicals, which can further react to form H2O. In addition, uncoupled radicals and hydrocarbons may undergo additional oxidation, forming CO and CO2,42 which appeared at 1756 and 2390 cm−1, respectively.However, a comparison between the mechanistic pathway proposed in this study and that of previously reported hybrid systems—specifically, the dual-layer catalyst comprising 5Ni/Al2O3 as the first layer and 4.6K–Co/Al2O3 as the second—reveals a fundamental distinction: in the earlier system, the reaction initiates as the reactant gases (CH4 and O2) pass through the first layer of the hybrid catalyst system, a portion of CH4 is transformed into CO, CO2, and H2 via the partial oxidation of methane (POM) reaction. These products, particularly CO and H2, serve as intermediate species for the subsequent Fischer–Tropsch synthesis occurring over the second catalyst layer. In the Fischer–Tropsch mechanism, which follows a chain-growth polymerization model, syngas components (CO and H2) undergo surface dissociation into C, O, and H atoms during the initiation phase. A surface-bound C atom subsequently reacts with H atoms to generate CH2 monomers, which then polymerize through successive coupling steps, ultimately leading to the formation of longer-chain hydrocarbons.
The 4.6K–Co/Al2O3 catalyst has several key components, each contributing to the catalytic process. The active components of this catalyst include K, Co, and Al2O3. The K component serves as a promoter, enhancing the number of basic sites on the catalyst that are essential for forming C2+ hydrocarbons.43 Cobalt oxides, particularly Co3O4 with a spinel structure, are highly efficient at methane adsorption. Additionally, cobalt-based catalysts have major activity and selectivity in producing long-chain hydrocarbons.44 Primarily, Al2O3 serves as a support material for the catalyst, owing to its advantageous properties, including a high surface area and well-distributed pore sizes, which facilitate superior metal dispersion and enhance catalyst stability.45 In the current study, these factors likely contributed to the catalyst's high activity and exceptional performance at relatively low temperatures during the OCM reaction.
3.8 Comparative performance of 4.6K–Co/Al2O3 with other catalysts
Several catalysts investigated previously—particularly those comprising Na2WO4 in combination with Mn—have been recognized for their superior reactivity in the OCM process. To evaluate the performance of the optimized K–Co/Al2O3 catalyst developed in this study, a comparative analysis was conducted against selected representatives from this category, as illustrated in Fig. 14 and detailed in Table S3.† Reported performance for Na2WO4–Mn catalysts varies widely, with C2+ yields ranging from 0.2% to 31.6%, C2+ selectivities between 4.0% and 80.2%, and CH4 conversion 1.0% to 45.4% at reaction temperatures of 650–850 °C. However, achieving both high conversion and high selectivity concurrently remains challenging. For commercial viability, a benchmark of at least 30% CH4 conversion and 80% C2+ selectivities is typically required (as highlighted by the gray zone in Fig. 14). While a few catalysts exceed one of these thresholds, simultaneous attainment is rarely observed, indicating limitations in current materials. Notably, the K–Co/Al2O3 catalyst presented in this work attained 32.1% CH4 conversion with 24.0% selectivity and 8.1%C2+ yield products at 640 °C, which lower than the operational temperatures of many high-performing systems. These findings underscore the importance of developing next-generation catalysts that can deliver high selectivity (>80%) while maintaining efficient CH4 conversion (>30%), which is a critical direction for future innovation in OCM catalysis. | ||
| Fig. 14 Comparison of the catalyst developed in this study with other catalysts previously reported for the OCM reaction. | ||
4. Conclusion
