
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure-based design, synthesis, computational screening and biological evaluation of novel pyrrole fused pyrimidine derivatives targeting InhA enzyme†
Deepshikha Singh a,
Praveen M. Parkalia,
Umme Hanib,
Riyaz Ali M. Osmanic,
Nazima Haiderd,
Jyothi Kumarie,
Dharmarajan Srirame,
Christian Lherbetf,
B. C. Revan Siddappag and
Sheshagiri R. Dixit*a
a,
Praveen M. Parkalia,
Umme Hanib,
Riyaz Ali M. Osmanic,
Nazima Haiderd,
Jyothi Kumarie,
Dharmarajan Srirame,
Christian Lherbetf,
B. C. Revan Siddappag and
Sheshagiri R. Dixit*a
aDepartment of Pharmaceutical Chemistry, JSS College of Pharmacy, JSS Academy of Higher Education and Research, Sri Shivarathreeshwara Nagar, Mysuru, Karnataka 570015, India. E-mail: sheshagiridixit@jssuni.edu.in; Tel: +91 721107207
bDepartment of Pharmaceutics, College of Pharmacy, King Khalid University, Guraiger, Abha, 62529, Saudi Arabia
cDepartment of Pharmaceutics, College of Pharmacy, King Khalid University, Al-Faraa, Abha, 61421, Saudi Arabia
dDepartment of Pathology, College of Medicine, King Khalid University, Guraiger, Abha, 62529, Saudi Arabia
eDepartment of Pharmacy, Birla Institute of Technology and Science-Pilani, Hyderabad Campus, Jawahar Nagar, Hyderabad, Telangana 500 078, India
fUniversité de Toulouse, CNRS, Laboratoire de Synthèse et Physico-Chimie de Molécules d'Intérêt Biologique (LSCPMIB), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France
gDepartment of Pharmaceutical Chemistry, NGSM Institute of Pharmaceutical Sciences (NGSMIPS), Nitte (Deemed to be University), Mangalore, Karnataka 575018, India
First published on 21st July 2025
Abstract
In this study, a series of 12 novel pyrrolyl chalcones and 22 pyrrole-fused pyrimidine derivatives were synthesized with good yields. Structural characterization was performed using FT-IR, NMR, and mass spectrometry techniques. The antitubercular potential of these compounds was evaluated using the microplate alamar blue assay (MABA). Among the synthesized compounds, compound 4g exhibited the highest potency, with a minimum inhibitory concentration (MIC) of 0.78 mg mL−1 demonstrating greater efficacy than the standard drug isoniazid. Several other analogues also showed moderate to good inhibitory activity. Selected compounds were further assessed for cytotoxicity using human lung cancer (A549) and normal RAW cell lines, revealing low toxicity profiles. Enzymatic assays indicated that compound 4g achieved 36% inhibition of InhA at a concentration of 50 μM. Additionally, molecular dynamics simulations were conducted to analyze the stability of the protein–ligand complexes, suggesting that these compounds hold potential for future development as InhA inhibitors in the fight against MDR-TB.
1. Introduction
Tuberculosis (TB) remains a leading cause of infectious disease-related mortality globally, with Mycobacterium tuberculosis infecting millions annually.1 In 2023 alone, over 10.8 million new TB cases and approximately 1.25 million related deaths were reported, surpassing even COVID-19 in prevalence.2 The growing emergence of multidrug-resistant (MDR-TB) and extensively drug-resistant (XDR-TB) strains has significantly hindered global eradication efforts.3 The World Health Organization (WHO) has set ambitious targets to reduce TB mortality by 95% by 2035.4 A critical barrier to effective TB management is the resistance of M. tuberculosis to first-line drugs such as isoniazid (INH), rifampicin, ethambutol, and pyrazinamide.5 INH, a prodrug, requires activation by the KatG enzyme to inhibit enoyl-ACP reductase (InhA) depicted in (Fig. 1), a key enzyme in the fatty acid synthase-II (FAS-II) system essential for mycolic acid biosynthesis and bacterial survival.6 However, mutations in KatG lead to resistance by preventing the activation of INH.7 This has spurred interest in developing direct InhA inhibitors that bypass KatG and target the enzyme more selectively.8,9 | ||
| Fig. 1 Isoniazid-induced inhibition of mycolic acid synthesis. | ||
Recent studies have explored various chemical scaffolds as InhA inhibitors, including antimycobacterial drugs such as triclosan, diphenyl ether derivatives, arylamide, pyrrolidine carboxamide analogues, 4-hydroxy-2-pyridones, chalcones, triazoles, quinolines, pyrroles, and pyrimidines.8,10–14 Chalcones, synthesized through aldol condensation, which is a precursor for the synthesis of many other biological compounds, is the end product of the process itself, which combines substituted aryl ketones with aromatic aldehydes15 and it have shown promising pharmacological properties such as anti-bacterial,16 anti-inflammatory,17 anti-cancer,18 osteoporosis activity,19anti-fungal,20 anti-malarial,21 anti-viral,22 anti-allergic,23 and estrogenic effects.24 Pyrimidines, another biologically active class, exhibit broad antimicrobial,25 anti-inflammatory,26 as anti-tubercular27,28 antibacterial agent & an anti-cancer agent.29 Kaur et al. (2020) was investigated potential effectiveness of using pyrimidine derivatives as antitubercular drugs by several research organizations.30 The maximum active compounds were examined used for their inhibition growth of M. tuberculosis.31 According to literature survey we explore chalcones and pyrimidine derivatives found potential inhibitors against InhA. The combination of these motifs offers a promising strategy for novel therapeutic design.
Inha inhibitors are primarily used to treat tuberculosis due to their ability to improve and inhibit the tuberculocidal growth, but some studies have also suggested pyrimidines may have potential benefits in TB. In the framework of such research efforts, a pyrrole ring in the head region that is responsible for the activation. Our recent study aimed to design novel InhA inhibitors by replacing the aromatic ring structure with a pyrrole ring in the head region and aromatic trunk region for activation. Pyrimidine cyclization with aldehyde substitutions as electron withdrawing and donating group. With the understanding of the structural features of under clinical trial InhA inhibitors, such as acidic head, aromatic region with pyrrole ring, and lipophilic tail with pyrimidine ring and aromatic aldehyde substitutions a newer fragment focused library of molecules was designed (Fig. 2).
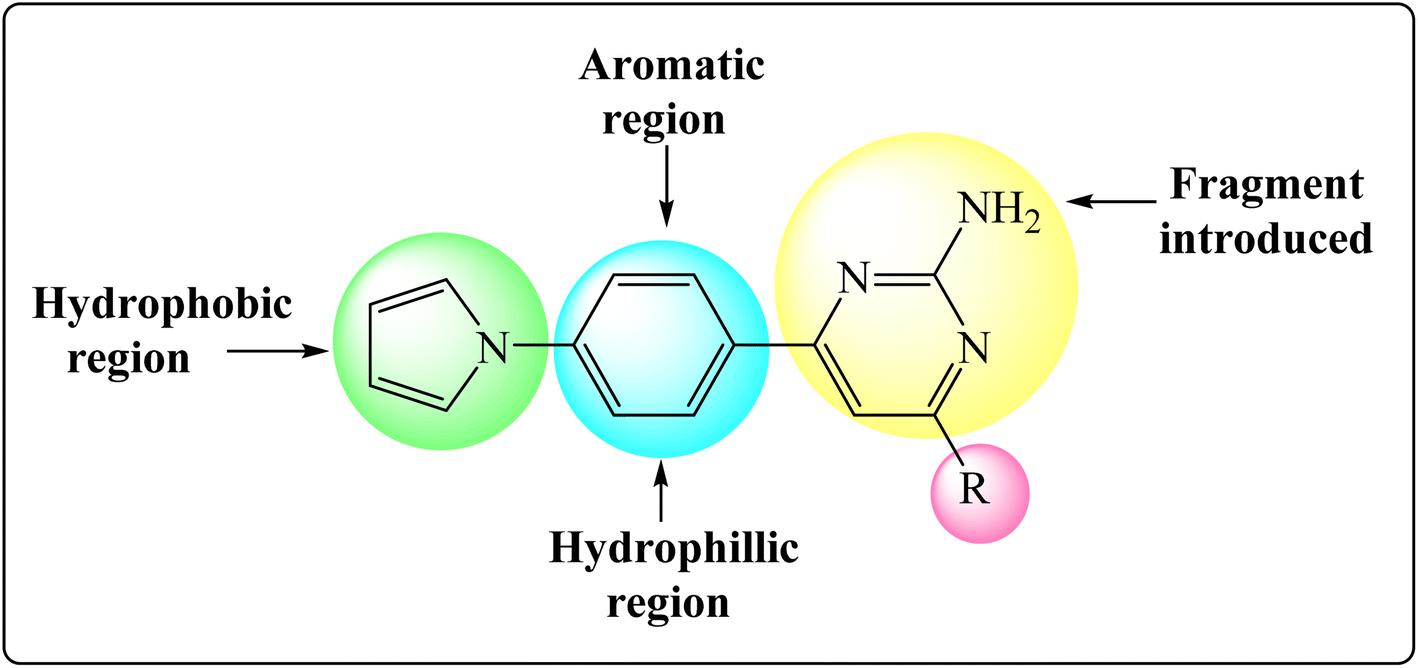 | ||
| Fig. 2 Designed InhA inhibitors based on the structural features of InhA inhibitors. | ||
Pyrrole, a heterocyclic compound present in biomolecules like chlorophyll and heme, has also been widely studied for its antimicrobial activity. Its derivatives have demonstrated antitubercular efficacy both in vitro and in vivo, and compounds like (Sudoterb)LL3858 have progressed into preclinical development for TB.32
Based on these findings, the fusion of pyrrole, chalcone, and pyrimidine frameworks presents an attractive route for drug development.33 Furthermore, several pyrimidine-containing compounds such as GSK2556286 and TBA-7371 are currently undergoing clinical trials for TB treatment depicted in (Fig. 3), showing efficacy without cross-resistance to current therapies. Similarly, SPR720, an oral antibiotic targeting bacterial DNA replication, is in clinical testing for mycobacterial infections.34
 | ||
| Fig. 3 Pyrimidine based anti-tb drugs candidate under clinical trials. | ||
In our study, we report the design, synthesis, and biological evaluation of a series of novel pyrrolyl chalcones and pyrimidine-fused pyrroles. Their antitubercular potential was assessed through in vitro assays, cytotoxicity studies, enzyme inhibition activity, and molecular docking and dynamics simulations, with the aim of identifying effective and safe inhibitors of InhA.
2. Results and discussion
2.1 Rationale of molecular design
The novelty of this work highlights in presenting a pioneering approach through the design and synthesis of a new class of pyrrole-fused pyrimidine derivatives as potential inhibitors of the enoyl-ACP reductase (InhA) enzyme, a validated drug target involved in the biosynthesis of mycolic acids essential for bacterial survival, by development of a unique chemical scaffold, pyrrole-fused pyrimidines remain largely unexplored in the context of InhA inhibition. The rational design of these hybrid molecules introduces structural diversity and expands the chemical space for anti-tubercular drug discovery. Employing structure-based drug design strategies, including molecular docking and molecular dynamics simulations, the study effectively validates the binding affinity and potential inhibitory mechanism of the compounds against the InhA enzyme.352.2 ADME prediction and drug-likeness evaluation
The selected compounds were evaluated for their pharmacokinetic properties, including absorption, distribution, metabolism, and excretion (ADME), using the QikProp module of Maestro Schrodinger. This analysis was essential to assess their drug-likeness and biological significance. The results of the ADME prediction are summarized in (Table 1). All compounds displayed pharmacokinetic descriptors within the acceptable ranges for drug-like molecules, showing favourable comparisons with the standard anti-TB drug, isoniazid. Interestingly, isoniazid itself exhibited notable deviations in certain parameters, including molecular weight, drug-likeness, and predicted human oral absorption, when compared to the new compounds. Despite these deviations, all test compounds were predicted to possess favourable ADME profiles. The analysis suggests that further structural optimization (e.g., derivatization) could lead to improvements in key parameters such as molecular weight, the number of hydrogen bond acceptors, predicted biological responses, blood–brain partition coefficient (QPlog, BB), and percent human oral absorption.36 Among the tested compounds, 4c and 4u demonstrated the highest enhancement in drug-likeness, as reflected in their QikProp #star values, outperforming 4g in terms of overall pharmacokinetic desirability. Therefore, after complete analysis of the structural features of available InhA inhibitors were designed, synthesized, analysed, and utilized in further in vitro studies. These results indicate their potential for further development as promising drug candidates against tuberculosis.| S. no | Compound | #Stars | MW | Dipole | SASA | Donor H bond | Acceptor H bond | QPlog P0/w | QPlog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) S S |
QPlogBB |
No. of metabolites | QPlogkhsa |
% Human oral absorption |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Key: #Stars: number of property or descriptor values that fall outside the 95% range of similar values for known drugs. A large number of stars suggests that a molecule is less drug-like than molecules with few stars. Dipole: computed dipole moment of the molecule. SASA: total solvent accessible surface area (SASA) in square angstroms using a probe with a 1.4 A radius. Donor H-bond: estimated number of hydrogen bonds that would be donated by the solute to water molecules in an aqueous solution. Acceptor H-bond: estimated number of hydrogen bonds that would be accepted by the solute from water molecules in an aqueous solution. QPlogPo/w: predicted octanol/water partition coefficient. QPlogS: predicted aqueous solubility, logS. QPlogkhsa: prediction of binding to human serum albumin, No. of metabolites: number of likely metabolic reactions. QPlogBB: predicted brain/blood partition coefficient. % Human oral absorption: predicted human oral absorption on 0 to 100% scale. |
|||||||||||||
| 1 | 3k | 1 | 315.41 | 0.00 | 658.09 | 0.000 | 2.000 | 5.890 | −6.585 | −0.242 | 1 | 1.172 | 100% |
| 2 | 3l | 1 | 279.35 | 0.00 | 569.92 | 0.000 | 2.000 | 4.920 | −6.827 | −0.019 | 1 | 0.676 | 100% |
| 3 | 4c | 0 | 372.42 | 0.00 | 372.42 | 2.000 | 2.000 | 4.858 | −6.471 | 0.661 | 2 | 0.812 | 100% |
| 4 | 4g | 1 | 342.39 | 0.00 | 651.07 | 2.000 | 2.000 | 4.685 | −6.105 | −0.581 | 1 | 0.768 | 100% |
| 5 | 4h | 2 | 381.26 | 0.00 | 651.45 | 2.000 | 2.000 | 5.469 | −7.047 | −0.194 | 0 | 0.958 | 100% |
| 6 | 4u | 0 | 358.39 | 0.00 | 660.53 | 2.000 | 2.000 | 4.006 | −5.670 | −1.052 | 2 | 0.551 | 100% |
| 7 | 4k | 1 | 354.45 | 0.00 | 700.42 | 2.000 | 2.000 | 5.556 | −7.231 | −0.615 | 1 | 1.158 | 100% |
| 8 | Isoniazid | 1 | 137.14 | 0.00 | 329.57 | 2.000 | 3.000 | −0.646 | −0.050 | −0.840 | 2 | −0.752 | 100% |
The synthesis of the compounds was initiated with the Paal–Knorr reaction of 4-aminoacetophenone and 2,5-dimethoxy tetrahydrofuran to yield (4-pyrrol-1-yl) acetophenone, which then underwent Claisen–Schmidt condensation with different substituted aldehydes to produce chalcones. These were subsequently cyclized with guanidine hydrochloride in sodium ethoxide and dry DMF to form pyrimidine derivatives. The final products were achieved after several rounds of refluxing, quenching, filtration, and column chromatographic purification.
