DOI:
10.1039/D5RA02813B
(Review Article)
RSC Adv., 2025,
15, 18034-18088
Vistas in the domain of 3-acetyl-4-hydroxy-2-quinolinone derivatives (AHQ) and their applications
Received
22nd April 2025
, Accepted 20th May 2025
First published on 4th June 2025
Abstract
This review discusses the significant advances in the current status and latest synthesis techniques for N-substituted 3-acetyl-4-hydroxyquinolinones. They are an important class of alkaloids possessing different electrophilic and nucleophilic centers. In this review, we comprehensively summarize the synthesis of 3-acetylquinolones via various reactions, emphasizing recent developments and challenges. Synthetic transformations of quinolone are focused on electrophilic substitution reactions supported by their mechanistic pathways, as well as nucleophilic substitution reactions and cycloaddition reactions, which have allowed access to an extensive scope of binary and fused heterocyclic scaffolds, including pyrazoles, imidazoles, tetrazoles, pyridines, pyrimidines, triazines, azepines, and pyranones. Also, the utilization of quinolone reactions as key intermediates in the ongoing and forthcoming marketed pharmaceutical syntheses is discussed. Additionally, this review covers various potential applications, including complexation, fluorescence sensing, chemical pH sensors, agricultural, and anticorrosion.
1. Introduction and scope
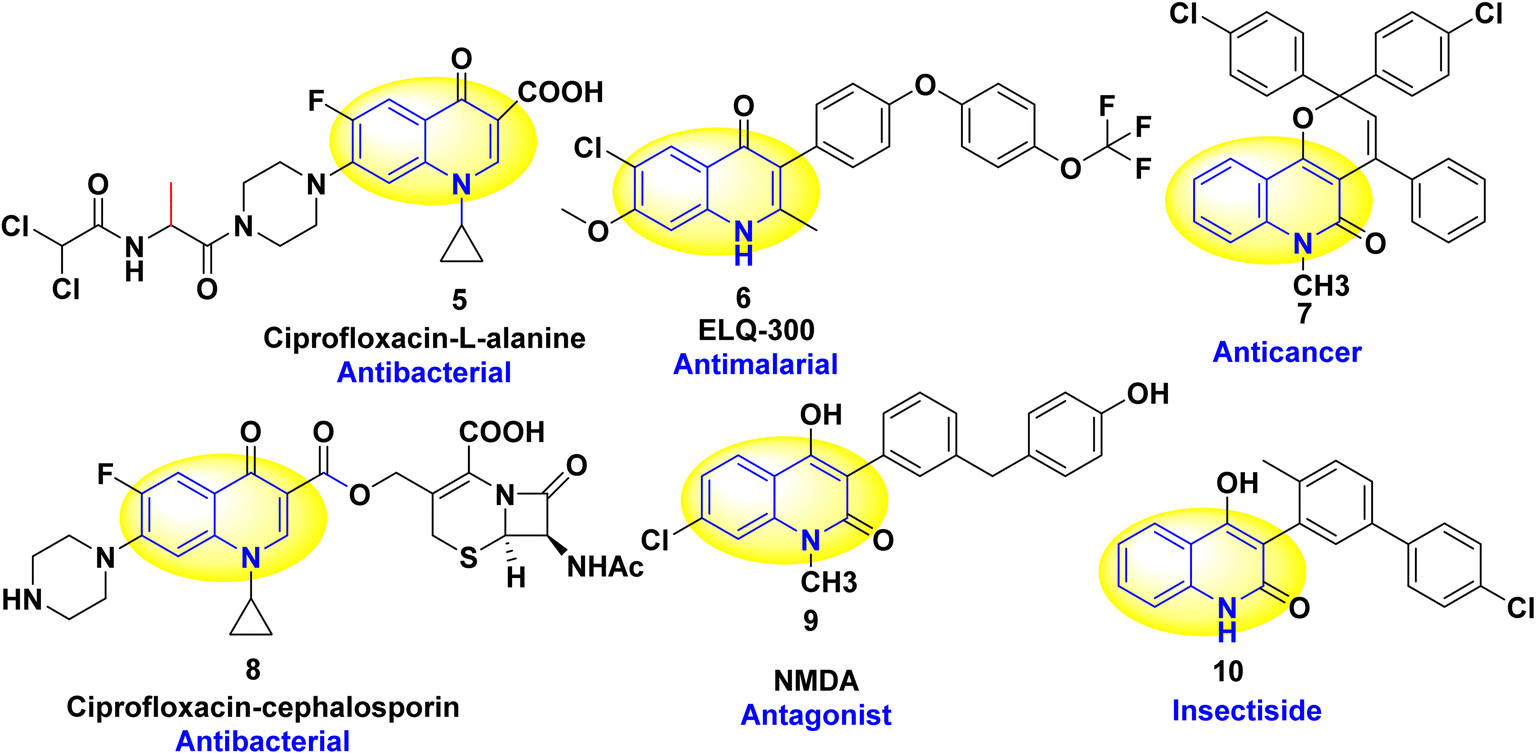
Heterocyclic skeletons which contain a nitrogen atom represent a common unit of a vast amount of marketed drugs,1 constituting the most extensive and diverse collection of natural, synthetic organic compounds, agrochemicals, and pharmaceuticals.2–4 Furthermore, the chemistry of heterocycles aggregates an essential branch of the field of drug design and the evolution of novel biologically active compounds,5–11 including antiparasitic,12,13 anti-HIV,14,15 antiepileptic drugs,16,17 analgesic,18,19 anthelmintic,20,21 inhibitors of platelet aggregation, anti-inflammatory,22–25 antibacterial and antiviral drugs,26–29 atherosclerosis,30,31 inhibitors for Parkinson's disease,32 antiproliferative, pulmonary antifibrotic agents33 and antitumor.34–36 Additionally, they provide one of the most fruitful sources for drug discovery and development, owing to synthesizing various scaffolds via robust synthetic approaches.37–40 These are important in synthesizing dyes, pigments, and polymeric materials41–43 due to their optical and fluorescence properties.44 The ongoing research in synthetic organic chemistry is dedicated to advancing these goals and addressing the demand for improved and sustainable approaches in synthesizing aromatic heterocyclic compounds.2–4 Easily accessible nitrogenous scaffolds are fundamental feedstocks; among these, quinoline and quinolones have piqued the interest of scientists since the 19th century, as over 600 quinoline derivatives have been extracted from natural sources to date.
Quinolinones have been perceived for their fascinating structural notions and versatile applications.45–56 Also, the quinolinones structures are pivotal building blocks,46 as they are commonly conspicuous scaffolds in a wide range of pharmacologically active synthetic and natural compounds.57–68 Quinolin-2-one, also referred to as benzo[b]pyridine, and 1-azanaphthalene, is important classes of natural product as N-based heterocycles which extracted from Toddalia asiatica leaves and plants, marine organisms and microorganisms which containing the quinolinone derivatives alkaloid findersine,52,69–75 consists of a benzene ring fused with a pyridine ring, sharing two carbon atoms between them and nitrogen atom not present as ring junction atom.
Quinoline is a weak base capable of forming salts with acids. It reacts similarly to pyridine and benzene and undergoes either nucleophilic or electrophilic substitution processes.47,76–81 The importance of the quinoline moiety is being a vital component in many naturally occurring heterocyclic and medicinal plant families. In addition, bucharidine 1 and foliosidine 2 are quinoline alkaloids that are extracted from Haplophyllum foliosum and Haplophyllum bucharicum, respectively (Fig. 1). Also, both of them have viral-RNA polymerase inhibitory action and estrogenic action, such as compounds 3 and 4, which strongly inhibit the replication of the hepacivirus C.82–84
 |
| | Fig. 1 Some natural alkaloids quinolinone-based molecules. | |
Additionally, quinolines have various potential biological and pharmacological activities including antimalarial,84–86 antibacterial,87 antifungal,88,89 anticancer,90–92 anti-HIV (Human Immunodeficiency Virus),93 antiviral,94 antitumor,95–97 anthelmintic,98 antioxidant,49 cardiotonic,76,99–101 anticonvulsant,102,103 anti-inflammatory and analgesic properties76,77,104,105 (Fig. 2).
 |
| | Fig. 2 Chemical structure of biologically active quinolinone scaffolds. | |
2. Chemical reactivity
2.1. 3-Acetyl-N-substituted/(un)-4-hydroxy-quinolin-2(1H)one derivatives (AHQ)
AHQ is the basic ring structure of numerous alkaloids and is a highly versatile scaffold with an extensive range of applications. Progress in synthetic technologies is leading to the production of various functionalized quinolinone derivatives as biosteric analogues to linomide (4-hydroxyquinolinone) derivatives.82,106–112
2.2. Structural feature of AHQ
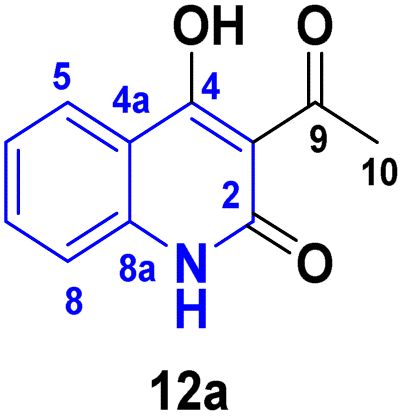
2.2.1. Tautomeric structures. Whereas, the β,β′-tricarbonyl (TC) group in AHQ compound provides an analogue of 3-acyl pyrrolidine-2,4-diones (tetramic acid), and has suitable sites for interaction with different nucleophilic reagents, with its regioselectivity of TC for AHQ [(A)–(D)] given below in a framework of molecular orbital perturbation.113Detsi and coworkers114 predicting the four tautomeric structures A–D. of AHQ [R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H] 12a (Fig. 3). The tautomeric equilibrium showed the presence of tautomeric forms that could be in one of four possible structures, A–D (Fig. 3). Whereas, semiempirical quantum calculations showed that the tautomer D was the most stable favorable one according to the total energy values.114,115 Whereas, the constitutions of AHQ derivatives were fully elucidated via different spectroscopic analyses as shown in Table 1.114,116–118
H] 12a (Fig. 3). The tautomeric equilibrium showed the presence of tautomeric forms that could be in one of four possible structures, A–D (Fig. 3). Whereas, semiempirical quantum calculations showed that the tautomer D was the most stable favorable one according to the total energy values.114,115 Whereas, the constitutions of AHQ derivatives were fully elucidated via different spectroscopic analyses as shown in Table 1.114,116–118
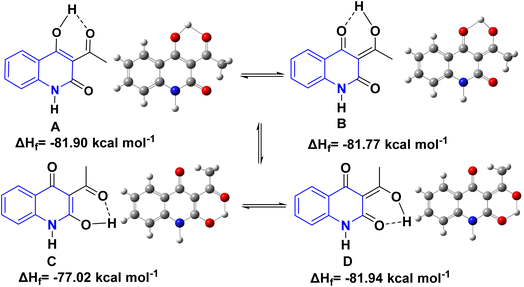 |
| | Fig. 3 Tautomeric form of AHQ 12a and its geometrical structures. | |
Table 1 Spectral data of AHQ 12a–c
| Spectroscopic technique |

|

|

|
| 1H NMR (DMSO-d6) |
2.72 (s, 3H, COCH3), 7.23 (t, 1H, H-6), 7.30 (d, 1H, H-8, J = 8.3 Hz), 7.65 (t, 1H, H-7), 7.99 (dd, 1H, H-5, J = 8.0, J = 1.2 Hz), 11.53 (s, 1H, NH) and 17.04 (s, 1H, OH)116 |
2.79 (s, 3H, COCH3), 3.52 (s, 3H, N–CH3), 7.30 (t, 1H, H-6), 7.50 (d, 1H, H-8, J = 8.1 Hz), 7.78 (t, 1H, H-7), 7.96 (dd, 1H, H-5, J = 8.4 Hz, J = 1.5 Hz) and 17.04 (s, 1H, OH)116 |
2.77 (3H, s, 3-COCH3), 7.12–7.41 (9HArom), 8.23 (s, OH)117 |
| Our work |
| 2.79 (s, 3H, COCH3, 6.58 (d, 1H, J = 9 Hz), 7.23 (m, 1H), 7.27 (d, 2H, J = 8 Hz), 7.45 (t, 1H, J = 8 Hz), 7.53 (t, 1H, J = 7 Hz), 7.61 (t, 2H, J = 8 Hz), 8.24 (d, 1H, J = 8 Hz), 17.16 (s, 1H, OH) |
| 13C NMR (DMSO-d6) |
205.7 (C-9), 174.7 (C-4), 161.1 (C-2), 140.5 (C-8a), 134.8 (C-7), 124.7 (C-5), 122.0 (C-6), 115.5 (C-8), 113.3 (C-4a), 105.7 (C-3) and 30.5 (C-10)116 |
206.7 (C-9), 173.3 (C-4), 160.6 (C-2), 141.6 (C-8a), 135.7 (C-7), 125.4 (C-5), 122.3 (C-6), 114.4 (C-8), 115.3 (C-4a), 105.7 (C-3), 31.3 (C-10) and 28.9 (N–CH3)116 |
206.9 (C-9), 175.0 (C-4), 161.9 (C-2), 142 (1C), 137.4 (1C), 134.4 (1C), 130.3 (2C), 129.0 (2C), 125.8 (1C), 122.3 (1C), 117.7 (1C), 116.0 (1C), 115.2 (1C), 106.0 (1C), 31.4 (1C) |
| IR (KBr, vmax/cm−1) |
3360 (OH), 3160 (NH), 1661 (CO, acetyl), 1622 (CO, amide), 1606 (CC)116 |
3250 (OH), 1658 (CO, acetyl), 1623 (CO, amide 1598 (CC)117 |
3072 (CHArom), 2924 (CHAliph), 1654(CO, acetyl, enol form), 1618 (CO, amide).114,118 |
2.2.2. X-ray crystallographic. The constitution of AHQ 12a was supported by X-ray analysis, as the non-hydrogen atoms in compound 12a are located in a single plane with an accuracy of 0.02 Å, this phenomena indicated the formation of an intramolecular hydrogen bond O(3)–H(3O)⋯O(2) (H⋯O 1.41 Å, O–H⋯O 157°). Also, this phenomenon displayed the re-estimation of the intramolecular electronic charge as shown by the bond lengths O(2)–C(10) 1.255(2) and C(7)–C(8) bonds 1.393(2) (Fig. 4).119,120
 |
| | Fig. 4 X-ray structural analysis 12a.120 | |
3. Aspects of the AHQ synthetic approach
Quinolines can be synthesized via different synthetic strategies involving [3 + 3], [4 + 2] and cyclization methodologies (I–III).121
(I) [3 + 3] Annulation: This strategy includes established procedures, for instance, the Skraup, Conrad-Limpach, Doebner-von Miller, Combes, and Gould-Jacobs syntheses. However, the drawback of these techniques is that they are restricted in regioselectivity for the synthesis of multi-substituted quinolines.121–123
(II) [4 + 2] Annulation: There are two techniques to build pyridine nuclei annulated to the benzene ring as shown in designed forms (II-a and II-b), as the first one has minimal regioselectivity with a narrow substrate range.121,124,125 While the other method is more prevalent and employs easily available substrates to produce products with high regioselectivity as Ptzinger and Friedlander reactions.121,122,126,127
(III) Cyclization: There are four distinct ways to generate the pyridine ring of quinolines (III-a to III-d) using cyclization. However, they have not been achieved due to the complicated reaction methodology and the limited availability of the starting substrates (Fig. 5).128,129
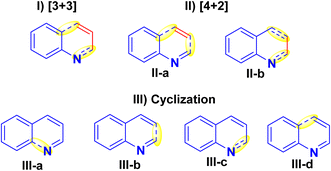 |
| | Fig. 5 Common synthetic methods of quinolines. | |
Synthetic approaches to construct AHQ derivatives have been achieved via many routes: aniline, alkaline hydrolysis of pyranoquinoline, anthranilic acid derivatives, heterocyclization of 3,1-benzoxazine-4-one and furo[3,2-c]quinoline.
3.1. Synthesis from amines/pyrano[3,2-c]quinolindione
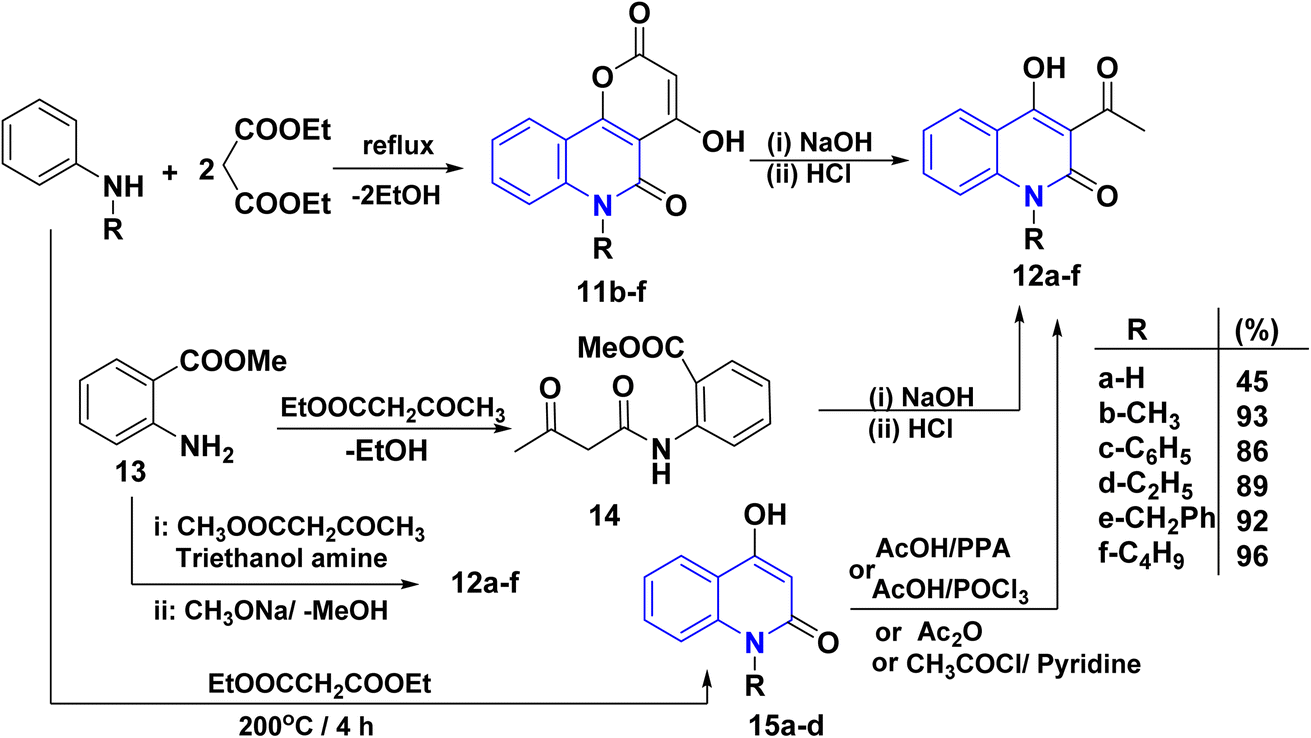
Quinoline derivatives 12a–f were prepared by cyclocondensation of substituted amines and diethyl malonate in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 for 3–6 h. Till the calculated amount of ethanol was collected using Dean Stark by a short Vigreux column as distillation system afforded the lactone 11, followed by alkaline hydrolysis [NaOH (2 N)], of lactone 11 and subsequent decarboxylation of the intermediate β-oxocarboxylate then acidified with HCl (2 N) to yield the products 12a–f in high yields (86–96%). In the case of 12b, compound 11 was synthesized following the methods of Kappe and Stadlbauer, which involved reacting N-methylaniline with diethyl malonate in diphenyl ether. However, Kappe's method utilized an excess of diethyl malonate, which served a dual purpose as both a reagent and a solvent (Scheme 1).115,130–135 Also, the ring opening of 11b–f by NaOH and subsequent spontaneous decarboxylation afforded 4-hydroxyquinolinone derivatives 12b–f (Scheme 1).112,133,135–137
:2 for 3–6 h. Till the calculated amount of ethanol was collected using Dean Stark by a short Vigreux column as distillation system afforded the lactone 11, followed by alkaline hydrolysis [NaOH (2 N)], of lactone 11 and subsequent decarboxylation of the intermediate β-oxocarboxylate then acidified with HCl (2 N) to yield the products 12a–f in high yields (86–96%). In the case of 12b, compound 11 was synthesized following the methods of Kappe and Stadlbauer, which involved reacting N-methylaniline with diethyl malonate in diphenyl ether. However, Kappe's method utilized an excess of diethyl malonate, which served a dual purpose as both a reagent and a solvent (Scheme 1).115,130–135 Also, the ring opening of 11b–f by NaOH and subsequent spontaneous decarboxylation afforded 4-hydroxyquinolinone derivatives 12b–f (Scheme 1).112,133,135–137
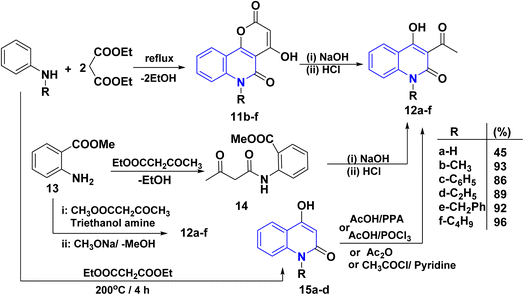 |
| | Scheme 1 Synthesis of AHQ 12a–f. | |
3.2. Synthesis from 4-hydroxyquinolin-2-ones
Cyclocondensation reaction of secondary amine and diethyl malonate afforded 4-hydroxyquinolin-2-ones 15a–d. After that, acetylation reaction of N-unsubstituted AHQ 15a–d with acetyl chloride using acetic acid or pyridine in the presence of polyphosphoric acid (PPA) or AcOH with phosphorus oxychloride (POCl3) yielded acetylquinoline derivatives 12a–d (Scheme 1).135–140
3.3. Synthesis from methyl 2-aminobenzoate/o-aminobenzaldehydes/o-aminoarylketones
Synthesis of acetylquinoline 12a was accomplished via acylating methyl anthranilate 13 using ethyl acetoacetate or methyl acetoacetate and triethanolamine by continuous removal of methanol or ethanol using Dean–Stark apparatus, resulting in the formation of 2-methoxycarbonyl anilide 13, followed by Dieckmann intramolecular cyclization of anilide derivative 14 (Scheme 1).120,141–143
3.4. Synthesis from 2-methyl-3,1-benzoxazin-4-one
The synthesis of 12a was performed through a two-step. First, 2-methyl-3,1-benzoxazin-4-one 16 was synthesized by [4 + 2] annulation of anthranilic acid or by using the iminium cation from a mixture of cyanuric chloride and dimethylformamide as a cyclizing agent, and was achieved under mild conditions.144 Next, C-acylated with ethyl acetoacetate by 3,1-benzoxazin-4-one 16 to form the ester 17. Finally, cyclization of ester 17 in a basic medium [aqueous Na2CO3/NaOH] at room temperature furnished the target compound 12a. This reaction pathway involves the nucleophilic addition of the active methylene of ethyl acetoacetate (EAA) to the carbonyl group of the benzoxazine ring 16, followed by intramolecular cyclization (Scheme 2).108,116,145–147
 |
| | Scheme 2 Synthesis of 12a from anthranilic acid. | |
N-Acylated anthranilic acid intermediate 18 is created from anthranilic acid and acid chloride in the presence of a catalytic amount of triethylamine (TEA) as the HCl scavenger via N-acylation reaction, affording N-acylated anthranilic acid. After that, intramolecular nucleophilic attack at intermediate 18 yielded benzoxazin-4-one ring 16. The cyclization reaction can be achieved by converting the carboxylic group of acetylated derivative 18 into a dynamic ester through either microwave irradiation or traditional heating (Scheme 3).144
 |
| | Scheme 3 Suggested mechanism for the synthesis of benzoxazinone derivatives 16. | |
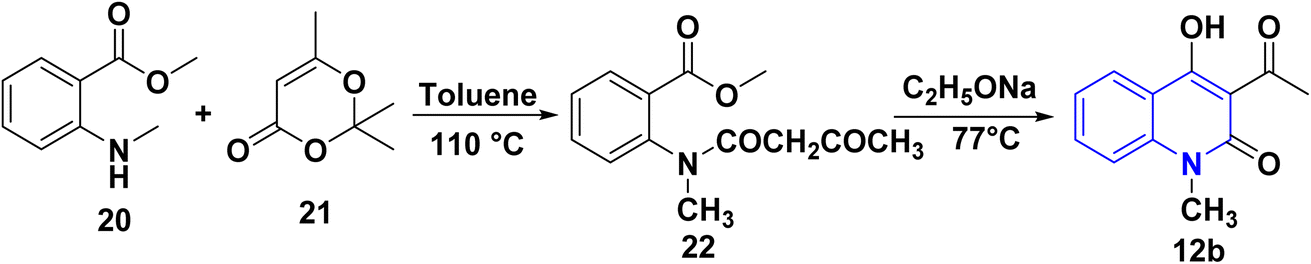
3.5. Synthesis from 1,3-dioxinone with methyl 2-(methylamino)benzoate
The synthesis of 12b was accomplished via an acylation reaction of secondary amine 20 with trimethyl-1,3-dioxinone 21, followed by a cyclization reaction of the acylation product 22 under basic conditions (Scheme 4).108
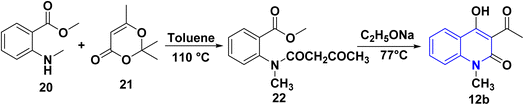 |
| | Scheme 4 Synthesis of 12b through acylation of 20 followed by cyclization of compound 22. | |
3.6. Synthesis from angular furo[3,2-c]quinolinone derivatives
In 2021, the group of Elgogary described the photooxygenation reaction of furo [3,2-c] quinolin-4(5H)-one derivatives 23 in CHCl3 led to the formation of the photocleaved product 24 via the intermediate 24A. Next, acid hydrolysis of quinolinone derivative 24 furnished 12b (Scheme 5).139
 |
| | Scheme 5 Photooxygenation reaction of 23. | |
3.7. Synthesis from ketene dithioacetal
Compound 12a was synthesized through in situ direct addition of carbon disulfide (CS2) to acetoacetanilide 25 in the presence of K2CO3 and tetrabutylammonium bromide (TBAB) as a green ionic liquid. The alkylation process was achieved using dimethyl sulfate, which yielded the ketene dithioacetal 26. In the same context, thermal cyclization of ketene 26 afforded the quinolinone 27 in fair yield. Finally, basic hydrolysis of the quinolone 27 furnished 3-acetyl-4-hydroxyquinolinone 12a (Scheme 6).140
 |
| | Scheme 6 Synthesis of 12a from acetoacetanilide. | |
3.8. Synthesis of dehydroxylated-3-acetylquinoline (AQ)
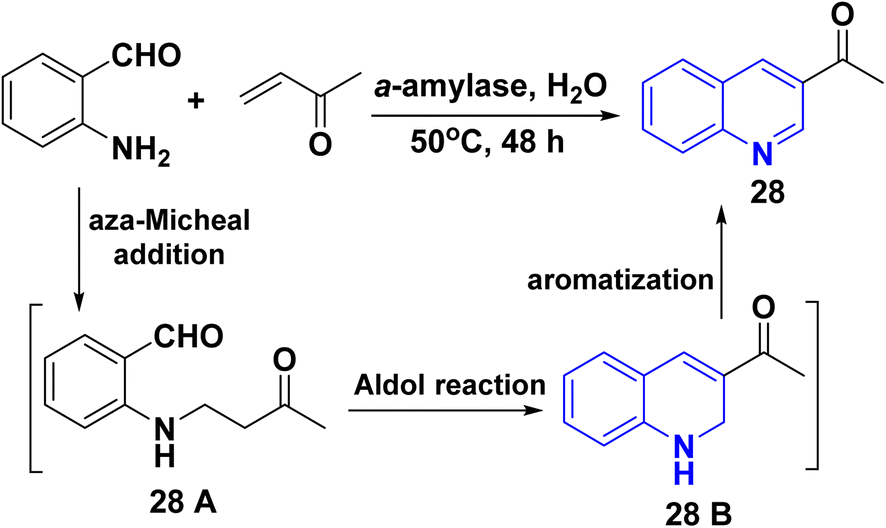
Also, synthesis of dehydroxylated-3-acetylquinoline (AQ) 28 via the reaction of 2-aminobenzaldehydes with α,β-unsaturated carbonyl compounds as methyl vinyl ketone in high catalytic efficiency (56–86% yield), through sequence process as aza-Michael reaction gave 28 A followed by Aldol reaction afforded 28 B then aromatized in presence of α-amylase catalyzed (Scheme 7).148
 |
| | Scheme 7 Synthesis of dehydroxylated-3-acetylquinoline (AQ) 28. | |
3.9. Synthesis of 2-acetylquinoline derivatives
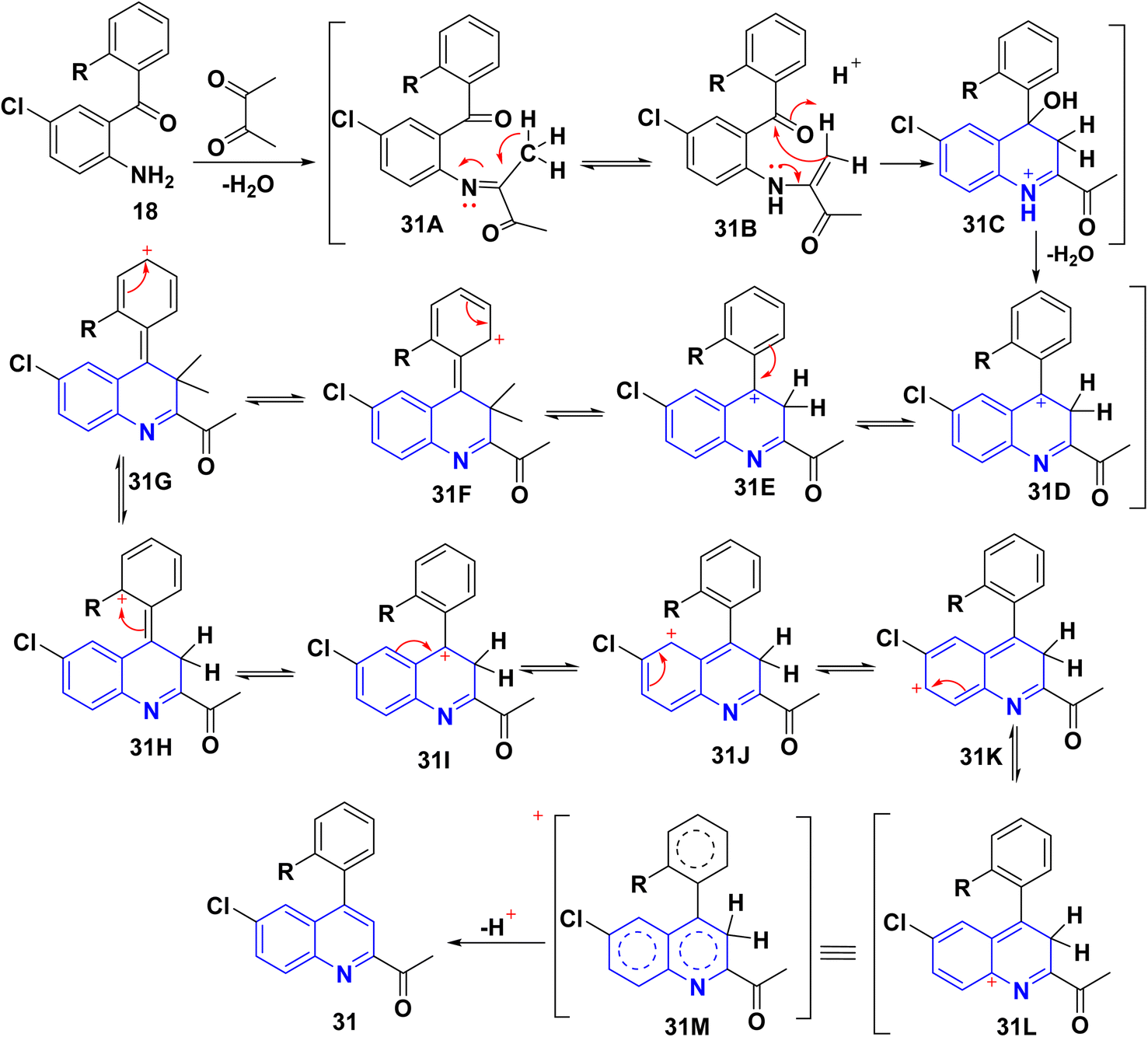
The reaction of o-aminoarylketones 29 with butanedione as a symmetrical 1,2-diketone 30 according to Friedländer synthesis. Similarly, o-aminoarylketones condensed with pentanedione (unsymmetrical 1,2-diketone) under acidic conditions led to the formation of regioselective 2-acetylquinoline derivatives 31 (Scheme 8). The suggested reaction mechanism (Scheme 9) is analyzed using density functional theory calculations at the B3LYP/6-311G (d,p) level. It was determined that all relative energy barriers and activation energies for the reaction steps are minimized at these theoretical levels.149
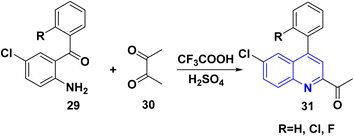 |
| | Scheme 8 Synthesis of 2-acetylquinoline 31. | |
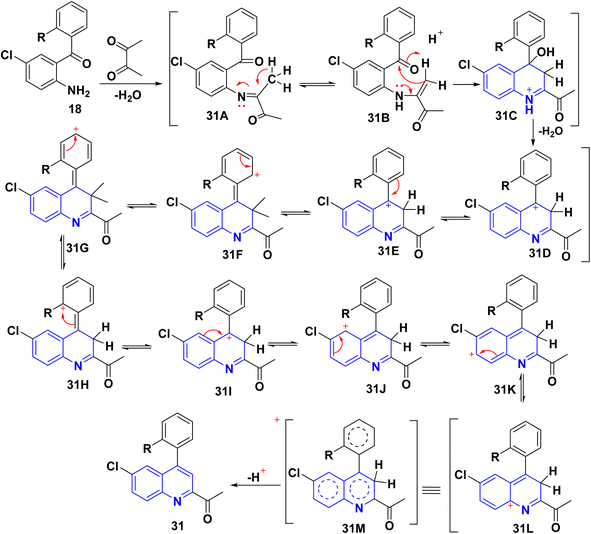 |
| | Scheme 9 Proposed mechanism for synthesis of 2-acetylquinoline 31. | |
A plausible mechanism for forming product 31 is shown in Scheme 9. Initially, o-aminoaryl ketone (29) condenses with butan-2,3-dione (30) in the presence of TFA with conc. H2SO4 to give an imine intermediate 31A as geometrical isomer E by eliminating H2O. Then, tautomerization of the 31A into a more activated form, the enamine intermediate 31B. Followed by nucleophilic attack of β-position of enamino group on the carbonyl carbon and this gives the benzylic carbocation (which stabilise through eight resonance hybrid structures 31(G-H) and subsequently loses a proton to form final product subsequently loses a proton via aromatization to form final product 31.