This study demonstrates the considerable influence of alkali metal promotion on the catalytic performance of Co/Al2O3 for the oxidative coupling of methane (OCM), a process crucial for sustainable methane utilization. Among the investigated alkali metal promoters (Li, Na, K, and Rb), K-promoted catalysts produced the most pronounced enhancement, with the optimized 4.6K–Co/Al2O3 catalyst achieving 8.1% C2+ yield, 24.0% C2+ selectivity, and 32.1% CH4 conversion at 640 °C. Characterization revealed that K increased the surface basicity and modified the electronic environment of active sites, facilitating selective methane activation and suppressing complete oxidation to CO and CO2. The mechanistic investigations, supported by in situ DRIFTS analysis, demonstrated that molecular O2 dissociated on the catalyst, generating O2− species that extracted hydrogen from CH4 to generate surface –OH groups and ˙CH3, which subsequently recombined to produce C2H6 and dehydrogenated into C2H4. While Rb demonstrated potential, Li and Na had comparatively lower efficacy, emphasizing the importance of promoter selection in optimizing catalytic performance. This research established that alkali metal-promoted Co/Al2O3, particularly the K-promoted variant, was a promising candidate for low-temperature OCM applications. These findings should provide valuable insights into the design of efficient, selective, and stable catalysts for the valorization of methane. Future research should focus on improving the properties of the catalyst and enhancing its performance to realize the full industrial potential of these systems.Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.Author contributions
Sarannuch Sringam: Writing – original draft, writing – review & editing, methodology, investigation, validation, formal analysis, data curation. Punyanut Thansiriphat: Methodology, investigation. Thongthai Witoon: Methodology, investigation conceptualization. Waleeporn Donphai: Methodology, investigation. Metta Chareonpanich: Resources, conceptualization. Chularat Wattanakit: Resources, methodology. Hiesang Sohn: Resources, review & editing. Nevzat Yigit: Resources, methodology. Günther Rupprechter: Resources, conceptualization. Anusorn Seubsai: Writing – original draft, writing – review & editing, methodology, investigation, validation, formal analysis, data curation, supervision, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Sringam, S. acknowledges the Center of Excellence on Petrochemical and Materials Technology for a PhD scholarship. This work was financially supported by the National Research Council of Thailand through the Fundamental Fund under the Kasetsart University Research and Development Institute (KURDI), Bangkok, Thailand, through Grant No. FF(KU)38.67; and the National Research Council of Thailand through the Research Team Promotion Grant/Senior Research Scholar (Grant No. N42A640324). Rupprechter, G. acknowledges support from the Austrian Science Fund (FWF; SFB TACO F81-P08).References
- C. Wei, M. Wang, Q. Fu, C. Dai, R. Huang and Q. Bao, Atmos. Res., 2020, 235, 104759 CrossRef CAS.
- C. Weng, X. Peng and Y. Han, Adv. Appl. Microbiol., 2023, 124, 119–146 CAS.
- N. Sun, J. Zhang, L. Ling, R. Zhang, L. Jia, D. Li and B. Wang, Appl. Catal., 2023, 650, 118998 CrossRef CAS.
- H. Zhang, Y. Su, N. Kosinov and E. J. M. Hensen, Chin. J. Catal., 2023, 49, 68–80 CrossRef CAS.
- Y. Wang, X. Yang, F. Yin, K. Zhang, H. Guo, G. Wang, G. Jiang, C. Li and X. Zhu, J. Energy Chem., 2022, 73, 49–59 CrossRef CAS.
- C. A. Ortiz-Bravo, C. A. Chagas and F. S. Toniolo, J. Nat. Gas Sci. Eng., 2021, 96, 104254 CrossRef CAS.
- A. D. Talpade, G. Canning, J. Zhuchen, J. Arvay, J. Watt, J. T. Miller, A. Datye and F. H. Ribeiro, Chem. Eng. J., 2024, 481, 148675 CrossRef CAS.
- Z. Guo, W. Chen, Y. Song, X. Dong, G. Li, W. Wei and Y. Sun, Chin. J. Catal., 2020, 41, 1067–1072 Search PubMed.
- Y. Yu, K. Obata, W. J. Movick, S. Yoshida, J. Palomo, S. T. B. Lundin, A. Urakawa, S. M. Sarathy and K. Takanabe, J. Catal., 2024, 432, 115414 CrossRef CAS.