This study focused on the development of novel pyrrole-based molecules, specifically 1-(4-(1H-pyrrol-1-yl)phenyl)-3-substituted prop-2-en-1-ones (Scheme 1) and 4-(4-(1H-pyrrol-1-yl)phenyl)-6-phenylpyrimidin-2-amines (Scheme 2), as potential antitubercular agents. Biological evaluation against Mycobacterium tuberculosis H37Rv revealed that several compounds, notably 3k, 3l, 4c, 4g, 4h, 4k, and 4u, demonstrated significant inhibitory effects. Among these, compound 4g stood out with a minimum inhibitory concentration (MIC) of 0.78 μg mL−1, indicating potent anti-TB activity. Cytotoxicity assessments using the A549 human lung cancer cell line showed that the most active compounds exhibited minimal toxicity, with cell viability remaining above 95% at tested concentrations. These results suggest that the synthesized compounds possess a favourable therapeutic index, supporting their potential for further pharmacological development.
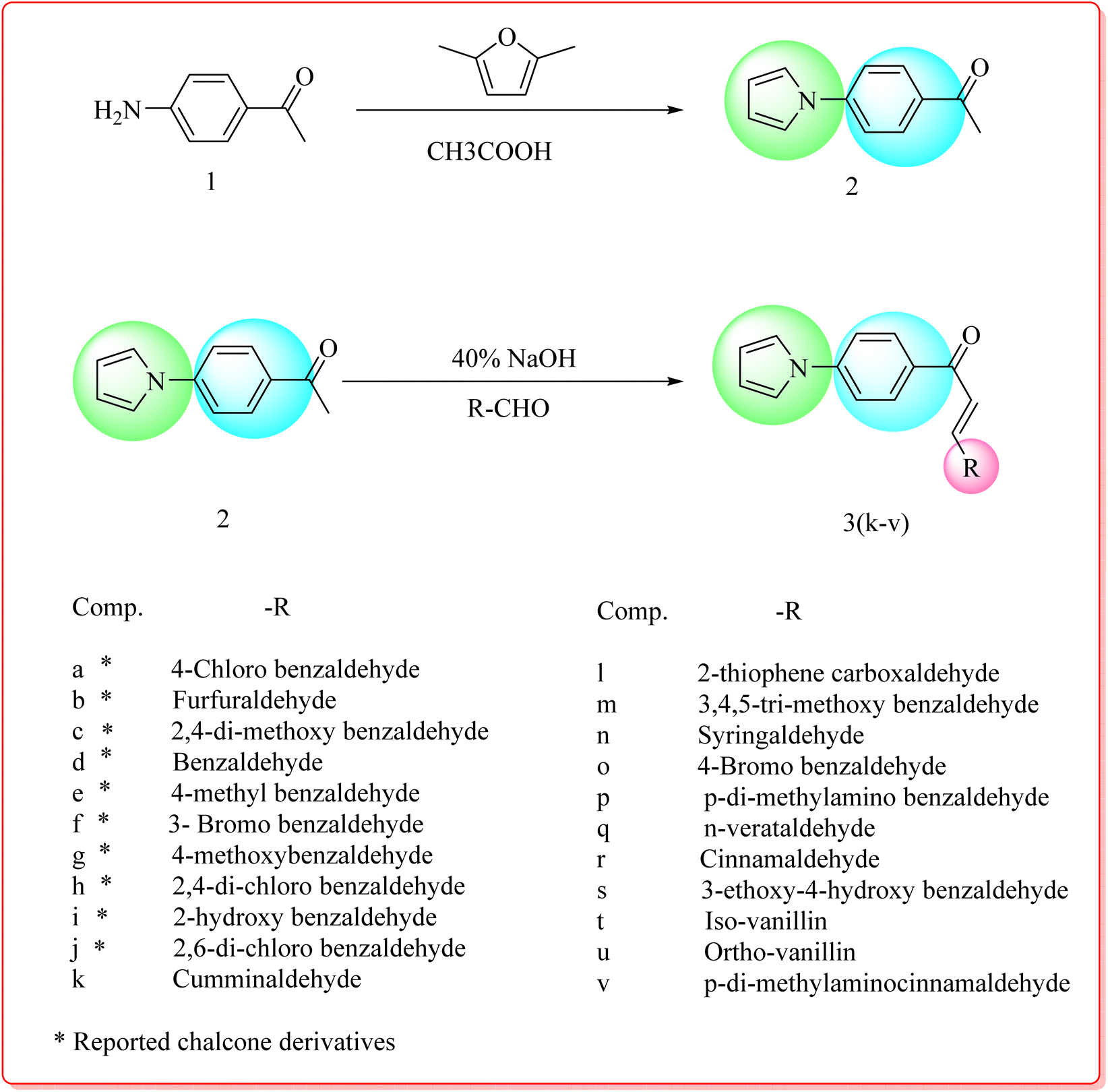 | ||
| Scheme 1 Synthetic route of a novel series of pyrrole chalcone derivatives. | ||
 | ||
| Scheme 2 Synthetic route of a novel series of pyrimidine derivatives. | ||
InhA enzyme inhibition assays further supported the antitubercular efficacy of these compounds, with the same set of molecules displaying notable inhibition. Computational docking studies revealed favourable binding conformations within the InhA active site, including key hydrogen bonds and hydrophobic interactions, which likely contribute to their biological activity. These interactions were consistent with those observed for the reference drug isoniazid, strengthening the hypothesis that these compounds act as direct InhA inhibitors. To further substantiate these findings, MM-GBSA (molecular mechanics/generalized born surface area) calculations were performed to estimate the binding free energies of the ligand–enzyme complexes.37 The calculated energies confirmed that the interactions were thermodynamically favourable, reinforcing the molecular docking results. Density functional theory (DFT) calculations were also carried out on the most promising compound, 4g, which revealed a narrow HOMO–LUMO energy gap. This indicates high electronic reactivity and supports its potential for biological interaction. Additionally, molecular dynamics simulations conducted over 200 ns provided insight into the stability of the 4g–InhA complex under physiological conditions. The complex remained stable throughout the simulation period, and analysis of the binding interactions identified key amino acid residues responsible for maintaining ligand–protein affinity. In silico ADMET predictions showed that the lead compounds adhered to Lipinski's rule of five and exhibited acceptable pharmacokinetic properties, suggesting good oral bioavailability and low toxicity risks. These findings highlight the drug-likeness of the synthesized molecules and their promise as lead scaffolds for further optimization.38–44
Overall, the combination of synthetic, biological, and computational approaches used in this study provides a solid foundation for the development of new antitubercular agents. The promising results, particularly for compound 4g, warrant further investigations aimed at enhancing efficacy and selectivity, as well as exploring their mechanism of action in greater detail. These efforts may ultimately contribute to the development of effective, low-toxicity treatments targeting drug-resistant tuberculosis.
2.3 Molecular docking
A molecular docking study was conducted on InhA enzyme. The incorporation of carbon atoms into the respective mycolic acid demonstrates the involvement of the type II fatty acid biosynthesis pathway. MAs are a class of compounds known as long-chain alpha-alkyl beta-hydroxy fatty acids, C74 to C90, and are the principal component of the mycobacterial cell wall which resists antibiotics and greatly increases mycobacterial virulency. Thus, the inhibition of InhA is considered one of the best ways to kill Mtb when it is both aerobic and anaerobic because of its lysis promoting action on cell walls, specifically on the mycolic acid that is crucial for the integrity of the mycobacterial cell wall.45The inhibitors attach to InhA and interact with the enzyme's lipophilic binding pockets, tyrosine amino acid, and cofactor (NADH). InhA's binding pocket contains three significant sites where inhibitors interact. The catalytic site I comprises the tyrosine amino acid and the NAD cofactor's ribose group. Hydrogen bonds are established between the ribose ring of NAD and the active hydroxyl group of Tyr by suppressing its chain reaction. The structure of the inhibitor can determine the formation of bonds with methionine. Site II, which is a hydrophobic pocket, can be found near the side chain of Tyr and allows for the binding of hydrophilic components of inhibitors. Site III has a limited amount of research, with inhibitor rings located near NAD's phosphate groups and Gly 96 and Phe 97 at approximately the van der Waals distance.46 These reported interactions of various ligands with the InhA enzyme show opportunities for designing new inhibitors that can attach to this area. In 2018, Prati et al. presented multiple crystal structures of new InhA NADH ligand complexes. One of them contains pyrimidine based co-crystallized ligand, which is obtained from the protein data bank (PDB ID: 5OIR),47check the interaction in between of protein & ligand by molecular docking study protocols. Docking results recommend that the developed compounds may serve as possible scaffolds for the development of anti-tubercular treatments. The glide score (kcal mol−1) and glide emodel score, along with the interactions involving amino acid residues such as hydrophobic, hydrogen, and pi–pi interactions-indicate strong binding interactions between the protein and the all ligands, and with isoniazid used as the standard drug showing in below (Table 2).
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S. no. | Compound id | –R | Glide score (kcal mol−1) | Glide emodel score | Amino acid residue and hydrophobic, hydrogen and pi–pi interaction | ||||||
| Hydrophobic | Hydrogen | Pi–pi interaction | Positive | Negative | Halogen bond | Polar | |||||
| 1 | 3k |  |
−7.794 | −53.064 | Met 98, Phe 97, Met 103, Phe 149, Met 155, Tyr 158, Met 161, Pro 193, Ala 198, Met 199, Ile 202, Ile 215, Leu 218, | — | — | — | — | — | |
| 2 | 3l |  |
−7.697 | −57.493 | Phe 97, Met 98, Met 103, Phe 149, Tyr 158, Met 161, Ala 198, Met 199, Pro 193, Ile 202, Ile 215, Leu 218, | Tyr 158 | Tyr 158 | Lys 165 | — | — | — |
| 3 | 4c |  |
−6.623 | −56.456 | Met 98, Phe 97, Met 103, Phe 149, Tyr 158, Ala 198, Pro 193, Met 199, Ile 215, Leu 218 | Gly 96 | Tyr 158 | — | — | Met 98 | |
| 4 | 4g |  |
−8.911 | −70.260 | Phe 97, met 98, pro-99, Met 103, Phe 149, Met 155, Tyr 158, Met 161, Pro 193, Ile 194, Ala 198, Met 199, Ile 202, Ile 215, Leu 218, | Gly 96 | — | — | — | — | Gln 100 |
| 5 | 4h |  |
−7.947 | −66.028 | Phe 97, Met 98, Pro 99, Met 103, Ala 198, Phe 149, Tyr 158, Ile 194, Pro 193, Ala 198, Met 199, Ile 202, Ile 215, Leu 218 | — | Tyr 158 | — | — | — | Gln 100 |
| 6 | 4k |  |
−8.470 | −63.035 | Phe 97, Met 98, Pro 99, Met 103, Met 103, Phe 149, Tyr 158, Pro 193, Ala 198, Met 199, Ile 202, Ile 215, Leu 218 | Gly 96 | Tyr 158 | — | — | — | Gln 100 |
| 7 | 4u |  |
−7.947 | −68.612 | Phe 97, Met 97, Met 98, Pro-99, Met 103, Phe 149, Tyr 158, Pro 193, Ala 198, Met 199, Ile 202, Ile 215, Leu 218 | Met 98 | Tyr 158 | — | — | — | Gln 100 |
| 8 | Standard drug | Isoniazid | −4.160 | −38.971 | Met 103, Phe 149, Tyr 158, Met 161, Ala 198, Met 199, Ile 202 | Tyr 158 | — | — | — | — | |

We docked all compounds into the active pocket of InhA and identified their binding methods to the enzyme in order to support experimental anti-tubercular data and InhA inhibition data using computational approaches. The enzyme's binding affinity amino acid residues that interact with the all compounds include in ESI.†
Detailed intermolecular interaction between the ligands and protein was studied using glide docking. All compounds showed better docking scores compared to standard drug. Compounds 4h and 4k exhibited showed H-bonding interactions with the amine group, amides present at 5th position of pyrimidine ring and with Gly96 amino acid residue. Mostly all compounds showed pi–pi stacking interaction with Tyr 158 amino acid residue and 4u compound showed the H-bonding interactions between OH group with Met 98 amino acid residue hydroxy group present at 2nd position of benzene ring, in compound 4l showing H-bonding interactions between carbonyl group in bridging phenyl ring with Tyr 158 amino acid residue and pi–pi stacking interaction with thiophene ring.
Here, based on the biological activity data we focus on compound 4g, which showed the most successful InhA inhibitors (Fig. 4). Displays the 2D, 3D image & spatial orientation and interaction patterns of compound 4g.
 | ||
| Fig. 4 (3D & 2D) interactions of compound 4g with active site residues of the target protein PDB ID-5OIR. | ||
2.4 MMGBSA
Here, the study was finalized to discover the free binding energies of the protein and ligand complexes using the (Molecular Mechanics, Generalized Born model and Solvent Accessibility). The complexes had their docking score (the lowest score) among all calculated and their optimal binding energy through prime module of Maestro Schrodinger software. OPLS-AA force field with electrostatics, such as the OPLS-AA force field was used for the analysis, in which the Vsbg 2.0 model employed a contained solvent model, and physics-based terms for pi–pi, hydrophobic interactions, and hydrogen bonding self-contact interactions were included in the present study. Docking was performed with the protein being static and ligands as stretchy.37 The docked pose of ligand was rescored using MMGBSA process under prime module option in Schrodinger software showed in (Table 3).| S. no | Compound IDs | MMGBSA-dG-binding energy | MMGBSA-dG-bind in coulomb | MMGBSA-dG-bind Co-valent | MMGBSA-dG-bind H bond |
|---|---|---|---|---|---|
| 1 | 3k | −38.05 | −1.33 | 9.75 | −0.00 |
| 2 | 3l | −54.86 | −18.17 | 5.25 | −0.71 |
| 3 | 4c | −47.47 | −12.94 | 6.36 | −2.01 |
| 4 | 4g | −44.23 | −7.99 | 9.85 | −1.63 |
| 5 | 4h | −49.14 | −7.81 | 7.34 | −1.64 |
| 6 | 4k | −52.78 | −14.78 | 10.77 | −1.76 |
| 7 | 4u | −49.16 | −9.46 | 6.44 | −2.87 |
| 8 | Isoniazid | −36.17 | −33.93 | 0.15 | −1.41 |
The docking complexes were carried out using MM-GBSA calculation for the energy minimization of the protein–ligand complexes (Ecomplex), the free protein (Eprotein), and the free ligands (Eligand). The binding free energy ΔGbind was determined according to the following equation:
| DGbind = Ecomplex(minimized) − Eligand(minimized) − Ereceptor(minimized) |
3. Chemistry and synthesis
All compounds were synthesized as per steps outlined in Scheme 1 depicted in (Fig. 4). Condensation of 4-amino acetophenone (1) with 2,5-dimethoxytetrahydrofuran produced (4-pyrrol-1-yl) acetophenone (2) via the Paal–Knorr reaction. By using a sodium hydroxide catalyst to speed up the Claisen Schmidt condensation of (4-pyrrol-1-yl) acetophenone (2) with the substituted aldehydes in ethanol, the necessary critical intermediates, namely, chalcones (3a–3v), were produced. Chalcones (3a–3v) were cyclized to pyrimidine by reacting with Guanidine hydrochloride (0.007 mol) in the presence of sodium ethoxide (0.02 mol) and dried DMF (15 mL) as a solvent as depicted in Scheme 2 depicted in (Fig. 5). Reaction mixture was refluxed at 80 °C for 30–35 h, and reaction monitored by TLC. The reaction mixture was quenched with crushed ice, yielding a ppt solid which was filtered off, washed with water, hydrated and purified by column-chromatography (2:1), ethyl acetate and n-hexane. FTIR, 1HNMR, 13CNMR, and mass spectroscopy were used to confirm the structures of all the synthesized compounds. IR spectra of compound 3k, peaks were observed at the carbonyl group from the α,β-unsaturated fragment was linked to an absorption band at 1657.65 cm−1 in the which is characteristic of the chalcone series (3a–3v). The presence of a methyl group is indicated by a singlet at d 1.23–1.22 ppm and doublets at d 7.96 and 8.26 ppm in compound 3k 1H NMR spectra. Two triplets were produced by the pyrrole moiety's protons at d 6.34 and 7.35 ppm, respectively. Compound 3k 13C NMR spectra revealed the signal at d 187.68 ppm. Every other aromatic carbon resonated within the anticipated d 111.56–143.98 ppm range. The synthesis of the needed product was confirmed by the molecular ion signal at m/z (M+ + 1) 316.15. The amino group and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching at 1676.37 cm−1 are represented by the aromatic peak at 3320.38 cm−1 in compound 4g IR spectra. Furthermore, two hydrogens were anticipated to be present at 5.25 (s, 2H) in the 1H NMR spectra, which is related to the aromatic NH2 group. Additionally, the mass spectrum supported this molecule by showing its molecular ion peak at m/z (M+ + 1) 343.14, which is in line with its molecular weight and validated the synthesis of the intended product.