3.10. Synthesis of quinolinones
Zhang et al.150 reported an efficient methodology for preparing of 3,4-dihydroquinolinone 34 and quinolinone derivatives 35 through intramolecular cyclization of N-aryl cinnamides 33. Compound 33 was synthesized through Knoevenagel condensation of β-oxo-amides 32 with various aryl aldehydes, which were then subjected to intramolecular cyclization. The optimized conditions for cyclization, including solvent, the reaction temperature, and various catalysts such as Tf2O and DDQ, were investigated, whereas the optimum temperature is 80 °C, and N,N-dimethyl trifluoroacetamide (DTA) as solvent afforded the most satisfactory results. Under the optimised conditions, the reaction of a wide range of substrates such as α-acetyl-N-aryl cinnamides and α-benzoyl N-aryl secondary cinnamides proceeded efficiently to afford the corresponding quinoline-2(1H)-ones 34 and 35 (Scheme 10).150
 |
| | Scheme 10 Triflic anhydride-mediated synthesis of quinolinone. | |
4. Reactivity of AHQ
According to the current literature survey, the reactivity of AHQ 12a–f is part of scientific research with many reagents, as illustrated below. AHQ is a quinolin-2-one derivative with a unique chemical structure consisting of a β,β′-tricarbonyl (TC) group and thus a quinolinone ring with a carbonyl (acetyl), hydroxyl (enolate), and carbonyl (lactamic) group attached to it.
4.1. Electrophilic substitution reactions
An electrophilic substitution reaction is a chemical reaction in which an electrophile replaces a functional group attached to a compound, typically displacing a hydrogen atom. These reactions usually follow a three-step mechanism that includes forming an electrophile, forming a carbocation (an intermediate), and removing a proton from this intermediate. AHQ derivatives can efficiently undergo electrophilic substitution reactions such as alkylation, halogenation, Friedel–Crafts, and formylation.151
4.1.1. Alkylation.
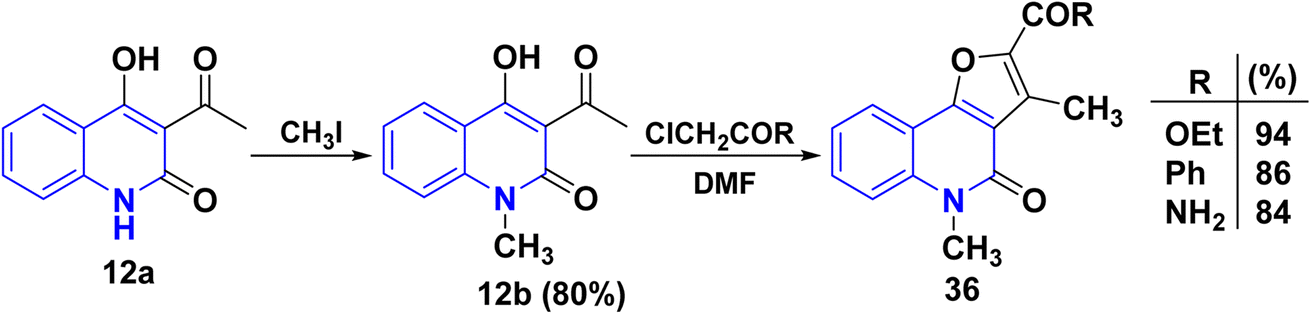
4.1.1.1. N1-Alkylation. Compound 12a can potentially undergo methylation at either the nitrogen atoms at positions 1 or the oxygen atom at C4. However, the methylation exclusively occurs at the nitrogen atom rather than the oxygen atom.115,116,143,145 Following that, angular N-methylfuro[3,2-c]quinolinone derivatives 36 were synthesized from 12b through the Rap–Stöermer reaction, with α-chlorocarbonyl substrates in the presence of K2CO3 in DMF (Scheme 11).
 |
| | Scheme 11 Formation of angular N-methylfuro[3,2-c]quinolinones 36. | |
Whereas, the reaction of 3-acetyl-6-chloro-4-phenylquinolinone 37 with different α-halocarbonyl compounds afforded linear tetrasubstituted furo[2,3-b]quinolines 38 (Scheme 12). Thus, angular fluoroquinolinones 36 have been synthesized via the reaction of 12b derivatives with α-chlorocarbonyl compounds, as applied to the Rap–Stöermer reaction through both conventional and/or microwave methods. Thus, the microwave irradiation provided higher yields (88–93%) compared to the conventional method (64–72%) (Scheme 12).143,152,153
 |
| | Scheme 12 Formation of linear furo[2,3-b]quinolinones 38. | |
 |
| | Scheme 13 Synthetic route of 41 from 12b,c. | |
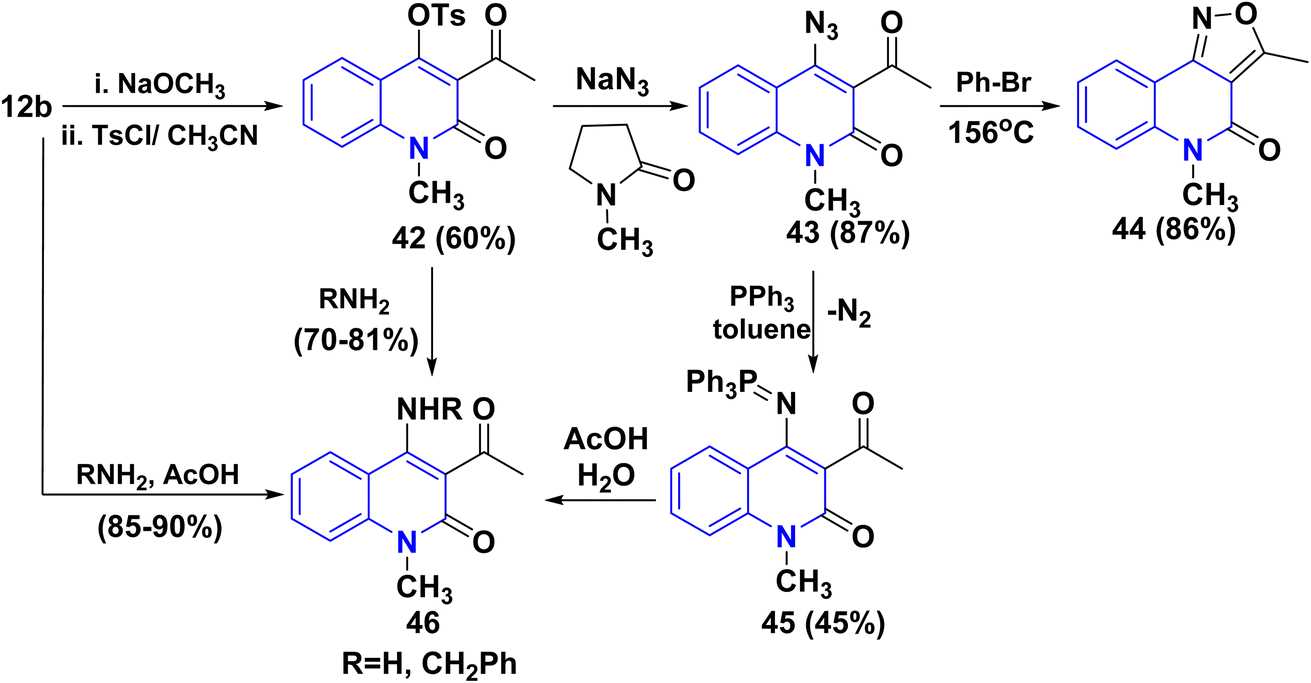
Tosylation of the sodium salt of 12b furnished a very reactive compound 3-acetyl-4-tosyloxyquinolone 42, which was used as the key compound to synthesize o-acetylazido-quinolinone 43 via transformation of the tosyloxy group to the azido group by using NaN3/N-methylpyrrolidone as solvent. Followed by ring closure through elimination of nitrogen gas by thermal heating in bromobenzene at 156 °C, 3-acetylazido-quinolinone afforded angular isoxazolo[4,3-c]quinolones 44. The azide 43 undergoes the Staudinger reaction with triphenylphosphane, yielding the phosphazene 45 via nitrogen loss. The hydrolysis of 45 in AcOH (80%) produced the 4-aminoquinolone 46. This amine can also be produced by reacting tosyloxyquinolone 42 with ammonia. Similarly, benzylamine combines with 42 to yield substituted quinoline 46 (Scheme 14).155–157
 |
| | Scheme 14 Transformation of the hydroxyl group of 12b. | |
Mechanism of Staudinger reaction via very mild reduction of the azide 43 with triphenylphosphane yields the phosphazene 43B by loss of nitrogenous gas. The hydrolysis of 43C/43D in AcOH (80%) produced the 4-aminoquinolone 46 (Scheme 15).155
 |
| | Scheme 15 Mechanism of the Staudinger reaction. | |
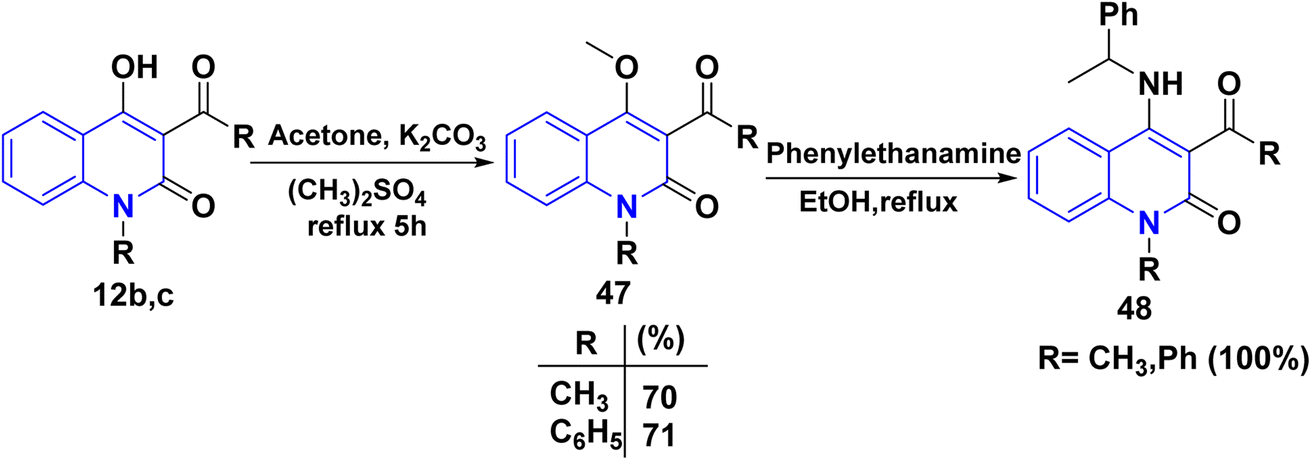
Whereas, methylation of compound 12b,c by dimethyl sulphate, a potent methylating agent, in acetone, which replaced a hydrogen atom in the hydroxyl group. This process yielded a single product 3-acetyl-4-methoxyquinolinone 47.158,159 Following this interpretation, the product reacted with a primary amine in EtOH, yielding phenylethylaminoquinolin-2-one 48 (Scheme 16).158
 |
| | Scheme 16 Synthesis of phenylethylaminoquinolinone 48. | |
Treatment of 3-acetyl-4-methoxyquinolinone 47 was observed by the Chimichi group158 with 1,2-bisnucleophiles, such as hydroxylamine, to synthesize fused isoxazolo[4,5-c]-and/or isoxazolo[4,3-c]quinolin-4(5H)ones, which gave three different products 49–51 depending on the conditions (Scheme 17). As a result, the utilization of either hydroxylamine hydrochloride or its free base leads to the formation of two distinct products. The obtained product was through a reaction of 47 with NH2OH·HCl is structurally assigned to the regioisomeric isoxazolo[4,5-c] or isoxazolo[4,3-c]quinolin-4(5H)one in the following ways:
 |
| | Scheme 17 Synthesis of isoxazoloquinolinone. | |
(a) The presence of a three-bond connection between C-3a and the methyl group at position 3 of compounds 49 and 50 led to the exclusion of the oxazole structure 51.
(b) The chemical shift of the C-3 atom being analyzed, which resembles that of a 3-substituted isoxazole more than a 5-substituted one, was used as a preliminary criterion to distinguish between the two regioisomeric isoxazole skeletons. In contrast, compound 47 exclusively and preferentially generates the regioisomeric isoxazolo[4,5-c]- or isoxazolo[4,3-c]quinolin-4(5H)on e (compounds 49 and 50, respectively).158
Whereby, synthesis of α,β-unsaturated ketones 52 was achieved through Claisen–Schmidt reaction of compound 47 with various aromatic aldehydes in glacial AcOH. Michael addition reaction of o-phenylenediamine to α,β-unsaturated ketone 52 afforded binary diazepin-quinolinones 53 (Scheme 18), whereas derivatives of 53 have shown gratifying sedative, anxiolytic, and muscle relaxant activities.159
 |
| | Scheme 18 Claisen–Schmidt reaction of compound 47. | |
Methylated compound 47 was reacted with substituted phenylhydrazines, which resulted in the formation of hydrazone 54, which were then cyclized with thioglycolic acid in methanol using zinc chloride (ZnCl2) as catalyst, affording 4-thiazolidinone derivatives 55 (Scheme 19).160
 |
| | Scheme 19 Cyclization reaction of 54 to yield 4-thiazolidinone derivatives 55. | |
The o-alkylation of 12b with ethyl bromoacetate in anhydrous acetone and K2CO3 led to the formation of ethyl oxoquinolinoxyacetate 56. Hydrazinolysis of 56 with hydrazine hydrate in methanol at room temperature (RT) yielded hydrazide compounds 57 in 80%, after which heating hydrazide compound 57 with different aromatic aldehydes in refluxing EtOH yielded derivatives 58. Finally, compounds 58 were refluxed with Ac2O to obtain the corresponding substituted 1,3,4-oxadiazolyl)methoxy)-1-methylquinolinone 59 (Scheme 20).161
 |
| | Scheme 20 Synthesis of oxadiazolylquinolinone derivatives 59. | |
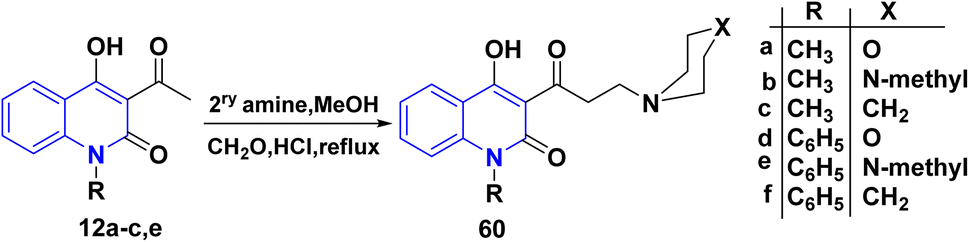
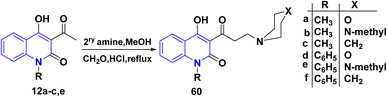 |
| | Scheme 21 Synthesis of Mannich base 4-hydroxy-3-(3-morpholinopropanoyl)quinolinone 60. | |
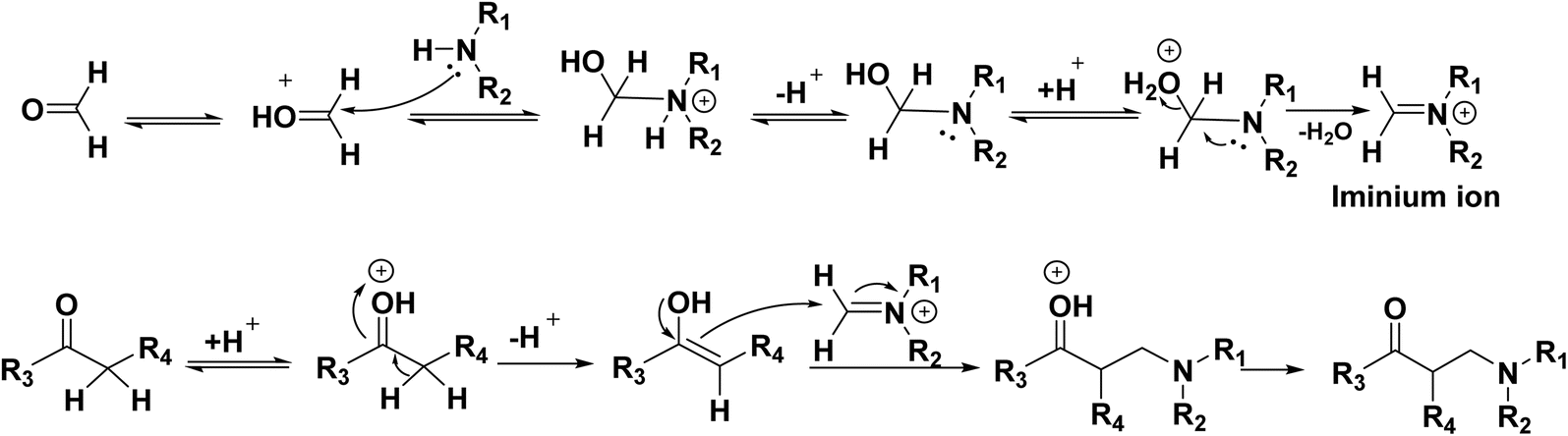
The Mannich reaction mechanism starts with the reaction between formaldehyde and amine, leading to the formation of the iminium ion. The enol form of the organic compound is obtained via tautomerization of the ketone form. The reactive iminium ion is attacked by this enol form, which ultimately produces the necessary β-amino-carbonyl molecule or Mannich base (Scheme 22).
 |
| | Scheme 22 The proposed Mannich reaction mechanism. | |
4.1.2. Halogenation and its synthetic value for constructing various heterocyclic systems.
4.1.2.1. Synthesis of 3-dichloroacetyl derivative and its utility. Among organic synthesis transformations, chlorination holds one of the most fundamental reactions that can be achieved directly by using molecular chlorine or chlorinating agents. Thus, the electrophilic chlorination reaction of 12b,d with sulfuryl chloride (SO2Cl2) in 1,4-dioxane yielding 3-dichloroacetyl derivative 61, also by heating pyranoquinolindione 11b with SO2Cl2 (Scheme 23).163,164
 |
| | Scheme 23 Chlorination of acetylquinolinone 12d and pyranoquinolindione 11b. | |
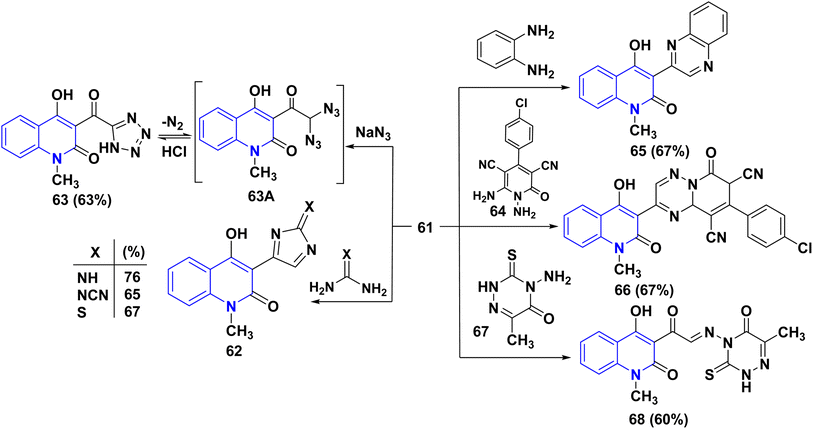
Treatment of compound 61 with some 1,3-binucleophilic reagents, namely, guanidine hydrochloride, cyanoguanidine, and thiourea, was carried out under reflux in DMF, to afford the corresponding 3-imidazolylquinolinones 62. While the reaction of 61 with NaN3 in DMF occurred at ambient temperature, the intermediate diazido compound 63A yielded the tetrazole 63 under loss of N2 gas. Condensation of compound 61 with 1,4-N,N-binucleophiles such as o-phenylenediamine and 1,6-diaminopyridine derivative 64 under reflux in DMF, afforded the quinoxalinylquinolinone 65 and the pyridotriazine derivative 66, respectively.163
Reaction of 61 with 4-aminotriazine derivative 67 was carried out in boiling pyridine. Interestingly, the only product that was obtained revealed the absence of chlorine; analysis results indicate that a nucleophilic replacement took place with the leaving of a chlorine atom, which is followed by elimination of hydrogen chloride, leading to the α-iminone 68 (Scheme 24).164
 |
| | Scheme 24 Treatment of 3-dichloroacetyl 61 with nucleophilic nitrogenous reagents. | |
Reaction of compound 61 with aminoguanidine and thiosemicarbazide as 1,4-binucleophiles, under reflux in DMF, yielded the corresponding 1,2,4-triazine derivatives 69. Hydrazinolysis of 69 in DMF produced the hydrazinotriazine 70, Thermal cyclocondensation reaction of 70 with CH(OEt)3 under fusion condition was carried out to get the triazolotriazine derivative 71 whereas, reaction with [bis(methylthio)methylene] malononitrile 72, in DMF under reflux, gave the compound 73 (Scheme 25).163
 |
| | Scheme 25 Hydrazinolysis of the 1,2,4-triazine derivative 69. | |
α-Iminone 68, as an interesting starting material, was subjected to interaction with some binucleophilic reagents to synthesize new quinolinone systems fused with pyrimidine, benzodiazepine, and pyrazole heterocycles. Therefore, treatment of α-iminone 68 with guanidine, o-phenylenediamine, and hydrazine hydrate, in DMF under reflux, afforded the pyrimidoquinolinone 74 and benzodiazepinoquinolinone 75, and pyrazoloquinolinone 76, respectively (Scheme 26).163
 |
| | Scheme 26 Synthesis of pyrimidine, diazepine, and pyrazole heterocycles from 68. | |
 |
| | Scheme 27 Bromination of N-substituted-3-acetyl-4-hydroxyquinolinones 12b–d. | |
The desired thiocyanate 78 was obtained from the reaction of 77 with potassium thiocyanate. Meanwhile, phenacyl halides were used for the alkylation of 2-aminopyridine, followed by cyclization to imidazo[1,2-a]pyridines. Thus, when 77 was heated with 2-aminopyridine in ethanol, 4-hydroxy-3-imidazoyl methylquinolinone 79 was formed.167 Moreover, condensation of bromo-derivatives 77 with o-vaniline gave 4-hydroxy-3-(7-methoxybenzofuran-2-carbonyl)-1-methylquinolin-2(1H)-one 80. Whereas, compound 77 is a versatile intermediate in synthesizing compounds 83 and 84. So that, the synthesis of 83 were achieved through condensation reaction of 77 with various thiadiazole 81 and the observed compound benzo[d]thiazol-2-yl-phenylglycylquinolinone 84 were synthesized in good yield (60–81%) through the reaction of disubstituted-2-benzo[d]thiazole 82 with 77 in the presence of AcOH (Scheme 28).166,168
 |
| | Scheme 28 Reactions of bromo-derivatives 77 with variable reagents. | |
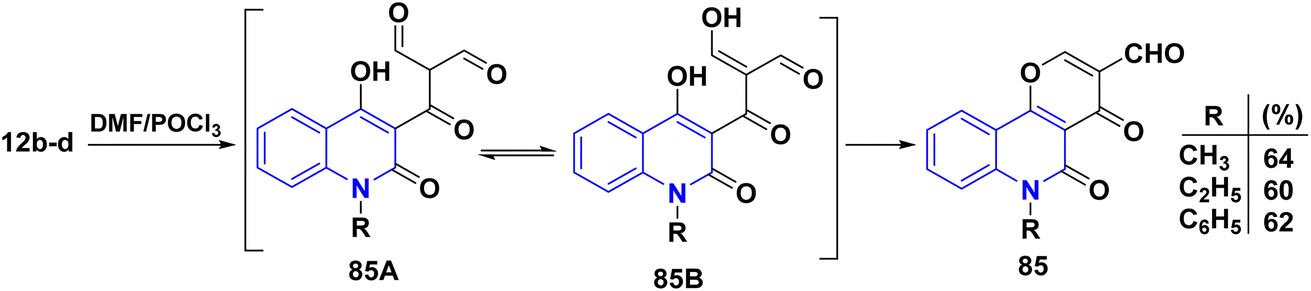
4.1.3. Formylation of AHQ and its uses. The Vilsmeier–Haack reaction was known since 1927,169 which uses DMF, an acid chloride and an aqueous work-up to change an aromatic ring with a lot of electrons into an aryl aldehyde. The “Vilsmeier reagent” is an iminium salt that is created when DMF reacts with acid chloride at the start of the mechanism (Scheme 30). Moreover, double formylation of the methyl group present in acetylquinolone 12b–d with subsequent in situ cyclization of intermediates 85A and 85B yielded pyrano[3,2-c]quinoline-3-carboxylaldehyde derivatives 85 via Vilsmeier–Haack reaction of acetylquinolinones 12b–d (Scheme 29).137,170
 |
| | Scheme 29 Vilsmeier–Haack reaction of skeletons 12b–d. | |
 |
| | Scheme 30 Plausible mechanism of Vilsmeier reaction. | |
Ibrahim et al.170 independently reported the treatment of 85 with various nucleophilic reagents affording pyrano[3,2-c]quinolines via ring-opening followed by ring closure (RORC).
