
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and multifaceted exploration of dibenzoxepinones: in vitro antimicrobial and ct-DNA binding, DFT/TD-DFT, molecular docking and simulation studies†
Shilpa Yadava,
Mansi a,
Priyanshub,
Pratibha Chananab,
Pankaj Khannac,
Asmita Singha and
Leena Khanna*a
a,
Priyanshub,
Pratibha Chananab,
Pankaj Khannac,
Asmita Singha and
Leena Khanna*a
aUniversity School of Basic & Applied Sciences, Guru Gobind Singh Indraprastha University, Dwarka, New Delhi-110078, India. E-mail: leenakhanna@ipu.ac.in
bUniversity School of Chemical Technology, Guru Gobind Singh Indraprastha University, Dwarka, New Delhi-110078, India
cSynthesis & In-Silico Drug Design Laboratory, Department of Chemistry, Acharya Narendra Dev College, University of Delhi, Kalkaji, New Delhi-110019, India
First published on 30th May 2025
Abstract
In the present study, ten novel derivatives of dibenzoxepine-11-one, have been synthesized and thoroughly characterized using spectroscopic techniques, including 1H and 13C NMR, FTIR, and HRMS. Density Functional Theory (DFT) calculations were carried out to analyze the geometrical structure and vibrational modes, enabling the identification of the most stable conformation of the lead compound. Time-Dependent DFT (TD-DFT) was employed to investigate the electronic transitions within the UV-Vis spectrum. Molecular docking studies were performed for all derivatives using various target proteins with PDB IDs:IKZN (for E. coli), 1IYL (for C. albicans), and the DNA dodecamer structure (PDB ID: 1BNA). The results revealed favorable binding interactions across all targets. Additionally, molecular dynamics (MD) simulations were conducted for 50 ns using the most promising compound, 7a, confirming the stability of its binding conformation. From in vitro studies, antibacterial activity was assessed for all the synthesized derivatives against Gram-positive strains (B. subtilis, L. rhamnosus) and a Gram-negative strain (E. coli). Compounds 7a, 7b, 7c, 7d, and 7f exhibited strong antibacterial efficacy, with minimum inhibitory concentration (MIC) values of 8 μg ml−1 for E. coli and L. rhamnosus and 16 μg ml−1 for B. subtilis bacterial strains. Additionally, compound 7a exhibited good antifungal activity, with a maximum zone of inhibition of 18 mm against the fungal strain C. albicans. Further UV-Vis absorption and fluorescence quenching studies, conducted for compounds 7a, 7b, and 7e with calf thymus DNA (ct-DNA), suggested a groove-binding interaction. Compound 7a demonstrated the strongest binding affinity, with a binding constant (Kb) of 3.61 × 105 M−1 and a Gibbs free energy change (ΔG) of −31.70 kJ mol−1. Therefore, these novel dibenzoxepine-11-ones derivatives are multifaceted in their action as potential antimicrobial and DNA-binding agents, and will be useful in developing new therapeutics.
1. Introduction
Dibenzoxepinone is a tricyclic heterocycle featuring two fused benzene rings and a seven-membered oxepine ring. This chemical structure serves as the foundation for various pharmacological applications such as antibacterial, antifungal, antitumor, antihistamine, antidepressant, and anti-inflammatory.1–3 A prominent example of a dibenzoxepinone derivative is doxepin, which treats depression, anxiety, and chronic skin conditions such as urticaria.4,5 Its other derivatives, such as Oxepinoquinones and Napthodibenzoxepine, disrupt bacterial membrane integrity, leading to bacterial cell death and also preventing bacterial DNA replication. Halogenated dibenzoxepinone inhibits fungal membrane integrity and reduces infection persistence.6 These compounds often disrupt bacterial cell walls, inhibit DNA replication, or target specific bacterial enzymes. Modifying the core dibenzoxepinone framework can improve antibacterial activity against various bacterial strains, including both Gram-negative and Gram-positive bacteria.7 Furthermore, additional derivatives of dibenzoxepinone are under investigation as potential anticancer agents, as their structure can be altered to interact with key processes in cancer cells. They have the potential to block cancer cell growth by interfering with DNA, inhibiting crucial enzymes, or triggering cell death.8,9Thus, motivated by the diverse importance of dibenzoxepinone and as active