N stretching at 1676.37 cm−1 are represented by the aromatic peak at 3320.38 cm−1 in compound 4g IR spectra. Furthermore, two hydrogens were anticipated to be present at 5.25 (s, 2H) in the 1H NMR spectra, which is related to the aromatic NH2 group. Additionally, the mass spectrum supported this molecule by showing its molecular ion peak at m/z (M+ + 1) 343.14, which is in line with its molecular weight and validated the synthesis of the intended product.
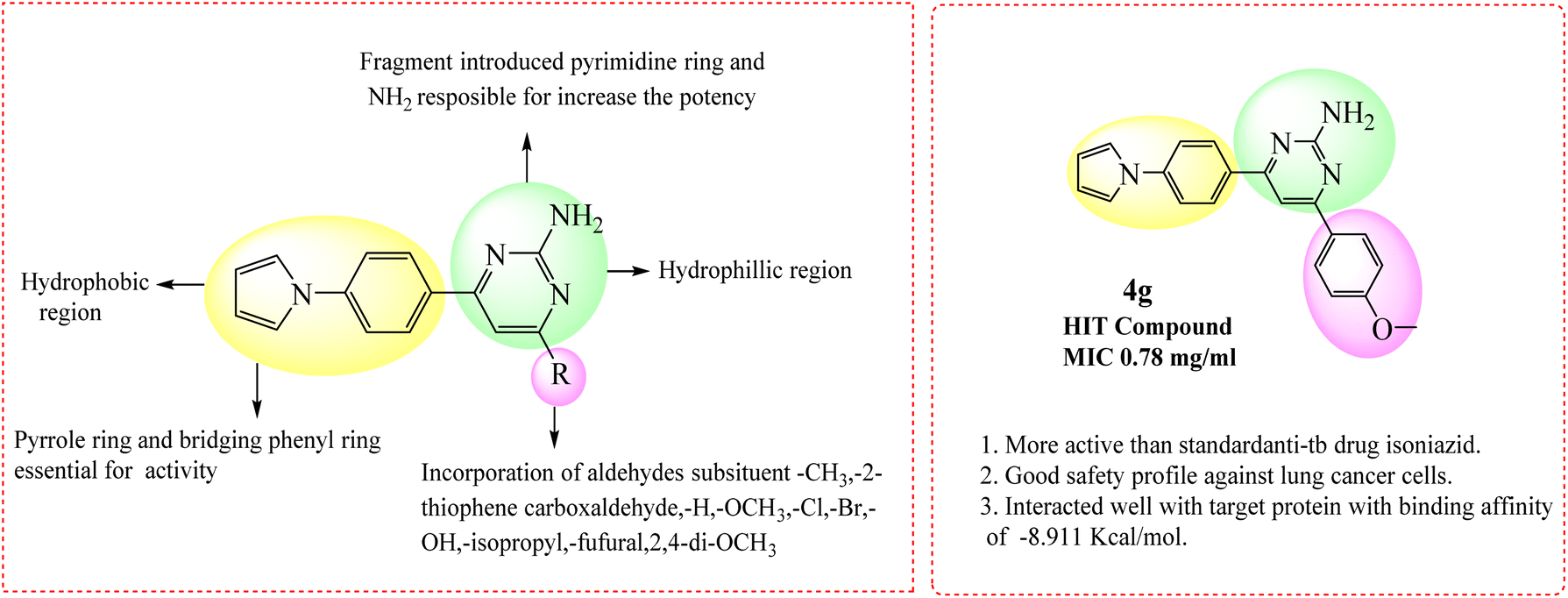 | ||
| Fig. 5 SAR of pyrimidine derivatives and more active compound 4g. | ||
3.1 Anti-tubercular activity
The described derivatives were examined for their anti-TB activity (Table 4). As a result of the preliminary anti-TB screening the most compounds own good activity. The activities of 3k, 3l, 3u, 4c, 4d, 4e, 4g, 4h, 4j, 4k, 4r are expressed of minimum inhibitory concentration values, is shortcut by drugs: isoniazid, ethambutol, rifampicin. In series 3l, 4h compounds were active at MIC of 6.25 mg mL−1, and better activity were shown with 3u, 4c, and 4k compounds with MIC value of 3.125 mg mL−1. Most active compound was 4g is with a MIC value of 0.78 mg mL−1. Better anti-tubercular activity was shown with this from the existence of biologically active hetero-aryl groups chalcone and aromatic ring link with pyrrole ring. The inhibitory activity of the pyrimidine derivatives was relatively higher than that of the chalcones. That's encouraging to observe that compounds 3u, 4c and 4k was revealed with good anti-tubercular activity against tuberculosis MIC value 3.125 mg mL−1 while 4g exhibited the capable activity MIC 0.78 mg mL−1. Based on the study structure–activity relationship of the molecule depicted in (Fig. 5 and 6, Table 4). | ||
| Fig. 6 Graphical representation of MIC for selected active compounds (MIC value range between 0.8 and 12.5 μg mL−1). | ||
| S. no. | Compound ID | MIC values (mg mL−1) |
|---|---|---|
| 1 | 3k | 12.5 |
| 2 | 3l | 6.25 |
| 3 | 3m | >25 |
| 4 | 3n | 25 |
| 5 | 3o | >25 |
| 6 | 3p | 25 |
| 7 | 3q | >25 |
| 8 | 3r | >25 |
| 9 | 3s | >25 |
| 10 | 3t | >25 |
| 11 | 3u | 3.125 |
| 12 | 3v | 25 |
| 13 | 4a | >25 |
| 14 | 4b | 25 |
| 15 | 4c | 3.125 |
| 16 | 4d | 12.5 |
| 17 | 4e | 12.5 |
| 18 | 4f | >25 |
| 19 | 4g | 0.78 |
| 20 | 4h | 6.25 |
| 21 | 4i | >25 |
| 22 | 4j | 12.5 |
| 23 | 4k | 3.125 |
| 24 | 4l | >25 |
| 25 | 4m | >25 |
| 26 | 4n | >25 |
| 27 | 4o | 25 |
| 28 | 4p | 25 |
| 29 | 4q | >25 |
| 30 | 4r | 12.5 |
| 31 | 4s | >25 |
| 32 | 4t | 25 |
| 33 | 4u | >25 |
| 34 | 4v | 25 |
| 35 | Isoniazid | 0.05 (μg mL−1) |
| 36 | Rifampicin | 0.1 (μg mL−1) |
| 37 | Ethambutol | 1.56 (μg mL−1) |
3.2 MTT cytotoxicity studies
000 units, and streptomycin at 10 mg per mL until attaining 80–90% confluency. The cells stayed then scraped and sowed into wells at a thickness of 5000 cells per well in poly-L-lysine coated plates. The microtiter plates remained kept on controlled environment of 37 °C, 5% CO2, and 100% relative humidity for 24 h already added the trial drugs. The trial compounds were added to the cells at a concentration of 50 μg per mL and the plate was then kept on an incubator at 37 °C for 48 h 10 μL of 0.5 mg mL−1 of MTT was then added to the solution and then incubated for 3 h at 37 °C and the ending produce formazan crystals stayed measured at 595 nm and 625 nm showed in (Table 5).
| S. no. | Comp. ID | % Toxicity at 25 μg mL−1 |
|---|---|---|
| 1 | 3k | 30.1 |
| 2 | 3l | 26.6 |
| 3 | 4c | 25.80 |
| 4 | 4g | 19.09 |
| 5 | 4h | 26.80 |
| 6 | 4k | 28.90 |
| 7 | 4u | 19.10 |
| 8 | Isoniazid | 0.05 (μg mL−1) |
| S. no. | Compound ID | IC50 (mM) |
|---|---|---|
| a IC50 – is half maximal inhibitory concentration-it is the half maximal (50%) inhibitory concentration (IC) of a substance (50% IC, or IC50). | ||
| 1 | 4c | 55.12 |
| 2 | 4g | 49.44 |
| 3 | 4h | 104.77 |
| 4 | 4k | 123.36 |
| 5 | 4u | 123.36 |
| 6 | Cisplatin | 9.90 |
 | ||
| Fig. 7 Overlaid IC50 values of given test compounds against A549 cells after the incubation period of 24 h. | ||
3.3 Enzyme inhibition studies
As in vitro anti-mycobacterial studies, four compounds selected for in vitro enzyme inhibition activity against M. tuberculosis at a concentration of 50 μM by using the routine method. A triclosan was the first to be tested at the same concentration and 50 mM was able to completely inhibit InhA enzyme. The pyrimidine compounds bearing methoxy group compound 4c, 4g, & 4u on the methoxy aldehyde aromatic group are responsible for inhibition of InhA enzyme. It showed inhibition at 50 μM and 2-thiophene carboxaldehyde substituted and isopropyl group substituted chalcone compound analogue 3k & 3l, 4g showed 36% and 4k showed 10% inhibition at 50 μM. The outcome shows an increase in the inhibitory action. In antagonist chalcone compound 3k and 3l, where thiophene ring at 2nd position substitution with isopropyl group in the 4th position and methoxy group in the pyrimidine ring compounds 3k, 3l, 4c, 4g, 4h & 4k displayed very good % inhibition at 50 μM and 4u displayed no inhibition, showed in Table 7.| S. no. | Compound ID | Solubility | % inhibition InhA at 50 μM of inhibitor |
|---|---|---|---|
| a NI: for no inhibition. | |||
| 1 | 3k | DMSO-d6 | 36 |
| 2 | 3l | DMSO-d6 | 36 |
| 3 | 4c | DMSO-d6 | 16 |
| 4 | 4g | DMSO-d6 | 36 |
| 5 | 4h | DMSO-d6 | <5 |
| 6 | 4k | DMSO-d6 | 10 |
| 7 | 4u | DMSO-d6 | NI |
| 8 | Triclosan | DMSO-d6 | >99 |
3.4 DFT calculation
Compound 4g showed total gas phase energy −1105.367894 kcal mol−1 and −0.211939 eV and −0.054720 eV for HOMO and LUMO orbitals respectively depicted in (Fig. 8). Overall electron distribution of 4g was found to be stable. Electrostatic potential was found to be asymmetrically distributed showed. Parallelly, isoniazid as standard drug showed a total gas-phase energy of −0.224326 eV. HOMO and LUMO orbitals of isoniazid showed −0.224326 eV and −0.053617 eV and the energy gap of 4g compound −0.157219 and isoniazid is −0.170716 depicted in (Table 8). A molecule is considered to be more stable if its energy gap and chemical hardness values are larger. Here 4g has optimum stability and isoniazid is not showing good stability. The results reveal that of the interplay between molecular geometry and electronic behaviour, shedding light on the chemical properties and potential reactivity of the studied substances. This detailed computational analysis not only contributed to elucidating the structural features but also facilitated the interpretation of the reactivity patterns, enhancing the overall comprehension of the molecular systems being investigated. | ||
| Fig. 8 DFT studies of compound 4g and standard drug isoniazid. | ||
| S. no. | Compound ID | Gas phase energy | Alpha HOMO | Alpha LUMO | Energy gap |
|---|---|---|---|---|---|
| 1 | 4g | −1105.367894 | −0.211939 | −0.054720 | −0.157219 |
| 2 | Isoniazid | −472.311674 | −0.224326 | −0.053617 | −0.170716 |
3.5 MD simulation