Also, reaction of carboxaldehyde 85 with hydrazine hydrate in refluxing EtOH yielded 4-hydroxypyrazol-4-ylcarbonylquinolin-2-one 86, via the non-isolable hydrazone intermediate, which underwent intramolecular nucleophilic attack of NH2 at the C-2 position with concomitant γ-pyrone ring opening in situ. In the same way, the condensation reaction of 85 with phenyl hydrazine in EtOH containing a catalytic amount of TEA afforded phenylpyrazole derivative 87. Whereby, the condensation of carboxaldehyde 85 with hydrazinylquinoline quinoline 88 and 3-hydrazinyl-1,2,4-triazine-1,2,4-triazine 89 under the same reaction conditions was achieved yielding quinolinylpyrazolylquinolinone 90 and triazinylpyrazolylquinolinone 91, respectively (Scheme 31).170
 |
| | Scheme 31 Synthesis of pyrazolylquinolinone skeletons. | |
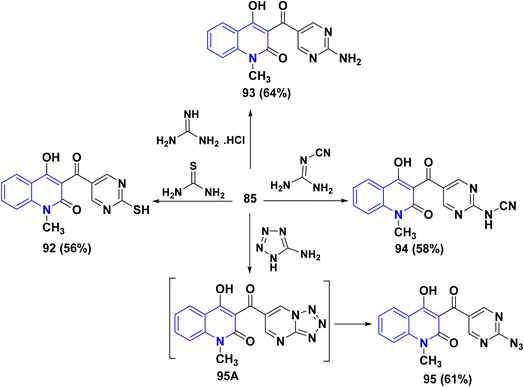
Furthermore, aldehyde 85 was subjected to react with some 1,3-N,N-binucleophiles such as thiourea, guanidine hydrochloride and cyanoguanidine in ethanolic KOH yielded the corresponding pyrimidine derivatives 92, 93 and 94, respectively. Whereby, refluxing of 85 with 5-aminotetrazole in EtOH afforded 3-[(2-azidopyrimidin-5-yl)carbonyl]4-hydroxyquinolin-2-one 95 through non-isolable tetrazolo[1,5-a]pyrimidine intermediate 95A (Scheme 32).
 |
| | Scheme 32 Synthesis of six-membered heterocyclic scaffolds. | |
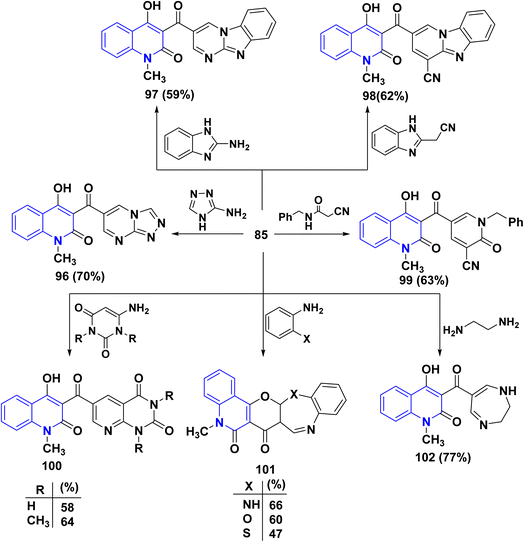
Meanwhile, condensation of aldehyde 85 with 3-aminotriazole in EtOH yielded triazolo[4,3-a]pyrimidine 96 containing the quinolinylcarbonyl moiety. Ultimately, condensation of 85 with 2-aminobenzimidazole in EtOH containing one crystal of p-toluenesulfonic acid afforded pyrimido[1,2-a]pyrimidine derivative 97. Also, reaction of 85 with 1H-benzimidazol-2-ylacetonitrile and N-benzyl-2-cyanoacetamide in EtOH containing catalytic drops of TEA yielded 98 and 99, respectively.
Next, condensation reaction 85 with o-phenylenediamine, o-aminophenol, and o-aminothiophenol in AcOH yielded the corresponding heteroannulated pyrano[3,2-c]quinoline derivatives 101. Whereby, reaction of 85 with 6-aminouracil and 6-amino-1,3-dimethyluracil in EtOH afforded pyrido[2,3-d]pyrimidines 100. Furthermore, the chemical reactivity of 85 was investigated towards different 1,4-binucleophiles. By the way, the condensation reaction of 85 with ethylenediamine in EtOH yielded 1,4-diazepinylcarbonylquinolin-2-one 102. The reaction was accomplished through the synthesis of the corresponding Schiff base intermediate, followed by an intramolecular nucleophilic addition at the C-2 position associated with γ-pyrone ring opening to afford 102 as the final product (Scheme 33).170
 |
| | Scheme 33 Synthesis of different heterocyclic systems containing quinolone scaffolds. | |
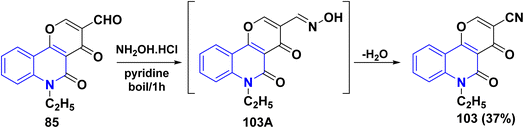
In refluxing pyridine, the reaction of tricyclic aldehyde 85 with NH2OH·HCl yielded the corresponding carbonitrile derivative 103, through the formation of non-isolable oxime 103A, after in situ dehydration step (Scheme 34).137
 |
| | Scheme 34 Synthesis of tricyclic carbonitrile derivative 103. | |
Reaction of aldehyde 85 with NH2OH·HCl, in AcOH, did not yield either oxime 103A or carbonitrile 103 but gave unexpected product showed that dioxopyrano[3,2-c]quinoline-3-carbonitrile 104 through two molecules of hydroxylamine reacted step-wisely with 85 yielded 103 which is regarded as a highly reactive cyclic push–pull system that quickly produces the adduct 104B, which exists in equilibrium due to ring-chain tautomerism. The dehydration of oxime tautomer readily afforded the corresponding intermediate 104C. Intramolecular 6-exo-dig cyclization type of intermediate 104C gave 2-iminopyranoquinoline 104D following that hydrolysis of 104D yielded the final product 104 (Scheme 35).137
 |
| | Scheme 35 Synthesis of 4-hydroxypyrano[3,2-c]quinoline-3-carbonitrile derivative 104. | |
Reaction of aldehyde 85 with NH2OH·HCl, in ethanolic KOH (1%) as a basic catalyst, instead of an acidic medium, gave 105 in 46% yield. The reaction gave the same intermediate 104D, in a basic catalyzed route, which underwent the addition of another molecule of NH2OH·HCl led to the formation of 2-imino-5-oxopyrano[3,2-c]quinoline-3-carboximidamide 105A. Whereas, the cyclization of imidamide through a condensation reaction yielded isoxazole derivative 105B. After that, hydrolysis of imine 105B was achieved, leading to the final product 105. Additionally, compound 105 was accurately prepared from the reaction of 104 with NH2OH·HCl in refluxing EtOH (Scheme 36).137
 |
| | Scheme 36 Formation of 1-amino-5-ethylisoxazolopyrano[3,2-c]quinolindione 105. | |
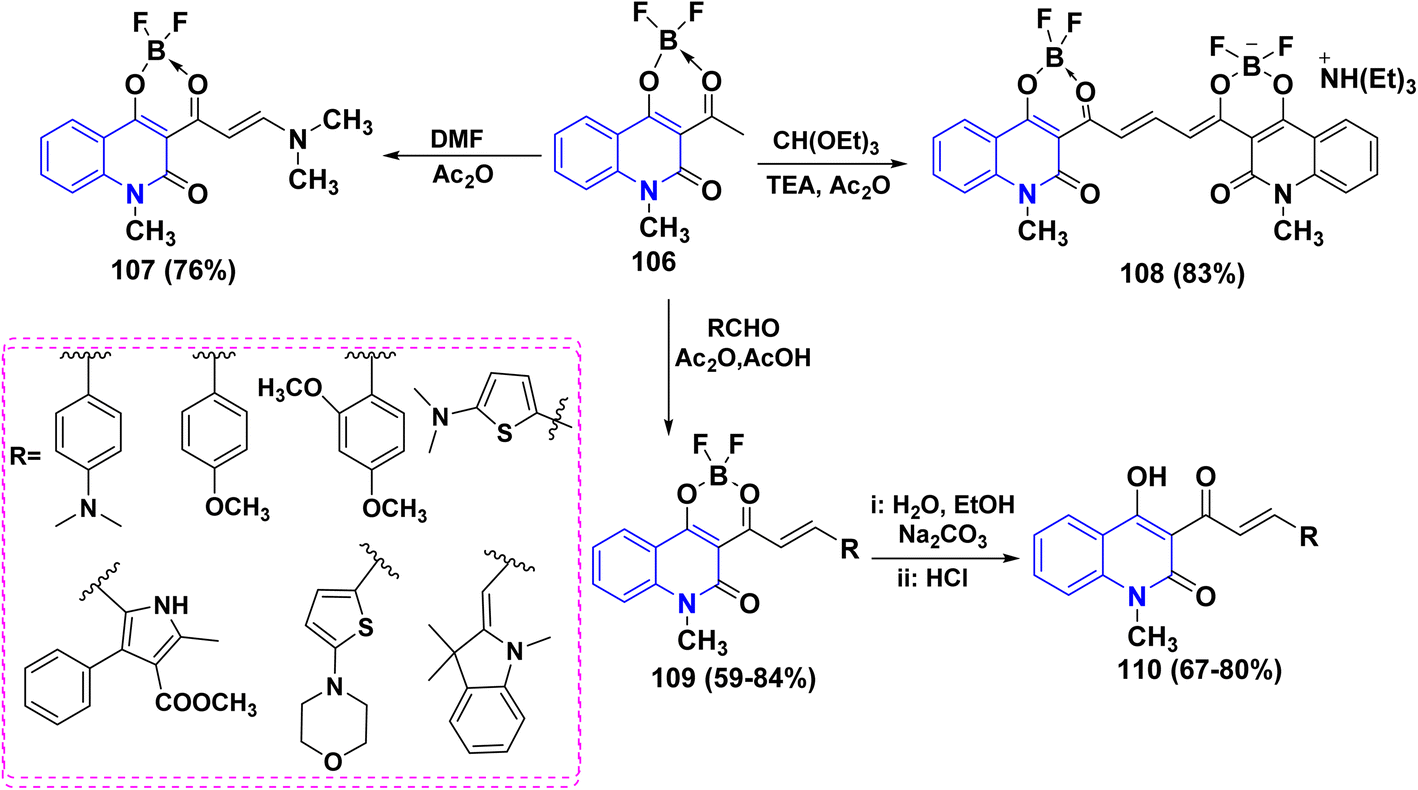
4.1.4. Synthesis of 1,3,2-dioxaborinino[5,4-c]quinolinone via metallation and its utility. The metallation reaction of acetylquinolinone 12b,d with boron trifluoride etherate BF3·(OC2H5)2 in benzene gave its boron difluoride chelate complex 106 (Scheme 37).171
 |
| | Scheme 37 Complexation of compounds 12b,d. | |
Reaction of complex 106 with DMF gave enamine 107, which can serve as a valuable synthon for transformations of the two-quinolinone fragment. Moreover, complex 106 was treated with different carbonyl compounds to yield the corresponding condensation products at the methyl group. For example, a reaction of two moles of complex 106 with CH(OEt)3 with a catalytic amount of TEA afforded symmetric polymethine dye 108. In acetonitrile, complex 108 absorbs at 581 nm, displaying noticeable fluorescence. Lastly, boron-containing styryl dyes 109 were produced by the condensation reaction of several aromatic and heterocyclic aldehydes with complex 106. Compounds 109 were hydrolyzed to produce novel 4-hydroxy-2-quinolinone derivatives 110, which exhibit fluorescence in both solid and solution states (Scheme 38).171
 |
| | Scheme 38 Condensation reactions on complex 106. | |
4.2. Nucleophilic substitution reactions
The substitution of a nucleophile at a tetrahedral or sp3 carbon is known as aliphatic nucleophilic substitution. Substitutions involving aliphatic nucleophiles are not glamorous or essential in the field of chemistry. The way carbonyl additions and carboxyloid replacements seem to occur in biochemistry, they do not occur in all significant processes. Rather, they are commonplace, minor reactions with significant, minor effects everywhere.
4.2.1. Reactions with nitrogen bases.
4.2.1.1. With amines. Condensation reaction of N-substituted (un)acetylquinolinone 12a–c,e with different amines under various conditions afforded enaminones 111.143,155,162,172,173 While, addition of a solution of substituted aniline in dimethylformamide (DMF) and pyridine to 60 and refluxed for 17–24 h to give 112 (Scheme 39).162,173
 |
| | Scheme 39 Synthesis of imine 111, Mannich base (QVIR) 60 and its imine analogue 112. | |
The reaction of quinolinone 12a with nitrogen bases derivatives such as guanidine nitrate, urea and thiourea in boiling ethanol using AcONa as a catalyst through nucleophilic addition of the amino group with the regioselectively at the CO of acetyl function of 12a accompanied by intramolecular cyclocondensation afforded regioisomer pyrimido[5,4-c]quinolinones 113a–c (Scheme 40).174
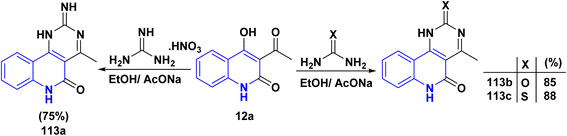 |
| | Scheme 40 Synthesis of pyrimido[5,4-c]quinolinones 113a–c. | |
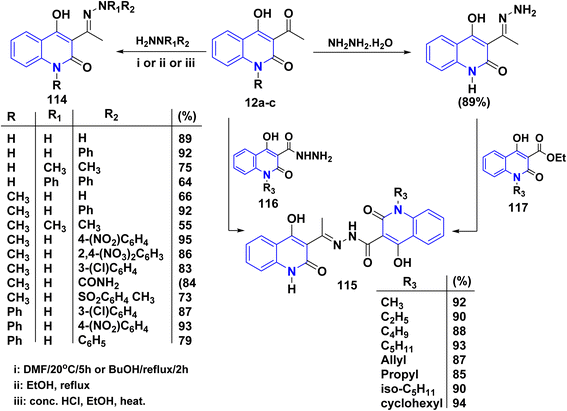 |
| | Scheme 41 Reactions of quinolinones 12a–c with variable hydrazines. | |
The formation of compound 115 can be accomplished through various methods. This reaction involves two straightforward and efficient approaches. The first method involves a one-step condensation of acetylquinoline 12a with hydroxyquinolone-3-carboxylic acids hydrazides 116, resulting in high yields. The second method consists of two stages: initially, treatment of acetylquinoline 12a to quinolinone, followed by acylation using ethyl hydroxyquinoline-3-carboxylates 117. This method also proves to be effective in the production of the hydrazones 115 described in this study; both methods were found to be equally valuable (Scheme 41).115,120,134
Similarly, compounds 119 and 120 were not achieved immediately by the condensation of 12a–c with the following hydrazines as hydrazine hydrate, semicarbazide, or thiosemicarbazide but gave acyclized hydrazone 114 and 118, respectively. These products were cyclized with Conc. H2SO4 to yield angular fused pyrazoloquinolinone 119 and angular triazepinoquinolinone 120 (Scheme 42).118,174
 |
| | Scheme 42 Condensation and cyclization of 114 and 118 to synthesize compounds 119 and 120. | |
Angular pyrazolo[4,3-c]quinoline derivatives 121 were synthesized under via enolizable 3-acetylquinolinone 12a,b with phenylhydrazine derivatives, utilizing InCl3 as an effective Lewis acid in providing greater yields of products 121 under microwave irradiation (Scheme 43).175
 |
| | Scheme 43 Synthesis of pyrazolo[4,3-c]quinoline 121. | |
Also, bis quinolinone 123 and 124 were obtained by reaction of 12b,d with hydrazinyl derivative 122 in different conditions, in hot ethanol or glacial acetic acid and sodium acetate (AcONa) (Scheme 44).176
 |
| | Scheme 44 Synthesis of compounds 123 and 124 through condensation and cyclization. | |
Initially, the reaction of hydrazone derivatives 114 with CH3ONa in diethyl ether converted the hydroxy group at position 4 to the sodium enolate group, affording 125. The reaction of sodium salts with toluenesulfonyl chloride in dry CH3CN yielded targeted toluenesulfonyloxy quinolones 126. Conversely, the attempts to obtain the toluenesulfonyloxy compounds 126 (RH) were not avail However, the separation of sodium salts 125 (RH) in good yields may be attributed to the high reactivity, in both cases, after reaction with TsCl yielded non-isolable mixture of products (Scheme 45).135
 |
| | Scheme 45 Synthesis of phenylhydrazonoquinolinylmethylbenzenesulfonate 126. | |
For the synthesis of pyrazole-4-carbaldehydes 127, the hydrazones 114 were exposed to Vilsmeier–Haack reagent (DMF-POCl3).177,178 While traditional condensation of pyrazole-4-carbaldehydes 127 with hydrazine hydrate or thiosemicarbazide furnished hydrazone derivatives 128 in acceptable yields. Another system in this area was created in two steps. Condensing acetylquinolinone 12b with aldehyde 127 (R = Ph) furnished the α, β-unsaturated ketone, which was then treated with NH2NH2, affording polynuclear heterocyclic skeleton 129 (Scheme 46).
 |
| | Scheme 46 Synthesis of functionalized quinolone derivatives 128–131. | |
Unexpectedly, the condensation of 127 (R = Ph) with NH2OH·HCl in AcOH did not produce the predicted oxime or the likely carbonitrile 130A but yielded phenylpyrazolopyrano[3,2-c]quinolindione 130, while the oxime transformed to carbonitrile via a dehydration process. The carbonitrile 130A was then cycloadditionally converted to an iminopyranoquinolinone, hydrolyzed to yield the isolated tetracyclic system 130. Another option is that the hydrolysis of the intermediate 130A into the equivalent carboxylic acid causes the pyrone ring to form via intramolecular condensation. Anyway, both routes are rational and feasible.178 Condensation of equimolar amounts of carbaldehyde 127 (R = Ph) with the 3-acetylcoumarin in the presence of piperidine as catalyst afforded the corresponding chalcone 131 (Scheme 46).177
Additionally, the synthesis of fused phosphorus heterocycles with quinolinone moieties was achieved through an efficient protocol via the reaction of hydrazone 114 with some phosphorus reagents. As the treatment of hydrazone 114 with Lawesson's reagent (LR) [which forms two reactive dithiophosphine ylides (R–PS2)], phosphorus pentasulfide (P2S5) and diethyldithiophosphoric acid in refluxing dioxane afforded the corresponding fused tricyclic oxadiazaphosphepino[6,7-c]quinolinones 132–134 (Scheme 47).179
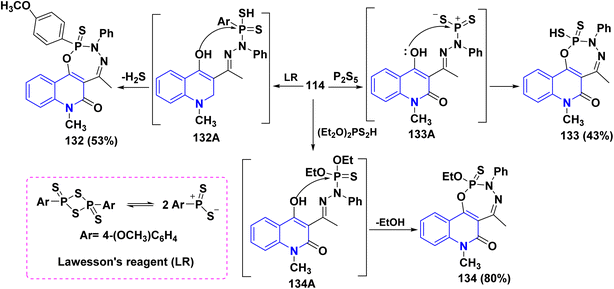 |
| | Scheme 47 Reaction of hydrazone 114 with sulfur-containing phosphorus reagents. | |
Synthesis of cyclic phosphonic ester 135 was accomplished through treatment of hydrazone 114 with tris(2-chloroethyl)phosphite P(OCH2CH2Cl)3. The credible mechanism for synthesis of compound 135 starting with Michael addition of the phosphite to the azomethine bond to produce the non-isolatable intermediate 135A, which undergoes cyclization through nucleophilic attack of NH functionality at position 4 of the quinoline ring to remove a water molecule yielding the pyrazolyl non-isolatable intermediate 135B which underwent cyclization through elimination of HCl, followed by acidic hydrolysis yielding 135 as final product (Scheme 48).179
 |
| | Scheme 48 Treatment of hydrazone 114 with P(OCH2CH2Cl)3. | |
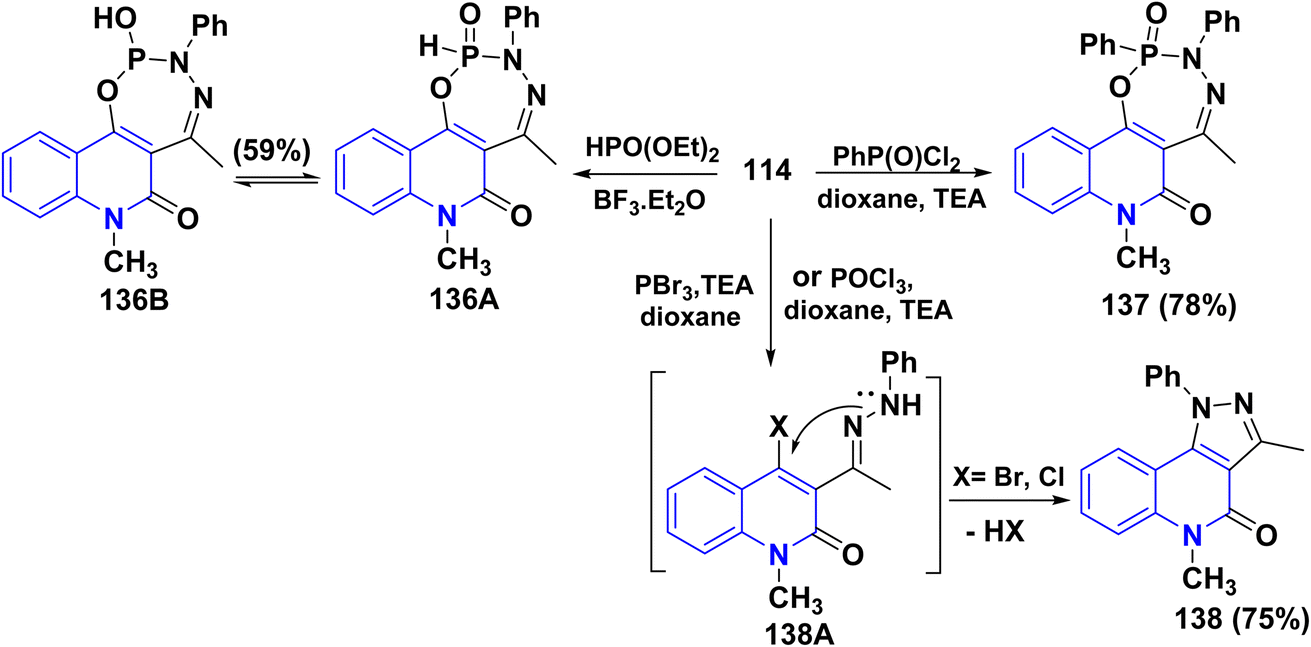
Furthermore, Pudovik reaction of hydrazone 114 with diethyl phosphite in the presence of BF3 etherate at 80–90 °C furnished oxadiazophosphepino[6,7-c]quinolinone 136A,B. The plausible mechanism involves a cyclization of hydrazone 114 through the nucleophilic attack of OH and NH groups at the phosphorus atom, followed by stripping off two molecules of EtOH. Additionally, heterocyclization of hydrazone 114 with phenylphosphonic dichloride (C6H5Cl2PO) in dioxane afforded the corresponding oxadiazaphosphepinoquinolinone 137(Scheme 49).179
 |
| | Scheme 49 Annulation of hydrazone 114 with diethyl phosphite and phosphohalogenating agents. | |
Finally, reaction of hydrazone 114 with equimolar amounts of with diethyl phosphite, PhPOCl2, phosphorus tribromide (PBr3) or POCl3 in dry dioxane in molar ratio (1:1) containing catalytic amount of TEA afforded 138, The proposed mechanism for the synthesis of fused tricyclic 138 may be attributed to halogenation of OH group process through the at C4. A phosphorus halide of the quinolinone ring, cascade by cyclization of intermediate 138A through dehydrohalogenation elimination of HX using TEA yielding 138 (Scheme 49).179
4.2.1.3. With hydroxylamine. Reaction of acetylquinolinone 12a with NH2OH·HCl was accomplished in an ethanolic solution in the presence of AcONa furnished the corresponding oxazole 139. Whereas, repeating the reaction in the presence of NaOH instead of AcONa furnished corresponding oxime 140 (Scheme 50).115,118,134,145,172,174
 |
| | Scheme 50 Condensation and thermal Beckmann rearrangement of oxime product. | |
3-Oximatoacyl-4-hydroxyquinolones 140 were synthesized by reacting AHQ 12a,b,e with excess hydroxylammonium chloride and hydrogencarbonate, yielding hydroxyiminoquinolones 140 (R1 = H). To obtain the oximethers 140 (R1 = methyl), quinolinone 12a,b were reacted with methyloxyammonium chloride and hydrogencarbonate. Both types of compounds 140 structurally resemble biologically active compounds like alloxydim or sethoxy. When oximes are thermolyzed according to Beckmann rearrangement and converted into isomeric oxazolo[5,4-c]quinolinones 141 in high-boiling solvents like 1,2-dichlorobenzene and diphenyl ether. Whereby, the hydrolysis process of oxazole derivatives 141 with dil. HCl led to formation of 3-acylamino-4-hydroxyquinolones 142 (Scheme 50).172
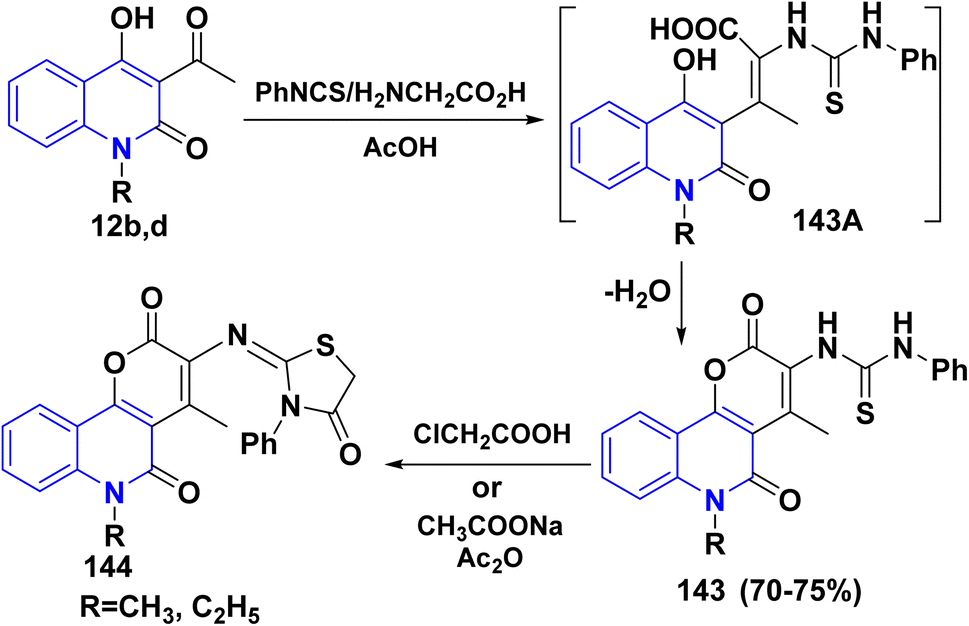
4.2.2. With phenylisothiocyante. Multicomponent reaction (MCR) of 12b,d, phenylisothiocyanate and glycine furnished N-pyrano[3,2-c]quinolinedion phenylthiourea 143 in acceptable yields via condensation of the carbonyl group of 12b,d with the active methylene group in glycine to yield the intermediates 143A which readily cyclized through stripping off water molecule to afford 143. Whereas, the condensation of 143 with chloroacetic acid and Ac2O afforded thiazolidine derivatives 144 (Scheme 51).151
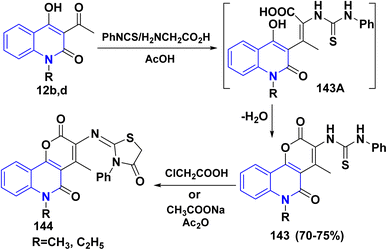 |
| | Scheme 51 Formation of thiazolidine derivatives 144. | |
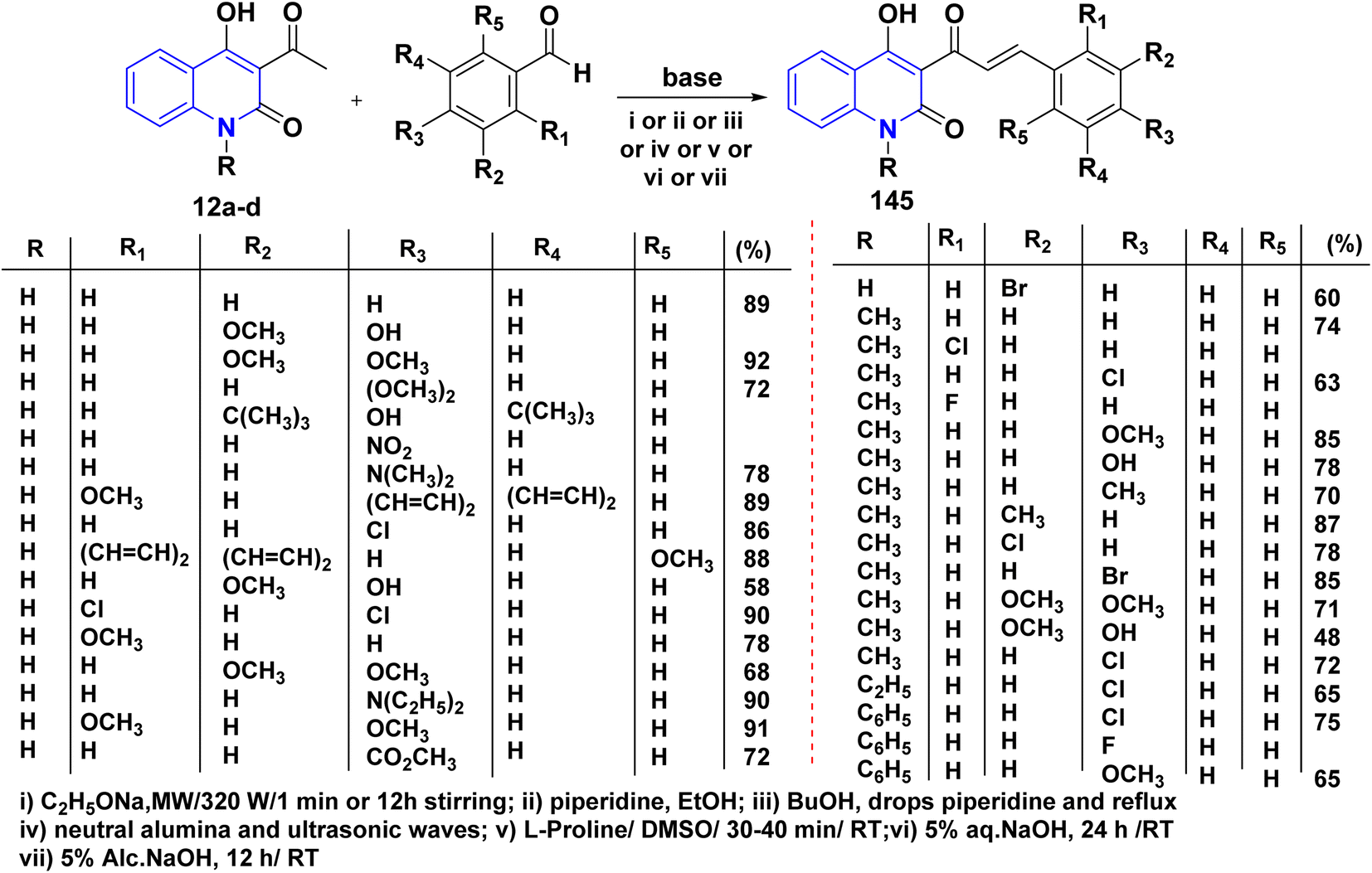
4.3. Condensation reactions
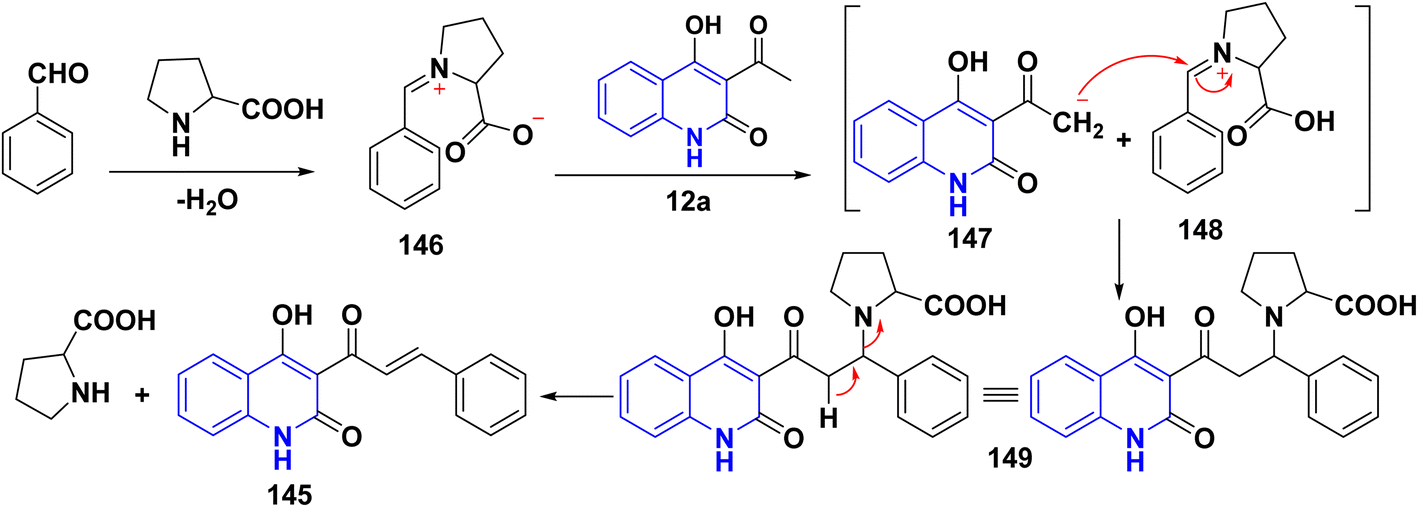
4.3.1. With aldehydes (synthesis of α,β-unsaturated ketones). A series of α,β-unsaturated ketones of quinolinone analogues 145 were synthesized through Claisen–Schmidt condensation of 12a–d with aldehydes under reflux or microwave irradiation.115,131,146,180–184 The mechanism of the reaction started by reacting of formyl group with L-proline to form an iminium carboxylate ion 146, after that this ion eliminating a proton from active methylene of 12a affording the carboxylic acid containing iminium ion 148 and quinoline anion 147 as ion pairs that combine to form the adduct 149 which loses proline to afford α,β-unsaturated ketone 145 (Schemes 52 and 53).183
 |
| | Scheme 52 Synthesis of α,β-unsaturated ketones 145. | |
 |
| | Scheme 53 Proposed mechanism for the synthesis of α,β-unsaturated ketone 145 in the presence of L-proline. | |
Similarly, reaction of quinolone 12a,e with cinnamaldehyde in the presence of piperidine in refluxing EtOH afforded α,β-unsaturated ketone 150. After that, treatment of 150 with hydrazine hydrate led to the formation of styrylpyrazolylquinolinone 151 (Scheme 54).146,185
 |
| | Scheme 54 Reaction of quinolinone 12a,e with cinnamaldehyde. | |
The predominant transformations for generating pyrazole rings typically involve the use of α,β-unsaturated carbonyl group derivatives as parent skeletons. Chalcones 145 were treated with appropriate hydrazines in AcOH, furnished functionalized pyrazoline analogues 152 (Scheme 55).108,146,184,186
 |
| | Scheme 55 Synthesis of pyrazolylquinolinones through the reaction of 145 with hydrazines. | |
Whereby, the hydrazinoquinolinone 122 underwent condensation followed by cycloaddition reaction with α,β-unsaturated ketone derivatives 145, affording binary quinolinylpyrazoloquinolinone derivatives 153 up to yield 59% (Scheme 56).176
 |
| | Scheme 56 Synthesis of binary pyrazolylquinolinone derivatives. | |
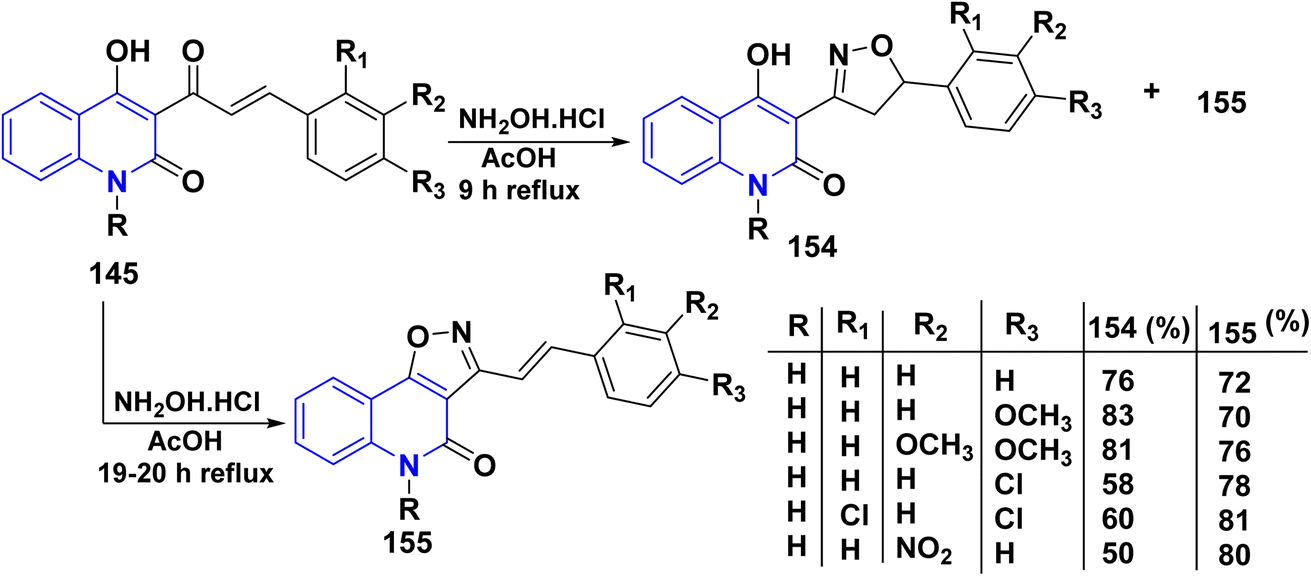