researchers of heterocyclic compounds,10–14 we propose synthesizing novel derivatives of dibenzoxepin-11-one. The methodology involves synthesizing dibenzoxepin-11-one, followed by introducing various amines to one of its aromatic rings via a plausible route to assess their biological applications. Ten novel compounds were prepared and characterized using various spectroscopic techniques. DFT studies provided a potential energy scan for different conformations of the best compound. UV and FTIR calculations were also performed to observe the electronic transitions and vibrations of the compound and compare the data with the experimental results.15,16 Molecular docking was conducted with potential targets of antibacterial, antifungal, and ct-DNA using PDB IDs: 1KZN, 1IYL, and 1BNA through AutoDock 4.2.17–19 This aimed to determine the binding energy and uncover how proteins interact with ligands, along with the types of bonds formed between proteins and ligands. An MD simulation study was carried out for 50 ns to evaluate the stability of the protein-ligand complex in a biological system.20,21 Antibacterial and antifungal in vitro studies were performed to inhibit the growth of bacterial strains (E. coli, L. rhamnosus and B. subtilis) and a fungal strain (C. albicans) by MIC and Zone of inhibition. Additionally, UV-vis absorption and fluorescence quenching in vitro experiments were conducted to evaluate the DNA binding abilities of the dibenzoxepinone derivatives.22,232. Experimental techniques
2.1 Materials and methods
Reagents used in the synthesis of products were purchased from Sigma Aldrich. Positive control drugs Ampicillin and Fluconazole were purchased from SRL. Solvents were purchased by Merck and SRL. TLC plates of silica gel mesh 60F254 (Merck) were used as a preliminary test to check the progress of the reaction by the spots located in UV and Iodine vapors. Jeol Spectrometer of 400 MHz was used for 1H and 13C (TMS as a reference). FTIR spectra were recorded by PerkinElmer Spectrum BX spectrophotometer. HRMS was recorded by ESI-MS Thermo LTQ Orbitrap XL in methanol. Absorbance for the ct-DNA binding activity and UV Fluorescence quenching study property was taken by UV-1900 Shimadzu UV VIS Spectrophotometer, Double Beam.2.2 Synthesis
Dibenzoxepinone derivatives were synthesized in four steps. First step involved the formation of aryl benzyl ether (3) via Williamson ether synthesis. The second step is the cyclodehydration or intramolecular Friedel–Crafts acylation reaction using lewis acid FeCl3 and DCME to form dibenzoxepin-11-one (4). Third step is bromination of methyl group present in the aromatic ring of dibenzoxepin-11-one via Wohl–Zeigler bromination reaction i.e. allylic bromination using with N-bromosuccinimide. Fourth step is the nucleophilic displacement of bromine group by various aliphatic and aromatic amines to get the desired novel dibenzoxepinone derivatives (7a–j) as the product.24–27 | ||
| Scheme 1 Plausible mechanism of intramolecular cyclization for step (ii). | ||
For this step we have proposed a mechanism to highlight the importance of Lewis acid FeCl2 over FeCl3 in this cyclization. This is an intramolecular cyclization via the Friedel–Crafts acylation mechanism in which a molecule containing both an aromatic ring and an acyl group forms cyclic ketone. The reaction begins with the conversion of the carboxylic group into an acid chloride group in the presence of DCME (dimethyl methyl ether) which is a well-known reagent for this conversion to form intermediate acid chloride (B) via A.30 In this process HCl is liberated which reacts with FeCl2 to generate FeCl3 in situ. The FeCl3 further acts as a lewis acid for B and helps to generate acylium ion (C).31 The acylium group then attacks the aromatic ring and intramolecular acylation occurs within the same molecule and creating a new cyclic structure as 4-methyl-dibenzoxepin-11-one (4).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–, oleyl chain), 7.12 (m, 3H, ArH), 7.53 (t, 2H, J = 8Hz, Ar–H), 7.82 (d, 1H, Ar–H, J = 8Hz), 8.11 (d, 2H, Ar–H, J = 8Hz). 13C-NMR: δ 11.42, 14.42, 19.73, 22.70, 26.85, 27.18, 29.38, 30.05, 32.76, 38.22 (aliphatic region), 121.3, 129.74, 129.98, 132.82, 161.20, 164.78 (carbonyl carbon). HRMS calcd [M + H]+ for C33H49NO2: 492.38, found: 492.3755.