Molecular dynamics simulation predicts the biological system's dynamic nature over a predetermined period of time. MD simulation was carried out in physiological settings to better understand the dynamic nature of the interactions and the stability of the protein–drug complex. Considering the bound free energy values and the XP docking score. Determination of the in silico stability of the drug–protein interaction for the docked complex at the molecular level was enhanced by performing MD simulations for 200 ns on all the drug–protein complexes docked (compound 4g and standard drug isoniazid). Together, these were put through SID (simulation interaction diagram) utility as a 200 ns analysis, which was additionally used by the semi-empirical Desmond module of the Maestro-Schrodinger suite. We analysed the steadiness of the docked compounds within the vigorous pocket of the (5OIR) protein by employing RMSD and its impact on the overall system stability.48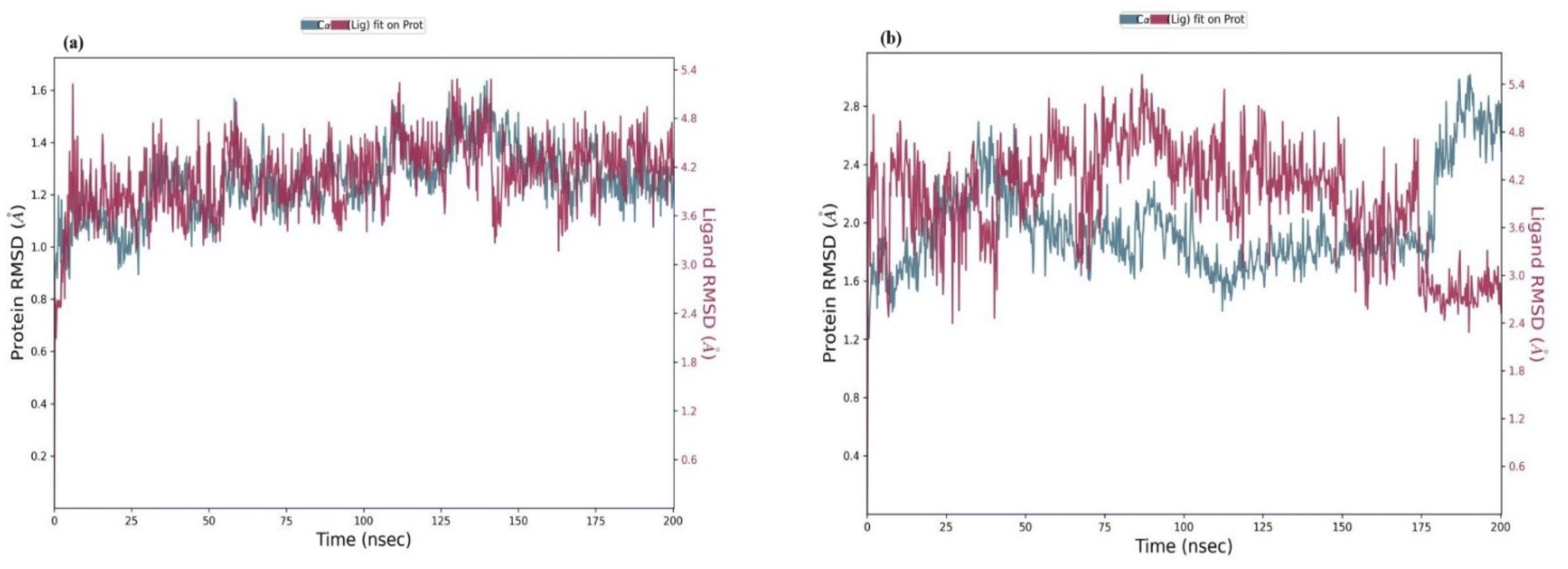 | ||
| Fig. 9 (a) Shows the RMSD of the 4g complex and (b) shows the RMSD of the isoniazid complex. | ||
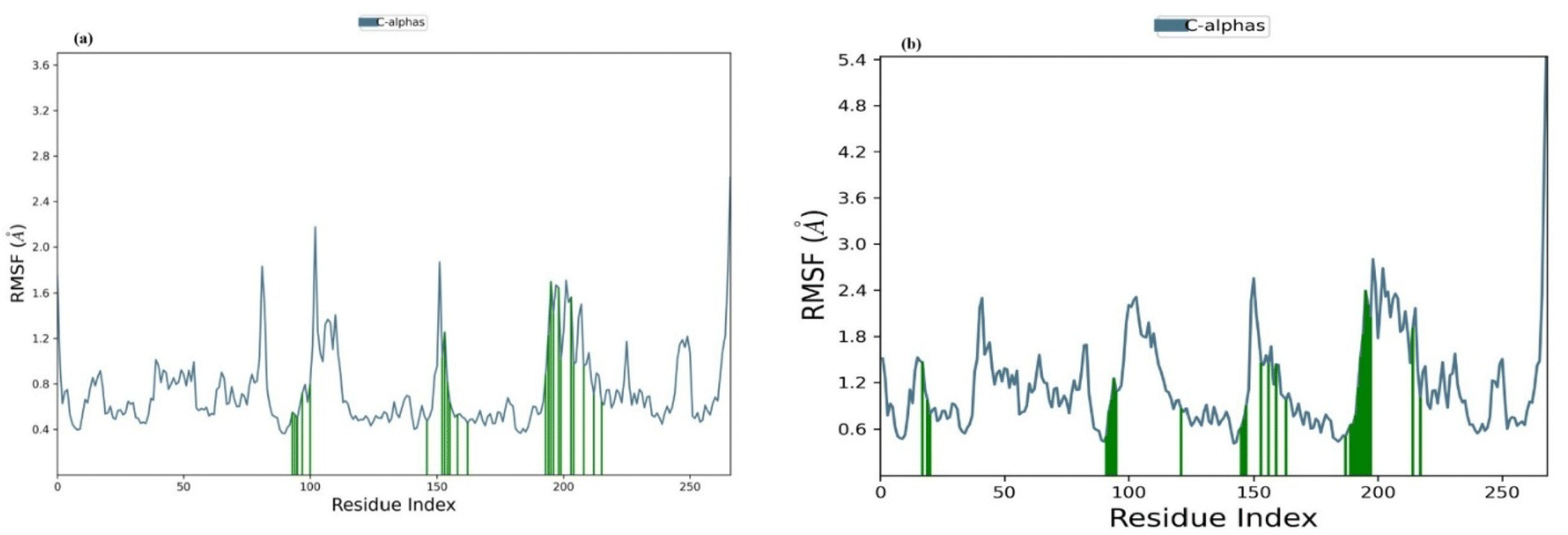 | ||
| Fig. 10 (a) Shows the RMSF of the 4g complex and (b) shows the RMSF of the isoniazid complex. | ||
Protein–ligand amino acid residue contacts analysis of MD trajectory.
Protein–ligand contact analysis amino acid residues which have positive hydrophobic interactions with the protein with ligands 4g are Phe 97, Met 103, Phe 149, Met 155, Pro 156, Ala 157, Tyr 158, Met 161, Thr 196, Leu 197, Ala 198, Met 199, Ala 201, Ile 202, Ala 206, Leu 207, Ala 211, Ile 215, and Leu 218. These interactions promote the stabilization of the protein–ligand complex. The water bridges with the ligand that play a significant role in facilitating the interactions among the protein and ligand and a few of the Gly 96, Met 98, Tyr 158, Lys 165, and Leu 197 residues were responsible for the formation of the protein–ligand complex. The hydrogen bonds to the ligand 4g in (Fig. 11) (a) were in part due to Gly 96 and Gln 100. The residues that facilitated the protein–ligand isoniazid interactions through hydrophobic bonds are: Ser 19, Met 147, Phe 149, Met 161, Val 189, Ala 191. These types of interactions are significant in the maintenance of the protein–ligand complex. The formation of water bridges to the ligand which are of crucial importance to protein–ligand interactions in the form of bridges are constructed by: Ile 21, Ala 22, His 93, Ser 94, Phe 97, Asp 148, Lys 165, Pro 193, Arg 195, Elu 219. The amino His 93, Ser 94, Met155, Lys 165, Ile 194, Thr 196, Leu 197, Ala 198, Met 199 were involved in isoniazid attachment to the protein with the aid of hydrogen bonds in (Fig. 11) (b). The 4g molecule interacts with 23 amino acids that are present in the 5OIR protein. The residues are constant since the majority of the variations are smaller than 2 Å. For the majority of residues interacting with ligands, the variations are extremely small at 3 nanoseconds. This indicates that the interactions are quite stable during the experiment. During the simulation, the interaction between the ligand and protein was observed. Tyr 158 builds water bridges within the time frame provided, while Gln 100 forms hydrogen bond interaction and Tyr 158 forms hydrophobic interaction for most of the time and water bridges for just a short period of time.
 | ||
| Fig. 11 (a) Protein–ligand amino acid residue contacts of 4g complex and (b) protein–ligand amino acid residue contacts of isoniazid complex. | ||
3.6 Protein–ligand contact analysis based on the two-dimensional interactions
The precise sequence correlations between the different amino acid residues found in the contact of 4g with 5OIR protein, 4g interacts along with the 5OIR protein residues. 51–54% of the simulation period is spent with these interactions steady. Protein–ligand contact analysis based on the two-dimensional interactions which are ligand-mediated is shown. There are also notable Tyr158 and Met103 amino acids that are involved in hydrophobic bonding accounting to some portion of 4g the compound 46% of the simulation time. Also, for Gln 100, each of 17% polar interactions were noted. For isoniazid protein–ligand contact interaction assessments, the interactions mediated by ligand. The amino acids Phe 149 and Ile 194 have a compound that incorporates 30% of their interaction for 70% of their simulation time.4. Conclusion
In summary, a series of novel pyrrolyl chalcones and pyrimidine-fused derivatives were successfully synthesized and evaluated for their antitubercular potential. Several compounds, particularly 4g, demonstrated strong activity against M. tuberculosis H37Rv, with minimal cytotoxicity and promising InhA enzyme inhibition. Molecular docking, MM-GBSA, DFT, and MD simulation studies further supported the binding stability and favourable drug-like properties of the active compounds. These findings highlight the potential of these scaffolds as promising leads for the development of new, safe, and effective anti-TB agents targeting InhA.5. Experimental section
The synthesis was carried out using Sigma-Aldrich® Lab production chemicals & analytical-grade solvents and reagents that were desiccated, purified in accordance with the protocols described in the Vogel's practical book of organic chemistry. To evaluate both the reaction rate and product purity, thin-layer chromatography precoated TLC sheets of silica gel 60 F254 (Merck, Darmstadt, Germany) visualized by long- and short-wavelength UV lamps. The mobile phase consisting of n-hexane and ethyl acetate in a 6:4 ratio. Synthesized compounds were purified via recrystallization and column chromatography using by Merck silica gel (70–230 mesh). Melting points were determined using ANALAB scientific melting/boiling point apparatus. FTIR spectra in KBr pellets were recorded on a Bruker FTIR spectrophotometer. The 1H and 13C NMR spectra were recorded using a 400 MHz FT-NMR spectrometer with DMSO-d6 as the solvent and tetramethyl silane (TMS) as the internal standard, with a sensitivity of δ ppm. The abbreviations used to describe the peak patterns are: (b) broad, (s) singlet, (d) doublet, (t) triplet, (q) quartet, and (m) multiplate. Mass spectra (MS) were recorded in a JEOL GCMATE II GC-mass spectrometer and Shimadzu QP 20105 GC-mass spectrometer.
5.1 General procedure for the synthesis of 1-(4-(1H-pyrrol-1-yl)phenyl)-3-substituted prop-2-en-1-ones (3k–3v)
Paal Knorr synthesis was used to constructed pyrrole ring from 4-aminoacetophenone 1 (4.05 g, 0.030 mol) (1) by condensing it with 2,5-dimethoxy tetrahydrofuran (4.23 g, 0.032 mol). Followed by the Claisen Schmidt reaction was used to form chalcones (3k–3v) by condensing (2) with different aldehydes using catalytic amount of ethanolic 20 mL NaOH as depicted in Scheme 1. The reaction mixture was stirred for 24–30 h. This was then plunged into cold water and neutralized with hydrochloric acid. The precipitated solid was filtered, washed with water, dried, and purified by column chromatography on silica gel (ethyl acetate/n-hexane (6:4) then eluent). Compound (3a–3j) are used from our earlier reported work49 and chalcones (3k–3v) are reported in this current work.