Whereas, the reaction of 145 with NH2OH·HCl in glacial AcOH yielded a mixture of two products 154 and 155. Conversely, repeating the same reaction in other solvents such as benzene, MeOH, and EtOH was unsuccessful. Since the isoxazolo[4,5-c]quinolin-4(5H)-ones 155 formation could not be controlled to get only isoxazolines 154, increasing the reaction duration to 19–20 h led to isoxazoles 155 as major products, and the minor products could not be isolated (Scheme 57).180
 |
| | Scheme 57 Synthesis of fused and binary isoxazolo and isoxazolyl quinolinone 154 and 155. | |
Pyrimidinylquinolinone quinolinyl pyrimidine derivatives 156 have been synthesized through both traditional methods and microwave irradiation. These compounds were obtained via condensing α,β-unsaturated ketone derivatives 145 with urea or thiourea in basic media (Scheme 58).182
 |
| | Scheme 58 Synthesis of pyrimidinylquinolinone 156. | |
Furthermore, substituted 1,5-benzothiazepinylquinolinones 157 were synthesized by cyclocondensation reaction followed by Michael addition. Whereas, a mixture of α,β-unsaturated ketone 145, o-aminothiophenol in absolute EtOH then added glacial AcOH (5 mol%). The obtained reaction mixture was refluxed for 5–7 h, furnishing the target product 1,5-benzothiazepinylquinolinones 157 in yields 62–82% (by the optimized condition) (Scheme 59).133
 |
| | Scheme 59 Synthesis of 1,5-benzothiazepin ylquinolinones 157 anchored on quinolone. | |
MCRs of α,β-unsaturated ketone 145, malononitrile, and ammonium acetate in DMF at 75 °C afforded functionalized pyridine binary with quinoline systems 158 (Scheme 60).187
 |
| | Scheme 60 Synthesis of 4-hydroxy-oxoquinolin-4-arylpyridine-3-carbonitriles 158. | |
The desired precursor chromenacryloylquinolinone 160 was obtained smoothly via a one-pot Aldol condensation dehydration reaction of 12b with 3-formylchromone 159 (Scheme 61).188
 |
| | Scheme 61 Reaction of quinolinone 12b with 3-formylchromone 159. | |
In refluxing EtOH, the treatment of compound 160 with an equimolar amount of hydrazine hydrate led to the opening of the pyrone ring and the enone side chain unaffected, affording a new product assigned as 4-hydroxy-3-[3-(2-hydroxyphenyl)pyrazolyl]acryloylg quinolineone 161. Whereas, using AcOH as the solvent oriented the reaction away from the γ-pyrone nucleus, yielding 3-[1-acetyl-5-(4-oxo-4H-chromen-3-yl)-4,5dihydropyrazolyl]-4-hydroxyquinolinone 162.
While by, compound 160 was reacted with of hydrazine hydrate (1:2) in refluxing DMF affording 4-hydroxy-3-[30-(2-hydroxyphenyl)bipyrazolyl]quinolinone 163 in 73% yield, through double nucleophilic attack at both the α,β-unsaturated ketone side chain and chromone ring, where by, the same product was prepared by reacting the pyrazolylacryloylquinolinone 161 with an excess amount of hydrazine hydrate in boiling DMF in 60% yield. Interestingly, the reaction of compound 160 with hydrazine (1:2) in AcOH did not give either compound 162 or compound 163 but yielded the N-acetyl derivative of compound 163 and formulated as binary bipyrazoles 164. Annotative synthesis of 164 was preceded by reaction of 161 with NH2NH2 in AcOH or treatment of compound 162 with NH2NH2 in boiling DMF (Scheme 62).188
 |
| | Scheme 62 Synthetic pathway to generate pyrazolo and bipyrazoloquinolone. | |
Reaction of equimolar amounts of 160 with NH2OH·HCl in a mixed solvent of ethanol–DMF led to the formation of 4-hydroxy[5-(2-hydroxyphenyl)isoxazolyl]acryloylgquinolinone 165. Additionally, the reaction of both compounds 165 with NH2OH·HCl in boiling DMF or pyridine afforded 4-hydroxy-3-[5-(2-hydroxypheny1)biisoxazoly1]quinolinone 166, which is obtained directly from 160, when using an excess of NH2OH·HCl (1:2) in refluxing pyridine. Additionally, polycyclic skeleton 167 was obtained upon reacting isoxazolylacryloy1quinolinone 165 with NH2NH2 in boiling DMF. Moreover, boiling compound 167 in AcOH led to acetylation, yielding acetyl-5-[5-(2-hydroxypheny1)isoxazol-4-y1]pyrazol-3-y1-4-hydoxyquinolinone 168, a compound that was also obtained during heterocyclization of the acryloy1 derivative 165 by means of hydrazine hydrate in AcOH. In the same line, the reaction of N-acetylpyrazoline 162 with NH2OH·HCl furnished the same compound 168 (Scheme 63).
 |
| | Scheme 63 Reaction of 160 with NH2OH·HCl in different conditions. | |
Whereas, the reaction of 160 with an equimolar amount of NH2OH·HCl in AcOH proceeds in a different manner, and afforded 4-hydroxy-1-methy1-3-[5-(4-oxochromeny1)isoxazol-3-yl)quinolinone] 169. After that, the reaction of chromeny1isoxazoliny1quinolinone 169 with NH2OH·HCl in boiling DMF afforded 4-hydroxy-3-[5-(2-hydroxypheny1)biisoxazol-y1]quinolinone 166. Also, chromeny1isoxazoliny1quinolinone 169 was treated with NH2NH2 in boiling DMF yielded 4-hydroxy[3-(2-hydroxy phenyl) -pyrazol-4-yl]isoxazol-3-y1-quinolinone 170 (Scheme 63).188
Equimolar ratio of 160 and thiourea was refluxed in the presence of sodium ethoxide, affording thioxopyrimidinquinolin-2-one 171 under various reaction conditions. In refluxing EtOH containing a catalytic amount of HCl or in refluxing AcOH, the same reaction resulted in the formation of chromenthioxopyrimidinquinolinone 172. The reaction of compound 172 with both hydrazine hydrate and NH2OH·HCl proceeded smoothly using ethanolic potassium hydroxide to afford pyrazolyl 173 and its isoxazolyl analogue 174, respectively (Scheme 64).188
 |
| | Scheme 64 Synthetic pathway to generate pyrimidine, pyrazole and oxazole scaffolds. | |
2-Thioxopyridine-3-carbonitriles have great interest due to their usage as intermediates for the synthesis of various medicines. Thus, synthesis of 4-hydroxy-1-methyl-2-oxoquinolin thioxo-3-carbonitrile 175 was achieved by reaction of an equimolar amount of cyano thioacetamide with compound 160, using sodium ethoxide as a catalyst. Whereas, in a relatively moderate basic medium using piperidinium acetate, the reaction approaching another possible product accessible by a Michael route led to the formation of 6-(4-hydroxy-2-oxo-1,2-dihydroquinolinyl)-4-(4-oxo-4H-chromen-3-yl)-2-thioxopyridine-3-carbonitrile 176. The heterocyclization reaction of both compounds 175 and 176 with NH2OH·HCl, in boiling pyridine and/or DMF, yielded two important triheterocyclic systems: 5-[3-(4-hydroxy-2-oxo-quinolin-3-yl)-4,5-dihydro-isoxzolyl]-6-(2-hydroxyphenyl)-2-thioxopyridine-3-carbonitrile 177 and 6-(4-hydroxyl-2-oxoquinolin-3-yl)-4-[5-(2-hydroxyphenyl)-isoxazolyl]-2-thioxopyridine-3-carbonitnile 178 (Scheme 65).188
 |
| | Scheme 65 Reaction of 160 with cyano thioacetamide in different reaction conditions. | |
2-Thiobarbituric acid is recognized as a reactive cyclic methylene compound of the 1,3-dione type, which is utilized to synthesize pyranopyrimidines when cyclized with enone systems or 1,3-dicarbonyl compounds, and more recently with 3-substituted chromones.189 When compound 160 was treated with 2-thiobarbituric acid in an equimolar ratio in the presence of sodium ethoxide, it yielded (4-hydroxy-2-oxoquinolin-3-yl)-3-oxopropenyl]thioxopyrano[2,3-d]pyrimidinone 179.
While by, the same reaction occurred in the presence of piperidinium acetate, a polycyclic molecule, thioxopyrano[2,3-d]pyrimidinone derivative 180 was obtained. Furthermore, both 179 and 180 were reacted with NH2OH·HCl in DMF affording the [3-(4-hydroxy-2-oxoquinolin-3-yl)isoxazol-5-yl]-7-(2-hydroxyphenyl)-2-thioxopyrano[2,3-d]-pyrimidinone 181 and (4-hydroxy-2-oxo-quinolin-3-yl)-5-[5-(2-hydroxyphenyl)isoxazol-4-yl]-2-thioxopyrano[2,3-d]pyrimidinone 182, respectively (Scheme 66).188
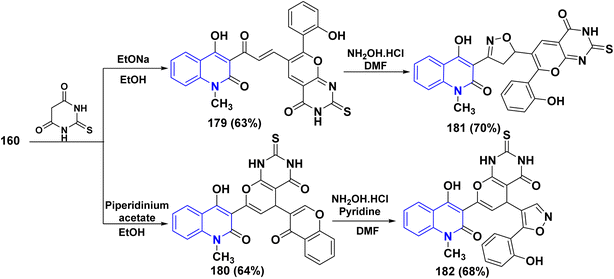 |
| | Scheme 66 Reaction of 160 with thiobarbituric acid in different reaction conditions. | |
4.3.2. With DMF/DMA. N,N-Dimethylformamide dimethyl acetal (DMFDMA), also known as 1,1-dimethoxy-N,N-dimethylmethanamine, The chemical structure of DMF/DMA has two significant sites: a partially positive carbon that functions as an electrophile in condensation processes and a partially negative nitrogen that acts as a nucleophile. Owing to its structure, DMF/DMA reacts with a wide range of organic groups and can be a useful reagent as a C1 synthon, particularly in the production of heterocycles. Also, it is used to formylate aromatic compounds. The process involves initial conversion of DMF to a chloroiminium ion, [(CH3)2NCH(Cl)]+, known as a Vilsmeier reagent, which attacks arenes. Thermal condensation of 12b,d with DMF/DMA, afforded the corresponding dimethylaminoacryloylquinolinone derivatives as (enaminone compound) 183.190 Reaction of enaminone 183 with 4-aminoantipyrine, furnished 3-(3-((1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)amino)acryloyl)-4-hydroxyquinolinone 184. Whereas, the reaction of the enaminone 183 with morpholine and piperidine as cycloaliphatic secondary amines smoothly proceeded, leading to the corresponding 4-hydroxy-1-alkyl-3-(3-morpholino/piperidinoacryloyl)quinolinone 185.While by, treating enaminone with hippuric acid (N-benzoylglycine) or aceturic acid (N-acetylglycine) 186 in Ac2O resulted in the production of 187. It is suggested that this process begins with the cyclization of 186 into oxazolone, which then adds to the activated double bond system of enaminone 183, followed by further spontaneous rearrangement yielding 187.177,191
Furthermore, compound 189 was synthesized through condensing the active methylene group in diethyl acetonedicarboxylate 188 with the carbonyl group in compound 183, via water elimination and the formation of an intermediate 189B. This intermediate is then cyclized in the presence of AcONH4, resulting in the stripping of a water molecule yielding angular tetracyclic compound 188 (Scheme 67).190
 |
| | Scheme 67 Reaction series of enaminone 183 with different heterocyclic reagents. | |
A probable mechanism for the synthesis of 187 started with the cyclization of hippuric acid into oxazolone 186A, which then adds to the activated double bond system of the enaminone 183, yielding 187A, followed by further rearrangement of this intermediate to give 187. Whereas, compound 189 is thought to be formed via condensation of the active methylene in 188 with the carbonyl function in 183 with water elimination, forming the intermediate 189A that was cyclized in the presence of ammonium acetate via elimination of a molecule of EtOH and water to afford the final product 189 (Scheme 68).
 |
| | Scheme 68 Plausible mechanism for synthesis of compond 187 and 189. | |
Biological activities of pyridazinone ring systems represent one of the most active classes of heterocycles. Consequently, enaminone 183 was employed as a promising building block for synthesizing pyridazinone 191. Compound 183 coupled with 4-nitrobenzenediazonium chloride, leading to the formation of the arylazo intermediate 190A, which undergoes hydrolysis during the reaction to produce α-hydrazono-β-oxopropanal 190. Cyclization of the hydrazone derivative 190 with hippuric acid 186 in Ac2O produced the 4-benzoylamino-6-[(quinolin-3-yl)carbonyl]-pyridazinone 191 in 66% yield. Whereas, the reaction of compound 183 with malononitrile in absolute ethanol in the presence of piperidine yielded carbonitrile derivative 192 in 55% yield (Scheme 69).178
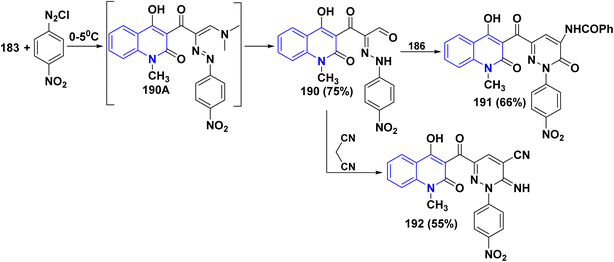 |
| | Scheme 69 Coupling of enaminone 183 with 4-nitrobenzenediazonium chloride followed by cyclization. | |
Additionally, treatment of the enaminone 183 with thiourea in ethanolic potassium hydroxide solution did not give the expected pyrimidinylquinolinone 193. Similarly, reaction of enaminone 183 with cyanoguanidine as amidine reagent gave another pyrimido[5,4-c]quinolinone derivative 194. Moreover, when enaminone 183 was heated in AcOH, it went through intramolecular cyclocondensation, resulting in the formation of 6-methylpyrano[3,2-c]quinolindione 195. Finally, when enaminone 183 was reacted with compounds containing active methylene like malononitrile and ethyl cyanoacetate in the presence of KOH, it yielded the pyranoquinolinone derivatives 196 (Scheme 70).178
 |
| | Scheme 70 Reaction series of enaminone 183 analogues synthesis. | |
Moreover, treatment of the enaminone 183 with thiosemicarbazide and thiocarbodihydrazide yielded 197. The cyclocondensation of 7-chloro-4-hydrazinoquinoline 88 with enaminone 183 in acetic acid resulted in the formation of 4-hydroxy-1-methyl-3-[1-(7-chloroquinolin-4-yl)-1H-pyrazol-3-yl]quinolinone 198. The proposed structure for compound 198 was further validated through an alternative synthesis starting from acetylquinolinone 12b. Specifically, treating compound 12b with compound 88 yielded hydrazone 199. Subsequently, the in situ thermal condensation of hydrazone 199 with DMF/DMA smoothly produced compound 198 (Scheme 71).178
 |
| | Scheme 71 Reaction of enaminone 183 with binucleophilic reagents. | |
Reaction of enaminone 183 with different hydrazines and hydrazides, such as hydrazine hydrate, phenylhydrazine, and 4-amino-5-hydrazineyl-4H-1,2,4-triazole-3-thiol afforded binary pyrazole 200 in good yield. Whereas, treatment of 183 with heterocyclic hydrazine as 2-hydrazineyl-1H-benzo[d]imidazole or 2-imino-2H-chromene-3-carbohydrazide yielded chemoselective fused pyrazole (E)-3-(2-(dimethylamino)-5-phenyl-4H-pyrazolo[4,3-c]quinolin-4-one (201) (Scheme 72).192
 |
| | Scheme 72 Reaction of enaminone 183 with different hydrazine derivatives. | |
Reaction of enaminone 183 with triethylenetetramine (TETA) as highly active linear polyamines in CH2Cl2 led to the formation of benzo[h][1,6]naphthyridine-4,5-dione derivative 202 instead of the anticipated transamination products. Furthermore, refluxing of 183 with malononitrile led to the formation of tetrahydrobenzo-[h][1,6]naphthyridine-3-carbonitrile 203 through 1,2-addition mechanism followed by the nucleophilic attack of OH group to the cyano function. After that, the Dimroth rearrangement was achieved instead of an alternative path involving the replacement of the active methylene with the dimethylamino group (Scheme 73). Refluxing of enaminone 183 with pyrazolone 204 in AcOH gave fused triazaindenoanthracenol 205 (Scheme 73). The mechanistic pathway for the formation of 205 was recognized through a condensation reaction afforded 1,5-dicarbonyl intermediate 205A, followed by cyclization reactions of intermediate 205A through double nucleophilic attacks of the both two OH groups to the endo-ketonic carbonyl group of quinolinone and β-position of enaminone led to affording the fused polycyclic system 205 in 68% yield (Scheme 73).192
 |
| | Scheme 73 Synthesis of fused heterocyclic scaffolds 202, 203, and 205. | |
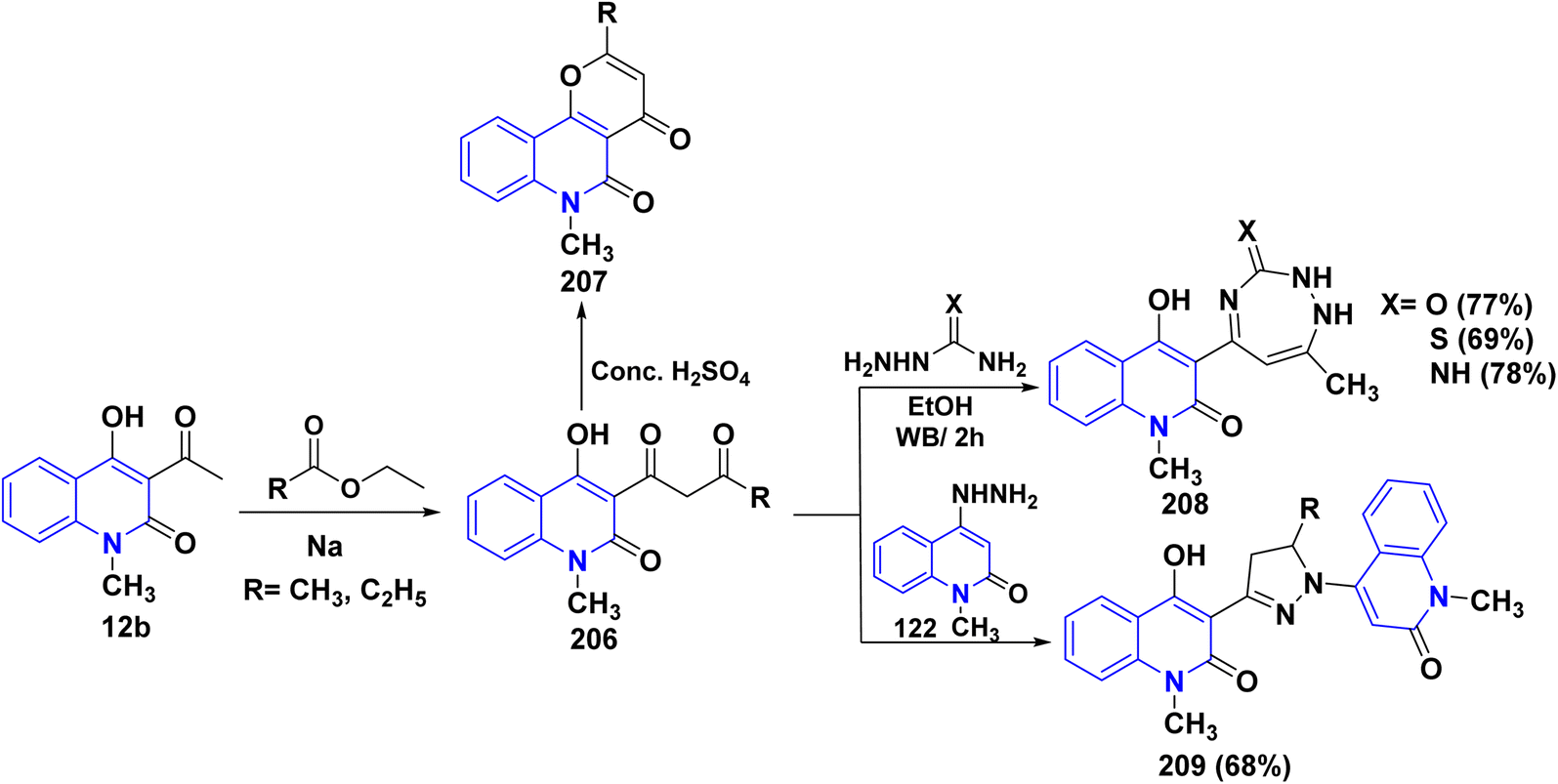
4.3.3. With esters. Compound 206 was synthesized through the condensation of quinolinone derivative 12b with methyl/ethyl propionate in the presence of Na metal. This reaction occurred through the nucleophilic attack of the active methylene group on the carbonyl group of the ester reagent. Cyclization of compound 206 with sulfuric acid gave 2-ethyl-6-methylpyrano[3,2-c]quinolindione 207 (Scheme 74).151 The bifunctional ammonia derivatives such as semicarbazide, thiosemicarbazide or aminoguanidine reacted with 206 in refluxing ethanol to furnish a binary triazepinone, triazepinthione and iminotriazepine 208, respectively. Furthermore, reaction of compound 206 with hydrazinoquinolinone 122 afforded the intended dihydromethylquinolinyl)-3-pyrazolyl)-4-hydroxyquinolinones 209 (Scheme 74).176
 |
| | Scheme 74 Synthesis and cyclization reactions of compound 206. | |
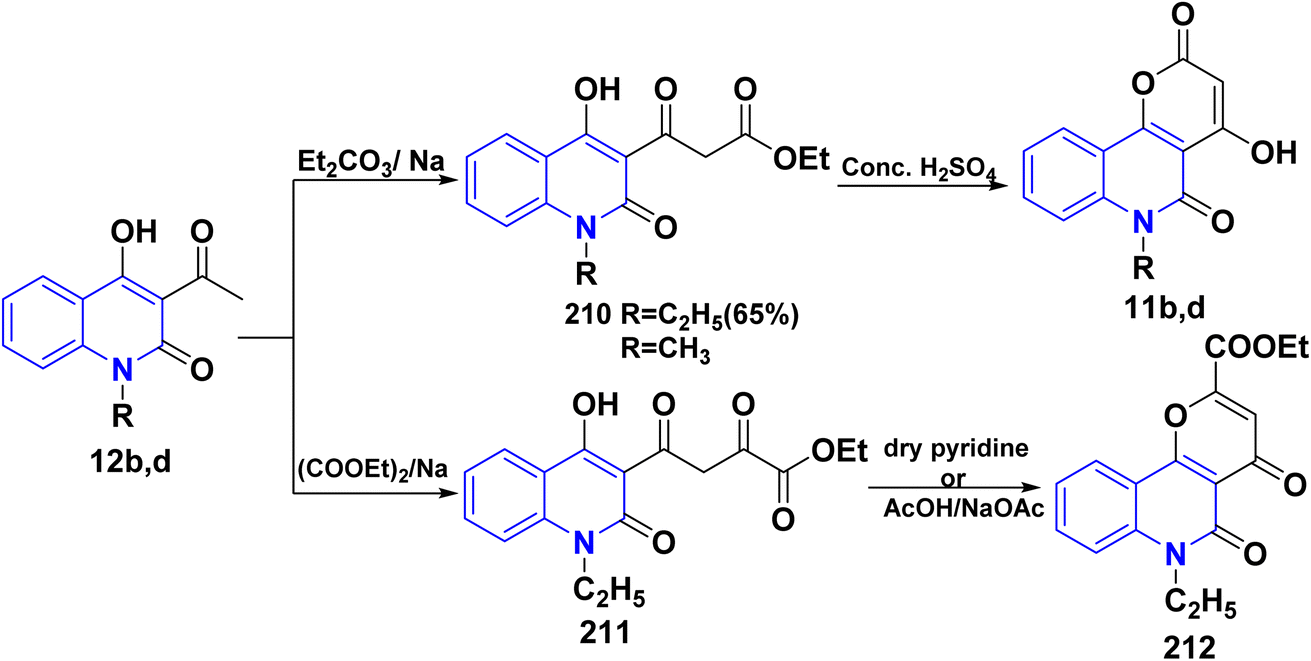
Additionally, Claisen condensation reaction of 12b,d with diethyl carbonate led to 1,3-diketone; 3-ethoxycarbonylacetyl-4-hydroxyquinolin-2-one 210 in the presence of sodium metal. Cyclization of compound 210 with sulfuric acid afforded 4-hydroxypyrano[3,2-c]quinolindione 11b,d. In a similar process, Claisen condensation of compound 12d with diethyl oxalate in the presence of Na metal produced the diketo-ester; (ethylhydroxy-1,2-dihydroquinolinyl)dioxobutyrate 211. Cyclization of 211, in dry pyridine, or AcOH/AcONa (freshly fused) yielded ethyl 6-ethyldihydropyrano[3,2-c]quinoline-2-carboxylate 212 (Scheme 75).151
 |
| | Scheme 75 Claisen condensation of compound 12b,d with different esters. | |
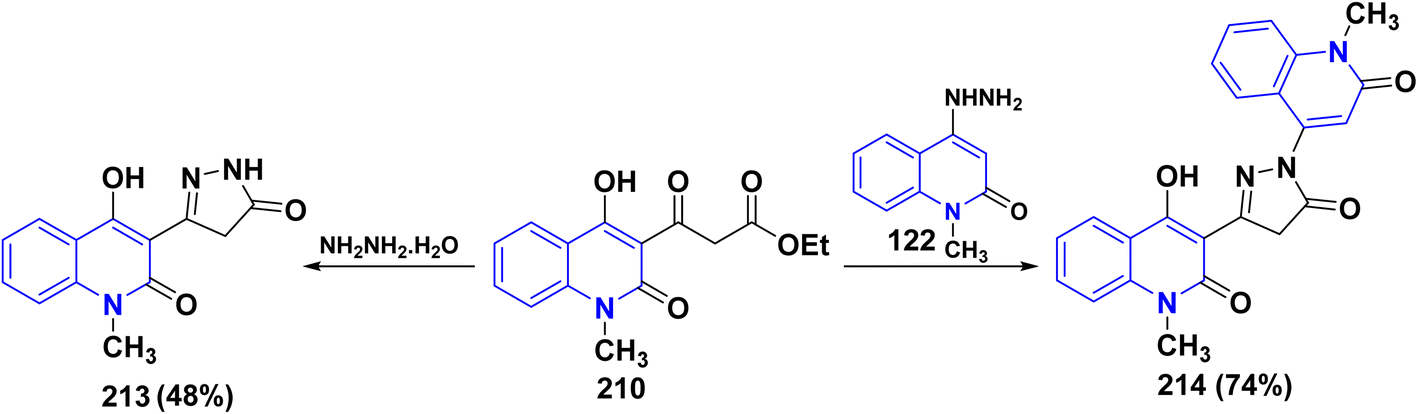
The synthesis of binary pyrazolylquinolinone 213 was achieved by reacting the β-ketoester 210 with hydrazine hydrate to give the target compound 213 in 48% yield.193a While, direct treatment of pyranoquinolindione 11 with hydrazine hydrate gave the desired pyrazolylquinoline 213 in 92% yield. The possibility of forming the isomeric quinolinylpyrazolin-3-yl)quinolinone 214 was ruled out by independently synthesizing this derivative through the cyclization of hydrazinoquinolinone 122 with 3-ethoxycarbonylacetyl-4-hydroxy-1-methylquinolinone 210 in 74% yield (Scheme 76).176b
 |
| | Scheme 76 Synthesis of pyrazolonyl moiety anchored on quinolone 213 and 214. | |
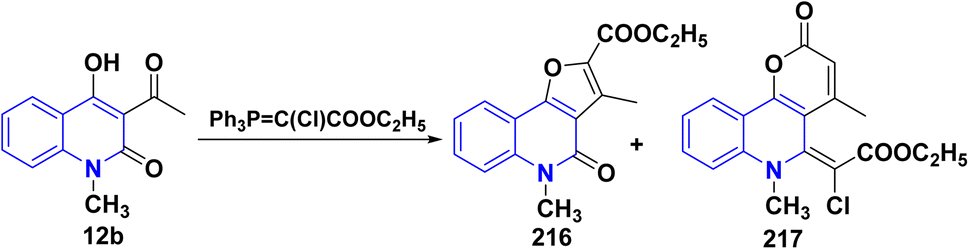
Whereas, the reaction of acetylquinolinone 12b with ethyl (triphenylphosphoranylidene)acetate in refluxing xylene afforded 215A followed by bromination to give compound 215B that underwent hydrolysis gave furo[3,2-c]quinolinone 215 (Scheme 77).193b
 |
| | Scheme 77 Synthesis of furo[3,2-c]quinolone 215. | |
The proposed reaction mechanism for the molecular rearrangement of 215B to acids 215 begins with the opening of the pyrane ring in a basic medium, resulting in the formation of dianion A. Tautomer B is considered more likely than A due to the greater distance between the two negatively charged functional groups. Following this, the elimination of a halogen atom from the sp2 hybridized carbon atom occurs alongside the migration of the substituent in a trans position relative to the departing halogen. This migrating group exhibits partial carbanionic character due to its potential tautomerism. This rearrangement, which can be classified as a Wagner–Meerwein type, leads to the formation of intermediate C, which subsequently yields compound D and ultimately product 215 after acidification (Scheme 78).