CH–, oleyl chain), 7.12 (m, 3H, ArH), 7.53 (t, 2H, J = 8Hz, Ar–H), 7.82 (d, 1H, Ar–H, J = 8Hz), 8.11 (d, 2H, Ar–H, J = 8Hz). 13C-NMR: δ 11.42, 14.42, 19.73, 22.70, 26.85, 27.18, 29.38, 30.05, 32.76, 38.22 (aliphatic region), 121.3, 129.74, 129.98, 132.82, 161.20, 164.78 (carbonyl carbon). HRMS calcd [M + H]+ for C33H49NO2: 492.38, found: 492.3755.3. In silico study
3.1 DFT study
A Density Functional Theory study is a computational quantum model used to investigate the electronic structure, energy levels, molecular orbitals, and mechanism of a compound. DFT study also helps to find out the theoretical NMR, FTIR, and UV-visible. The desired structure is first made in Gauss view 6.0 and set the calculation for optimization in Gaussian 09W at B3LYP/6-31+G(d,p) basis set. After optimizing the structure, the Potential energy surface (PES) study was performed on the flexible bonds by changing the bond angle from 90° to 120° by a transformation of 0.5° and the bond length of the NH bond by 0.05 Å each time of compound 7a.15 The frequency calculation was also done on the same basis set for analyzing the vibrations in a compound as FTIR spectra.33,34 No imaginary frequency in the output file approves the stationary state of the compound. Further, a UV-visible theoretical study was performed via the TD-SCF method at B3LYP/6-31+G(d,p) basis set.35 UV-visible and FTIR theoretical results were compared with experimental data.3.2 Molecular docking
3.3 ADME and physicochemical (drug likeliness) properties
ADME study analyses the chemical behavior of pharmacological compounds in a living organism. All the pharmacokinetic parameters were calculated from the PreADMET online server. This study helps to find out the detailed behavior of compounds by absorption, distribution, metabolism, and excretion in the body. These factors are crucial in drug development and its therapeutic effects and minimizing the side effects. Numerous parameters such as Blood–brain Barrier (BBB), Plasma Protein Binding (PPB), Skin permeability, etc., are used to describe drug toxicity.40Physicochemical parameters help to predict whether a compound has some features to become an effective drug or not. The parameters such as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, PSA (Polar Surface Area), molecular weight, no. of heavy atoms, etc., were calculated from https://www.molinspiratiom.com. All these parameters are a set of guidelines for Lipinski's rule that assess the drug likeliness of a compound. Any compound with only one violation of Lipinski's rule becomes an orally active drug.41
P, PSA (Polar Surface Area), molecular weight, no. of heavy atoms, etc., were calculated from https://www.molinspiratiom.com. All these parameters are a set of guidelines for Lipinski's rule that assess the drug likeliness of a compound. Any compound with only one violation of Lipinski's rule becomes an orally active drug.41
Bioactivity scores, such as those of GPCR ligands, kinase or protease inhibitors, and ion exchange modulators, illustrate the relationship between drugs and receptors. The mol-inspiration online server (https://www.molinspiration.com) helps to calculate the bioactivity score of ligands. Any drug is considered to be active if its bioactivity score is greater than 0.0, moderately active if its score lies between −5.0 and 0.0, and inactive if the score is lesser than −5.0.42
3.4 MD simulation
The optimal ligand 7a was subjected to molecular dynamics (MD) simulations by GROMACS (Version 2020) to assess the stability of docked complexes.43 The PRODRG server-generated ligand topology files, and GROMOS96_43a1 applied the force field for the simulations, which were conducted over 50 nanoseconds. The protein–ligand combination was solubilized in a cubic box utilizing the SPC (single point charge) water model. The system was neutralized by the addition of 4 Na+ ions.To reduce solvent fluctuations and eliminate any residual strain, energy minimization was performed using the steepest descent algorithm with a maximum of 50000 steps and a step size of 0.01. Following this, equilibration was conducted at 300 K using the NVT and NPT ensembles. The LINCS algorithm was employed to constrain protein bond lengths. The velocity-Verlet method, along with the Macro model, was used for the Particle-Mesh Ewald (PME) approach to compute long-range electrostatic interactions, with a cutoff distance of 1.2 nm for short-range electrostatic forces.
Temperature control at 300 K was maintained using the Berendsen thermostat, and pressure was held at 1 atm with the Parrinello–Rahman method to avoid any system clashes. Trajectory analysis was carried out using VMD, yielding RMSD, RMSF, hydrogen bond count, and the radius of gyration.44
4. In vitro studies
4.1 Anti-microbial study by zone of inhibition method
Antimicrobial activity refers to a substance's capability to prevent, kill, or inhibit the growth of microorganisms such as bacteria and fungi. The Kirby Bauer Disk Diffusion method was used to demonstrate this activity.45Compound 7a was tested against Gram-positive Bacillus subtilis at the following six dilutions (0, 2.5 μg, 5 μg, 12.5 μg, 25 μg, and 50 μg). For E. coli, compound 7a was prepared in three sets of dilutions (0, 25 μg, and 50 μg) for E. coli, prepared in DMSO of compound 7a, and added to the well of the Petri plates. After 50 μL of each was added to the well, the plates were incubated at 37 °C for 24 h. The diameter of clear zones (mm) around each well was measured. Ampicillin (5 mg ml−1) and DMSO were used as controls.46
4.2 Antibacterial study by RMDA method
The antibacterial activity of the synthesized compounds was evaluated using the Resazurin Microtiter Assay (RMDA). The assay was performed under aseptic conditions using a 96-well microtiter plate. In the first row of the plate, 100 μL of each test compound (7a–j) was added at an initial concentration of 1024 μg mL−1 in DMSO. Subsequently, 100 μL of nutrient broth was added to each well. A twofold serial dilution was carried out along each column by transferring 100 μL from one well to the next, resulting in a final minimum concentration of 0.5 μg mL−1.47Following dilution, 10 μL of bacterial suspension adjusted to 0.5 McFarland standard (approximately 5 × 106 CFU mL−1) was introduced into each well. The bacterial strains tested were Lacto bacillus rhamnosus (MTCC1408), Bacillus subtilis (MTCC 441), and Gram-negative Escherichia coli (MTCC 443), each prepared separately. To prevent dehydration of the cultures, the plates were sealed and gently wrapped with cling film. 30 μL of 5 mf per ml stock solution of ampicillin was injected as a positive control on each plate, and a negative control containing all reagents except the test compound was used.48
The plates were incubated at 37 °C for 18–24 hours to allow bacterial growth. After incubation, 100 μL of 0.01% resazurin solution was added to each well as a viability indicator, and the plates were incubated for an additional 30 minutes. Antibacterial activity was assessed visually based on a color change in the wells. Ampicillin was used as the reference antibacterial agent.