O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ ppm: 8.23–8.26 (s, 2H, –CHCH–), 7.92–8.96 (s, 1H), 7.71–7.84 (m, 5H, bridging phenyl-C2, C3, C5, C6–H), 7.56–7.57 (s, 2H), 7.33–7.35 (d, J = 8.0 Hz, 2H, pyrrole-C2, C4–H), 6.33–6.34 (s, 2H, pyrrole-C1, C5–H), 2.92–2.94 (s, 1H), 1.22–1.23 (d, J = 4 Hz, 6H, isopropyl methyl). 13C NMR (100 MHz, DMSO-d6) δ ppm: 187.68, 151.50, 143.98, 143.21, 134.09, 132.48, 130.53, 129.18, 126.96, 120.95, 119.18, 118.59, 111.56, 33.50, 23.68; MS (ESI): m/z = found 316.15 [M+ + 1]; calcd. 315.16. Anal. calcd. for C22H21NO: C, 83.78; H, 6.71; N, 4.44. Found: C, 83.99; H, 6.80; N, 4.50.O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ ppm: 8.18–8.20 (d, J = 8 Hz, 2H, –CHCH–), 7.90–8.94 (s, 1H), 7.71–7.80 (m, 3H) and 7.60–7.64 (d, J = 16 Hz, 1H, bridging phenyl-C2, C3, C5, C6–H), 7.55–7.56 (s, 1H), 7.52–7.53 (s, 2H, pyrrole-C2, C4–H) and 7.19–7.21 (m, 1H, thiophene ring, C2, C3, C4), 6.33–6.34 (s, 2H, pyrrole-C1, C5–H). 13C NMR (100 MHz, DMSO-d6) δ ppm: 187.17, 143.14, 139.75, 136.51, 133.90, 132.77, 130.47, 130.34, 128.71, 120.24, 119.10, 118.59, 111.49; MS(ESI): m/z = found 280.06 [M+ + 1]; calcd. 279.07. Anal. calcd. for C17H13NOS:C, 73.00; H, 4.69; N, 5.05. Found: C, 73.15; H, 4.75; N, 5.19.O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ ppm: 8.25–8.27 (d, J = 8.8 Hz, 2H, –CHCH–), 8.03–8.11 (q, 2H), 7.92–8.01 (s, 1H), 7.80–7.81 (d, J = 4 Hz, 2H), 7.78–7.79 (d, J = 4 Hz, 2H), 7.69–7.74 (m, 2H), 7.51–7.57 (m, 4H, bridging phenyl-C2, C3, C5, C6–H), 7.23–7.25 (s, 2H, pyrrole-C2, C4–H), 6.31–6.35 (m, 2H, pyrrole-C1, C5–H), 3.83–3.87 (s, 9H, 3-OCH3). 13C NMR (100 MHz, DMSO-d6) δ ppm:153.11, 144.32, 130.49, 119.12, 119.07, 118.55, 111.48, 106.60, 60.13, 56.14; MS(ESI): m/z = found 364.08 [M+ + 1]; calcd. 363.15. Anal. calcd. for C22H21NO4: C, 72.71; H, 5.82; N, 3.85. Found: C, 72.79; H, 5.90; N, 3.91.O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ ppm: 9.055 (s, 1H, –OH), 8.23–8.25 (d, J = 8 Hz, 2H, –CHCH–), 8.06–8.08 (d, J = 8 Hz, 1H) and 7.67–7.85 (m, 3H, bridging phenyl-C2, C3, C5, C6–H), 7.52–7.55 (d, J = 12 Hz, 2H), 7.22 (s, 2H, pyrrole-C2, C4–H), 6.34 (s, 2H, pyrrole-C1, C5–H), 3.85 (s, 6H, 2-OCH3); 13C NMR (100 MHz, DMSO-d6) δ ppm: 198.30, 186.32, 149.21, 143.05, 142.41, 135.30, 133.30, 129.88, 119.06, 118.48, 111.45, 111.25, 55.87; MS(ESI): m/z = found 350.11 [M+ + 1]; calcd. 349.13. Anal. calcd. for C21H19NO4: C, 72.19; H, 5.48; N, 4.01. Found: C, 72.32; H, 5.50; N, 04.10.O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ ppm: 8.25–8.27 (d, J = 8 Hz, 2H, –CHCH–), 8.02–8.06 (d, J = 15.6 Hz, 1H), 7.87–7.89 (d, J = 8 Hz, 2H, bromo phenyl C2, C4, –H) and 7.78–7.71 (d, J = 9 Hz, 2H, bridging phenyl-C2, C3, C5, C6–H), 7.70–7.74 (d, J = 15 Hz, 1H) and 7.66–7.68 (d, J = 8 Hz, 1H, bromo phenyl C1, C5, –H), 7.56–7.57 (t, 2H, pyrrole-C1, C5–H), 6.33–6.34 (t, 2H, pyrrole-C1, C5–H). 13C NMR (100 MHz, DMSO-d6) δ ppm: 143.35, 142.43, 134.04, 131.88, 130.84, 130.58, 123.96, 122.72, 119.14, 118.57, 111.53; MS (ESI): m/z = found 353.11 [M+ + 2]; calcd. 351.03. Anal. calcd. for C19H14BrNO: C, 64.79; H, 4.01; N, 3.08. Found: C, 64.80; H, 4.12; N, 3.12.O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ ppm: 8.19–8.21 (d, J = 8.4 Hz, 2H, –CHCH–), 7.67–7.77 (m, 6H, bridging phenyl-C2, C3, C5, C6–H), 7.54–7.55 (t, 2H, pyrrole-C2, C4–H), 6.74–6.80 (m, 2H), 6.33–6.33 (t, 2H, pyrrole-C1, C5–H), 3.01–3.04 (s, 6H, dimethyl-amino phenyl). 13C NMR (100 MHz, DMSO-d6) δ ppm: 187.17, 151.99, 145.00, 142.76, 134.80, 130.81, 130.11, 122.04, 119.08, 118.52, 115.90, 111.74, 111.36, 111.06; MS (ESI): m/z = found 317.14 [M+ + 1]; calcd. 316.16. Anal. calcd. for C21H20N2O: C, 79.72; H, 6.37; N, 8.85. Found: C, 79.80; H, 6.37; N, 8.89.O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ ppm: 8.24–8.26 (d, J = 8 Hz, 2H, –CHCH–), 7.85–7.89 (d, J = 15 Hz, 1H), 7.77–7.80 (d, J = 8 Hz, 4H, bridging phenyl-C2, C3, C5, C6–H), 7.70–7.73 (d, J = 15 Hz, 1H), 7.55–7.56 (t, 3H), 7.40–7.42 (dd, J = 10 Hz, 1H) and 7.02–7.04 (d, J = 8.4 Hz, 1H, pyrrole-C2, C4–H), 6.33–6.34 (t, 2H, pyrrole-C1, C5–H), 3.82–3.87 (s, 6H, 2-OCH3). 13C NMR (100 MHz, DMSO-d6) δ ppm: 187.55, 151.29, 149.02, 144.34, 143.04, 134.29, 130.38, 127.55, 123.95, 119.46, 119.11, 118.54, 111.56, 111.44, 110.87, 55.75, 55.60; MS (ESI): m/z = found 334.11 [M+ + 1]; calcd. 333.14. Anal. calcd. for C21H19NO3: C, 75.66; H, 5.74; N, 4.20. Found: C, 73.70; H, 5.78; N, 4.22.O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ ppm: 8.06–8.08 (d, J = 8 Hz, 2H, –CHCH–), 7.68–7.77 (m, 2H, pyrrole-C2, C4–H), 7.32–7.57 (m, 4H, bridging phenyl-C2, C3, C5, C6–H), 7.24–7.21 (m, 2H, Ar-phenyl), 7.08–7.10 (m, 3H), 6.27–6.30 (m, 2H, pyrrole-C1, C5–H). 13C NMR (100 MHz, DMSO-d6) δ ppm:144.16, 143.08, 141.61, 130.12, 128.95, 127.30, 127.25, 119.08, 118.6.3, 111.49, 111.44; MS (ESI): m/z = found 314.11 [M+ + 1]; calcd. 313.15. Anal. calcd. for C22H19NO: C, 83.31; H, 6.11; N, 4.47. Found: C, 83.35; H, 6.15; N, 6.15.O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ ppm: 8.21–8.23 (d, J = 8 Hz, 2H, –CHCH–), 7.75–7.77 (d, J = 9 Hz, 2H, CHCH), 7.64–7.74 (m, 4H, bridging phenyl-C2, C3, C5, C6–H), 7.54–7.55 (t, 1H), 7.51–7.52 (m, 1H) and 7.46–7.47 (d, J = 4 Hz, 1H, pyrrole-C2, C4–H), 7.25–7.27 (d, J = 8 Hz, 1H), 6.77–6.79 (d, J = 8 Hz, 1H), 6.31–6.34 (t, 2H, pyrrole-C1, C5–H), 1.35–1.38 (t, 3H, CH3). 13C NMR (100 MHz, DMSO-d6) δ ppm:187.31, 147.46, 145.03, 142.86, 134.61, 130.23, 124.62, 119.10, 118.53, 117.47, 115.96, 113.05, 111.38, 63.93; MS(ESI): m/z = found 334.11 [M+ + 1]; calcd. 333.14. Anal. calcd. for C21H19NO3:C, 75.66; H, 5.74; N, 4.20. Found: C, 75.69; H, 5.76; N, 4.23.O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ ppm: 9.16 (d, 1H, –OH), 8.21–8.23 (d, J = 8 Hz, 2H, –CHCH–), 8.01–8.03 (q, 1H), 7.78 (s, 1H), 7.76 (s, 1H) and 7.72–7.74 (d, J = 8 Hz, 2H, bridging phenyl-C2, C3, C5, C6–H), 7.65 (s, 1H), 7.54–7.56 (m, 2H), 7.29–7.35 (m, 2H, pyrrole-C2, C4–H,), 6.91–7.01 (d, 1H) 6.31–6.34 (s, 2H, pyrrole-C1, C5–H), 3.36–3.86 (s, 3H, –OCH3). 13C NMR (100 MHz, DMSO-d6) δ ppm: 187.54, 150.37.146.78.144.38, 143.02, 134.34, 130.35, 129.89, 127.69, 122.13, 119.25, 119.11, 119.07, 118.57, 118.57, 118.45, 114.93, 111.91, 111.46, 55.69; MS (ESI): m/z = found 320.10 [M+ + 1]; calcd. 319.12. Anal. calcd. for C20H17NO3: C, 75.22; H, 5.37; N, 4.39. Found: C, 75.28; H, 5.40; N, 4.42.O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ ppm: 9.16 (d, 1H, –OH), 8.09–8.12 (t, 2H, –CHCH–), 8.06 (s, 1H, –CH), 7.84–7.88 (d, J = 15 Hz, 1H), 7.73–7.75 (d, J = 8 Hz, 2H, bridging phenyl-C2, C3, C5, C6–H), 7.51–7.52 (t, 2H, pyrrole-C2, C4–H), 7.21–7.22 (d, J = 7.6 Hz, 1H), 6.74–6.76 (d, J = 7 Hz, 1H) and 6.39 (s, 1H, Ar-phenyl), 6.32–6.33 (t, 2H, pyrrole-C1, C5–H), 3.83 (s, 3H 2-OCH3). 13C NMR (100 MHz, DMSO-d6) δ ppm: 174.64, 150.37.146.78.144.38, 143.02, 134.34, 130.35, 129.86, 119.06, 118.53, 111.28, 155.69; MS (ESI):m/z = found 320.09 [M+ + 1]; calcd. 319.12. Anal. calcd. for C22H21NO: C, 75.22; H, 5.37; N, 4.39. Found: C, 75.25; H, 5.41; N, 4.43.O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ ppm: 8.09–8.010 (q, 4H, –CHCH–), 7.73–7.78 (t, 4H, bridging phenyl-C2, C3, C5, C6–H), 7.43–7.54 (q, 4H, pyrrole-C2, C4–H and Ar-phenyl), 6.32–6.33 (t, 4H, pyrrole-C1, C5–H and CHCH), 2.58–2.97 (s, 6H, –2CH3). 13C NMR (100 MHz, DMSO-d6) δ ppm: 196.65, 143.15, 133.33, 130.07, 129.90, 128.88, 119.07, 118.60, 118.44, 112.00, 111.45, 26.60; MS (ESI): m/z = found 357.15 [M+ + 1]; calcd. 356.19. Anal. calcd. for C24H24N2O: C, 80.87; H, 6.79; N, 7.86. Found: C, 80.92; H, 6.83; N, 7.88.5.2 General procedure to synthesis of 4-(4-(1H-pyrrol-1-yl)phenyl)-6-phenylpyrimidin-2-amine (4a–4v)
Chalcones (3a–3v) were cyclized to pyrimidine by reacting with Guanidine hydrochloride (0.007 mol) in the presence of sodium ethoxide (0.02 mol) and dried DMF (15 mL) as a solvent as depicted in scheme −2. Reaction mixture was refluxed at 80 °C for 30–35 h, and reaction progress monitored by TLC. The reaction was quenched with crushed ice, yielding a ppt solid which was filtered off, washed with water, hydrated and purified by column-chromatography (2:1), ethyl acetate and n-hexan.
C). 1H NMR (400 MHz, DMSO-d6) δ 8.29–8.31 (d, J = 8 Hz, 1H), 8.24–8.26 (d, J = 8 Hz, 2H) and 7.92 (s, 2H, bridging phenyl-C2, C3, C5, C6–H), 7.73–7.76(d, J = 12, 2H, Ar-phenyl), 7.58–7.71 (t, 2H, Ar-phenyl), 7.56 (s, 2H, pyrrole-C2, C4–H), 7.49 (s, 2H, –NH2), 6.29–6.76 (s, 2H, pyrrole-C1, C5–H); 13C NMR (100 MHz, DMSO-d6) δ 16, 4.02, 164.41, 164.41, 164.62, 141.92, 136.66, 135.68, 134.20, 129.27, 129.12, 129.27, 128.94, 119.46, 119.32, 111.43, 101.88. MS (ESI): m/z = found (M+ + 1) 347.19, calc 346.10. Anal. calcd. for C20H15ClN4: C, 69.26; H, 4.36; N, 16.15. Found: C, 69.30; H, 4.40; N, 16.20.C). 1H NMR (400 MHz, DMSO-d6) δ 8.14–8.16 (d, J = 8 Hz, 2H), and 7.44–7.61 (s, 2H, bridging phenyl-C2, C3, C5, C6–H), 7.27 (s, 2H, pyrrole-C2, C4–H), 7.21–7.22 (d, J = 3, 1H), 7.18–7.19 (d, J = 4 Hz, 3H, –furfural ring), 6.58–6.59 (q, 1H, –CHCH, pyrimidine ring), 6.39–6.40 (t, J = 4 Hz, 2H, pyrrole-C1, C5–H), 5.22 (s, 2H, –NH2). 13C NMR (100 MHz, DMSO-d6) δ 164.75, 164.39, 163.57, 141.96, 140.15, 133.55, 134.14, 131.2, 130.00, 129.02, 128.86, 127.09, 127.54, 119.33, 119.45, 111. 40, 24.2. MS (ESI): m/z = found (M+ + 1) 303.12, calc. 302.12. Anal. calcd. For C18H14N4: C, 71.51; H, 4.67; N, 18.53. Found: C, 71.56; H, 4.40; N, 18.58.O), 1563.58 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 8.12–8.14 (d, J = 8.4 Hz, 1H, CHCH), 7.87–7.89 (d, J = 8 Hz, 1H), and 7.34–7.75 (m, 2H,) and 6.68–6.70 (s, 1H, bridging phenyl-C2, C3, C5, C6–H), 6.61–6.67 (d, J = 2 Hz, 2H, pyrrole-C2, C4–H), 6.48–6.56 (s, 3H, Ar-phenyl), 6.31–6.32 (s, 2H, pyrrole-C1, C5–H), 6.25–6.26 (d, J = 4 Hz, 2H, –NH2), 3.84–3.90 (s, 3H–OCH3), 3.41–3.51 (s, 3H, –OCH3). 13C NMR (100 MHz, DMSO-d6) δ 164.20, 163.11, 162.46, 159.59, 141.65, 134.68, 131.85, 130.58, 128.60, 128.39, 126.65, 119.71, 119.53, 119.38, 119.24, 118.99, 111.96, 111.44, 110.83, 106.01, 99.14, 56.26–55.69; MS (ESI): m/z = found (M+ + 1) 373.15, calc. 372.16. Anal. calcd. For C22H20N4O2: C, 70.95; H, 5.41; N, 15.04. Found: C, 70.98; H, 5.45; N, 15.09.O), 1565.41 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 8.31–8.29 (d, J = 8 Hz, 1H, Ar phenyl) and 8.26–8.24 (d, J = 8 Hz, 1H, –CHCH pyrimidine ring), 7.92 (s, 2H, bridging phenyl-C2, C6–H), 7.73–7.71 (t, 2H, bridging phenyl- C3, C5, –H), 7.58–7.56 (d, J = 8.4 Hz, 3H, Ar phenyl), 7.49 (s, 2H, pyrrole-C2, C4–H) 6.76 (s, 2H, NH2), 6.29 (s, 2H, pyrrole-C1, C5–H). 13C NMR (100 MHz, DMSO-d6) δ 165.33, 164.45, 164.38, 141.85, 137.85, 134.34, 130.91, 129.07, 128.90, 127.48, 119.45, 119.33, 111.93, 111.42, 101.97. MS (ESI): m/z = found (M+ + 1) 313.16, calc. 312.14. Anal. calcd. for C20H16N4: C, 76.90; H, 5.16; N, 17.94. Found: C, 76.91; H, 5.18; N, 17.97.O), 1575.64 cm−1 (Ar CC). 1H NMR (400 MHz, CDCl3) δ 8.13–8.15 (d, J = 8.4 Hz, 2H, –CHCH, pyrimidine ring), 7.97–7.99 (d, J = 8 Hz, 2H bridging phenyl-C2, C6–H), 7.51–7.53 (d, J = 8 Hz, 2H, bridging phenyl-C3, C5, –H), 7.45 (s, 1H, Ar phenyl), 7.31–7.33 (d, J = 8 Hz, 2H), 7.12 (s, 1H, Ar phenyl), 7.10–7.12 (d, J = 6.4 Hz, 2H, pyrrole-C2, C4–H), 7.063 (s, 1H, –CHCH pyrimidine ring) 6.34–6.41 (s, 2H, pyrrole-C1, C5–H), 5.35 (s, 2H, –NH2), 2.26–2.45 (s, 3H, –CH3). 13C NMR (100 MHz, DMSO-d6) δ 165.23, 164.40, 164.24, 141.81, 140.72, 130.82, 129.86, 129.67, 128.87, 127.40. MS (ESI): m/z = found (M+ + 1) 327.15, calc. 326.15. Anal. calcd. for C21H18N4: C, 77.28; H, 5.56; N, 17.17. Found: C, 77.31; H, 5.59; N, 17.20.O), 1562.43 cm−1 (Ar CC). 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 1H, –CHCH, pyrimidine ring), 8.12–8.15 (d, J = 8.8 Hz, 2H, bridging phenyl-C2, C6–H), 7.96–7.98 (d, J = 8 Hz 2H, bridging phenyl-C3, C5, –H), 7.60–7.62 (d, J = 7 Hz 1H, Ar phenyl), 7.50–7.52 (d, J = 8, 2H, pyrrole-C2, C4–H), 7.34–7.42 (q, 3H, Ar phenyl), 7.25 (s, 2H), 6.39 (s, 2H, pyrrole-C1, C5–H), 5.23 (s, 2H, –NH2). 