 |
| | Scheme 78 Plausible mechanism for the synthesis of 215. | |
Treatment of 12b with ethyl (triphenylphosphoranylidene)chloroacetate in refluxing xylene afforded a complex mixture of compounds 216 and 217. Compound 216 was isolated by repeated column chromatography and identified as an ethyl ester. The second reaction product 217 contained an unsaturated lactone moiety (Scheme 79).193,194
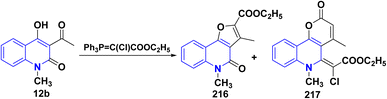 |
| | Scheme 79 Reaction of quinolone 12b with ethyl (triphenylphosphoranylidene)chloroacetate. | |
4.3.4. With activated olefins as synthetic routes to pyrano skeletons and their benefits (ylidene nitriles). A Michael reaction refers to a type of organic chemistry reaction involving the addition of a nucleophile (Michael donor) to an activated olefin or alkyne (Michael acceptor), such as an acrylate. This reaction is known for its rapid occurrence, tolerance of various functional groups, and its utility in synthesizing novel step-growth compounds with tailored macromolecular architectures. Derivatives 219 are similarly synthesized via Michael type addition of the anion ion of the acetyl group in 12c,e to the activated double bond in ylidenenitriles 218 in (1:1) or (1:2) molar ratios in ethanol/piperidine to yield the acyclic intermediate 219A, which dehydrogenated and undergo cyclization into the adducts 219B which afford compounds 219 under basic conditions by adding one molecule of CH2(CN)2, which exists in equilibrium with two moles (Scheme 80).191
 |
| | Scheme 80 Michael addition of compounds 12c,e. | |
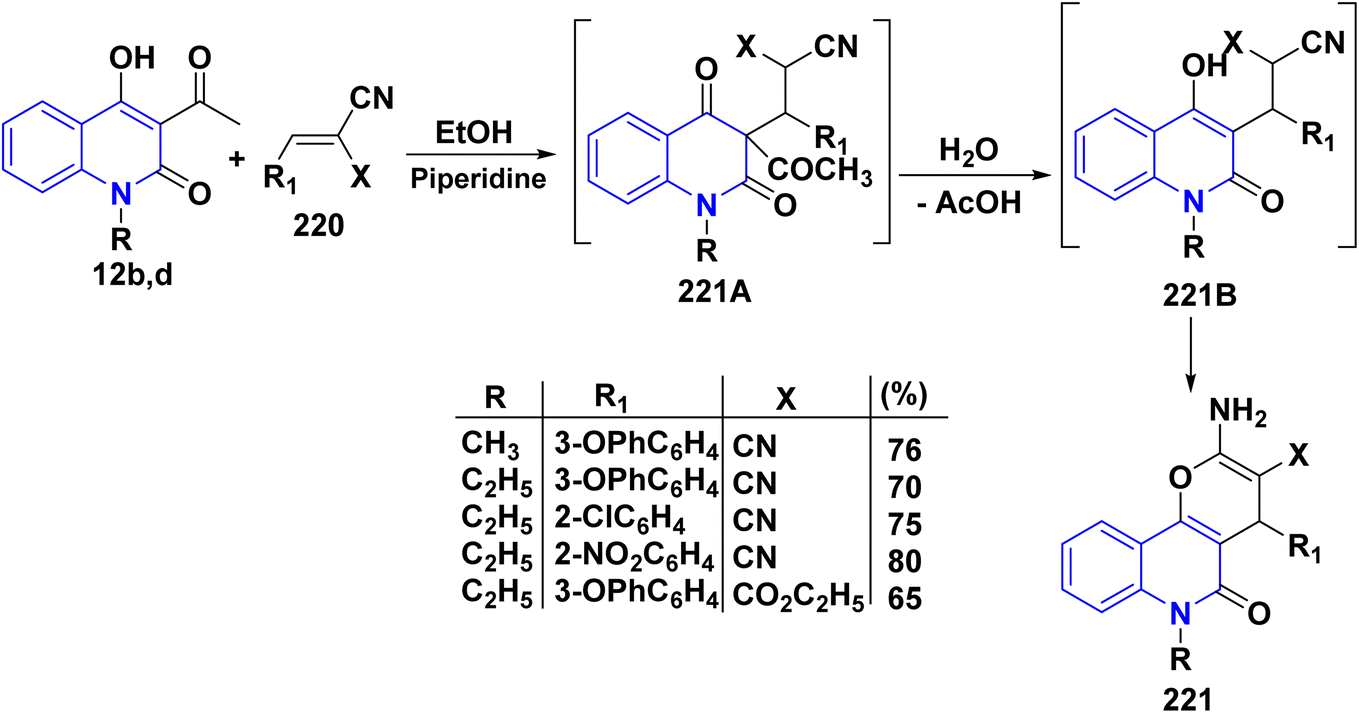
On the other hand, the nucleophilic addition of 3-acetylquinolinones 12b,d to 220 in EtOH, using piperidine as a catalyst, produced intermediate 221A. This intermediate underwent hydrolysis, losing the acetyl group, to form quinolone intermediate 221. Subsequently, cyclization of this intermediate yielded 2-amino-5-oxopyrano[3,2-c]quinoline derivatives 221 (Scheme 81).115,134,195
 |
| | Scheme 81 Catalyzed nucleophilic addition of 3-acetylquinolinones 12b,d. | |
2-Amino-4-(2-chlorophenyl)oxopyrano[3,2-c]quinolinecarbonitrile 221, as a typical enaminonitrile derivative, reacted with HCOOH to afford tetracyclic 5-methylpyrimidopyrano[3,2-c]quinolindione 222. Compound 221 also reacted with Ac2O to yield 5,10-dimethylpyrimidopyrano[3,2-c]quinolindione 223 (Scheme 82).195
 |
| | Scheme 82 Synthetic pathway to access tetracyclic systems 222 and 223. | |
6-Methyldioxodihydrospiro[indoline-3,4′-pyrano[3,2-c]-quinoline]-3′-carboxylate 225 was synthesized through the reaction of compound 12b with 224 in EtOH, pyridine (Scheme 83).115
 |
| | Scheme 83 Synthetic approach of spiro compound 225. | |
4.4. Miscellaneous reactions
4.4.1. Deacetylated quinolinone (HQ) and its utility. Hydrolysis reaction of acetylquinolinone 12b–f with sulfuric acid, accompanied by deacetylation produced 4-hydroxyquinoline derivatives (HQ) 15 (Scheme 84).115,130,132,141,145,155
 |
| | Scheme 84 Deacetylation of 3-acetyl-4-hydroxyquinolone derivatives 12b–f. | |
Refluxing of 4-hydroxyphenylquinolinone 15 with POCl3 for 2 h yielded the corresponding 4-chloroquinolinone derivative 226, reaction of 226 with three equivalent piperazine in DMF in the presence of TEA was heated at 80 °C for 6 h yielded 4-piperazinylquinolinone 227, heating 227 with formaldehyde and the appropriate amine in EtOH at reflux then leave overnight afforded the target N-Mannich bases 228. Alkylation of the piperazinyl nitrogen of compounds 227 with 2-bromo-N-phenylacetamide derivatives 229 was achieved by stirring overnight in DMF using K2CO3 as a base to yield the target compounds 230 (Scheme 85).194
 |
| | Scheme 85 Chloronation of 15 followed by synthesis of N-Mannich base. | |
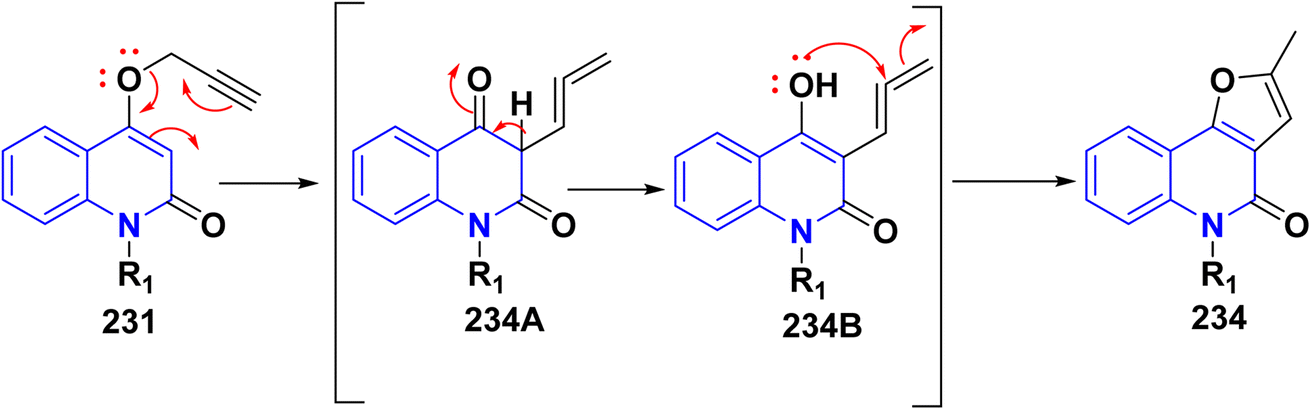
Refluxing of 15 with propargyl bromide in acetone in the presence of anhydrous K2CO3 for 14–16 h afforded a mixture of 231, 232, and 233, which were separated by HPLC.196 Base-catalysed Claisen rearrangement of propynyloxyquinolone 231 in hexamethylphosphoric triamide (HMPT) in the presence of NaHCO3 (2 equiv), refluxed for 10–18 h afforded angular furo[3,2-c]quinolinone 234 according to the shown mechanism (Schemes 86 and 87). Whereas, 231 products followed by Sonogashira coupling yielded 4-((3-arylprop2-yn-1-yl)oxy)quinolin-2-onederivatives 235 in good yield. Then, electrophilic cyclization between 235 and iodine to obtain 236, the reaction was initially performed in various solvents (DMF, CH3CN, CH3OH, THF, DCM, and CH3NO2) at 25 °C, using sodium bicarbonate as a base (Scheme 86).197
 |
| | Scheme 86 O-alkylation followed by Suzuki coupling reaction. | |
 |
| | Scheme 87 Mechanistic pathway for the synthesis of 234. | |
Benzoylation reaction of 15a–c with benzoyl chloride and a catalytic amount of TEA afforded 4-benzoyloxy-2-quinolinones 237.198 Compound 237 underwent Fries rearrangement with AlCl3, yielding 3-benzoyl-4-hydroxy-2-quinolinones 238 in excellent yields. Reaction of β-hydroxyketone compound 238 with phenylhydrazines in dimethylformamide (DMF) at room temperature and also at 0 °C yielded mixtures of compounds that can be separated, which contained cyclized pyrazolo[4,3-c]quinolinones 239. To obtain pyrazolo[4,3-c]quinolinones 239 directly from 3-benzoyl-4-hydroxy-2-quinolinones 240 pure and in good yield (87–99%). A suspension of 3-benzoyl compounds 238 was treated in AcOH with a few drops of conc. H2SO4 to enhance the reaction rate and obtain good to excellent yields. Heating for 2 h yielded pyrazolo[4,3-c]quinolones 239 (Scheme 88).135
 |
| | Scheme 88 Synthesis of pyrazolo[4,3-c]quinolinones 239 through o-benzoylation. | |
The synthetic pathway for compound 242 begins from 3-phenylaminomethylene quinolindiones 240, which were synthesized from 4-hydroxy 2-quinolones, aniline and triethyl orthoformate (CH(OEt)3), after that converted to 4-chloro-3-formylquinolinones 241.135,199 The reaction of carboxaldehydes 241 with a specific amount of phenylhydrazines in a solution of DMF at room temperature or in refluxing BuOH afforded 4-chloro-3-arylhydrazinonoquinolinones 242 in good to excellent yields (Scheme 89).135
 |
| | Scheme 89 Synthesis of chlorohydrazonoquinolinones 242. | |
The synthesis of a new series of triazolylquinolinones, specifically 3,3′–(oxoquinolin-4-yl)-1,2,3-triazol-4-yl)methoxy)phenyl)methylene)bis(4-hydroxyquinolinones) 247, was achieved by reacting 4-azido-2-quinolinones 246 with 3,3′–(prop-2-yn-1-yloxy)phenyl)methylene)bis(4-hydroxyquinolinones) 245 through a Cu-catalyzed [3 + 2] cycloaddition (Huisgen–Meldal–Sharpless reaction). Initially, the synthesis of terminal alkynes 245 was performed via interaction between 4-hydroxy-quinoline-2-ones 15 and p-hydroxybenzaldehyde in different molar ratios under reflux to afford 3,3′–(hydroxy-phenyl)methylene)bis(4-hydroxyquinolin-2-ones) 243. Phenol compound 243 reacts with propargyl bromide in DMF to produce alkynes 245. On the other hand, 4-hydroxy-2-quinolinones 15, interacted with aldehyde 244 in different molar ratios (2:1) to produce the desired terminal alkynes 245 (Scheme 90).200
 |
| | Scheme 90 Synthesis of triazolyl-bis(4-hydroxy-quinolin-2(1H)-ones) 247. | |
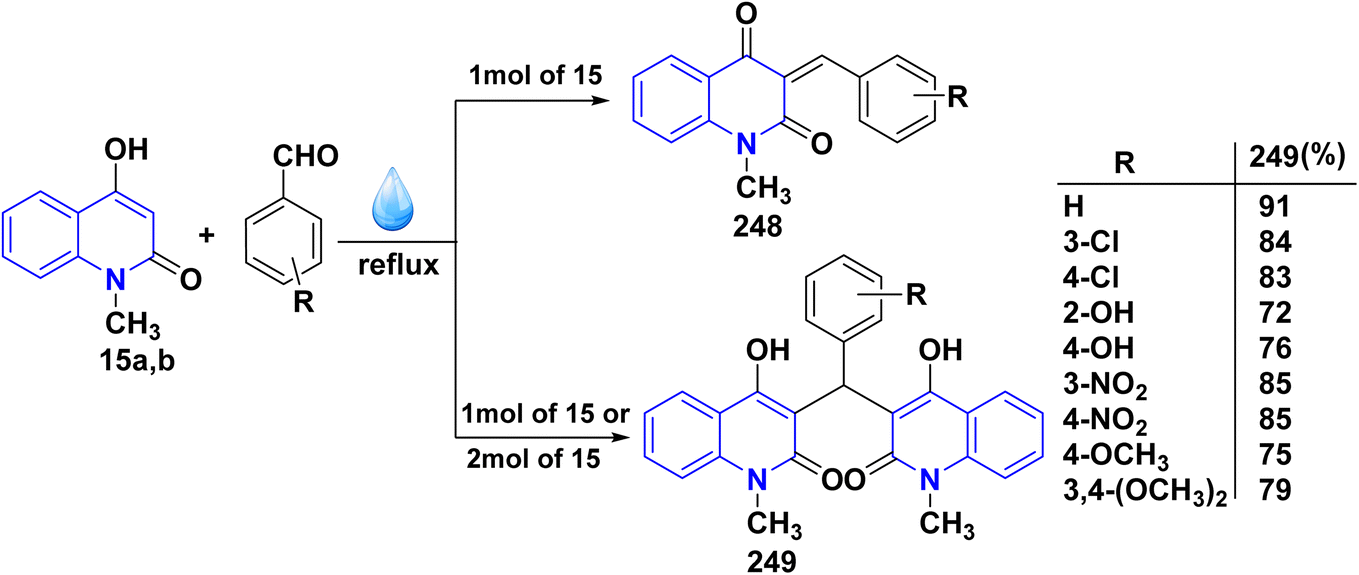
Reaction of compound 15 with several aromatic and heterocyclic aldehydes, which contain both electron-withdrawing and electron-donating groups, was investigated under catalyst-free conditions using water as the eco-medium to produce compound 249, through Knoevenagel condensation followed by a Michael-type addition with compound 15, leading to the smooth formation of a wide range of substituted bisquinolinones. In these cases, aldehydes with electron-withdrawing groups produced higher product yields, ranging from 85% to 90%. The study revealed that compound 249 was the primary product, rather than compound 248, when using different moles of compound 15 (Scheme 91).201–204
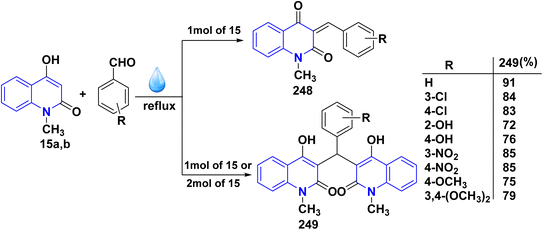 |
| | Scheme 91 Reaction of 4-hydroxyquinoline derivatives 15 with various aromatic aldehydes. | |
A believable mechanism for the formation of compound 15 has been suggested. Initially, one equivalent of HQ 15b undergoes a straightforward Knoevenagel condensation with aromatic and heterocyclic aldehydes to produce carbonyl intermediate 249A. This intermediate serves as a strong Michael acceptor and reacts with another equivalent of 15b through Michael addition, resulting in the keto–enol intermediate 249B, followed by isomerization to yield the final product (Scheme 92).201
 |
| | Scheme 92 Mechanistic pathway for the synthesis of 249. | |
Furthermore, treatment of 15b with the aromatic and heterocyclic aldehydes in different molar ratios (2:1) led to the formation of 250. Whereas, the three-component Knoevenagel–Michael reaction of 15b, aldehyde, and aminocyclohex-2-enone 251 produced skeleton 252. But, when the reactants were added sequentially, only a small amount of the desired product 252 was obtained, with compound 250 being the major product (Scheme 93).203
 |
| | Scheme 93 Synthesis of 250 and 252 via Knoevenagel–Michael reaction. | |
A plausible mechanism for the formation of compounds 250 and 252 started with simple Knoevenagel condensation of 15 with the aromatic or heterocyclic aldehydes (known to occur under solvent and catalyst-free conditions) to generate an adduct 253, which acts as a strong Michael acceptor. After that, another molecule of HQ 15 (two-component reaction) or 3-aminocyclohex-2-enone (three-component reaction) attacks the electron-deficient β position of 253 in a Michael addition fashion to afford 250 and 252 (Scheme 94).203
 |
| | Scheme 94 Suggested mechanism for the synthesis of 250 and 252. | |
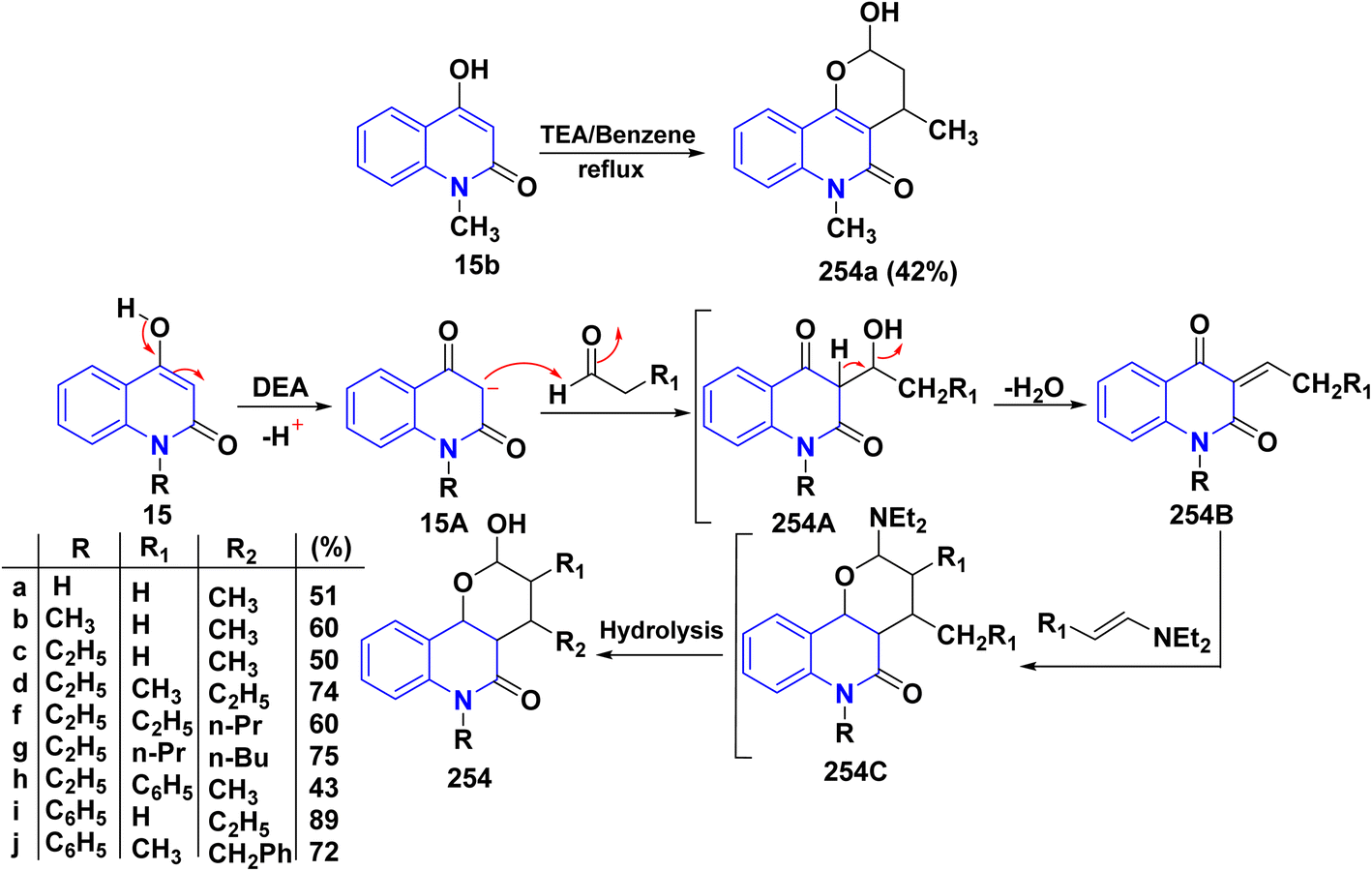
Pyrano[3,2-c]quinolinone derivatives represent a significant category of naturally occurring alkaloids. As their synthetic counterparts are currently the focuses of research due to their diverse biological activities with potential medicinal applications. Additionally, these compounds are frequently employed as synthetic precursors in producing dimeric quinoline and polycyclic heterocycles.205 While by, 2-hydroxydimethylpyrano[3,2-c]quinolin-5-one (254a) was synthesized by a photoinduced reaction of 15b in the presence of TEA, which generated DEA and acetaldehyde derivatives in situ, whereas, photoinduced electron–transfer reactions with TEA as an monodentate electron donating through a redox reactions were achieved according sketeched mechanism in Scheme 93.206 Reaction of 4-hydroxyquinolinone 15 with aliphatic aldehyde gave the corresponding quinolone methide, base-catalyzed condensation of a quinolinone 15 with an aldehyde yields the corresponding 4-hydroxy-3-(1-hydroxyethyl)quinolin-2-one 254A which underwent dehydration in basic medium to furnish the highly electrophilic quinone methide intermediate 254B. After that, Michael's addition reaction at the exocyclic methylene carbon of quinone methide 254B by the generated enamine (from DEA and the aldehyde) and the carbanion 15A derived from the deprotonation of 15. The Michael-type addition of the enamine proceeds in a 1,4-fashion and results in an intramolecular cyclization to yield 2-(diethylamino)pyrano[3,2-c]quinolin-5-one 254C which hydrolyzed to afford 254 (Scheme 95).206
 |
| | Scheme 95 Photoinduced reaction of compound 15b with TEA producing 254a and the Plausible mechanism. | |
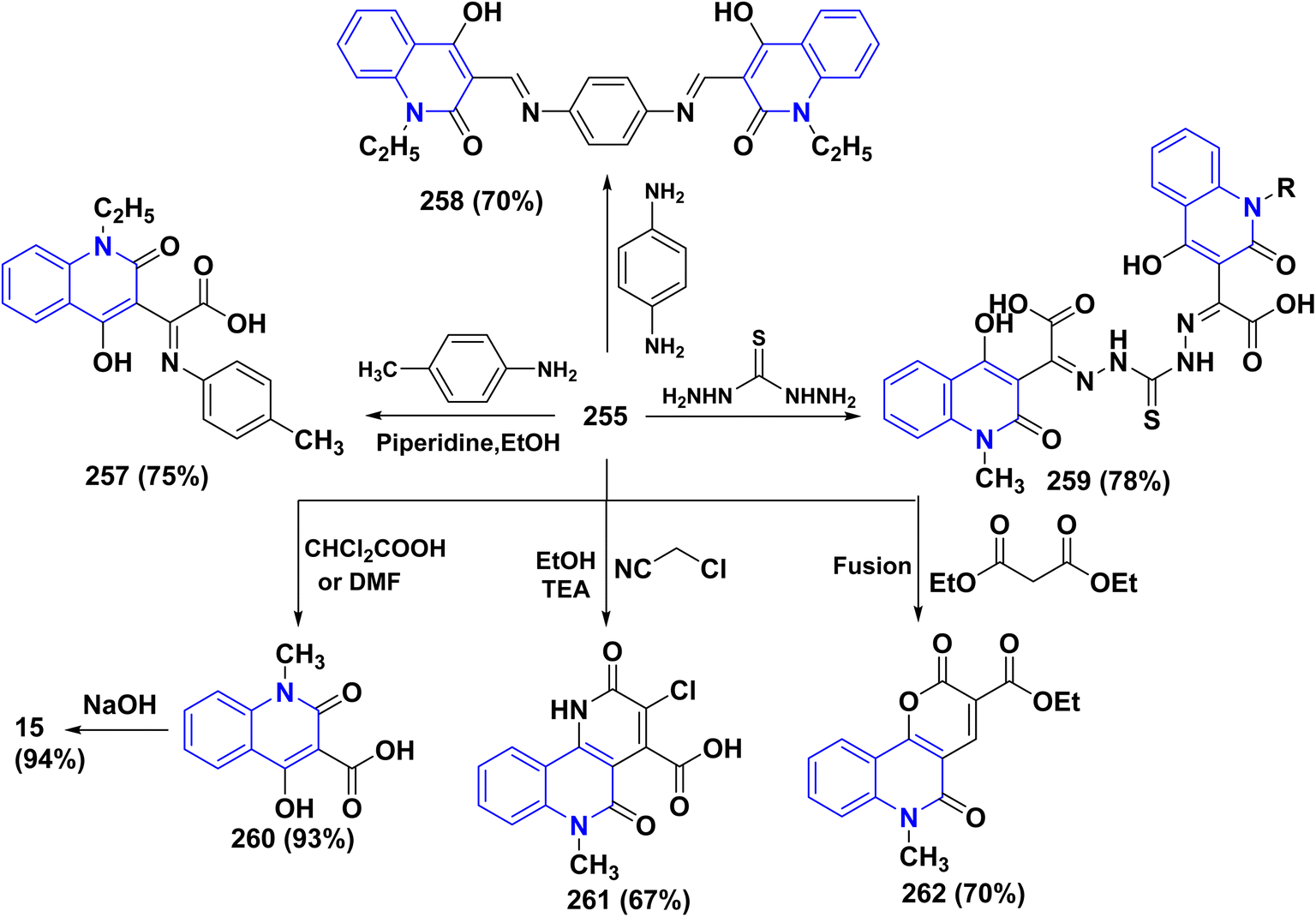
4.4.2. Oxidation. Oxidation of quinolone derivatives 12b,d under the classic Riley conditions afforded a mixture of two products: α-keto acid 255 and its dehydrated dimer derivatives 256. The major product is soluble in aq. Na2CO3; besides, it can be isolated after neutralization to yield the α-keto acid 255, whereby the minor product is insoluble in aq. Na2CO3 solution, but soluble in boiling NaOH and crystallizable from AcOH and identified as spiropolyketonic scaffold 256 (Scheme 96).207
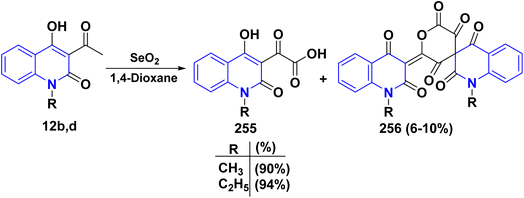 |
| | Scheme 96 Oxidation of quinolone derivatives 12b,d. | |
A probable mechanism for the synthesis of the α-keto acids 255 can be simply explained by the fast oxidation of quinolone derivatives 12b,d to α-keto aldehyde intermediates 255A, which cyclize into the hemiacetal intermediates 255B, which can afford the corresponding 5-alkylfuro[3,2-c]quinolinetriones 255C. These furoquinolinetriones 255C and 255D are easily hydrolyzed, furnishing the formation of 255 as a major product and 256 as a minor product (Scheme 97).