- E. G. Maulidanti, M. Awaji and K. Asami, Gas Sci. Eng., 2023, 116, 205057 CrossRef CAS.
- L. Xu, A. Zanina, K. Wu, J. Li, J. Chen, Y. Li, G. Jiang and E. V. Kondratenko, Chem. Eng. J., 2023, 473, 145372 CrossRef CAS.
- K. Zhao, J. Huang, Z. Huang, Y. Lin, M. Zheng, D. Song, A. Liu, X. Wang, A. Zheng and Z. Zhao, J. Energy Inst., 2022, 105, 273–281 CrossRef CAS.
- Y. Wang, W. Kong, Y. Fu, L. Zheng, B. Pan, H. Zhu, S. Li, J. Li, J. Zhang and Y. Sun, J. Environ. Chem. Eng., 2024, 12, 112757 CrossRef CAS.
- A. Zanina, D. Makhmutov and E. V. Kondratenko, Catal. Today, 2024, 439, 114829 CrossRef CAS.
- P. Somchuea, T. Sukprom, S. Sringam, S. Ampansang, T. Witoon, M. Chareonpanich, K. Faungnawakij, G. Rupprechter and A. Seubsai, Top. Catal., 2023, 66, 1553–1568 Search PubMed.
- T. Sukprom, P. Somchuea, S. Sringam, T. Witoon, M. Chareonpanich, P. Iamprasertkun, K. Faungnawakij, G. Rupprechter and A. Seubsai, React. Chem. Eng., 2023, 8, 1868–1881 RSC.
- J. Deng, P. Chen, S. Xia, M. Zheng, D. Song, Y. Lin, A. Liu, X. Wang, K. Zhao and A. Zheng, Atmosphere, 2023, 14, 1538 CrossRef CAS.
- P. Kidamorn, W. Tiyatha, T. Chukeaw, C. Niamnuy, M. Chareonpanich, H. Sohn and A. Seubsai, ACS Omega, 2020, 5, 13612–13620 CrossRef CAS PubMed.
- B. Małecka, A. Łącz, E. Drożdż and A. Małecki, J. Therm. Anal. Calorim., 2015, 119, 1053–1061 CrossRef.
- H. Zhao, H. Song, F. Wang, Z. Miao and L. Chou, Mol. Catal., 2020, 495, 111141 CrossRef CAS.
- S. Alkhursani, N. Aldaleeli, A. Elbasiony, M. Ghobashy, S. Al-Gahtany and A. Sharshir, Polym. Bull., 2024, 81, 15841–15864 CrossRef CAS.
- J. Liu, J. Yue, M. Lv, F. Wang, Y. Cui, Z. Zhang and G. Xu, Carbon Resour. Convers., 2022, 5, 1–14 CrossRef CAS.
- L. Xu, J. Zhang, J. Ding, T. Liu, G. Shi, X. Li, W. Dang, Y. Cheng and R. Guo, Minerals, 2020, 10, 72 CrossRef CAS.
- J. Yang, F. Wei, Y. Sui, J. Qi, Y. He, Q. Meng and S. Zhang, RSC Adv., 2016, 6, 61803–61808 RSC.
- J. Acharya, B. G. S. Raj, T. H. Ko, M. S. Khil, H. Y. Kim and B. S. Kim, Int. J. Hydrogen Energy, 2020, 45, 3073–3085 CrossRef CAS.
- Q. Zhang, L. Pastor-Pérez, W. Jin, S. Gu and T. R. Reina, Appl. Catal., B, 2019, 244, 889–898 CrossRef CAS.
- Z. Wei, W. J. Movick, K. Obata, S. Yoshida, D. Asada, T. Ikeda, A. Nakayama and K. Takanabe, J. Phys. Chem. C, 2024, 128, 12969–12977 CrossRef CAS.