4.3 ct-DNA study
Ct-DNA activity is a biochemical and biophysical phenomenon that encompasses the interaction of ct-DNA with ligands to study the binding affinity. UV-visible absorption spectroscopy helps to identify the binding behavior of ct-DNA with compounds, and their interaction influences the absorbance property of the solution. A stock solution of ct-DNA (300 μM) was prepared in a Tris–HCl buffer solution containing 0.6 M HCl and 50 mM NaCl at pH 7.33. The UV absorbance ratio at 260 and 280 nm (A260/A280) was 1.9, confirming that the ct-DNA was free of protein contamination.49 The concentration of ct-DNA was determined from its absorbance at 260 nm, using a molar extinction coefficient of 6600 cm−1. A fixed volume (1 mL) of compounds 7a, 7b, and 7e of concentration 10 μM was titrated with increasing concentrations (20–100 μM) of ct-DNA. The binding constant Kb was calculated by the Benesi–Hilderbrand equation.50where Kb is the binding constant, A and A0 are the absorbance taken of ligands with ct-DNA at different concentrations and ligands without ct-DNA, respectively. εG and εH–G are the absorption coefficients taken of ligands with ct-DNA at different concentrations and ligands without ct-DNA, respectively. The binding energy was calculated by the ratio of intercept to the slope from the straight-line graph between A0/(A − A0) vs. 1/DNA.

5. Results and discussion
We successfully synthesized ten different derivatives of dibenzoxepin-11-one, the synthetic protocol includes four steps (Scheme 2). In the first step, the intermolecular cyclization of the C–O bond or Williamson ether synthesis reaction was performed between phthalide (1) and o-cresol (2) to form 2-((o-tolyloxy)methyl)benzoic acid (3). The synthesized compound was characterized by 1H NMR and compared the peaks already given in the literature.28 The peaks at 5.52 ppm and 2.34 ppm, which correspond to carbon–oxygen bond formation as O–CH2 and –CH3 group of tolyl moiety respectively, confirmed the formation of 3. In the next step, the intramolecular cyclization of compound (3) in the presence of Lewis acid (FeCl2 or CeCl3) and DCME was done and the product obtained as dibenzoxepin-11-one (4), its spectral proton NMR data was compared with the given literature data, the peaks of O–CH2 at 5.52 ppm get shifted to the upfield range and observed at 5.22 ppm which confirmed that intramolecular cyclization has occurred. In the third step, the Wohl–Zeigler allylic bromination reaction of the methyl group present in tolyl ring was done. The peak for methyl group peak in 4, which was initially at 2.34 ppm get shifted to the downfield at 3.22 ppm due to the substitution of a bromine atom in place of hydrogen in compound 5, validating the progress of the reaction. The formation of 5 was further confirmed by 13CNMR and HRMS analysis. In the last step, the bromo group of 5 was treated with various substituted amines 6a–j. During this the CH2–Br peak was replaced by CH2–N– and the 1HNMR showed a downfield shift of methylene protons at 4.45 ppm in compound 7a. In all other compounds from (7b–j), this peak was observed between 4.0 ppm to 5.0 ppm. In FTIR spectrum of 7a, showed a signal at 3390 cm−1 frequency for the N–H bond, 1750 cm−1 represents the carbonyl group, 1290 cm−1 represents the N–CH2, 990 cm−1 represents C–O bond/ether linkage, and the other two peaks at 3010 cm−1 and 1590 cm−1 also verify the formation of compound 7a. Further, 13CNMR and HRMS confirmed the formation of the desired product. | ||
| Scheme 2 Synthesis of dibenzoxepinone derivatives. | ||
5.1 DFT study
 | ||
| Fig. 1 Potential energy scan of 7a. | ||
 | ||
| Fig. 2 Potential energy scan grid of 7a. | ||
 | ||
| Fig. 3 FTIR Data (a) theoretical data (b) experimental data. | ||
| Experimental | Theoretical/TD-DFT/6-31+G(d,p) | |||
|---|---|---|---|---|
| λcal (nm) | Band gap (eV) | λcal (nm) | Band gap (eV) | F (frequency) |
| 330.10 | 3.2146 | 332.44 | 3.7295 | 0.0367 |
| 373.56 | 2.9752 | 350.30 | 3.5393 | 0.0057 |
| 457.29 | 2.7113 | 0.0039 | ||
 | ||
| Fig. 4 UV-visible spectrum (a) theoretical (b) experimental. | ||
 | ||
| Fig. 5 Electronic transition orbitals in UV-visible (a) first excitation (b) second excitation (c) third excitation. | ||
5.2 Molecular docking
 | ||
| Fig. 6 Validation of the docking methodology by superimposing re-docked chlorobiocin (yellow color) with co-crystallized protein complex (red) with a RMSD: 1.54 Å. | ||
 | ||
| Fig. 7 Validation of the docking methodology by superimposing re-docked N-myristoyl transferase (yellow color) with co-crystallized protein complex (dark red) with a RMSD: 0.98 Å. | ||
This study revealed that all the compounds showed good binding interaction with different amino acids in the protein with different bonds. The binding energy range of the compounds with protein lies between −6.0 to −9.2 kcal mol−1. The maximum binding energy −9.2 kcal mol−1 was observed for compound 7a, as mentioned in Table S1,† which has a Pi-donor hydrogen bond with ASN 46, a Pi-sigma bond with ILE 78, and THR 165. Some of the interactions with bond length greater than 5 Å due to hydrophobic interactions but they still contribute to ligand binding in long range binding interactions particularly between the charged residues and ligands. All other binding interactions are shown in Fig. 8. The interaction diagram of the remaining compounds was shown in Fig. S1† to S9.