 |
| | Scheme 97 Proposed mechanism for the synthesis of 255 and 256. | |
Specifically, the reactivity of 255 with various nucleophilic reagents was investigated, as the reaction of 255 with p-toluidine in refluxing EtOH with a few drops of piperidine produced (4-hydroxy-2-oxoquinolin-3-yl)-p-tolyliminoacetic acid 257. Similarly, reaction of 255 with p-phenylenediamine in DMF yielded bis[(hydroxy-1,2-dihydroquinolinyl)methylidene]-1,4-phenylenediamine 258. While condensing α-ketoacid 255 with thiocarbohydrazide in different molar ratios in boiling EtOH containing TEA afforded bis[2-(4-hydroxy-2-oxoquinolin-3-yl)-2-oxoacetic acid]thiocarbohydrazone 259. Additionally, the reaction of α-ketoacid 255 with various carbon nucleophiles was explored through its reaction with active methylene such as dichloroacetic acid, chloroacetonitrile, and diethyl malonate, which led to the synthesis of quinoline-3-carboxylic acids 260 and fused tricyclic scaffolds (3-chloro-2-hydroxymethylbenzo) carboxylic acid 261 and 2,5-dioxopyrano[3,2-c]quinoline 262, respectively. Whereas, the hydrolysis process of quinoline-3-carboxylic acids 260 by NaOH was continued for a day, yielding 4-hydroxy-2-oxoquinoline 15 (Scheme 98).208
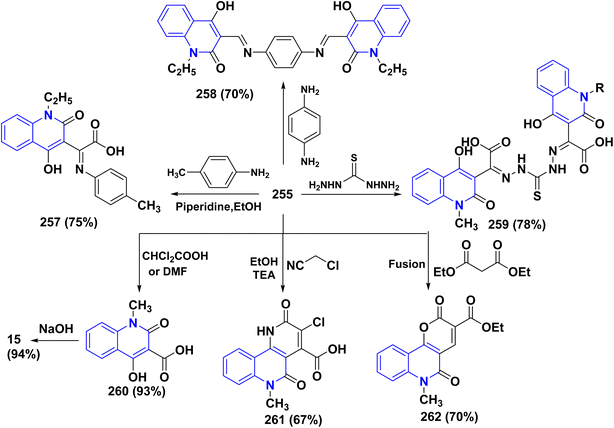 |
| | Scheme 98 Reaction of α-keto acids 255 with different reagents. | |
4.4.3. Reduction of AHQ. Whereas, the reduction is one of the most effective chemical transformation processes in organic chemistry209 as the development in this field has been tremendous, progressing from the use of stoichiometric reagents to get novel organic systems, whereas, Clemmensen-type reduction of acetylquinolinone 12b in the presence of zinc dust (particle size <45 μm) afforded 3-ethyl-4-hydroxyquinolinone 263 in acceptable yield 75% (Scheme 99).210 The Clemmensen reduction is a chemical reaction used to convert a carbonyl group (CO) into a methylene group directly. Firstly, the carbonyl group is protonated by acidic hydrogen to be more electrophilic and easily attacked. Then, nucleophilic Zn attacks the protonated carbocation, leading to the formation of a tetrahedral carbinol containing the carbon–zinc bond, which is protonated by HCl through a carbenoid mechanism. After that, the reduction process happens on the surface of the zinc metal, leading to the formation of the corresponding alkane through undergoing two sequential 2e− reduction steps involving a dehydration step and anionic intermediates (Scheme 99).211
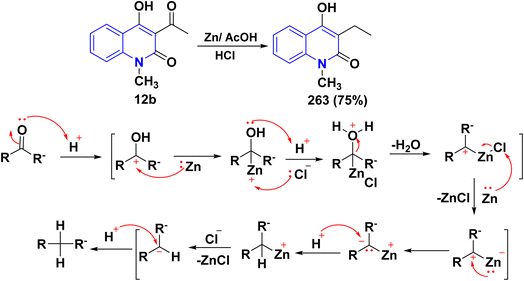 |
| | Scheme 99 Reduction of the acetyl group of 12b to form compound 263, in addition to the plausible mechanism of the Clemmensen reduction. | |
Reduction of derivatives 12b,c via using sodium borohydrate (NaBH4) as hydrogen source and electron donor afforded 4-hydroxy-3-(hydroxyethyl)quinolinones 264. Whereby, a chemoselective reduction of 264 was achieved by the treatment of 264 with substituted phenylhydrazines, yielding hydrazono-3-(1-hydroxyethyl)quinolin-2-one derivatives 265 (71–92%) (Scheme 100).212
 |
| | Scheme 100 Reduction of compounds 12b,c using NaBH4. | |
5. Applications
5.1. Medicinal perspective of quinolinones
Quinolinone derivatives are significant heterocyclic systems with multiple medicinal applications.84 As these compounds possess various pharmacological properties,213,214 including analgesic effects,106,215 anti-inflammatory,108,216–219 antiallergenic,107,220,221 diuretic,222,223 cardiovascular agents,47,224 orally active antagonists,225–228 antimicrobial,161,229–232 anticonvulsant,110,233–235 acetylcholinesterase reactivators,236–239 antitumor, anticancer,136,240 Farnesyl transferase inhibitor,241 antioxidant,107,192,216,242 anti-tubercular activity106,243–246 and other potential biological applications 247–252 as shown in Fig. 6.
 |
| | Fig. 6 Biologically active compounds containing quinolinone scaffolds. | |
Globally, tuberculosis (TB) is a highly lethal infectious disease caused by the bacterium Mycobacterium tuberculosis, which is responsible for approximately three million deaths. The recent report from the World Health Organization (WHO) identifies TB as the second leading cause of death from infectious diseases worldwide. About one-third of the global population is at risk due to latent infections with Mycobacterium tuberculosis, according to the WHO.106,253,254 On the other hand, quinolines are a crucial active pharmaceutical ingredient that plays a vital role in discovering new drug candidates. Many quinoline-based compounds are currently in clinical and preclinical development for tuberculosis treatment. Additionally, several quinoline-derived medications, such as moxifloxacin, gatifloxacin, and TMC207, are utilized to treat tuberculosis.255–257 Interestingly the incorporation of pyrazole derivatives into the quinolinone scaffold enhances its biological efficacy, as it alters modes of action, improves selectivity profiles, and reduces unwanted side effects. Those compounds may be further changed to have better pharmacokinetics and oral bioavailability (Fig. 7).106,258
 |
| | Fig. 7 Design an approach to increase anti-tuberculosis activity. | |
The accidental discovery of nalidixic acid during chloroquine synthesis and its role in the development of numerous quinolinone analogues, including flumequine, rosoxacin, ofloxacin, ciprofloxacin, moxifloxacin, levofloxacin, trovafloxacin, and marbofloxacin. All of the aforementioned pharmaceutical candidates demonstrated significant antibiotic activities.259–261 Over the years, the history of quinolones is characterized by numerous iterations, innovations, and expansions, as evidenced by the many potent drugs available today. In 1962, nalidixic acid (1st generation) was discovered and then approved in 1967 for treating uncomplicated urinary tract infections (UTIs), but resistance quickly developed among various species.262 Due to adverse bioeffects as low serum concentrations and high minimum inhibitory concentrations, nalidixic acid was largely abandoned till the emergence of fluoroquinolones in the 1970s and 1980s.263,264 Modifying quinolones to fluoroquinolone scaffolds improved their pharmacokinetics and expanded their antimicrobial spectrum.263,264 Interestingly, certain second-generation marketed antibiotics, such as ofloxacin, ciprofloxacin, and norfloxacin, are still in use today (Fig. 8).250
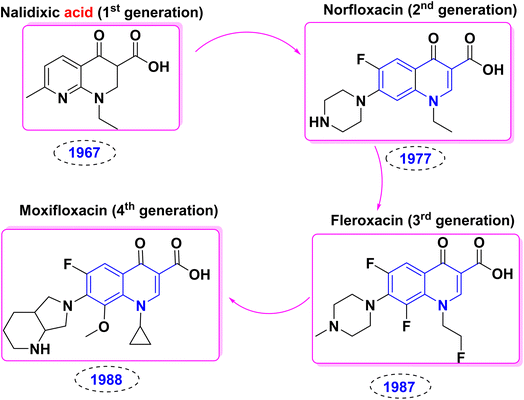 |
| | Fig. 8 The history of quinolinones and modifications to improve their pharmacokinetics. | |
Fleroxacin signifies the start of the “3rd generation” of quinolinones, which demonstrated meaningfully enhanced antimicrobial activity due to various modifications in its chemical structure, as shown to be photocarcinogenic and photomutagenic.265 Finally, Moxifloxacin, the “4th generation” of quinolones, is characterized by robust antimicrobial activity, including high efficacy against pathogens due to improved anaerobic coverage.250,263,266,267
Also synthesized N-(3-acetyl-2-oxoquinolin-1(2H)-yl)benzamide derivatives 266 were tested for their antitubercular and antimicrobial activities. The majority of the tested derivatives exhibited encouraging antitubercular efficacy in comparison to the standard drugs (isoniazid and streptomycin). The inclusion of electron-donating groups such as methyl, amino, hydroxy, and dimethylamino has enhanced its antitubercular activity. Notably, most of the investigated derivatives of 266 displayed considerable antibacterial efficacy against both Gram-positive and Gram-negative microorganisms. They also showed marked antifungal efficacy against C. albicans and A. niger (Fig. 9).268
 |
| | Fig. 9 Quinolinone scaffolds with biological efficacy. | |
Krishnakumar et al.269 reported the evaluation of in vitro antibacterial properties for ethyl-2-oxoquinoline-3-carboxylate 267, showing moderate activity against the Vibrio cholerae and Bacillus subtilis. Likewise, 1-methyl-3-(3-oxo-3-phenylprop-1-enyl) quinoline-2-(1H)-ones 268 were examined in vitro as antimicrobial agents exhibiting significant antibacterial activity against S. Typhi,S. aureus, P. aeruginosa, and B. subtilis (Fig. 10).270
 |
| | Fig. 10 Quinolinone skeletons with antibacterial activity. | |
Whereas, the next generation of anticancer, antimicrobial, and anti-HIV-1 drugs is expected to be characterized by quinolinone-pyrimidine-based. Anticancer evaluation of pyrimidoquinolinone 269 and pyrimidotetrazinoquinoline 270 was examined in vitro against the human liver cancer cell line (HEPG2). The anticancer activity results indicated that these compounds showed inhibitory activity against the tested cell line with IC50 values of 38.30 μM for 269 skeleton and 39.8 μM for 270 (Fig. 11).271
 |
| | Fig. 11 Quinolone-pyrimidine based molecular hybrids as potential anticancer agents. | |
Compound anilinouracil-fluoroquinolone 271 demonstrated Gram-positive antibacterial potency at least 15 times that of the corresponding [3-(4-hydroxybutyl)-6-(3-ethyl-4-methylanilino)uracil], effectively inhibited pol IIIC and topoisomerase/gyrase, inhibited gyrase and B. subtilis topoisomerase IV with IC50 of 31 and 43.6 μM, respectively, and, as anticipated, had a selective effect on bacterial DNA. Additionally, this compound showed the ability to attack both of its possible targets in the bacterium by being active against a wide range of Gram-positive pathogens that were resistant to antibiotics, as well as a number of Gram-negative organisms. It was also active against Gram-positive organisms that were resistant to fluoroquinolones and anilinouracils. Moreover, it lacked toxicity in vitro and was bactericidal for Gram-positive bacteria. With a unique dual mechanism of action and strong activity against Gram-positive bacteria that are both susceptible to and resistant to antibiotics, this class of anilinouracil–fluoroquinolone hybrids offers a promising new pharmacophore (Fig. 12).272
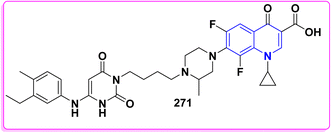 |
| | Fig. 12 Quinolone-pyrimidine based molecular hybrids as potential antibacterial agents. | |
Furthermore, the synthesized derivatives of 3-(2-amino-6-arylpyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-ones 272 had shown the best activity in the series, with anticancer efficacy against HepG2 cell lines (IC50 values of 1.32 and 1.33 μM, respectively) (Fig. 13).131
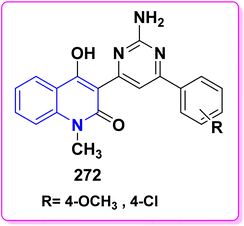 |
| | Fig. 13 Pyrimidinylquinolinones based molecular hybrids as potential anticancer agents. | |
5.2. Agricultural applications
In recent years, the discovery of agrochemicals based on the quinoline scaffold structure has led to great progress. Some new quinoline pesticides that have been commercialized or are under development not only inject new vitality into the market but also can replace unfriendly old pesticides.51 For example, ethyl-4-hydroxy-1-(4-methoxyphenyl)quinolinone-3-carboxylate 273, has been investigated for the first time as a sensitization chromophore for Tb(III) to improve selectivity and sensitivity for organophosphorus pesticide detection.273
The insecticidal activity of pyrazoloquinolone compounds was investigated, both in vitro and in vivo, against the cotton leafworm, S. littoralis, and cotton aphids. The most effective compounds were (E)-4-(2-(dimethylamino)phenyl)-2,5-dioxo-6-phenyl-1,2,5,6-tetrahydrobenzo[h][1,6] naphthyridin-3-carbonitrile 203 and 3-(1-(4-amino-5-mercapto-4H-1,2,4-trizol-3-yl)-1H-pyrazol-3-yl)-4-hydroxy-1-phenylquinoline-2(1H)-one 200, with LC50s of 119.79 and 164.63 mg L−1 against S. littoralis.192 Additionally, 1,4-dihydro-quinolinecarboxylate 275 is isolated from Beauveria sp. for the first time and has insecticidal activity against Bemisia tabaci with remarkable toxicity in contact and feeding assays, as its LC50 values were 10.59 μg mL−1 (contact) and 5.66 μg mL−1 for feeding. Whereas, no adverse effect on plant height/growth or phytotoxicity was detected on pepper, tomato, cotton, and cucumber during the treatment.274 Also, Liu et al.275 reported that the utilization of Schinifoline 276, the fruit pericarp of Zanthoxylum schinifolium possessed remarkable feeding toxicity against Sitophilus zeamais and Tribolium castaneum through reducing their food consumption and growth rate, leading to weakness of their reproductive ability and low resistance (Fig. 14).275
 |
| | Fig. 14 Agrochemical compounds with quinolinone scaffolds. | |
Additionally, a series of (E)-3-acyl-quinoline-2,4-(1H,3H)-dione imine derivatives 277 displayed remarkable herbicidal activity against monocot and dicot species by affecting PSII electron transport inhibitors and inhibiting the electron transport chain, leading to the prevention of the production of NADPH and ATP. As the majority of the compounds showed good to excellent herbicidal activities against a number of dicot or monocot species, with an inhibition percentage of over 50% at a dosage of 94 g per ha or even lower. Notably, the compounds featuring short straight-chain alkyl groups display the highest activity, comparable with the longer alkyl chains; insecticidal activity diminishes with increasing chain length (Fig. 15).276
 |
| | Fig. 15 Quinolinone scaffolds with agrochemical efficacy. | |
A series of substituted 4-hydroxy-1H-quinolin-2-one derivatives 278–280 were analyzed using RP-HPLC to determine lipophilicity and photosynthesis-inhibiting activity (the inhibition of photosynthesis in Spinacia oleracea L. As the synthesized quinolinones displayed high to moderate inhibitory effects on the photosynthesis activity process. In addition, in vitro antifungal screening of these scaffolds was evaluated against various fungal strains, displaying moderate efficacy (Fig. 16).277
 |
| | Fig. 16 Quinolinone scaffolds with photosynthesis-inhibiting activity. | |
Quinolactacide 281 is structurally analogous to quinolactacins derived from Penicillium species, which are recognized for exhibiting various biological activities, particularly insecticidal properties. This compound exhibited 88% mortality against the green peach aphid (Myzus persicae) at a concentration of 250 ppm. The insecticidal and miticidal efficacy of quinolactacide was assessed against five distinct insects and one mite at a concentration of 500 ppm. The insects utilized for the experiments included the green peach aphid, silverleaf whitefly (Bemisia argentifolii), diamondback moth (Plutella xylostella), common cutworm (Spodoptera litura), western flower thrips (Frankliniella occidentalis), and two-spotted spider mite (Tetranychus urticae). The synthetic quinolactacide exhibited no activity against the other insects, but it demonstrated 100% and 42% mortality against the green peach aphid and diamondback moth, respectively (Fig. 17).278
 |
| | Fig. 17 Quinolinone scaffolds with insecticidal activity. | |
5.3. Complex formation
Quinolinones bind metal ions to create complexes where they can function as bidentate, unidentate, or bridging ligands. Quinolinone molecules have a basic side nucleus that becomes protonated and shows up as cations in the ionic complexes under highly acidic conditions. Quinolone solubility, pharmacokinetics, and bioavailability are all significantly impacted by interaction with metal ions, which also plays a role in the bactericidal agents' mode of action. Many metal complexes have shown various biological activities, including antifungal, anticancer, antiparasitic, and antimicrobial (Fig. 18).279
 |
| | Fig. 18 Coordination modes of quinolinone complexes with high biological activity. | |
A series of Cu(II) 282 and Co(II) 283 complexes were designed and synthesized by Khalaf et al.280 as 4-hydroxy-2H-pyrano[3,2-c]quinolin-2,5(6H)-dione acts as monobasic didentate ligand and form tetrahedral and octahedral complexes at molar ratio of 1:2. Interestingly, the synthesized complexes displayed greater efficacy as antibacterial, antifungal and antioxidant candidates in comparison with the free pyrano[3,2-c]quinolin-2,5(6H)-dione as ligand (Fig. 19).280
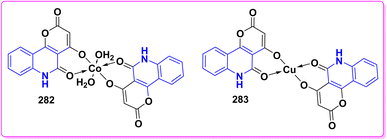 |
| | Fig. 19 Bioactive complexes with quinolinone scaffolds. | |
The potential antibacterial and antifungal properties of the quinoline ligands (SL1–SL4) and their Cu(II) and Zn(II) complexes (Fig. 20) were evaluated in vitro. The ligands (SL1–SL4) demonstrated moderate antibacterial activity against Gram-positive bacteria but exhibited no effectiveness against Gram-negative bacteria or fungal strains. Among them, the SL3 ligand displayed the highest activity, achieving a zone of inhibition of 24 mm against S. aureus. However, the antibacterial activity of these ligands was significantly lower than that of the standard drugs amoxicillin and fluconazole. These metal complexes generally enhanced antibacterial activity against both Gram-positive S. aureus and E. faecalis strains. On comparing the metal complexes, it was found that [Cu(SL1)2] was the most harmful substance to Gram-positive bacteria and had moderate antifungal properties.281
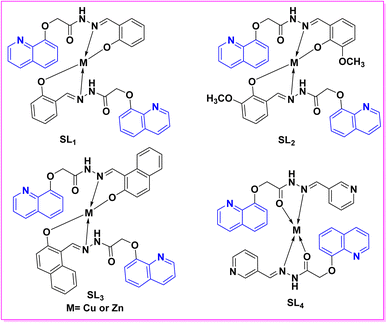 |
| | Fig. 20 Hydroxyquinoline functionalized Schiff base copper and zinc complexes SL1–4. | |
Fouad et al.282 studied lanthanide nano-complexes (Gd3+, Er3+, Pr3+, Y3+, Dy3+, and La3+) containing quinolinone. The obtained results reveal that the quinolinone 284 acts as a bidentate via OO donor sites, creating octahedral complexes. The complexes are nanoscale, having crystalline or amorphous structures. The synthesized nano-complexes have acceptable anticancer efficacy against the hepatocellular carcinoma cell line. Whereby, the maximum anticancer activity of Pr3+ nano-complex (Fig. 21).282
 |
| | Fig. 21 The chemical structures of lanthanide complexes. | |
A series of quinolinone-derived Schiff bases 286 were synthesized via the condensation of 7-amino-4-methylquinolinone 285 with different aromatic aldehydes. Then, complexation of Schiff ligands with cupric salts involving Cu(II) acetate or Cu(II) perchlorate hexahydrate was achieved, affording Cu(II) complexes of the corresponding ligands 287 (Scheme 101). These complexes showed significant antifungal effects, suppressing C. albicans growth by 50% at a dosage of 4 μM.283
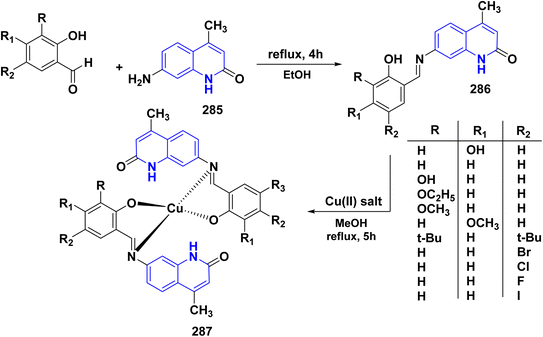 |
| | Scheme 101 Complexation of ligand 286 with Cu(II) salts. | |
5.4. Corrosion inhibitors
Hussein et al.185 reported the utilization of 4-hydroxyquinolinone derivatives as antioxidant agents for various lubricating oils through using the ASTM D-942 and ASTM D-664 tests. The compositions that they synthesized are: dichlorophenyl-pyrazol-4-hydroxy-1-methylquinolinone 288, 4-hydroxy-8-methyl-2-oxoquinolin-1-phenyl-2-hydroxyhydropyrimidin-4,6-dione 289, and 1-butyl-4-hydroxy-3-1H-pyrazol-3-yl)quinolinone 290 (Fig. 22). The obtained results indicate that the hydroxyquinolinone scaffolds reduce both the total acid number and the oxygen pressure drop in lubricating oils. On the other hand, it has been discovered that the antioxidant activity is most efficient when quinolinones have both butyl and hydroxyl groups. These quinoline scaffolds (288–290) inhibit radical processes during oil oxidation, as well as their continuation and production. A comparison of the antioxidant activity of compounds 288, 289, and 290 reveals that the efficacy and antioxidant benefits of the third compound.184,284
 |
| | Fig. 22 Quinolinone scaffolds with antioxidant effect in lubricating oils. | |
Quinolinone chalcone (PPQ) 291 showed effective corrosion inhibition for high carbon steel (HCS) in 1.0 M HCl, and the inhibition efficiency improved as the inhibitor concentration was raised. The compound's efficiency is attributed to the presence of heteroatoms and phenyl rings with delocalized π electrons that can act as adsorption centers. These factors support both physical and chemical interactions and adsorption of inhibitor scaffolds with the metal surface according to the Langmuir adsorption isotherm (Fig. 23).285,286
 |
| | Fig. 23 Quinolinone scaffolds as corrosion inhibitors. | |
The dihydroquinoline-3-carboxylate derivatives 292 and 293 (NODC, AODC, CODC, and MODC) display potential corrosion inhibition for carbon steel in 1 M HCl, due to the presence of a substituent attached to the benzene portion of the 4-quinolinone (Fig. 24).287
 |
| | Fig. 24 Quinolinones scaffolds as potential corrosion inhibitors. | |
Imino quinolinone IQ 294 acts as a corrosion inhibitor on low carbon steel (LCS) in 0.5 M HCl solution with inhibition efficiency 93.2% at the optimal concentration of IQ (30 × 10−3 Mm), adhered to the Langmuir adsorption model, signifying a monolayer adsorption mechanism. The characterization of IQ as a mixed-type inhibitor was evidenced due to its capacity to impede both cathodic and anodic processes, predominantly functioning as an anodic inhibitor (Fig. 25).288
 |
| | Fig. 25 Imino quinolinone IQ 294. | |
5.5. Sensors
5.5.1. Chemosensors. Quinolinone derivative-based chemosensors designed to detect various metal ions were discussed. There is considerable interest in improving these chemosensors due to their ease of synthesis, high sensitivity, and stability. Nonetheless, there remains substantial potential for enhancement in their in vivo applications, particularly regarding water solubility, selectivity, and fluorescent bio-imaging capabilities. Consequently, the development of receptors tailored for different ions is essential. For instance, skeleton 295 employed an alternative bonding model to identify Zn2+ and Cd2+. Also, 296 act as a Cr3+ chemosensor. Additionally, two-photon laser sources can be used to excite an extended conjugated quinoline system (Fig. 26).289
 |
| | Fig. 26 Quinolinone/hydroxyquinoline scaffolds as chemosensors. | |
5.5.2. Fluorescence sensors. Fluorescence-based techniques have become essential tools in metalworking because of their sensitivity and selectivity benefits.290–292 A lot of work has gone into creating fluorescent Fe3+ indicators that are highly sensitive and selective. The majority of documented fluorescent sensors for Fe3+ are based on a fluorescence quenching mechanism due to the paramagnetic nature of Fe3+,293,294 and the majority of these have interference issues brought on by transition metal cations like Cu2+, Co2+, or Hg2+. Thus, looking for highly selective chemosensors for Fe3+ based on the fluorescence enhancement is crucial. While using the ether derivative 297 as a starting material, the synthesis of Schiff base 299 was utilized. Whereby, quinoline 299 is a chemosensor that made up of the quinoline fluorophore 298 and p-anisidine binding site led to formation of bearing polarized C–C, C–N and C–O bonds conjugated to the quinoline moiety, it displays a strong fluorescence increase of Fe3+ ions, demonstrating excellent sensitivity and good selectivity for Fe3+ over a broad spectrum of other ecologically and physiologically significant metal ions (Schemes 102 and 103).295
 |
| | Scheme 102 Hydroxyquinoline scaffolds as fluorescence sensors. | |
 |
| | Scheme 103 Complexation mechanism of sensor 299 and Fe3+ ions. | |
Sensor 299 is an excellent sensitive chemosensor for the Fe3+ ion as it exhibits no fluorescence in MeOH, but when Fe3+ is added, it affords a highly effective fluorescence sensor 300, and there is a noticeable increase in fluorescence of about 44-fold (Scheme 103). As a result, the fluorescence response is explained by Fe3+ blockage of electron transfer to the quinoline group, which encouraged sensor 299 to produce high fluorescence. A color shift from colorless to brown that is readily apparent to the unaided eye resulted from the identification of Fe3+. Consequently, the Fe3+ ion can be readily distinguished in visible light from all other metal ions. Additionally, under light from a 365 nm UV lamp, Fe3+ shows an increase in the intensity of fluorescence, vivid green, while other metal ions show no change under the same circumstances.295
6. Conclusion
With the ever-increasing importance of 3-acetyl-4-hydroxyquinolinone and its N-substituted derivatives as structurally decisive scaffolds in bioactive natural products and pharmaceutical drugs, so in this review enormous efforts have been made to summarize the synthesis, structural features and chemical reactivity of 3-acetyl-4-hydroxy-2-quinolinone entities and its N-substituted derivatives as structurally decisive scaffolds in bioactive natural products and pharmaceutical drugs, accredited by reaction mechanisms. Also, we highlight the most important breakthroughs of 3-acetyl-4-hydroxy-2-quinolinone derivatives as an auspicious class displaying a wide range of potential pharmacological activities and their developments in the various clinical stages. Generally, this review supplies an overview of the chemistry for 3-acetyl-4-hydroxy-2-quinolinone derivatives and documents more than two hundred references over the last decade of research, covering mainly the period from 2020 to the beginning of 2025.