- G. I. Siakavelas, N. D. Charisiou, A. AlKhoori, V. Sebastian, S. J. Hinder, M. A. Baker, I. V. Yentekakis, K. Polychronopoulou and M. A. Goula, J. Environ. Chem. Eng., 2022, 10, 107259 CrossRef CAS.
- S. Sringam, T. Witoon, C. Wattanakit, W. Donphai, M. Chareonpanich, G. Rupprechter and A. Seubsai, Carbon Resour. Convers., 2024, 100261 Search PubMed.
- T. Wu, Y. Wei, J. Xiong, Y. Yang, Z. Wang, D. Han, Z. Zhao and J. Liu, J. Energy Chem., 2024, 91, 331–344 CrossRef CAS.
- N. F. Khairudin, M. Mohammadi and A. R. Mohamed, Environ. Sci. Pollut. Res., 2021, 28, 29157–29176 CrossRef CAS PubMed.
- H. Lin, Y. Liu, J. Deng, L. Jing and H. Dai, Catalysts, 2023, 13, 427 CrossRef CAS.
- J. Hao, F. Cai, J. Wang, Y. Fu, J. Zhang and Y. Sun, Chem. Phys. Lett., 2021, 771, 138562 CrossRef CAS.
- A. H. K. Owgi, A. A. Jalil, M. A. A. Aziz, M. Alhassan, H. U. Hambali, W. Nabgan, R. Saravanan and A. H. Hatta, Fuel, 2023, 340, 127592 CrossRef CAS.
- S. Ampansang, S. Sringam, P. Somchuea, T. Witoon, C. Wattanakit, M. Chareonpanich, H. Sohn and A. Seubsai, Top. Catal., 2024, 67, 394–408 CrossRef CAS.
- J. Xu, Y. Zhang, X. Xu, X. Fang, R. Xi, Y. Liu, R. Zheng and X. Wang, ACS Catal., 2019, 9, 4030–4045 CrossRef CAS.
- R. Singh Pal, S. Rana, S. Kumar Sharma, R. Khatun, D. Khurana, T. Suvra Khan, M. Kumar Poddar, R. Sharma and R. Bal, Chem. Eng. J., 2023, 458, 141379 CrossRef CAS.
- Y. Zhang, J. Xu, X. Xu, R. Xi, Y. Liu, X. Fang and X. Wang, Catal. Today, 2019, 355, 518–528 CrossRef.
- T. Shen, Z. Wang, S.-M. Xu, X. Sun, G. Liu, S. Bai, J. Li, Z. Song, L. Zheng and Y.-F. Song, Cell Rep. Phys. Sci., 2023, 4, 101478 CrossRef CAS.
- Y. Wang, B. Wang, S. Sourav, R. Batchu, Z. Fang, M. R. Kunz, G. Yablonsky, E. Nikolla and R. Fushimi, Catal. Today, 2023, 417, 113739 CrossRef CAS.
- D. Kiani, S. Sourav, J. Baltrusaitis and I. E. Wachs, ACS Catal., 2019, 9, 5912–5928 CrossRef CAS.
- T. Chukeaw, W. Tiyatha, K. Jaroenpanon, T. Witoon, P. Kongkachuichay, M. Chareonpanich, K. Faungnawakij, N. Yigit, G. Rupprechter and A. Seubsai, Process Saf. Environ. Prot., 2021, 148, 1110–1122 CrossRef CAS.
- Y. Xie, W. Wang, J. Cui, H. Li, K. Cheng, Q. Zhang and Y. Wang, Chem. Eng. Sci., 2024, 294, 120119 CrossRef CAS.
- S. Guo, Z. Ma, J. Wang, B. Hou, L. Jia, B. Wang and D. Li, Fuel, 2021, 292, 120398 CrossRef CAS.
- S. Khan, S. S. Shah, N. K. Janjua, A. B. Yurtcan, M. T. Nazir, K. M. Katubi and N. S. Alsaiari, Chemosphere, 2023, 315, 137659 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02408k |
| This journal is © The Royal Society of Chemistry 2025 |