 | ||
| Fig. 8 3D and 2D interaction diagram of compound 7a with E. coli bacterial protein (PDB ID:1KZN). | ||
Fig. 9 represents that all the ligands superimposing in the active site of the target after docking, it indicates a consistent binding mode and a well-defined binding pocket. This suggests that all derivatives interact with the same key residues, implying a common mechanism of action. Such consistency strengthens the reliability of the docking predictions and suggests that the ligands may exhibit similar biological activity.
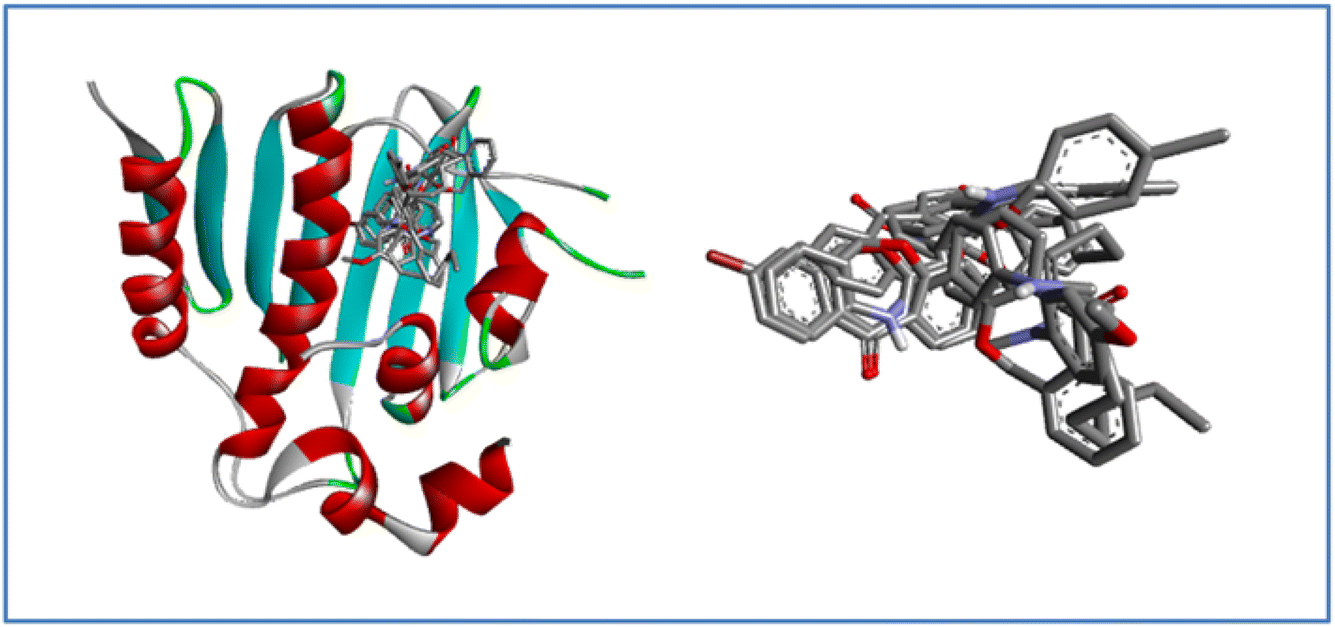 | ||
| Fig. 9 Superimposition of docked ligand 7a–j conformations within the active site of the E. coli bacterial protein. | ||
Its single-subunit enzyme facilitates the attachment of the fatty acid myristate from myristoyl-CoA to the N-terminal glycine residue of various eukaryotic and viral proteins. PDB 1D:1IYL represents the crystal structure N-myristoyl transferase, which is an essential enzyme in ergosterol biosynthesis, and is a crucial component of fungal cell membranes. Inhibiting this enzyme can hinder ergosterol, weakening cell membrane integrity and ultimately causing cell death in fungi. The binding energy of the compounds with the protein falls in the range of −8.0 to −11.0 kcal mol−1 noted in Table S1.