Abbreviations
| AcOH | Acetic acid |
| AHQ | 3-Acetyl-4-hydroxyquinolinone |
| BQB | 1,4-Bis-(quinolin-6-ylimino methyl)benzene |
| BF3·(OC2H5)2 | Boron trifluoride etherate |
| CS2 | Carbon disulfide |
| CCl4 | Carbon tetrachloride |
| DMF | Dimethylformamide |
| DMA | Dimethylamine |
| DMSO | Dimethyl sulfoxide |
| EAA | Ethyl acetoacetate |
| HCS | High carbon steel |
| NH2OH·HCl | Hydroxylamine hydrochloride |
| HMPT | Hexamethylphosphoric triamide |
| HQ | 4-Hydroxyquinoline |
| LCS | Low-carbon steel |
| LR | Lawesson's reagent |
| MCR | Multicomponent reaction |
| DMF/DMA | N,N-Dimethylformamide dimethyl acetal |
| DTA | N,N-Dimethyl trifluoroacetamide |
| NBS | N-Bromosuccinimide |
| POCl3 | Phosphorous oxychloride |
| P2S5 | Phosphorus pentasulfide |
| PBr3 | Phosphorus tribromide |
| PhPOCl2 | Phosphonic dichloride |
| PPA | Polyphosphoric acid |
| RORC | Ring opening and ring closure |
| RT | Room temperature |
| AcONa | Sodium acetate |
| SO2Cl2 | Sulfuryl chloride |
| NaBH4 | Sodium borohydrate |
| CH(OEt)3 | Triethyl orthoformate |
| TEA | Triethylamine |
| P(OCH2CH2Cl)3 | Tris(2-chloroethyl)phosphite |
| TB | Tuberculosis |
| TC | β,β-Tricarbonyl |
| TBAB | Tetrabutylammonium bromide |
| TsCl | Toluenesulfonyl chloride |
| WHO | World Health Organization |
| ZnCl2 | Zinc chloride |
Data availability
No primary research results, software or code have been included, and no new data were generated or analyzed as part of this review.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this review.
Acknowledgements
The authors are thankful to Mansoura University, Egypt, for their support under project ID: MU-SCI-23-64.
References
- A. Gulraiz, M. Sohail, M. Bilal, N. Rasool, M. Usman Qamar, C. Ciurea, L. G. Marceanu and C. Misarca, Molecules, 2024, 29, 2232 CrossRef PubMed.
- X. Zhang, M. Liu, W. Qiu and W. Zhang, Molecules, 2024, 29, 1241 CrossRef PubMed.
- W. S. Hamama, M. E. Ibrahim, A. A. Gooda and H. H. Zoorob, RSC Adv., 2018, 8, 8484 RSC.
- P. Borah, S. Hazarika, A. Chettri, D. Sharma, S. Deka, K. N. Venugopala, P. Shinu, N. A. Al-Shar’i, S. K. Bardaweel and P. K. Deb, In Viral, Parasitic, Bacterial, and Fungal Infections, 2023, pp. 781–804 Search PubMed.
-
(a) D. A. Sabbah, H. H. Samarat, E. Al-Shalabi, S. K. Bardaweel, R. Hajjo, K. Sweidan, R. Abu Khalaf, A. M. Al-Zuheiri and G. Abushaikha, ChemistrySelect, 2022, 7, e202200662 CrossRef;
(b) R. S. Keri, S. Budagumpi and V. Adimule, ACS Omega, 2024, 9, 42630–42667 CrossRef PubMed.
- M. Rana, R. Ranjan, N. S. Ghosh, D. Kumar and R. Singh, Curr. Cancer Ther. Rev., 2024, 20, 372–385 CrossRef.
-
(a) M. Ramanathan and Z. Moussa, Org. Chem. Front., 2025, 12, 256–327 RSC;
(b) S. M. Alsafy and N. Abd Alrazzak, Eng. Proc., 2024, 59, 178 Search PubMed.
- W. S. Hamama, A. E. Hassanien, M. G. El-Fedawy and H. H. Zoorob, J. Heterocycl. Chem., 2016, 53, 945–952 CrossRef.
- N. Bhusare and M. Kumar, Oncol. Res., 2024, 32, 849–875 CrossRef PubMed.
- K. Kajal, R. Shakya, M. Rashid, V. Nigam, B. D. Kurmi, G. D. Gupta and P. Patel, Sustain. Chem. Pharm., 2024, 37, 101374 CrossRef.
- R. B. Bakr, I. H. El Azab and N. A. A. Elkanzi, Chem. Biodivers., 2024, 21, e202400200 CrossRef PubMed.
- A. Darque, A. Dumètre, S. Hutter, G. Casano, M. Robin, C. Pannecouque and N. Azas, Bioorg. Med. Chem. Lett., 2009, 19, 5962–5964 CrossRef CAS PubMed.
- D. Dize, M. B. T. Tali, C. A. N. Ngansop, R. Keumoe, E. A. M. Kemgne, L. R. T. Yamthe, P. V. T. Fokou, B. P. Kamdem, K. Hata and F. F. Boyom, Future Pharmacol., 2024, 4, 188–198 CrossRef.
- J. Sun and J. J. Kessl, Viruses, 2024, 16, 200 CrossRef CAS.
- D. Shi, S. Xu, D. Ding, K. Tang, Y. Zhou, X. Jiang, S. Wang, X. Liu and P. Zhan, Expert Opin. Drug Discovery, 2024, 19, 139–146 CrossRef CAS PubMed.
- W. S. Hamama, A. E. Hassanien, M. G. El-Fedawy and H. H. Zoorob, J. Heterocycl. Chem., 2015, 52, 492–496 CrossRef CAS.
- B. A. Mısır, Y. Derin, S. Ökten, A. Aydın, Ü. M. Koçyiğit, H. Şahin and A. Tutar, J. Biochem. Mol. Toxicol., 2024, 38, e23706 CrossRef PubMed.
- M. M. Hammouda, M. M. Rashed, W. S. Abo El-Yazeed and K. M. Elattar, ChemistrySelect, 2024, 9, e202401384 CrossRef CAS.
- S. Siddique, K. R. Ahmad, S. K. Nawaz, A. R. Raza, S. N. Ahmad, R. Ali, I. Inayat, S. Suleman, M. A. Kanwal and M. Usman, Sci. Rep., 2023, 13, 8639 CrossRef CAS.
- P. Kołodziej, M. Wujec, M. Doligalska, A. Makuch-Kocka, D. Khylyuk, J. Bogucki, M. Demkowska-Kutrzepa, M. Roczeń-Karczmarz, M. Studzinska, K. Tomczuk, M. Kocki, P. Reszka-Kocka, S. Granica, R. Typek, A. L. Dawidowicz, J. Kocki and A. Bogucka-Kocka, J. Adv. Res., 2024, 60, 57–73 CrossRef PubMed.
- R. Das, D. K. Mehta, S. Gupta, S. Mujwar, V. Sharma, A. Goyal, S. Patel and A. Patel, Lett. Org. Chem., 2023, 20, 1182–1191 CrossRef CAS.
- Y. M. Abdel Aziz, M. S. Nafie, P. A. Hanna, S. Ramadan, A. Barakat and M. Elewa, Pharmaceuticals, 2024, 17, 710 CrossRef.
- M. C. Costas-Lago, P. Besada, R. Mosquera, E. Cano and C. Terań, Bioorg. Chem., 2024, 150, 107615 CrossRef CAS.
- T. M. Ramsis, M. A. Ebrahim and E. A. Fayed, Med. Chem. Res., 2023, 32, 2269–2278 CrossRef.
- S. Siddique, K. Hussain, N. Shehzadi, M. Arshad, M. N. Ashad, S. Iftikhar, F. Saghir, A. Shaukat, M. Sarfraz and N. Ahmed, Org. Biomol. Chem., 2024, 22, 3708–3724 RSC.
- H. E. Hashem, S. Ahmad, A. Kumer and Y. El Bakri, Sci. Rep., 2024, 14, 1152 CrossRef.
- Y. U. Cebeci, O. O. Batur and H. Boulebd, J. Mol. Struct., 2024, 1299, 137115 CrossRef.
- S. K. Verma, S. Rangappa, R. Verma, F. Xue, S. Verma, K. S. S. Kumar and K. S. Rangappa, Bioorg. Chem., 2024, 145, 107241 CrossRef PubMed.
- K. B. Gangurde, R. A. More, V. A. Adole and D. S. Ghotekar, J. Mol. Struct., 2024, 1299, 136760 CrossRef.
- Y. Jia, Y. Zhao, M. Niu, C. Zhao, X. Li and H. Chen, Anim. Models Exp. Med., 2024, 7, 419–432 CrossRef.
- M. El Mesky, H. Zgueni, Y. Rhazi, O. El-Guourrami, O. Abchir, M. Jabha, A. Nakkabi, S. Chtita, S. Achamlale, M. Chalkha, D. Chebabe and El. Mabrouk, J. Mol. Struct., 2024, 1313, 138705 CrossRef.
-
(a) Swati, A. Raza, B. Singh and Dr. Pankaj Wadhwa, ChemistrySelect, 2025, 10, e202404978 CrossRef;
(b) I. Ahmad, H. Khalid, A. Perveen, M. Shehroz, U. Nishan, F. Ur Rahman, Sheheryar, A. A. Moura, R. Ullah, E. A. Ali, M. Shah and S. C. Ojha, ACS Omega, 2024, 9, 16262–16278 CrossRef.
- M. A. Fawzy, K. H. Ibrahim, A. A. Aly, A. H Mohamed, S. M. N. Abdel Hafez, W. Y. Abdelzaher, E. B Elkaeed, A. A Alsfouk and El. MN Abdelhafez, Future Med. Chem., 2024, 16, 2211–2230 CrossRef PubMed.
- A. M. Mohassab, H. A. Hassan, H. A. Abou-Zied, M. Fujita, M. Otsuka, H. A. M. Gomaa, B. G. M. Youssif and M. Abdel-Aziz, J. Mol. Struct., 2024, 1297, 136953 CrossRef.
- W. S. Hamama, M. E. Ibrahim, A. A. Gooda and H. H. Zoorob, J. Heterocycl. Chem., 2018, 55, 2623–2634 CrossRef CAS.
- M. Mashhadinezhad, M. Mamaghani, M. Rassa and F. Shirini, ChemistrySelect, 2019, 4, 4920–4932 CrossRef CAS.
- G. L. Monica, A. Bono, F. Alamia, A. Lauria and A. Martorana, Bioorg. Med. Chem., 2024, 109, 117791 CrossRef PubMed.
- E. A. Ghaith, H. H. Zoorob, M. E. Ibrahim, M. Sawamura and W. S. Hamama, ChemistrySelect, 2020, 5, 14917–14923 CrossRef CAS.
- S. Nadar and T. Khan, Chem. Biol. Drug Des., 2022, 100, 818–842 CrossRef CAS.
- W. S. Hamama, A. E. Hassanien, M. G. El-Fedawy and H. H. Zoorob, J. Heterocycl. Chem., 2017, 54, 859–863 CrossRef CAS.
- X. Guo, Q. Zhou, X. Fan, Q. Zhu and M. Jin, ACS Appl. Polym. Mater., 2024, 6, 5566–5575 CrossRef CAS.
- M. Olesiejuk, A. Kudelko and M. Świątkowski, Dyes Pigm., 2023, 220, 111721 CrossRef CAS.
- W. S. Hamama, M. A. Waly, I. I. EL-Hawary and H. H. Zoorob, J. Heterocycl. Chem., 2016, 53, 953–957 CrossRef CAS.
- S. Bozorgnia, M. Pordel, A. Davoodnia and S. A. Beyramabadi, Opt. Mater., 2024, 148, 114762 CrossRef CAS.
- M. Escolano, D. Gaviña, G. Alzuet-Piña, S. Díaz-Oltra, M. Sánchez-Roselló and C. del Pozo, Chem. Rev., 2024, 124, 1122–1246 CrossRef CAS.
-
(a) A. A. Aly, H. A. Abd El-Naby, E. Kh. Ahmed, S. A. Gedamy, M. B. Alshammari, A. Ahmad and S. Bräse, Curr. Org. Chem., 2025, 29, 181–212 CrossRef CAS;
(b) A. Mandal and A. T. Khan, Org. Biomol. Chem., 2024, 22, 2339–2358 RSC.
- O. O. Ajani, K. T. Iyaye and O. T. Ademosun, RSC Adv., 2022, 12, 18594–18614 RSC.
- S. Rajendran, K. Sivalingam, R. P. K. Jayarampillai, W. Wang and C. O. Salas, Chem. Biol. Drug Des., 2022, 100, 1042–1085 CrossRef CAS PubMed.
- L. F. Hernández-Ayala, E. G. Guzmán-López and A. Galano, Antioxidants, 2023, 12, 1853 CrossRef.
- R. Kumar, A. Thakur, Sachin, D. Chandra, A. K. Dhiman, P. K. Verma and U. Sharma, Coord. Chem. Rev., 2024, 499, 215453 CrossRef.
- Q. Cai, H. Song, Y. Zhang, Z. Zhu, J. Zhang and J. Chen, J. Agric. Food Chem., 2024, 72, 12373–12386 CrossRef.
- M. Owais, A. Kumar, S. M. Hasan, K. Singh, I. Azad, A. Hussain, Suvaiv and M. Akil, Mini-Rev. Med. Chem., 2024, 24, 1238–1251 CrossRef.
- S. Ghosh, S. Mallick, D. Karolly and S. D. Sarkar, ACS Org. Inorg. Au, 2024, 5, 492–497 CrossRef.
-
(a) W. Al Zoubi and Y. G. Ko, J. Colloid Interface Sci., 2020, 565, 86–95 CrossRef PubMed;
(b) Y. Senpradit, S. Wacharasindhu and M. Sukwattanasinitt, Spectrochim. Acta, Part A, 2025, 326, 125128 CrossRef.
- H. M. Abd El-Lateef, A. G. A. Gaafar, A. S. Alqahtani, A. A. Al-Mutairi, D. S. Alshaya, F. G. Elsaid, E. Fayadg and N. A. Farouk, RSC Adv., 2024, 14, 24781–24790 RSC.
- P. A. Jagtap, V. R. Sawant and B. M. Bhanage, ChemCatChem, 2024, 16, e202400979 CrossRef.
- S. Sharma, K. Singh and S. Singh, Curr. Org. Synth., 2023, 20, 606–629 CrossRef PubMed.
- M. Mohasin, M. Z. Alam, Q. Ullah, A. Ahmad, P. F. Rahaman and S. A. Khan, Polycycl, 2024, 44, 6369–6398 Search PubMed.
- A. Dorababu, Arch. Pharm., 2021, 354, 2000232 CrossRef.
- D. Mabire, S. Coupa, C. Adelinet, A. Poncelet, Y. Simonnet, M. Venet, R. Wouters, A. S. J. Lesage, L. V. Beijsterveldt and F. Bischoff, J. Med. Chem., 2005, 48, 2134–2153 CrossRef.
- T. L. Viveka, G. Angajala, V. Aruna, M. Nakka and Y. Aparna, J. Mol. Struct., 2024, 1303, 137482 CrossRef.
- F. H. Al-Ostoot, Zabiulla, S. Salah and S. A. Khanum, J. Iran. Chem. Soc., 2021, 18, 1839–1875 CrossRef.
- J. Bergwik, J. Liu, M. Padra, R. K. V. Bhongir, L. Tanner, Y. Xiang, M. Lundblad, A. Egesten and M. Adner, Respir. Res., 2024, 25, 146 CrossRef PubMed.
- P. Y. Wang, H. Chen, Y. Wanga and Y. K. Lyu, J. Chem. Technol. Biotechnol., 2020, 95, 2171–2179 CrossRef.
- C. M. Al-Matarneh, A. Nicolescu, I. C. Marinas, M. D. Găboreanu, S. Shova, A. Dascălu, M. Silion and M. Pinteală, Molecules, 2024, 29, 772 CrossRef.
- G. Shumi, T. B. Demissie, R. Eswaramoorthy, R. F. Bogale, G. Kenasa and T. Desalegn, ACS Omega, 2024, 9, 25014–25026 CrossRef.
- J. C. Coa, A. Yepes, M. Carda, L. Conesa-Milián, Y. Upegui, S. M. Robledo, Dr. and W. Cardona-G, ChemistrySelect, 2020, 5, 2918–2924 CrossRef.
- I. A. Bala, O. F. Al Sharif, A. M. Asiri and R. M. El-Shishtawy, Results Chem., 2024, 7, 101529 CrossRef.
- M. H. El-Shershaby, K. M. El-Gamal, A. H. Bayoumi, K. El-Adl, M. Alswah, H. E. A. Ahmed, A. A. Al-Karmalamy and H. S. Abulkhair, New J. Chem., 2021, 45, 13986–14004 RSC.
- S. Kondaparla, A. Soni, A. Manhas, K. Srivastava, S. K. Purib and S. B. Katti, RSC Adv., 2016, 6, 105676–105689 RSC.
- A. Rezvanian, B. Khodadadi, S. Tafreshi and P. Shiri, Mol. Diversity, 2024, 28(1), 197–207 CrossRef CAS PubMed.
- A. D. Sonawane, D. R. Garud, T. Udagawa and M. Koketsu, Org. Biomol. Chem., 2018, 16, 245–255 RSC.
- K. V. Belyaeva, L. P. Nikitina, L. A. Oparina, V. S. Saliy, D. N. Tomilin, A. V. Kuzmin, A. V. Afonin and B. A. Trofimov, New J. Chem., 2024, 48, 1336–1349 RSC.
- X. An, N. Li, L. Zhang, Z. Xu, S. Zhang and Q. Zhang, J. Hazard. Mater., 2024, 465, 133158 CrossRef CAS.
- http://www.africanplants.senckenberg.de/root/index.php?page_id=78%26id=4191#.
- R. Kaur and K. Kumar, Eur. J. Med. Chem., 2021, 215, 113220 CrossRef CAS.
- P. Yadav and K. Shah, Bioinorg. Chem., 2021, 109, 104639 CrossRef CAS.
- D. Talwar, A. Gonzalez-de-Castro, H. Y. Li and J. Xiao, Angew. Chem., 2015, 127, 5312–5316 CrossRef.
- B. S. Matada, R. Pattanashettar and N. G. Yernale, Bioorg. Med. Chem., 2021, 32, 115973 CrossRef CAS.
- A. D. Arboleda, L. M. Moreno and R. Abonia, Curr. Org. Chem., 2024, 28, 595–635 CrossRef CAS.
- M. Kischkewitz, B. Marinic, N. Kratena, Y. Lai, H. B. Hepburn, M. Dow, K. E. Christensen and T. J. Donohoe, Angew. Chem., Int. Ed., 2022, 61, e202204682 CrossRef CAS.
- k. Proisl, S. Kafka and J. Kosmrlj, Curr. Org. Chem., 2017, 21, 1949–1975 CrossRef CAS.
- S. S. Nazrullaev, I. A. Bessonova and Kh. S. Akhmedkhodzhaeva, Chem. Nat. Compd., 2001, 37, 551–555 CrossRef CAS.
-
(a) S. S. Anjanikar and S. S. Chandole, Orient. J. Chem., 2023, 39, 197 CAS;
(b) K. Gach-Janczak, J. Piekielna-Ciesielska, J. Wáskiewicz, K. Krakowiak, K. Wtorek and A. Janecka, Molecules, 2025, 30, 163 CrossRef CAS PubMed.
- M. A. Barmade, P. Agrawal, S. R. Rajput, P. R. Murumkar, B. Rana, D. Sahal and M. R. Yadav, RSC Med.Chem., 2024, 15, 572–594 RSC.
- A. Uddin, S. Gupta, R. Shoaib, B. Aneja, I. Irfan, K. Gupta, N. Rawat, J. Combrinck, B. Kumar, M. Aleem, P. Hasan, M. C. Joshi, Y. S. Chhonker, M. Zahid, A. Hussain, K. Pandey, M. F. Alajmi, D. J. Murry, T. J. Egan, S. Singh and M. Abid, Eur. J. Med. Chem., 2024, 264, 115969 CrossRef CAS.
- J. Gao, H. Hou and F. Gao, Eur. J. Med. Chem., 2023, 247, 115026 CrossRef CAS PubMed.
- B. Ç
![[i with combining dot above]](https://www.rsc.org/images/entities/char_0069_0307.gif) ftc, S. Ökten, Ü. M. Koçyğt, V. E. Atalay, M. Ataş and O. Çakmak, Eur. J. Med. Chem. Rep., 2024, 10, 100127 Search PubMed.
ftc, S. Ökten, Ü. M. Koçyğt, V. E. Atalay, M. Ataş and O. Çakmak, Eur. J. Med. Chem. Rep., 2024, 10, 100127 Search PubMed. - S. Guin, K. M. Alden, D. J. Krysan and M. J. Meyers, ACS Med. Chem. Lett., 2024, 15, 822–827 CrossRef CAS.
- S. Verma, S. Lal and R. Narang, Future Med. Chem., 2024, 16, 1283–1286 CrossRef.
- T. Arasakumar, S. Mathusalini, S. Gopalan, S. Shyamsivappan, A. Ata and P. S. Mohan, Bioorg. Med. Chem. Lett., 2017, 27, 1538–1546 CrossRef CAS.
- E. A. E. El-Helw, M. Asran, M. E. Azab, M. H. Helal, A. Y. A. Alzahrani and S. K. Ramadan, Sci. Rep., 2024, 14, 15522 CrossRef CAS.
- S. G. Azimi, G. Bagherzade, M. R. Saberi and Z. A. Tehranizadeh, Bioinorg. Chem. Appl., 2023, 2881582 CAS.
- M. Beus, L. Persoons, D. Daelemans, D. Schols, K. Savijoki, P. Varmanen, J. Yli-Kauhaluoma, K. Pavić and B. Zorc, Mol. Diversity, 2022, 26, 2595–2612 CrossRef CAS.
- H. Abdelmegeed, L. M. A. Abdel Ghany, A. Youssef, A. S. El-Etrawycd and N. Ryadc, RSC Adv., 2024, 14, 22092–22112 RSC.
- X. He, J. Chen, M. Kandawa-Shultz, G. Shao and Y. Wang, Dalton Trans., 2023, 52, 4728–4736 RSC.
- G. Jina, Z. Lib, F. Xiaoa, X. Qia and X. Sun, Bioorg. Chem., 2020, 99, 103837 CrossRef.
- N. D. Shashikumar, G. Krishnamurthy, H. S. Bhojyanaik, M. R. Lokesh and K. S. Jithendrakumara, J. Chem. Sci., 2014, 126, 205–212 CrossRef CAS.
- R. P. Pakhariya, A. Bhatnagar and G. Pemawat, RSC Adv., 2025, 15, 3646–3663 RSC.
- J. C. d. Santos, J. E. F. Alves, R. D. S. de Azevedo, M. L. de Lima, M. R. de Oliveira Silva, J. G. da Silva, J. M. da Silva, A. C. de Carvalho Correia, M. do Carmo Alves de Lima, J. F. de Oliveira, R. O. de Moura and S. M. V. de Almeida, Int. J. Biol. Macromol., 2024, 254, 127651 CrossRef.
- A. Saral, R. Shahidha, M. Thirunavukkarasu and S. Muthu, Chem. Phys. Impact., 2023, 6, 100193 CrossRef.
- S. B. Wang, X. Q. Deng, Y. Zheng, H. J. Zhang and Z. S. Quan, Arch. Pharmacal Res., 2013, 36, 32–40 CrossRef CAS.
- K. Shabana, Salahuddin, A. Mazumder, H. Singh, R. Kumar, S. Tyagi, V. Datt, A. S. Sharma, M. S. Yar, M. J. Ahsan and R. K. Yadav, ChemistrySelect, 2023, 8, e202300209 CrossRef CAS.
- J. Song, Y. Zhu, W. Zu, C. Duan, J. Xu, F. Jiang, X. Wang, S. Li, C. Liu, Q. Gaoa, H. Li, Y. Zhang, W. Tang, T. Lu and Y. Chen, Bioorg. Med. Chem., 2021, 29, 115856 CrossRef CAS.
- S. K. Suthar, V. Jaiswal, S. Lohan, S. Bansal, A. Chaudhary, A. Tiwari, A. T. Alex and A. Joesph, Eur. J. Med. Chem., 2013, 63, 589–602 CrossRef CAS.
- S. H. Pattanashetty, K. M. Hosamani and D. A. Barretto, Chem. Data Collect., 2018, 15, 184–196 CrossRef.
- A. Krishna, V. Vijayakumar and S. Sarveswari, ChemistrySelect, 2020, 5, 7967–7972 CrossRef CAS.
- I. Kostopoulou, A. Diassakou, E. Kavetsou, E. Kritsi, P. Zoumpoulakis, E. Pontiki, D. Hadjipavlou-Litina and A. Detsi, Mol. Diversity, 2021, 25, 723–740 CrossRef CAS.
- M. Azad, M. A. Munawar and M. Athar, J. Appl. Sci., 2007, 7, 1620–1625 CrossRef CAS.
- M. Rowley, P. D. Leeson, G. I. Stevenson, A. M. Moseley, I. Stansfield, I. Sanderson, L. Robinson, R. Baker, J. A. Kemp, G. R. Marshall, A. C. Foster, S. Grimwood, M. D. Tricklebank and K. L. Saywell, J. Med. Chem., 1993, 36, 3386–3396 CrossRef CAS PubMed.
- T. S. Tunna, I. S. M. Zaidul, Q. U. Ahmed, K. Ghafoor, F. Y. Al-Juhaimi, M. S. Uddin, M. Hasan and S. Ferdous, S. Afr. J. Bot., 2015, 99, 144–152 CrossRef CAS.
- M. M. Hassan and H. M. Hassanin, J. Heterocycl. Chem., 2017, 54, 3321–3330 CrossRef CAS.
- V. M. Krokhalev, V. I. Saloutin, A. D. Romas, B. A. Ershov and K. I. Pashkevich, Div. Chem. Sci., 1990, 39, 316–322 Search PubMed.
- A. Detsi, V. Bardakos, J. Markopoulos and O. Igglessi-Markopoulou, J. Chem. Soc., Perkin Trans. 1, 1996, 1, 2909–2913 RSC.
- M. M. Abdou, Z. Seferoğlu, M. Fathy, T. Akitsu, M. Koketsu, R. Kellow and E. Amigues, Res. Chem. Intermed., 2019, 45, 919–934 CrossRef CAS.
- G. Athanasellis, E. Gavrielatos, O. Igglessi-Markopoulou and J. Markopoulos, J. Heterocycl. Chem., 2003, 40, 645–648 CrossRef CAS.
- S. M. Abu-elwafa, E. E. Mohamed, R. M. Issa and M. Gaber, Indian J. Chem., 1985, 24A, 407–411 CAS.
- P. G. Dessai, S. P. Dessai, R. Dabholkar, P. Pednekar, S. Naik, S. Mamledesai, M. Gopal, P. Pavadai, B. K. Kumar, S. Murugesan, S. Chandavarkar, P. Theivendren and K. Selvaraj, Mol. Diversity, 2023, 27, 1567–1586 CrossRef CAS.
- H. B. Bürgi and J. D. Dunitz, Structure Correlation, John Wiley & Sons, 2008 Search PubMed.
- I. V. Ukrainets, A. A. Tkach and L. Y. Yang, Chem. Heterocycl. Compd., 2009, 45, 169–175 CrossRef CAS.
- V. V. Kouznetsov, L. Y. V. Méndez and C. M. M. Gómez, Curr. Org. Chem., 2005, 9, 141–161 CrossRef CAS.
- L. M. Nainwal, S. Tasneem, W. Akhtar, G. Verma, M. F. Khan, S. Parvez, M. Shaquiquzzaman, M. Akhter and M. M. Alam, Eur. J. Med. Chem., 2019, 164, 121–170 CrossRef CAS PubMed.