13C NMR (100 MHz, DMSO-d6) δ164.75, 164.39, 163.57, 141.46, 140.15, 134.14, 133.55, 131.25, 130.00, 129.02, 126.48, 122.69, 119.54, 119.46, 119.30, 118.92, 111.93, 111.44, 102.08. MS (ESI): m/z = found (M+ + 2)393.04, calc. 390.05. Anal. calcd. For C20H15BrN4: C, 61.39; H, 3.86; N, 14.32. Found: C, 61.41; H, 3.89; N, 14.36.O), 1537.69 cm−1 (Ar CC). 1H NMR (400 MHz, CDCl3) δ 8.13–8.15 (d, J = 8.8 Hz, 2H) and 8.05–8.07 (d, J = 8.8 Hz, 2H, bridging phenyl-C2, C3, C5, C6–H), 7.50–7.52 (d, J = 8.4 Hz 2H, Ar phenyl), 7.42 (s, 1H CHCH pyrimidine ring), 7.18–7.27 (s, 2H, pyrrole-C2, C4–H), 7.01–7.03 (d, J = 8.0 Hz, 2H, Ar phenyl), 6.40 (s, 2H, pyrrole-C1, C5–H), 5.25 (s, 2H, –NH2), 3.70–3.89 (t, 3H, –OCH3). 13C NMR (100 MHz, DMSO-d6) δ 164.89, 164.34, 164.07, 161.71, 141.76, 134.49, 130.11, 129.06, 128.85, 119.44, 119.32, 114.41, 111.40, 101.20, 55.79. MS (ESI): m/z = found (M+ + 1) 343.14, calc. 342.40. Anal. calcd. for C21H18N4O: C, 75.67; H, 5.30; N, 16.36. Found: C, 75.70; H, 5.32; N, 16.38.O), 1589.71 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 8.09–8.11 (d, J = 8 Hz, 2H) and 7.57–759 (d, 2H, bridging phenyl-C2, C3, C5, C6–H), 7.48–7.50 (d, 2H), 7.17–7.25 (s, 2H pyrrole-C2, C4–H), 7.16–7.17 (t, 2H, -Ar phenyl), 6.32–6.38 (s, 2H, pyrrole-C1, C5–H), 2.86–2.95 (s, 2H, NH2). 13C NMR (100 MHz, DMSO-d6) δ 163.32, 162.02, 140.82, 135.00, 130.16, 129.30, 128.55, 124.36, 119.36, 119.21, 110.53, 101.83. MS (ESI): m/z = found (M+ + 1) 381.03, calc. 380.06. Anal. calcd. for C20H14Cl2N4: C, 63.01; H, 3.70; N, 14.70. Found: C, 63.03; H, 3.73; N, 14.76.O), 1577.36 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 8.11–8.13 (d, J = 8 Hz, 1H, –CHCH, Ar phenyl), 8.01–8.03 (d, 2H, J = 8 Hz, 1H, –CHCH, pyrimidine ring), 7.86–7.88 (d, J = 8 Hz, H, Ar phenyl), 7.43–7.55 (m, 4H, bridging phenyl-C2, C3, C5, C6–H), 7.35–7.39 (m, 2H pyrrole-C2, C4–H), 7.25 (s, 1H, –OH), 7.17–7.18 (t, 3H, Ar phenyl), 7.03–6.92 (m, 2H, pyrrole-C1, C5–H), 6.39 (s, 2H, NH2). 13C NMR (100 MHz, DMSO-d6) δ 164.70, 164.67, 163.40, 141.76, 140.25, 134.10, 133.44, 131.20, 130.00, 129.01, 126.39, 122.48, 119.55, 119.43, 119.29, 118.90, 111.83, 111.84, 102.09. MS (ESI): m/z = found (M+ + 1) 329.11, calc. 328.13. Anal. calcd. for C20H16N4O: C, 73.15; H, 4.91; N, 17.06. Found: C, 73.18; H, 4.94; N, 17.09.O), 1569.58 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 8.03–8.10 (, 2H, d, J = 8 Hz, 1H, –CHCH, pyrimidine ring), 7.59–7.507 (d, J = 8 Hz, 1H, –CHCH, Ar phenyl), 7.48–7.50 (d, J = 8 Hz, 2H bridging phenyl-C3, C5, –H), 7.37–7.36,(d, J = 8 Hz, 2H, bridging phenyl-C2, C6), 7.25 (s, 1H, –CHCH, Ar phenyl), 7.08–7.09, (s 2H, pyrrole-C2, C4–H), 6.38 (s, 2H, pyrrole-C1, C5–H), 5.24–5.28 (s, 2H, NH2). 13C NMR (100 MHz, DMSO-d6) δ164.82, 164.69, 141.43, 137.30, 134.50, 132.30, 129.52, 128.65, 124.76, 119.50, 119.30, 111.03, 110.33. MS (ESI): m/z = found (M+ + 1) 381.02, calc. 380.06. Anal. calcd. for C20H14Cl2N4: C, 63.01; H, 3.70; N, 14.70. Found: C, 63.05; H, 3.72; N, 14.73.C). 1H NMR (400 MHz, DMSO-d6) δ 8.31–8.33 (d, J = 8 Hz, 2H, Ar phenyl) and 8.18–8.61 (d, J = 7 Hz, 2H, bridging phenyl-C2, C3, C5, C6–H), 7.72–7.75 (t, 4H Ar phenyl), 7.54 (s, 1H, –CHCH pyrimidine ring), 7.51 (s, 1H, –CHCH), 7.38–7.40 (d, J = 8 Hz, 2H), 7.25–7.27 (d, J = 9 Hz, 2H, pyrrole-C2, C4–H), 6.26 (s, 2H, pyrrole-C1, C5–H), 6.70 (s, 2H, NH2), 1.07–1.26 (m, 6H, isopropyl 2-CH3). 13C NMR (100 MHz, DMSO-d6) δ 164.14, 164.29, 157.00, 152.51, 145.72, 141.91, 119.39, 112.90, 112.14, 111.45, 99.9. MS (ESI): m/z = found (M+ + 1) 355.17, calc. 354.18. Anal. calcd. for C23H22N4: C, 77.94; H, 6.26; N, 15.81. Found: C, 77.96; H, 6.28; N, 15.83.C). 1H NMR (400 MHz, DMSO-d6) δ 8.29–8.31 (d, J = 8 Hz, 2H, –CHCH, pyrimidine ring), 8.159–8.151 (d, J = 3 Hz, 2H, –CHCH, thiophene ring), 7.74–7.78 (s, 4H, bridging phenyl-C2, C3, C5, C6–H), 7.51–7.52 (s, 2H, pyrrole-C2, C4–H), 7.23–7.25 (d, J = 8 Hz 1H, –CHCH, thiophene ring), 6.32 (s, 2H, pyrrole-C1, C5–H), 6.76 (s, 2H, NH2). 13C NMR (100 MHz, DMSO-d6) δ164.13, 164.00, 160.54, 143.76, 141.89, 134.10, 130.30, 128.87, 128.82, 128.45, 119.31, 111.43, 100.30. MS(ESI): m/z = found (M+ + 1) 319.07, calc. 318.09. Anal. calcd. for C18H14N4S: C, 67.90; H, 4.43; N, 17.06. Found: C, 67.92; H, 4.44; N, 17.08.C). 1H NMR (400 MHz, DMSO-d6) δ 8.33–8.35 (d, J = 8 Hz, 2H, Ar phenyl), 7.46–7.74 (t, 4H, bridging phenyl-C2, C3, C5, C6–H), 7.50–7.53 (q, 2H, pyrrole-C2, C4–H, 1H, –CHCH pyrimidine ring), 6.74 (s, 2H, pyrrole-C1, C5–H), 6.33 (s, 2H, NH2), 3.75–3.92 (s, 6H, 2OCH3), 3.35 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ 164.28, 153.49, 128.8, 119.47, 119.31, 111.42, 104.98, 60.61, 56.61. MS (ESI): m/z = found (M+ + 1) 403.16, calc. 402.17. Anal. calcd. for C23H22N4O3: C, 68.64; H, 5.51; N, 13.92. Found: C, 68.66; H, 5.53; N, 13.94.O), 1569.38 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 8.11–8.13 (d, J = 8 Hz, 2H, Ar phenyl), 8.08–8.10 (t, OH), 7.46–7.52 (m, 4H, bridging phenyl-C2, C3, C5, C6–H), 7.34–7.37 (J = 8 Hz, 2H, pyrrole-C2, C4–H), 7.25 (s, H, –CHCH pyrimidine ring), 7.16–7.17 (s, 2H, pyrrole-C1, C5–H), 6.38–6.39 (s, 3H, Ar phenyl), 5.16 (s, 2H, NH2), 4.00 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ165.24, 164.22, 148.44, 143.62, 141.75, 138.81, 134.58, 130.36, 128.91, 127.87, 119.55, 119.45, 119.40, 119.29, 118.97, 118.91, 111.94, 111.39, 105.26, 56.71, 27.10. MS (ESI): m/z = found (M+ + 1) 389.11, calc. 388.15. Anal. calcd. for C22H20N4O3: C, 68.03; H, 5.19; N, 14.42. Found: C, 68.05; H, 5.21; N, 14.45.O), 1564.81 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 8.11–8.13 (d, J = 8 Hz, 2H, Ar-phenyl), 7.93–7.95 (d, J = 8 Hz, 2H, bridging phenyl-C2, C6–H), 7.61–7.63 (d, J = 8 Hz, 2H, bridging phenyl-C3, C5–H), 7.49–7.51 (d, J = 8 Hz, 2H, pyrrole-C2, C4–H), 7.41 (s, 1H, CHCH, pyrimidine ring), 7.16–7.24 (s, 2H, Ar phenyl), 6.38 (s, 2H, pyrrole- C1, C5–H), 5.24 (s, 2H, NH2). 13C NMR (100 MHz, DMSO-d6) δ 164.42, 164.09, 141.92, 137.02, 134.19, 132.06, 129.52, 128.95, 124.56, 119.46, 119.31, 111.43, 101.83. MS (ESI): m/z = found (M+ + 1) 391.91, calc. 390.05. Anal. calcd. for C20H15BrN4: C, 61.39; H, 3.86; N, 14.32. Found: C, 61.43; H, 3.87; N, 14.34.O), 1561.02 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 8.02–8.12 (d, J = 8 Hz, 2H, Ar phenyl), 7.48–7.50 (d, 2H, pyrrole-C2, C4–H, and 1H, –CHCH, pyrimidine ring), 7.39-(s, 2H, bridging phenyl-C2, C3, C5, C6–H), 7.17 (s, 2H, Ar phenyl), and 6.76–6.78 (d, 2H, Ar phenyl), and 6.38 (s, 2H, NH2), 5.22 (s, 2H, pyrrole-C1, C5–H), 3.04 (s, 6H, 2CH3); 13C NMR (100 MHz, DMSO-d6) δ 166.02, 164.29, 163.54, 152.12, 141.97, 135.23, 128.38, 128.70, 128.27, 124.69, 120.05, 119.13, 111.75, 110.90, 102.40, 77.35, 77.03, 76.72. MS (ESI): m/z = found (M+ + 1) 356.91, calc. 355.00. Anal. calcd. for C22H21N5: C, 74.34; H, 5.96; N, 19.70. Found: C, 74.36; H, 5.99; N, 19.72.O), 1596.79 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 8.18–8.20 (d, J = 8 Hz, 2H, Ar phenyl), 7.78 (s, 1H, –CHCH, pyrimidine ring), 7.69–7.71 (d, 1H, Ar phenyl), 7.55–7.57 (d, 2H, pyrrole-C2, C4–H), 7.01–7.47 (m, 4H, bridging phenyl-C2, C3, C5, C6–H), 6.45 (s, 2H), 7.07–7.17 (s, 2H), 6.262–6.32 (s, 2H, pyrrole-C1, C5–H).5.39 (s, 2H, NH2), 4.01–4.07 (s, 6H, 2-OCH3). 13C NMR (100 MHz, DMSO-d6) δ 165.72, 164.83, 163.52, 151.27, 149.23, 134.77, 130.29, 128.47, 120.17, 120.05, 119.10, 111.03, 110.90, 109.97, 103.18, 77.37, 77.04, 76.73, 56.04, 56.00. MS (ESI): m/z = found (M++) 372.91, calc. 372.16. Anal. calcd. for C22H20N4O2: C, 70.95; H, 5.41; N, 15.04. Found: C, 70.97; H, 5.42; N, 15.06.O), 1564.46 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 8.11–8.14 (d, J = 8 Hz, 2H, Ar phenyl), 7.86 (s, 1H, –CHCH, pyrimidine ring), 7.82 (s, 2H, pyrrole-C2, C4–H), 7.61–7.63 (d, J = 7 Hz, 3H, Ar phenyl), 7.12–7.50 (m, 4H, bridging phenyl-C2, C3, C5, C6–H), 7.01–7.05 (s, 2H, –CHCH, aliphatic chain), 6.40–6.41 (s, 2H, pyrrole- C1, C5–H), 5.17 (s, 2H, NH2). 13C NMR (100 MHz, DMSO-d6) δ 164.96, 164.11, 163.35, 142.27, 136.14, 135.94, 134.50, 129.12, 128.43, 127.56, 126.49, 120.06, 119.10, 111.03, 105.81. MS (ESI): m/z = found (M++) 338.90, calc. 338.15. Anal. calcd. for C22H18N4: C, 78.06; H, 5.36; N, 16.56. Found: C, 78.08; H, 5.39; N, 16.59.O), 1566.53 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 9.45 (s, 1H, OH), 8.30–8.32 (d, 2H, –CH, Ar), 7.49–7.80 (m, 7H, bridging phenyl-C2, C3, C5, C6–H and -Ar), 7.82 (s, 2H, pyrrole-C1, C5–H), 6.91–6.93 (d, J = 8.0 Hz, 1H), 6.62 (s, 2H, pyrrole-C2, C4–H), 6.31 (s, 2H, NH2), 4.14–4.16 (s, 2H, –CH), 3.36 (s, 1H, –CH), 1.37–1.41 (s, 3H–CH); 13C NMR (100 MHz, DMSO-d6) δ 165.21, 164.26, 149.96, 147.28, 141.68, 134.58, 129.00, 128.82, 121.10, 119.41, 119.29, 115.93, 112.71, 111.36, 101.15, 64.61, 15.28. MS (ESI): m/z = found (M+ + 1) 372.91, calc. 372.16. Mass calc 372.16, Found (M++)372.91. Anal. calcd. for C22H20N4O2: C, 70.95; H, 5.41; N, 15.04. Found: C, 70.99; H, 5.43; N, 15.06.O), 1568.87 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 8.27–8.29 (s, 1H, OH), 8.03–8.14 (d, J = 8.0 Hz, 1H, Ar-phenyl), 7.68–7.73 (t, 3H) and 7.60 (s, 1H, bridging phenyl-C2, C3, C5, C6–H), 7.03–7.05 (d, J = 8 Hz, 1H, –CH pyrimidine), 6.60–6.74 (d, 2H, pyrrole-C1, C5–H), 6.32 (s, 4H, pyrrole-C2, C 4–H), 5.22–5 (s, 2H, NH2), 3.46 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ 164.89, 164.34, 164.07, 161.71, 130.57, 130.36, 128.81, 119.54, 119.43, 119.34, 119.19, 118.92, 111.95, 111.40. MS (ESI): m/z = found (M+ + 1) 359.12, calc. 358.14. Anal. calcd. for C21H18N4O2: C, 70.38; H, 5.06; N, 15.63. Found: C, 70.40; H, 5.09; N, 15.66.O), 1573.74 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 14.25 (s, 1H, OH), 8.33–8.35 (d, J = 8.4 Hz, 2H) and 7.82–7.83 (d, J = 6.0 Hz, 2H, Ar-phenyl), 7.74–7.76 (d, J = 8.4 Hz, 2H), 7.51 (s, 1H–CH-pyrimidine), 7.22 (s, 2H, pyrrole-C2, C4–H), 7.06–7.08 (d, J = 8 Hz, 1H, –Ar phenyl) and 6.85–6.89 (s, 2H, NH2), 6.32 (s, 2H, pyrrole-C1, C5–H), 3.46–3.81 (s, 3H, OCH3 Benz). 13C NMR (100 MHz, DMSO-d6) δ 165.91, 164.85, 161.62, 151.41, 149.36, 142.10, 129.15, 119.99, 119.54, 119.46, 119.29, 118.93, 118.26, 117.90, 111.51, 100.31, 56.34. MS (ESI): m/z = found (M+ + 1) 359.12, calc. 358.14. Anal. calcd. for C21H18N4O2: C, 70.38; H, 5.06; N, 15.63. Found: C, 70.42; H, 5.08; N, 15.66.O), 1562.84 cm−1 (Ar CC). 1H NMR (400 MHz, DMSO-d6) δ 8.19–8.21 (d, J = 8.0 Hz, 1H), 7.71–7.77 (t, 3H, CHCH), 7.48–7.51 (t, 5H, pyrrole-C2, C4–H), 7.30 (s, 1H, –CH–pyrimidine), 6.74–6.76 (dd, J = 10.4 Hz, 4H, bridging phenyl-C2, C3, C5, C6–H), 6.48 (s, 1H, NH2) and 6.28–6.31 (d, J = 12 Hz, 2H, pyrrole-C1, C5–H), 3.56–2.46 (s, 6H, 2CH3). 13C NMR (100 MHz, DMSO-d6) δ 164.90.164.75, 163.42, 155.61, 148.26, 141.10, 128.75, 119.79, 119.64, 11.56, 119.38, 118.56, 117.80, 111.71, 100.31, 41.03. MS (ESI): m/z = found (M+ + 1) 382.15, calc. 381.20. Anal. calcd. for C24H23N5: C, 75.56; H, 6.08; N, 18.36. Found: C, 75.59; H, 6.12; N, 18.40.5.3 ADMET, molecular docking, MMGBSA, DFT calculation & MD simulation
The crystal structures of Inha protein (PDB ID: 5OIR) from the PDB Database were used for docking studies. The InhA complexed protein structures were ready by Protein preparation wizard in Maestro Schrodinger Suite Release 2023-2 before performing docking. The absent hydrogens were added during the preparation and partial charges were given using OPLS-3E force field. Hydrogen and heavyweight atoms were minimized with constraints. Two-dimensional assemblies of 34 ligands with standard drugs prepared using Ligprep module in Maestro Schrodinger. With regard to this, Ligprep controls the protonation, and ionization conditions of the ligands, and given suitable bond commands.Acceptable ADMET profile with effectiveness of the most important benchmarks for the conclusion of a drug. Found possible chalcone and pyrimidine derivatives based on molecular docking study. Further selected for In silico ADMET prediction to check its draggability using QikProp module in Schrodinger. The results are shown in (Table 1).