† Affluence the capacity of cell death in fungi. 7a compound has the maximum binding energy (−11.0 kcal mol−1) with some imperative bonds such as conventional hydrogen bonds with TYR 225 and HIS 227, carbon–hydrogen bond with GLU 109, Pi-sigma bond with VAL 108, alkyl and Pi-alkyl bond with LEU 394 and TYR 107 as shown in Fig. 10. Interactions of compounds 7b–j with protein are depicted in Fig. S10 to S18.†
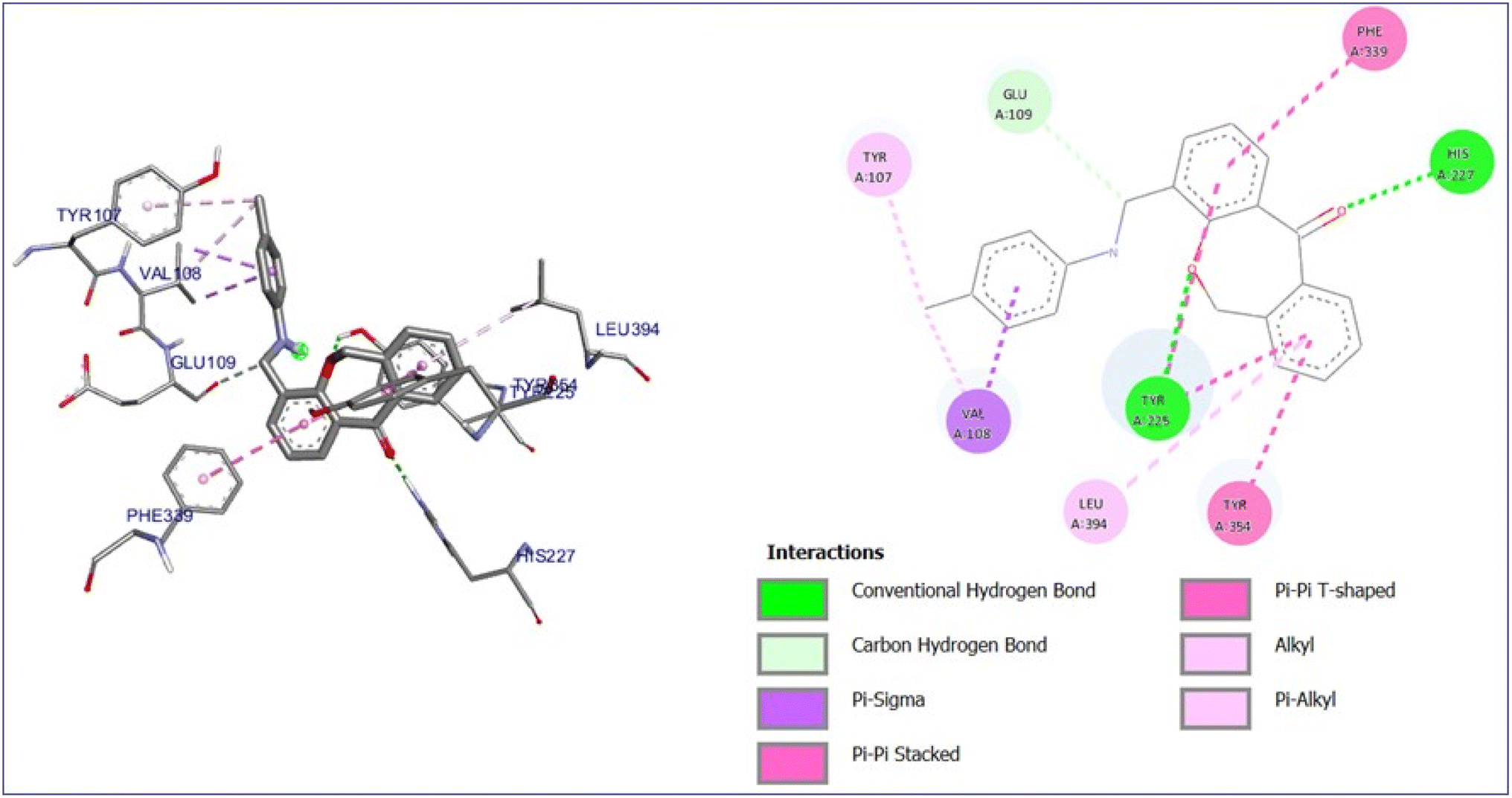 | ||
| Fig. 10 3D and 2D interaction diagram of compound 7a with antifungal protein (PDB ID:1IYL). | ||
Fig. 11 illustrates the superimposition of all ligands within the active site of the target after docking, demonstrating a consistent binding mode and a well-defined binding pocket. This indicates that all derivatives interact with the various amino acid residues, suggesting a similar mechanism of action. The high degree of overlap reinforces the reliability of the docking predictions and implies that the ligands may possess similar biological activity.