- G. A. Ramann and B. J. Cowen, Molecules, 2016, 21, 986 CrossRef.
- S. M. Prajapati, K. D. Patel, R. H. Vekariya, S. N. Panchal and H. D. Patel, RSC Adv., 2014, 4, 24463–24476 RSC.
- J. Barluenga, F. Rodriguez and F. J. FaÇanas, Chem.–Asian J., 2009, 4, 1036–1048 CrossRef CAS.
- A. Weyesa and E. Mulugeta, RSC Adv., 2020, 10, 20784–20793 RSC.
- J. B. Bharate, R. A. Vishwakarma and S. B. Bharate, RSC Adv., 2015, 5, 42020–42053 RSC.
- C. Teja and F. R. N. Khan, Chem.–Asian J., 2020, 15, 4153–4167 CrossRef CAS PubMed.
- E. Vessally, L. Edjlali, A. Hosseinian, A. Bekhradnia and M. D. Esrafil, RSC Adv., 2016, 6, 49730–49746 RSC.
- T. Kappe, R. Aigner, P. Hohengassner and W. Stadlbauer, J. Prakt. Chem., 1994, 336, 596–601 CrossRef CAS.
- D. N. Toan, N. D. Thanh, M. X. Truong and D. T. Van, Arabian J. Chem., 2020, 13, 7860–7874 CrossRef CAS.
- T. Razzaq and C. O. Kappe, Tetrahedron Lett., 2007, 48, 2513–2517 CrossRef CAS.
- D. N. Toan, N. D. Thanh, M. X. Truong, D. N. Bang, M. T. Ngaa and N. T. T. Huong, New J. Chem., 2020, 44, 20715–20725 RSC.
- D. Abdel-Kadera, M. Abass and S. Shawkat, Russ. J. Org. Chem., 2024, 60, 294–328 CrossRef.
- W. Stadlbauer and G. Hojas, J. Heterocyclic Chem., 2004, 41, 681 CrossRef CAS.
- R. E. Bowman, A. Campbell and E. M. Tanner, J. Chem. Soc., 1959, 81, 444–447 RSC.
- M. A. Ibrahim, H. M. Hassanin, M. Abass and S. Badran, Arkivoc, 2013, 4, 424–431 Search PubMed.
- K. Tomita, Yakugaku Zasshi, 1951, 71, 1100–1112 CrossRef CAS.
- S. Elgogary, H. Abd Elghafar and M. Mashaly, J. Chin. Chem. Soc., 2021, 68, 1082–1089 CrossRef CAS.
- M. M. Hassan, E. S. Othman and M. Abass, Res. Chem. Intermed., 2013, 39, 1209–1225 CrossRef CAS.
- K. Alla and S. Sarveswari, Iran J. Sci. Technol. Trans. Sci., 2019, 43, 465–475 CrossRef.
- S. Sarveswari, V. Vijayakumar, R. Siva and R. Priya, Appl. Biochem. Biotechnol., 2015, 175, 43–64 CrossRef CAS PubMed.
- J. Brawley, E. Etter, D. Heredia, A. Intasiri, K. Nennecker, J. Smith, B. M. Welcome, R. K. Brizendine, T. W. Gould, T. W. Bell and C. Cremo, J. Med. Chem., 2020, 63, 11131–11148 CrossRef CAS PubMed.
- M. Shariat, M. W. Samsudin and Z. Zakaria, Chem. Cent. J., 2013, 7, 58 CrossRef.
- C. Mitsos, J. Petrou, O. Igglessi-Markopoulou and J. Markopoulos, J. Heterocycl. Chem., 1999, 36, 881–887 CrossRef CAS.
- M. Roussaki, B. Hall, S. C. Lima, A. C. da Silva, S. Wilkinson and A. Detsi, Bioorg. Med. Chem. Lett., 2013, 23, 6436–6441 CrossRef CAS PubMed.
- B. Baruah, K. Dasu, B. Vaitilingam, A. Vanguri, S. R. Casturi and K. R. Yeleswarapu, Bioorg. Med. Chem. Lett., 2004, 14, 445–448 CrossRef CAS.
- S. Dutt and V. Tyagi, Tetrahedron Lett., 2021, 87, 153527 CrossRef CAS.
- R. Satheeshkumar, R. Shankar, W. Kaminsky and K. J. R. Prasad, ChemistrySelect, 2016, 21, 6823–6829 CrossRef.
- Q. Zhang, J. Yuan, M. Yu, R. Zhang, Y. Liang, P. Huang and D. Dong, Synthesis, 2017, 49, 4996–5002 CrossRef CAS.
- H. M. Hassanin, M. A. Ibrahim, Y. A. Gabr and Y. A. Alnamer, J. Heterocycl. Chem., 2012, 49, 1269–1289 CrossRef CAS.
- D. K. Kumar, R. Rajkumar and S. P. Rajendran, Chem. Heterocycl. Compd., 2016, 52, 322–325 CrossRef CAS.
- M. Abass, A. R. A. Alzandi, M. M. Hassan and N. Mohamed, Polycyclic Aromat. Compd., 2021, 41, 2120–2209 CrossRef CAS.
- S. D. Naik, S. K. Chandavarkar, S. S. Tawade, S. G. Shingade, M. B. Palkard and S. N. M. Desai, Indian J. Chem., 2022, 61, 544–550 CAS.
- P. Roschger and W. Stadlbaue, Liebigs Ann. Chem., 1990, 821–823 CrossRef CAS.
- W. Steinschifter, W. Fiala and W. Stadlbauer, J. Heterocycl. Chem., 1994, 31, 1647–1652 CrossRef CAS.
- G. Hojas, W. Fiala and W. Stadlbauer, J. Heterocycl. Chem., 2000, 37, 1559–1569 CrossRef CAS.
- S. Chimichi, M. Boccalini and A. Matteucci, Tetrahedron, 2007, 63, 11656–11660 CrossRef CAS.
- R. R. Raiturkara, S. N. M. Desaia, V. M. Graciasa, A. D. S. Sangavkara, C. Fernandesa, B. S. Biradarb and S. K. Chandavarkar, Indian J. Chem., 2022, 61, 849–857 Search PubMed.
- M. G. Reis, S. M. Desai, S. Naik, J. Fernandes and P. Tari, Indian J. Chem., 2016, 55B, 1254–1258 CAS.
- G. A. Salman, Al-Mustansiriyah J. Sci., 2018, 28, 73–79 CrossRef.
- S. N. M. Desai, R. N. S. Priolkar, H. A. N. Karmali, P. D. Amambe and B. S. Birdar, Int. J. Pharm. Pharm. Sci., 2017, 9, 240–244 CrossRef CAS.
- H. M. Hassanin and S. M. El-edfawy, Heterocycles, 2012, 85, 2421–2436 CrossRef CAS.
- K. Faber and T. Kappe, J. Heterocycl. Chem., 1984, 21, 1881–1883 CrossRef CAS.
- S. Goswami, K. Ghosh, R. Mukherjee, A. K. Adak and A. K. Mahapatra, J. Heterocycl. Chem., 2001, 38, 173–177 CrossRef CAS.
- B. P. Nandeshwarappa, S. K. Manjunatha, D. K. Ramesh, M. Suchitra and S. O. Sadashiv, J. Pharm. Sci., 2020, 10, 33–38 Search PubMed.
- M. M. Girges, M. A. Hanna, H. M. Hassan and E. B. Moawad, Collect. Czechoslov. Chem. Commun., 1988, 53, 3179–3183 CrossRef CAS.
- G. Bolakatti, M. Palkar, M. Katagi, G. Hampannavar, R. V. Karpoormathd, S. Ninganagouda and A. Badiger, J. Mol. Struct., 2021, 1227, 129413 CrossRef CAS.
- A. Vilsmeier and A. Haack, Ber. Dtsch. Chem. Ges., 1927, 60, 119–122 CrossRef.
- M. A. Ibrahim, H. M. Hassanin and Y. A. Alnamer, Synth. Commun., 2014, 44, 3470–3482 CrossRef CAS.
- A. V. Manaev, I. N. Okhrimenko, K. A. Lyssenko and V. F. Traven, Russ. Chem. Bull., 2008, 57, 1734–1739 CrossRef CAS.
- T. kappe, R. Aigner, M. Jobstl, P. Hohengassner and W. Stadlbauer, Heterocycl. Commun., 1995, 1, 341–352 CAS.
- M. Islamuddin, O. Afzal, W. H. Khan, M. Hisamuddin, A. S. A. Altamimi, I. Husain, K. Kato, M. A. Alamri and S. Parveen, ACS Omega, 2021, 6, 9791–9803 CrossRef CAS.
- M. Sankaran, C. Kumarasamy, U. Chokkalingam and P. S. Mohan, Bioorg. Med. Chem. Lett., 2010, 20, 7147–7151 CrossRef CAS.
- T. Arasakumar, S. Mathusalini, K. Lakshmi, P. S. Mohan, A. Ata and C. H. Lin, Synth. Commun., 2016, 46, 232–241 CrossRef CAS.
-
(a) S. S. Ibrahim, Z. M. El-Gendy, H. A. Allimony and E. S. Othman, Chem. Pap., 1999, 53, 53–64 CAS;
(b) M. Abass, Synth. Commun., 2000, 30, 2735–2757 CrossRef CAS.
- M. M. Hassan, A. R. A. Alzandi and M. M. Hassan, Arabian J. Chem., 2020, 13, 6184–6190 CrossRef CAS.
- M. Abdel-Megid, M. Abass and M. Hassan, J. Heterocycl. Chem., 2007, 44, 315–322 CrossRef CAS.
- M. M. Hassan, S. M. Abdel-Kariem and T. E. Ali, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 866–873 CrossRef CAS.
- S. Sarveswari and V. Vijayakumar, Arabian J. Chem., 2016, 9, S835–S839 CrossRef CAS.
- G. V. Kalechits, V. K. Ol’khovik, I. I. Kalosha, E. D. Skakovskii, A. A. Pap, A. A. Zenyuk and Y. V. Matveenko, Russ. J. Gen. Chem., 2001, 71, 1257 CrossRef CAS.
- M. A. Munawar, M. Azad, H. L. Siddiquia and F. Nasim, J. Chin. Chem. Soc., 2008, 55, 394–400 CrossRef CAS.
- R. S. Bhupathi, B. R. Devi and P. K. Dubey, Indian J. Chem., 2012, 51, 855 Search PubMed.
- H. Maruthesh, M. S. Katagi and B. P. Nandeshwarappa, Russ. J. Org. Chem., 2023, 49, 1059–1067 CAS.
- M. F. Hussein, M. A. Ismail and R. A. El-Adly, Int. J. Org. Chem., 2016, 6, 207–219 CrossRef CAS.
- V. F. Traven, A. V. Manaev, I. V. Voevodina and I. N. Okhrimenko, Russ. Chem. Bull., 2008, 57, 1508–1515 CrossRef CAS.
- A. Krishna and S. Sarveswari, ChemistrySelect, 2019, 4, 9987–9992 CrossRef CAS.
- M. Abass, M. Abdel-Megid and M. Hassan, Synth. Commun., 2007, 37, 329–352 CrossRef CAS.
- M. Abass and A. Hassan, Chem. Pap., 2003, 57, 267–277 CAS.
- M. Abass and B. B. Mostafa, Bioorg. Med. Chem., 2005, 13, 6133–6144 CrossRef CAS.
- F. M. A. A. El-Taweel, J. Heterocycl. Chem., 2005, 42, 943–946 CrossRef CAS.
- N. N. Elnaggar, W. S. Hamama, M. Abd El Salam and E. A. Ghaith, RSC Adv., 2025, 15(8), 6050–6067 RSC.
-
(a) M. Abass and E. S. Othman, Synth. Commun., 2001, 31, 3361–3376 CrossRef CAS;
(b) A. Klásek, K. Kořistek, P. Sedmera and P. Halada, Heterocycles, 2003, 60, 799–815 CrossRef.
- A. Hassan, M. Badr, D. Abdelhamid, H. A. Hassan, M. A. S. Abourehab and G. A. Abuo-Rahma, Bioinorg. Chem., 2022, 120, 105631 CrossRef CAS.
- A. A. Elagamey, F. M. A. El-Taweel and M. H. M. Khalil, Sci. J. Damietta Fac. Sci., 2012, 1, 33–41 Search PubMed.
- V. S. Rao and M. Darbarwar, Synthesis, 1989, 139–141 CrossRef CAS.
- Z. Chen and Z. Wang, Tetrahedron, 2016, 72, 4288–4293 CrossRef CAS.
- T. Kappe and B. Schnell, J. Heterocycl. Chem., 1996, 33, 663–670 CrossRef CAS.
- W. Fiala and W. Stadlbauer, J. Prakt. Chem., 1993, 335, 128–134 CrossRef CAS.
- E. M. El-Sheref, M. A. I. Elbastawesy, A. B. Brown, A. M. Shawky, H. A. M. Gomaa, S. Bräse and B. G. M. Youssif, Molecules, 2021, 26, 6798 CrossRef CAS.
- B. Madhu, C. V. R. Reddy and P. K. Dubey, Synth. Commun., 2017, 47, 421–427 CrossRef CAS.
- B. Madhu, B. R. Sekar, C. V. R. Reddy and P. K. Dubey, Res. Chem. Intermed., 2017, 43, 6993–7012 CrossRef CAS.
- S. I. Bhat and D. R. Trivedi, RSC Adv., 2014, 4, 11300–11304 RSC.
- R. Bhupathi, B. Madhu, B. R. Devi, C. V. R. Reddy and P. K. Dubey, J. Heterocyclic Chem., 2016, 53, 1911–1916 CrossRef CAS.
- S. A. Barr, C. F. Neville, M. F. Grundon, D. R. Boyd, J. F. Malone and T. A. Evans, J. Chem. Soc., Perkin Trans. 1, 1995, 1, 445 RSC.
- J. H. Ye, K. Q. Ling, Y. Zhang, N. Li and J. H. Xu, J. Chem. Soc., Perkin Trans. 1, 1999, 14, 2017–2024 RSC.
- M. Abass, H. A. Allimony and H. Hassan, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 1799–1810 CrossRef CAS.
- J. M. Morsy, H. M. Hassanin, M. M. Ismail and M. M. A. Abd-Alrazk, J. Chem. Res., 2016, 40, 239–246 CrossRef CAS.
- T. Ali, H. Wang, W. Iqbal, T. Bashir, R. Shah and Y. Hu, Adv. Sci., 2023, 10, 2205077 CrossRef CAS.
- T. Kappe, R. Aigner, P. Roschger, B. Schnell and W. Stadlbauer, Tetrahedron, 1995, 51, 12923–12928 CrossRef CAS.
- Q. R. Luca and A. Q. Fenwick, J. Photochem. Photobiol., B, 2015, 152, 26–42 CrossRef PubMed.
- V. Fonsecaa, S. Chandavarkar, R. Dabholkara, P. G. Dessaia, M. Deshpandec and S. N. M. Desai, Indian J. Chem., 2021, 60B, 267–272 Search PubMed.
- N. Azzman, S. Anwar, W. A. S. Mohamed and N. Ahemad, Med. Chem., 2024, 24, 1134–1157 CAS.
- A. Pervaiza, M. M. Athara, M. A. Khana, M. Pervaizb, M. Sagirc and M. Y. Naz, Sci. Int., 2014, 26, e202300209 Search PubMed.
- Y. Al-Majedy, A. A. Kadhum, H. Ibraheem, A. Al-Amiery, A. A. Moneim and A. B. Mohamad, Sys. Rev. Pharm., 2018, 9, 49–54 CrossRef CAS.
- B. A. Subhasrao, Master's thesis, Rajiv Gandhi University of Health Sciences, India, 2010 Search PubMed.
- K. C. Prousis, A. Tzani, N. Avlonitis, T. Calogeropoulou and A. Detsi, J. Heterocycl. Chem., 2013, 50, 1313–1321 CrossRef CAS.
- I. V. Ukrainets, N. L. Bereznyakova and E. V. Mospanova, Chem. Heterocycle. Cmpd., 2007, 43, 856–862 CrossRef CAS.
- X. Collin, J. M. Robert, M. Duflos, G. Wielgosz, G. Le Baut, C. Robin-Dubigeon, N. Grimaud, F. Lang and J. Y. Petit, J. Pharm. Pharmacol., 2001, 53, 417–423 CrossRef CAS.
- A. Zaman, I. Ahmad, M. Pervaiz, S. Ahmad, S. Kiran, M. A. Khan, T. Gulzar and T. Kamal, J. Mol. Struct., 2019, 1180, 227–236 CrossRef CAS.
- V. Vijayakumar, Int. J. Chemtech Res., 2016, 9, 629–634 CAS.
- Y. A. M. M. Elshaier, A. A. Aly, M. Abd El-Aziz, H. M. Fathy, A. B. Brown and M. Ramadan, Mol. Diversity, 2021, 26, 2341–2370 CrossRef.
- I. V. Ukrainets, M. Y. Golik, L. V. Sidorenko, V. I. Korniyenko, L. A. Grinevich, G. Sim and O. V. Kryvanych, Sci. Pharm., 2018, 86, 31 CrossRef PubMed.
- V. Yadav, J. Reang, V. Sharma, J. Majeed, P. C. Sharma, K. Sharma, N. Giri, A. Kumar and R. K. Tonk, Chem. Biol. Drug Des., 2022, 100, 389–418 CrossRef CAS PubMed.
- N. Priya, A. Gupta, K. Chand, P. Singh, A. Kathuria, H. G. Raj, V. S. Parmar and S. K. Sharma, Bioorg. Med. Chem., 2010, 18, 4085–4094 CrossRef CAS.
- T. Shiro, T. Fukaya and M. Tobe, Eur. J. Med. Chem., 2015, 97, 397–408 CrossRef CAS PubMed.
- S. H. Kwak, S. Shin, J. H. Lee, J. K. Shim, M. Kim, S. D. Lee, A. Lee, J. Bae, J. H. Park, A. Abdelrahman, C. E. Müller, S. K. Cho, S. G. Kang, M. A. Bae, J. Y. Yang, H. Ko, W. A. Goddard III and Y. C. Kim, Eur. J. Med. Chem., 2018, 151, 462–481 CrossRef CAS.
- L. Senerovic, D. Opsenica, I. Moric, I. Aleksic, M. Spasić and B. Vasiljevic, Adv. Exp. Med Bio., 2020, 14, 37–69 Search PubMed.
- A. A. Aly, E. M. El-Sheref, A. E. Mourad, M. E. M. Bakheet and S. Bräsem, Mol. Diversity, 2020, 24, 477–524 CrossRef CAS.
- K. Arya and M. Agarwal, Bioorg. Med. Chem. Lett., 2007, 17, 86–93 CrossRef CAS PubMed.
- M. S. Katagi, G. S. Bolakatti, A. M. Badiger, D. Satyanarayana, S. N. Mamledesai and M. L. Sujatha, Drug Res., 2013, 63, 53–59 CrossRef CAS.
- V. Sharma, R. Das, D. K. Mehta, D. Sharma, S. Aman and M. U. Khan, Mol. Diversity, 2024, 1–27 Search PubMed.
- Z. S. Quan, J. M. Wang, J. R. Rho, K. C. Kwak, H. C. Kang, C. S. Jun and K. Y. Chai, Bull. Korean Chem. Soc., 2005, 26, 1757–1760 CrossRef CAS.
- A. Pradhanand and S. K. Vishwakarma, Chem. Int., 2020, 6, 224–231 Search PubMed.
- S. Smita, G. Anand, S. Ranjit and V. Vikrant, Int. J. Pharm. Sci. Res., 2011, 3, 164–171 Search PubMed.
- M. S. Katagi, J. Fernandes, D. Satyanarayana, G. Bolakatti and S. N. Mamledesai, J. Pharm. Sci., 2014, 4, 57–61 Search PubMed.
- S. M. Katagi, J. Fernandes, S. Mamledesai, D. Satyanarayana, P. Dabadi and G. Bolakatti, J. Pharm. Res., 2015, 14, 51–56 CrossRef.
- H. Bi, Q. Ouyang, Z. Wei and Z. Zheng, Bioinorg. Chem., 2020, 100, 103902 CrossRef CAS.
- M. S. Katagi, S. Mamledesai, G. Bolakatti, J. Fernandes, S. ML and P. Tari, Chem. Data Collect., 2020, 30, 100560 CrossRef CAS.
- D. N. Toan, N. D. Thanh, M. X. Truong and D. T. Van, Med.Chem., 2022, 18, 36–50 CrossRef CAS PubMed.
- Q. Li, K. W. Woods, W. Wang, N. H. Lin, A. Claiborne, W. Gu, J. Cohen, V. S. Stoll, C. Hutchins, D. Frost, S. H. Rosenberg and H. L. Sham, Bioorg. Med. Chem. Lett., 2005, 15, 2033–2039 CrossRef CAS PubMed.
- A. Detsi, D. Bouloumbasi, K. C. Prousis, M. Koufaki, G. Athanasellis, G. Melagraki, A. Afantitis, O. Igglessi-Markopoulou, C. Kontogiorgis and D. J. Hadjipavlou-Litina, J. Med. Chem., 2007, 50, 2450–2458 CrossRef CAS.
- A. S. Ginsburg, J. H. Grosset and W. R. Bisha, Lancet Infect. Dis., 2003, 3, 432–442 CrossRef CAS.
- A. Aubry, N. Veziris, E. Cambau, C. Truffot-Pernot, V. Jarlier and L. M. Fisher, Antimicrob. Agents Chemother., 2006, 50, 04–112 CrossRef.
- G. C. Muscia, M. Bollini, A. M. Bruno and S. E. Asís, J. Chil. Chem. Soc., 2006, 51, 859–863 CAS.
- A. F. B. Cheng, W. W. Yew, E. W. C. Chan, M. L. Chin, M. M. M. Hui and R. C. Y. Chan, Antimicrob. Agents Chemother., 2004, 48, 596 CrossRef CAS PubMed.
- I. Dine, E. Mulugeta, Y. Melaku and M. Belete, RSC Adv., 2023, 13, 8657–8682 RSC.
- H. Yin, Y. Wu, X. Gu, Z. Feng, M. Wang, D. Feng, M. Wang, Z. Chenga and S. Wang, RSC Adv., 2022, 12, 21066–21078 RSC.
- M. A. Elanany, E. E. A. Osman, E. M. Gedawy and S. M. Abou-Seri, Sci. Rep., 2023, 13, 4144 CrossRef CAS PubMed.
- F. Thurner and F. A. Alatraktchi, Chemosensors, 2023, 11, 493 CrossRef CAS.
- C. Ji, I. Sharma, D. Pratihar, L. L. Hudson, D. Maura, T. Guney, L. G. Rahme, E. C. Pesci, J. P. Coleman and D. S. Tan, ACS Chem. Biol., 2016, 11, 3061–3067 CrossRef CAS PubMed.
- K. Alla, V. Vijayakumar and S. Sarveswari, Polycycl, 2023, 43, 2844–2865 CrossRef CAS.
- S. Khasnobis, V. E. Escuyer and D. Chatterjee, Expert. Opin. Ther. Targets, 2002, 6, 21–40 CrossRef CAS.
- M. J. Ahsan, M. Y. Ansari, S. Yasmin, S. S. Jadav, P. Kumar, S. K. Garg, A. Aseri and H. Khalilullah, Infect. Disord. Drug Targets, 2015, 15, 32–41 CrossRef CAS.
- N. Minovski, M. Vracko and T. Šolmajer, Mol. Diversity, 2011, 15, 417–426 CrossRef CAS PubMed.
- A. Lilienkampf, J. Mao, B. Wan, Y. Wang, S. G. Franzblau and A. P. Kozikowski, J. Med. Chem., 2009, 52, 2109–2118 CrossRef CAS PubMed.
- B. Villemagne, C. Crauste, M. Flipo, A. R. Baulard, B. Déprez and N. Willand, Eur. J. Med. Chem., 2012, 51, 1–16 CrossRef CAS.
- D. S. Reddy, K. M. Hosamani and H. C. Devarajegowda, Eur. J. Med. Chem., 2015, 101, 705–715 CrossRef CAS PubMed.
- M. K. Sharma, M. K. Kumawat, A. Diwan, S. Sardana, N. Yadav and B. Kumar, Herb. Med. J., 2024, 9 Search PubMed.
- V. T. Andriole, Infect. Dis. Clin. Pract., 1994, 3, S211–S218 CrossRef.
- P. S. Dube, L. J. Legoabe and R. M. Beteck, Mol. Diversity, 2023, 27, 1501–1526 CrossRef CAS.
- P. C. Appelbaum and P. A. Hunter, Int. J. Antimicrob. Agents, 2000, 16, 5–15 CrossRef CAS PubMed.
- T. D. M. Pham, Z. M. Ziora and M. A. T. Blaskovich, Med. Chem. Commun., 2019, 10, 1719–1739 RSC.
- A. M. Emmerson and A. M. Jones, J. Antimicrob. Chemother., 2003, 51, 13–20 CrossRef CAS PubMed.
- R. Stahlmann, Toxicol. Lett., 2002, 127, 269–277 CrossRef CAS.
- F. M. E. Wagenlehner and K. G. Naber, Curr. Infect. Dis. Rep., 2005, 7, 9–16 CrossRef CAS.
- Y. Pommier, E. Leo, H. L. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS PubMed.
- A. Kumar, A., J. Fernandes and P. Kumar, Orient. J. Chem., 2014, 4, 1993–1997 CrossRef.
- V. Krishnakumar, F. R. N. Khan, B. K. Mandal, S. Mitta, R. Dhasamandha and V. N. Govindan, Res. Chem. Intermed., 2012, 38, 1819–1826 CrossRef CAS.
- C. P. Kumar, M. S. Katagi and B. P. Nandeshwarappa, Chem. Data Collect., 2022, 42, 100955 CrossRef.
- S. A. Al-Issa, Saudi Pharm. J., 2013, 21(3), 305–316 CrossRef CAS PubMed.
- M. M. Butler, W. A. LaMarr, K. A. Foster, M. H. Barnes, D. J. Skow, P. T. Lyden, L. M. Kustigian, C. Zhi, N. C. Brown, G. E. Wright and T. L. Bowlin, Antimicrob. Agents Chemother., 2007, 51(1), 119–127 CrossRef CAS PubMed.
- H. A. Azaba, I. A. Ibrahim, N. Hassan, A. M. Abbas and H. M. Darwish, J. Lumin., 2017, 192, 376–384 CrossRef.
- R. An, M. Ahmed, H. Li, Y. Wang, A. Zhang, Y. Bi and Z. Yu, Sci. Rep., 2021, 11(1), 12020 CrossRef CAS PubMed.
- Z. L. Liu, S. S. Chu and G. H. Jiang, J. Agric. Food Chem., 2009, 57(21), 10130–10133 CrossRef CAS.
- Y. X. Liu, H. P. Zhao, Z. W. Wang, Y. H. Li, H. B. Song, H. Riches, D. Beattie, Y. C. Gu and Q. M. Wang, Mol. Diversity, 2013, 17, 701–710 CrossRef CAS PubMed.
- J. Jampilek, R. Musiol, M. Pesko, K. Kralova, M. Vejsova, J. Carroll, A. Coffey, J. Finster, D. Tabak, H. Niedbala, V. Kozik, J. Polanski, J. Csollei and J. Dohnal, Molecules, 2009, 14(3), 1145–1159 CrossRef CAS.
- M. Abe, T. Imai, N. Ishii and M. Usui, Biosci., Biotechnol., Biochem., 2006, 70(1), 303–306 CrossRef CAS.
- V. Uivarosi, Molecules, 2013, 18(9), 11153–11197 CrossRef CAS PubMed.
- M. M. Khalaf, M. Gouda, K. Shalabi, S. Shaaban and H. M. Abd El-Lateef, ACS Omega, 2024, 9, 6466–6481 CrossRef CAS.
- H. A. Althobiti and S. A. Zabin, Open Chem., 2020, 18, 591–607 CAS.
- R. Fouad and H. F. El-Shafiy, J. Mol. Struct., 2019, 1190, 68–76 CrossRef CAS.
- B. S. Creaven, B. Duff, D. A. Egan, K. Kavanagh, G. Rosair, V. R. Thangella and M. Walsh, Inorg. Chim. Acta, 2010, 363(14), 4048–4058 CrossRef CAS.
- K. Habibi, M. Mamaghani and M. Nikpassand, Bulg. Chem. Commun., 2018, 50, 76–82 Search PubMed.
- M. Azad, J. Appl. Sci., 2007, 7, 2485–2489 CrossRef CAS.
- G. Ayyannan, K. Karthikeyan, S. S. Vivekananthan, M. Gopiraman and A. Rathinavelu, Ionics, 2013, 19, 919–932 CrossRef CAS.
- C. M. Fernandes, A. R. Costa, M. C. Leite, V. Martins, H. S. Lee, F. da CS Boechat, M. C. B. V. de Souza, P. N. Batalha, H. Lgaz and E. A. Ponzio, J. Mol. Liq., 2023, 375, 121299 CrossRef.
- M. M. Abdou and M. N. EL-Haddad, J. Mol. Struct., 2025, 1324, 140889 CrossRef CAS.
- X. M. Meng, S. X. Wang and M. Z. Zhu, Quinoline-Based Fluorescence Sensors, Molecular Photochemistry - Various Aspects, 2012, ch. 2, DOI:10.5772/31771.
- B. Bag and A. Pal, Org. Biomol. Chem., 2011, 9, 4467–4480 RSC.
- I. Grabchev, J. M. Chovelon and C. Petkov, Spectrochim. Acta, Part A, 2008, 69, 100–104 CrossRef.
- L. Zhang, J. Fan and X. Peng, Spectrochim. Acta, Part A, 2009, 73, 398–402 CrossRef PubMed.
- U. Diwan, A. Kumar, V. Kumar and K. K. Upadhyay, Dalton Trans., 2013, 42, 13889–13896 RSC.
- Y. Yang, K. Yu, L. Yang, J. Liu, K. Li and S. Luo, Sensors, 2014, 15, 49–58 CrossRef PubMed.
- B. Li, J. Tian, D. Zhang and F. Tian, Lumin., 2017, 32, 1567–1573 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
Eslam A. Ghaith
*a and
Eslam A. Ghaith