Docking 3D in addition 2D pose of highest anti-tubercular activity showing compound 4g and involving the interaction with essential amino acids showing in (Fig. 7). Remaining compound showing better antitubercular activity glide docking score data and 2D, 3D images have been attached in ESI file.† The docking glide score shown in (Table 2).
The MMGBSA technique is a popular way to calculate a ligand's binding free energy to proteins or other macromolecules. It makes docking more efficient. Binding free energy calculate by the Prime module in Schrodinger software. For certain targets, MM-GBSA rescoring can be used to rank inhibitors and determine the proper binding poses score shown in (Table 3).
Schrodinger software's quantum physics suite, especially (Jaguar module), which is the company's tool for modelling electronic structures and quantum mechanics, may be used to do (DFT) computations and this module used for study the motion of electrons in atoms and molecules. It's based on the idea that the energy of a system is a function of its electron density, energy difference between the HOMO LUMO showed in (Table 8). After those molecular dynamics simulation was done (Desmond module of Schrodinger, LLC, New York, NY, version 2023-3). Molecular dynamics simulation was performed using Desmond module of Schrodinger suite (Desmond, Schrödinger, LLC, New York, NY, 2017). Performing MD simulation using OPLS4 forcefield. Molecular system was placed in an orthorhombic box in such a way that a buffer region of 10 Å was maintained between protein atoms and boundaries of the box. TIP3P water molecules was used to solvate the system and it is neutralized by adding 25 Na+ and 17 Cl− ions. The system was minimized and subjected to 200 ns MD simulation with NPT ensemble at 300 K temperature and 1.013 bar pressure while keeping other settings to their default values. In the MD simulation, a time step of 2 fs was used, while the energy and trajectory coordinates were recorded for every 5 ps. Maestro was used for analysing trajectories and calculating RMSDs from initial structures.
5.4 Biological activity
:20, and adjusted to an OD590 of 1.0. Stock solutions of the test compounds were serially diluted in 7H9-S medium to achieve two-fold concentration gradients in 96-well microtiter plates. Each plate included a sterility control, a growth control without drug, and wells containing the test compounds. To minimize evaporation, sterile water was added to peripheral wells. Plates were sealed and incubated at 37 °C for 7 days. Afterward, 30 μL of Alamar Blue reagent was added to each well and plates were further incubated overnight. A color change from blue to pink indicated bacterial growth, and the minimum inhibitory concentration (MIC) was defined as the lowest concentration of the compound that prevented this color shift.515.4.2.1 MTT-based cytotoxicity activity on normal cell line. Cytotoxicity was evaluated using the RAW 264.7 mouse macrophage cell line (Mouse macrophage cell line (RAW 264.7) procured from BITS-Pilani Hyderabad campus itself) at a test concentration of 50 μg mL−1. Cell viability was determined via the MTT assay, based on mitochondrial reduction of MTT to formazan after 48 hours of exposure. Cells were cultured in T25 flasks with RPMI medium containing 10% fetal bovine serum (FBS), (FBS was purchased from gibco, Thermo Fisher Scientific, USA). And 10
000 U per mL penicillin, and 10 mg per mL streptomycin, until they reached 80–90% confluency. Approximately 5000 cells were seeded per well in poly-L-lysine-coated 96-well plates and pre-incubated for 24 hours at 37 °C with 5% CO2 and 100% humidity. Test compounds were then added, and the cells were incubated for an additional 48 hours. Subsequently, 10 μL of MTT solution (0.5 mg mL−1) was added to each well, followed by 3 hours of incubation. The resulting formazan crystals were quantified by measuring absorbance at 595 nm and 625 nm.52,53 Cytotoxicity on mouse macrophage cell line RAW 264.7 of the synthesized compounds was carried out at Birla Institute of Technology and Science-Pilani, Hyderabad Campus, Jawahar Nagar, Hyderabad, Telangana, India. Cytotoxicity assay was performed under standard protocols by Jyothi Kumari, Dharmarajan Sriram.
5.4.2.2 MTT-based cytotoxicity activity on lung cancer cell line. The MTT assay is a colorimetric method that uses the reduction of the yellow dissolved in water tetrazolium dye to formazan crystals to measure cell growth and cytotoxicity. MTT is reduced to insoluble formazan crystals by mitochondrial lactate dehydrogenase, which is produced by living cells. Crystals dissolve in an appropriate solvent and show a purple blue, the intensity of which is related to volume of viable cells and detected by spectrometer at 570 nm.54,55 Lung Cancer cells procured from NCCS, Pune, India.
5.5 Enzyme inhibition studies
Sigma Aldrich provided the triclosan and NADH. The chosen compounds' stock solutions were made in DMSO so that, for all kinetic processes, the co-solvent's final concentration remained constant at 5% (v/v) in a final volume of 1 mL. As previously mentioned, trans-2-dodecenoyl-coenzyme A (DDCoA) and naturally occurring the protein InhA were used in kinetic tests. In summary, reactions were carried out at 25 °C in a buffer composed of water (30 mM PIPES (piperazine-N,N′-bis(2-ethanesulfonic acid)) and 150 mM NaCl pH 6.8) that included the test chemical (at 50 mM), 250 mM cofactor (NADH), and 50 mM substrate (DDCoA). InhA (final concentration of 100 nM) was added to start the reactions, and the oxidation of NADH was observed at a constant wavelength of 340 nm.56 The % of inhibition of InhA activity (the reaction's starting velocity) relative to control the reaction without an inhibitor was used to indicate the activity of each selected derivative.Abbreviations
| ADME | Absorption, distribution, metabolism, and excretion |
| Ar | Aromatic |
| B3LYP | Becke, 3-parameter, Lee–Yang–Parr |
| d | Doublet |
| DDCoA | Trans-2-dodecenoyl-coenzyme A |
| DFT | Density functional theory |
| DMSO | Dimethyl sulfoxide |
| DNA | Deoxyribonucleic acid |
| DprE1 | Decaprenyl phosphoryl-β-D-ribose 2′-epimerase |
| FAS | Fatty acid synthesis |
| FBS | Fetal bovine serum |
| HOMO | Highest occupied molecular orbital |
| INH | Isoniazid |
| IR | Infra-red |
| KAT | G enzyme catalase-peroxidase |
| LUMO | Lowest unoccupied molecular orbital |
| m | Multiplate |
| MD | Molecular dynamic |
| MDR | TB multidrug-resistant tuberculosis |
| MIC | Minimum inhibitory concentration |
| MM/GBSA | Molecular mechanics with generalized born and surface area |
| MS | Mass spectra |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay |
| NAD | Nicotinamide adenine dinucleotide |
| NMR | Nuclear magnetic resonance |
| NTM | Non-tuberculosis mycobacterial |
| OPLS-AA | Optimized potentials for liquid simulations in all atom version |
| PIPES | Piperazine-N,N′-bis(2-ethanesulfonic acid) |
| q | quadrate |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| RPMI | Roswell Park Memorial Institute Medium |
| TB | Tuberculosis |
| TIP 3P | Transferable intermolecular potential with 3 points |
| TLC | Thin-layer chromatography |
| TMS | Tetramethyl silane |
| vdW | Van der Waals |
| WHO | World health organization |
| XDR | TB extensively drug-resistant tuberculosis |
Data availability
The ESI† contain the spectral data, in silico ESI† and biological activity data.Author contributions
Deepshikha Singh: writing – review & editing, writing – original draft, software, resources, methodology, investigation, data curation, conceptualization, funding acquisition. Praveen M. Parkali: methodology & editing. Umme Hani, Riyaz Ali M. Osmani, Nazima Haider: visualization and formal analysis. Jyothi Kumari: performed anti-tubercular activity (MABA assay). Dharmarajan Sriram: supervision of anti-Tb activity and cytotoxicity assay. Christian Lherbet: provided INHA enzyme inhibition activity. Sheshagiri R. Dixit: conceptualization, supervision, validation, review & editing.Conflicts of interest and competing interest
The authors declare no financial conflicts of interest or personal affiliations that could have influenced the research conducted in this study or any kind of conflict of interest. The authors declare the following financial interests/personal relationships which may be considered as potential competing interests. An Indian patent entitled “Synthetic strategies: methods for producing pyrimidine as anti-tubercular agent” has been granted related to this work (Indian Patent No. 568101 granted on 30th June-2025).Acknowledgements
This research work was funded by Department of Science and Technology (DST), New Delhi by conferring the award of DST-WOMEN SCIENTIST Fellowship & Grant via DST Sanction Order No. DST-WOS-A (File No.: CS-13/WOS-A/2021) as financial support. The authors are thankful to the Management and Authorities of JSS Academy of Higher Education and Research (JSS AHER), Mysuru, India, for providing all computer aided drug design laboratory facilities. Authors also express gratitude to Mr Parthasarathi S., Analyst USIC, JSS College of Pharmacy, Mysuru, and the Director of SAIF, Karnataka University, Dharwad, and Vijnana Bhawan Mysore University, Mysuru, Karnataka, India, who have provided some of the NMR and mass spectral data. The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through a Large Group Research Project under grant number RGP.2/259/46. The biological activity (cytotoxicity on A549 human lung cancer cell line) was carried out at Averin Biotech Labs, Bangalore, India and cytotoxicity assay was performed under standard protocols by Shiva Shankar Reddy G.References
- G. B. Migliori, C. W. M. Ong, L. Petrone, L. D’ambrosio, R. Centis and D. Goletti, The definition of tuberculosis infection based on the spectrum of tuberculosis disease, Breathe, 2021, 17(3), 210079, DOI:10.1183/20734735.0079-2021.
- Report GT, WHO Reports Alarming Rise in Tuberculosis Cases in 2023, 2024, pp. 2021–2023 Search PubMed.
- R. Villar-Hernández, A. Ghodousi, O. Konstantynovska, R. Duarte, C. Lange and M. Raviglione, Tuberculosis: current challenges and beyond, Breathe, 2023, 19(1), 220166, DOI:10.1183/20734735.0166-2022.
- Global tuberculosis report 2024 by World Health Organization – Books on Google Play, n.d., https://play.google.com/store/books/details?id=cPwuEQAAQBAJ%26rdid=book-cPwuEQAAQBAJ%26rdot=1%26source=gbs_vpt_read%26pcampaignid=books_booksearch_viewport, accessed December 19, 2024 Search PubMed.
- V. A. Dartois and E. J. Rubin, Anti-tuberculosis treatment strategies and drug development: challenges and priorities, Nat. Rev. Microbiol., 2022, 20(11), 685–701, DOI:10.1038/s41579-022-00731-y.
- D. Singh, V. Singh, S. P. Mandal, K. Dsouza, B. R. P. Kumar and S. R. Dixit, Drug targets, current and future therapeutics for the treatment of multi drug resistant tuberculosis with their clinical applications: a critical review, Curr. Drug Ther., 2024, 19, 317–326, DOI:10.2174/1574885519666230830125139.
- A. N. Unissa, S. Subbian, L. E. Hanna and N. Selvakumar, Overview on mechanisms of isoniazid action and resistance in Mycobacterium tuberculosis, Infect. Genet. Evol., 2016, 45, 474–492, DOI:10.1016/J.MEEGID.2016.09.004.
- S. L. Parikh, G. Xiao and P. J. Tonge, Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid, Biochemistry, 2000, 39, 7645–7650, DOI:10.1021/BI0008940.
- K. Rožman, I. Sosič, R. Fernandez, R. J. Young, A. Mendoza and S. Gobec, et al., A new “golden age” for the antitubercular target InhA, Drug Discov. Today, 2017, 22, 492–502, DOI:10.1016/J.DRUDIS.2016.09.009.