 | ||
| Fig. 11 Superimposition of docked ligand 7a–j conformations within the active site of the antifungal protein N-myristoyltransferase. | ||
 | ||
| Fig. 12 3D and 2D interaction diagram of compound 7a with ct-DNA (PDB ID:1BNA). | ||
Conventional hydrogen bonding played a significant role in the interaction of ligand 7a with DNA through a maximum binding energy of −8.1 kcal mol−1. The conventional hydrogen bonds were formed between carboxylic acid and guanosine of A strand with a bond length of 2.24 Å and the NH group of the ligand and guanosine of A strand with a bond length of 1.80 Å as noted in the 2D diagram. All other ligands also showed groove binding with different bonding interactions and binding energies as mentioned in Table S1 and Fig. S19 to S27.†
Fig. 13 depicts the superimposition of all ligands within the target's active site after docking, highlighting a consistent binding mode and a well-defined binding pocket. This suggests that all derivatives interact with the same key residues, indicating a shared mechanism of action. The significant overlap enhances the credibility of the docking predictions and suggests that the ligands may exhibit similar biological activity. Additionally, the superimposition reinforces the strong affinity of the binding site for these ligands, making it a promising candidate for further drug optimization.
 | ||
| Fig. 13 Superimposition of docked ligand 7a–j conformations within the active site of DNA protein dodecamer. | ||
The docking results show that all the synthesized derivatives were showing good binding interaction with all the PDB IDs:1KZN, 1IYL, AND 1BNA having range lie between −6.0 to 9.2 kcal mol−1 for antibacterial target protein, −8.5 to −11.0 kcal mol−1 for antifungal target protein, and −5.3 to −8.3 kcal mol−1 for DNA gyrase, but compound 7a has maximum binding energy with all target proteins that include different interactions like conventional hydrogen bond, carbon hydrogen bond, pi-sigma, and pi-alkyl, etc. Maximum bonding interactions of compound 7a were observed with the target protein 1IYL, and 1BNA shows the best binding with it and good biological activity.
5.3 ADME and physicochemical parameters
For drug development, synthesized derivatives should have a good ADME profile. ‘Lipinski's rule of five’ parameters such as (i) MW under 500, (ii) no. of hydrogen bond less than 5, (iii) no of hydrogen bond acceptors less than 10, (iv) logP value less than 5, (v) total polar surface area (TPSA) should not be greater than 140 Å2, indicates that the molecule is likely to be an orally active drug or not. Additionally, one violation is acceptable. All the molecules obey Lipinski's rule except 7b and 7c, they both have one violation. Other ADME parameters like blood–brain barrier (BBB), CaCo2, HIA, and protein plasma binding lie in the range of active drug noted in Table S3,† which means that compounds 7a–j behave as orally active drug.
A bioactivity score of less than −5.0 suggests the compound is inactive, a score between −5.0 and 0.0 indicates slight activity, and a score greater than 0.0 signifies high activity. All synthesized derivatives fall in the range and exhibit good bioactivity scores in Table S4.†
5.4 MD simulation
The dynamic stability of the optimal dibenzoxepine complex with the antibacterial protein and antifungal protein (PDB ID:1KZN and 1IYL) was assessed through MD simulation using Gromacs. Ligand topology was generated via the free online ATB server, and the GROMOS96 force field was applied to the simulated complex. The dibenzoxepine–protein complex was initially positioned within a cubic simulation box, followed by energy minimization in a solvated system. After energy minimization, MD simulations were conducted for 50 ns under constant conditions of 300 K temperature and 1 atm pressure.5.4.1.1 Root mean square deviation (RMSD). The interaction between the backbone atoms of the native 1KZN protein and its complex with 7a was analyzed using the Root Mean Square Deviation (RMSD) method. A stable RMSD pattern was observed for 50 ns. However, variations in RMSD values were noted, ranging from 0.1 nm to 0.39 nm for the native 1KZN protein (Fig. 14a). These minor variations reflect the stability of the protein–ligand complex.