- P. Kamsri, N. Koohatammakun, A. Srisupan, P. Meewong, A. Punkvang and P. Saparpakorn, et al., Rational design of InhA inhibitors in the class of diphenyl ether derivatives as potential anti-tubercular agents using molecular dynamics simulations, SAR QSAR Environ. Res., 2014, 25, 473–488, DOI:10.1080/1062936X.2014.898690.
- M. E. Boyne, T. J. Sullivan, C. W. AmEnde, H. Lu, V. Gruppo and D. Heaslip, et al., Targeting fatty acid biosynthesis for the development of novel chemotherapeutics against Mycobacterium tuberculosis: evaluation of A-ring-modified diphenyl ethers as high-affinity InhA inhibitors, Antimicrob. Agents Chemother., 2007, 51, 3562–3567, DOI:10.1128/AAC.00383-07.
- X. He, A. Alian and P. R. Ortiz de Montellano, Inhibition of the Mycobacterium tuberculosis enoyl acyl carrier protein reductase InhA by arylamides, Bioorg. Med. Chem., 2007, 15, 6649–6658, DOI:10.1016/J.BMC.2007.08.013.
- X. He, A. Alian, R. Stroud and P. R. Ortiz De Montellano, Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis, J. Med. Chem., 2006, 49, 6308–6323, DOI:10.1021/JM060715Y.
- U. H. Manjunatha, S. P. S. Rao, R. R. Kondreddi, C. G. Noble, L. R. Camacho and B. H. Tan, et al., Direct inhibitors of InhA active against Mycobacterium tuberculosis, Sci. Transl. Med., 2015, 7, 269ra3, DOI:10.1126/SCITRANSLMED.3010597.
- B. Ardiansah, Chalcones bearing N, O, and S-heterocycles: recent notes on their biological significances, J. Appl. Pharm. Sci., 2019, 9, 117–129, DOI:10.7324/JAPS.2019.90816.
- A. Bavishi, H. Vala, A. Radadiya, S. Swami, S. Thakrar and D. Sarkar, et al., Synthesis, biological screening, and molecular docking of hybrid pyrazole scaffolds for antitubercular and antimicrobial activity, ChemistrySelect, 2025, 10, e202404830, DOI:10.1002/SLCT.202404830.
- A. Yadav, V. Sharma and G. Singh, Anti-inflammatory potential of chalcone related compounds: an updated review, ChemistrySelect, 2024, 9, e202401321, DOI:10.1002/SLCT.202401321.
- B. Chaithanya, D. P. Chary and V. R. Anna, Synthesis and biological evaluation of chalcone incorporated thaizole-isoxazole derivatives as anticancer agents, Chem. Data Collect., 2025, 55, 101177, DOI:10.1016/J.CDC.2024.101177.
- C. X. yin, Z. Z. jia, X. J. lin, M. Yang and Z. Wang, Experimental and molecular docking studies of estrogen-like and anti-osteoporosis activity of compounds in Fructus Psoraleae, J. Ethnopharmacol., 2021, 276, 114044, DOI:10.1016/J.JEP.2021.114044.
- D. U. Dave, H. Patel, N. C. Bhoraniya, P. Pankhaniya and D. M. Purohit, Synthesis, spectral studies and antimicrobial screening of some new chalcone and isoxazole derivatives bearing furan nucleus, World J. Pharm. Res., 2024, 13(12), 747–756, DOI:10.20959/wjpr202412-32783.
- R. Gaur, K. S. Jyoti, H. S. Cheema, F. Khan and M. P. Darokar, et al., Synthesis, molecular modelling studies of artemisinin-chalcone derivatives and their antimalarial activity evaluation, Nat. Prod. Res., 2024, 1–11, DOI:10.1080/14786419.2024.2375784.
- D. Elkhalifa, I. Al-Hashimi, A. E. Al Moustafa and A. Khalil, A comprehensive review on the antiviral activities of chalcones, J. Drug Target., 2021, 29, 403–419, DOI:10.1080/1061186X.2020.1853759.
- R. Segawa, H. Takeda, T. Yokoyama, M. Ishida, C. Miyata and T. Saito, et al., A chalcone derivative suppresses TSLP induction in mice and human keratinocytes through binding to BET family proteins, Biochem. Pharmacol., 2021, 194, 114819, DOI:10.1016/J.BCP.2021.114819.
- R. C. Choudhari, K. Kaur, A. Das and V. Jaitak, Synthesis, and in-silico studies of indole-chalcone derivatives targeting estrogen receptor alpha (ER-α) for breast cancer, Curr. Comput. Aided Drug Des., 2023, 20, 640–652, DOI:10.2174/0115734099263650230926053750.
- M. N. Gomes, E. N. Muratov, M. Pereira, J. C. Peixoto, L. P. Rosseto and P. V. L. Cravo, et al., Chalcone derivatives: promising starting points for drug design, Molecules, 2017, 22(8), 1210, DOI:10.3390/molecules22081210.
- N. Jeelan Basha and K. T. Akshay, Design, synthesis, drug-likeness, anti-inflammatory, antimicrobial activity, and molecular docking studies of pyrimidine analogs, Polycyclic. Aromat. Compd., 2024, 24(10), 6957–6969, DOI:10.1080/10406638.2024.2331519.
- W. Shehta, N. A. Alsaiari, B. Farag, M. M. Abdel-Aziz, S. Youssif, S. El-Kalyoubi, et al., Novel phthalimide-pyrimidine hybrids as potent anti-tubercular agents, 2024, DOI:10.21203/rs.3.rs-4397392/v1.
- M. S. Raghu, C. B. Pradeep Kumar, K. Yogesh Kumar, M. K. Prashanth, F. Alharethy and B. H. Jeon, Synthesis, biological evaluation and molecular docking study of pyrimidine linked thiazolidinedione derivatives as potential antimicrobial and antitubercular agents, Bioorg. Med. Chem. Lett., 2024, 103, 129707, DOI:10.1016/j.bmcl.2024.129707.
- A. Bhatnagar and G. Pemawat, Anticancer and antibacterial activeness of fused pyrimidines: newfangled updates, Bioorg. Chem., 2024, 153, 107780, DOI:10.1016/J.BIOORG.2024.107780.
- H. Kaur, L. Singh, K. Chibale and K. Singh, Structure elaboration of isoniazid: synthesis, in silico molecular docking and antimycobacterial activity of isoniazid–pyrimidine conjugates, Mol. Divers., 2020, 24, 949–955, DOI:10.1007/S11030-019-10004-1/METRICS.
- M. Y. Lone, M. Athar, V. K. Gupta and P. C. Jha, Identification of Mycobacterium tuberculosis enoyl-acyl carrier protein reductase inhibitors: a combined in silico and in vitro analysis, J. Mol. Graph. Model., 2017, 76, 172–180, DOI:10.1016/J.JMGM.2017.07.005.
- S. Chinnamulagund, A. S. Joshi, S. Avunoori, V. H. Kulkarni, S. D. Joshi and C. D. Shrinivas Joshi Professor, Synthesis and antitubercular evaluation of certain pyrrole derivatives, Indo Am. J. Pharm. Res., 2023, 13(03) DOI:10.5281/zenodo.7755252.
- F. Cerreto, A. Villa, A. Retico and M. Scalzo, Studies on anti-candida agents with a pyrrole moiety. Synthesis and microbiological activity of some 3-aminomethyl-1,5-diaryl-2-methyl-pyrrole derivatives, Eur. J. Med. Chem., 1992, 27, 701–708, DOI:10.1016/0223-5234(92)90090-N.
- D. Deidda, G. Lampis, R. Fioravanti, M. Biava, G. C. Porretta and S. Zanetti, et al., Bactericidal activities of the pyrrole derivative BM212 against multidrug-resistant and intramacrophagic Mycobacterium tuberculosis strains, Antimicrob. Agents Chemother., 1998, 42, 3035–3037, DOI:10.1128/AAC.42.11.3035.
- R. Shah, P. K. Verma, M. Shah and S. Kumar, Design, synthesis, biological evaluation and molecular docking studies of thiophene derivatives, J. Iran. Chem. Soc., 2024, 21, 2501–2515, DOI:10.1007/S13738-024-03088-6/FIGURES/9.
- G. Dheeraj, P. Kumar, K. Apte, H. Ashtekar and S. R. Dixit, Molecular docking, ADME analysis, and pharmacophore modelling of benzoxazole fused azetidinone derivatives as antibreast cancer agents, Ann. Phytomed. Int. J., 2023, 12(1), 318–324, DOI:10.54085/AP.2023.12.1.37.
- A. Fatima, G. Khanum, S. K. Srivastava, P. Bhattacharya, A. Ali and H. Arora, et al., Exploring quantum computational, molecular docking, and molecular dynamics simulation with MMGBSA studies of ethyl-2-amino-4-methyl thiophene-3-carboxylate, J. Biomol. Struct. Dyn., 2023, 41, 10411–10429, DOI:10.1080/07391102.2023.2180667.
- M. Khanal, A. Acharya, R. Maharjan, K. Gyawali, R. Adhikari and M. D. Das, et al., Identification of potent inhibitors of HDAC2 from herbal products for the treatment of colon cancer: molecular docking, molecular dynamics simulation, MM/GBSA calculations, DFT studies, and pharmacokinetic analysis, PLoS ONE, 2024, 19, e0307501, DOI:10.1371/JOURNAL.PONE.0307501.
- M. I. Choudhary, M. Shaikh, M. Tul-Wahab and M. Ur-Rahman, In silico identification of potential inhibitors of key SARS-CoV-2 3CL hydrolase (Mpro) via molecular docking, MMGBSA predictive binding energy calculations, and molecular dynamics simulation, PLoS One, 2020, 15, e0235030, DOI:10.1371/journal.pone.0235030.
- S. Jain, S. Sharma and D. J. Sen, Virtual screening, docking, ADMET and molecular dynamics: a study to find novel inhibitors of mycobacterium tuberculosis targeting QcrB, Jordan J. Chem. (JJC), 2021, 16, 131–146, DOI:10.47014/16.3.4.
- I. Ahmad, H. Jadhav, Y. Shinde, V. Jagtap, R. Girase and H. Patel, Optimizing bedaquiline for cardiotoxicity by structure based virtual screening, DFT analysis and molecular dynamic simulation studies to identify selective MDR-TB inhibitors, In Silico Pharmacol., 2021, 9(1), 23, DOI:10.1007/S40203-021-00086-X.
- D. Panigrahi and S. K. Sahu, Computational approaches: atom-based 3D-QSAR, molecular docking, ADME-Tox, MD simulation and DFT to find novel multi-targeted anti-tubercular agents, BMC Chem., 2025, 19, 39, DOI:10.1186/S13065-024-01357-2.
- A. Krishnan, F. I. Khan, S. Sukumar and M. K. A. Khan, Identification of potential molecular targets and repurposed drugs for tuberculosis using network-based screening approach, molecular docking, and simulation, J. Biomol. Struct. Dyn., 2025, 43(1), 73–91, DOI:10.1080/07391102.2023.2279699.
- S. Thapa, M. S. Biradar, S. L. Nargund, I. Ahmad, M. Agrawal and H. Patel, et al., Synthesis, molecular docking, molecular dynamic simulation studies, and antitubercular activity evaluation of substituted benzimidazole derivatives, Adv. Pharmacol. Pharm. Sci., 2024, 2024, 9986613, DOI:10.1155/2024/9986613.
- I. Pauli, R. N. Dos Santos, D. C. Rostirolla, L. K. Martinelli, R. G. Ducati and L. F. S. M. Timmers, et al., Discovery of new inhibitors of Mycobacterium tuberculosis InhA enzyme using virtual screening and a 3D-pharmacophore-based approach, J. Chem. Inf. Model., 2013, 53, 2390–2401, DOI:10.1021/CI400202T.
- A. Chollet, L. Maveyraud, C. Lherbet and V. Bernardes-Génisson, An overview on crystal structures of InhA protein: apo-form, in complex with its natural ligands and inhibitors, Eur. J. Med. Chem., 2018, 146, 318–343, DOI:10.1016/J.EJMECH.2018.01.047.
- F. Prati, F. Zuccotto, D. Fletcher, M. A. Convery, R. Fernandez-Menendez and R. Bates, et al., Screening of a novel fragment library with functional complexity against Mycobacterium tuberculosis InhA, ChemMedChem, 2018, 13, 672–677, DOI:10.1002/CMDC.201700774.
- A. Hospital, J. R. Goñi, M. Orozco and J. L. Gelpí, Molecular dynamics simulations: advances and applications, Adv. Appl. Bioinform. Chem., 2015, 8, 37, DOI:10.2147/AABC.S70333.
- S. D. Joshi, S. R. Dixit, M. N. Kirankumar, T. M. Aminabhavi, K. V. S. N. Raju and R. Narayan, et al., Synthesis, antimycobacterial screening and ligand-based molecular docking studies on novel pyrrole derivatives bearing pyrazoline, isoxazole and phenyl thiourea moieties, Eur. J. Med. Chem., 2016, 107, 133–152, DOI:10.1016/J.EJMECH.2015.10.047.
- S. Sahana, G. R. Vijayakumar, R. Sivakumar, D. Sriram and D. V. Saiprasad, Synthesis, docking study and in vitro evaluation of anti-tuberculosis activity of tri substituted imidazoles containing quinoline moiety, J. Korean Chem. Soc., 2022, 66, 194–201, DOI:10.5012/JKCS.2022.66.3.194.
- L. A. Collins and S. G. Franzblau, Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium, Antimicrob. Agents Chemother., 1997, 41, 1004–1009, DOI:10.1128/AAC.41.5.1004.
- T. M. Dhameliya, K. I. Patel, R. Tiwari, S. K. Vagolu, D. Panda and D. Sriram, et al., Design, synthesis, and biological evaluation of benzo[d]imidazole-2-carboxamides as new anti-TB agents, Bioorg. Chem., 2021, 107, 104538, DOI:10.1016/J.BIOORG.2020.104538.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63, DOI:10.1016/0022-1759(83)90303-4.
- C. M. Tsai, R. P. Perng, K. T. Chang, D. Venzon and A. F. Gazdar, Evaluation of the relative cytotoxic effects of anticancer agents in serum-supplemented versus serum-free media using a tetrazolium colorimetric assay, Jpn. J. Cancer Res., 1996, 87, 91–97, DOI:10.1111/j.1349-7006.1996.tb00205.x.
- P. Hoffmann, J. Azéma-Despeyroux, F. Goncalves, A. Stamilla, N. Saffon-Merceron and F. Rodriguez, et al., Imidazoquinoline derivatives as potential inhibitors of InhA enzyme and Mycobacterium tuberculosis, Molecules, 2024, 29, 3076, DOI:10.3390/MOLECULES29133076/S1.
- S. D. Joshi, S. R. Dixit, J. Basha, V. H. Kulkarni, T. M. Aminabhavi and M. N. Nadagouda, et al., Pharmacophore mapping, molecular docking, chemical synthesis of some novel pyrrolyl benzamide derivatives and evaluation of their inhibitory activity against enoyl-ACP reductase (InhA) and Mycobacterium tuberculosis, Bioorg. Chem., 2018, 81, 440–453, DOI:10.1016/J.BIOORG.2018.08.035.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra03004h |
| This journal is © The Royal Society of Chemistry 2025 |