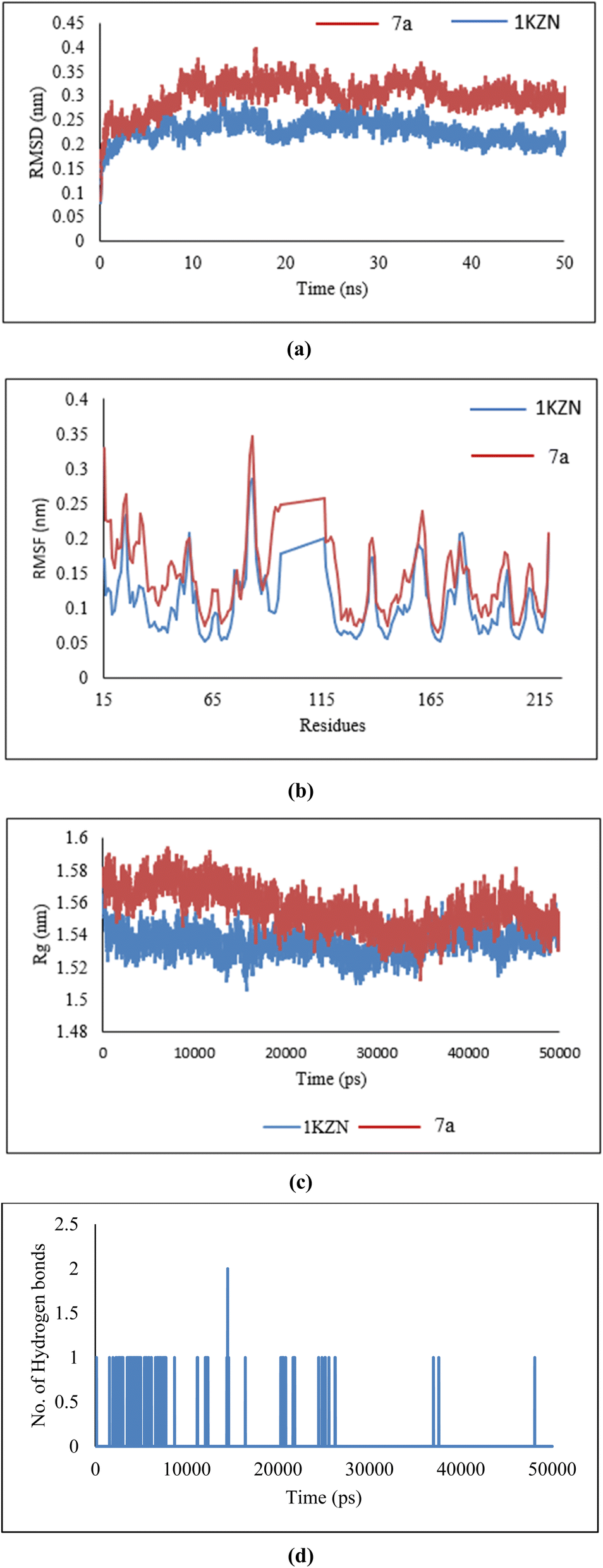 | ||
| Fig. 14 (a) RMSD, (b) RMSF, (c) radius of gyration, and (d) hydrogen bonds of 7a ligand in complex with 1KZN protein. | ||
5.4.1.2 Root mean square fluctuation (RMSF). The residue-wise variations were analyzed using Root Mean Square Fluctuation (RMSF). As shown in Fig. 14b, the 7a–protein complex exhibited fluctuations comparable to those of the native 1KZN structure, suggesting that the complex maintained significant stability due to ligand interactions.
5.4.1.3 Radius of gyration (Rg). The structural compactness of the protein was assessed using the Radius of Gyration (Rg). A consistent pattern was observed, with Rg values ranging from 1.51 nm to 1.61 nm (Fig. 14c) when comparing the native 1KZN protein with its ligand-bound complex. These results indicate the formation of a stable complex characterized by high structural compactness.
5.4.1.4 Hydrogen bonds. The analysis of antibacterial activity demonstrated a consistent and significant number of hydrogen bonds within the 7a–1KZN complex throughout the 50 ns MD simulation (Fig. 14d). The presence of these hydrogen bonds confirmed the structural integrity of the protein, strong protein–ligand interactions, and overall stability of the complex.
5.4.2.1 Root mean square deviation (RMSD). The interaction between the backbone atoms of the native 1IYL protein and its complex with 7a was evaluated using the Root Mean Square Deviation (RMSD) method. A stable RMSD pattern was observed over the 50 ns simulation period, with variations ranging from 0.1 nm to 0.34 nm for the native IYL protein (Fig. 15a). These minor deviations highlight the stability of the protein–ligand complex.
 | ||
| Fig. 15 (a) RMSD, (b) RMSF, (c) radius of gyration, and (d) hydrogen bonds of 7a ligand in complex with 1IYL protein. | ||
5.4.2.2 Root mean square fluctuation (RMSF). Residue-wise variations were analyzed using the Root Mean Square Fluctuation (RMSF) method. As depicted in Fig. 15b, the 7a–protein complex displayed fluctuations similar to those of the native 1IYL structure, indicating significant stability of the complex due to ligand interactions.
5.4.2.3 Radius of gyration (Rg). The structural compactness of the protein was analyzed using the Radius of Gyration (Rg). A consistent trend was observed, with Rg values ranging from 2.01 nm to 2.75 nm (Fig. 15c) for both the native 1IYL protein and its ligand-bound complex. These findings confirm the formation of a stable complex with high structural compactness.
5.4.2.4 Hydrogen bonds. The evaluation of antibacterial activity revealed a stable and substantial number of hydrogen bonds within the 7a–1IYL complex during the 50 ns MD simulation (Fig. 15d). These hydrogen bonds underscore the structural integrity of the protein, robust protein–ligand interactions, and overall stability of the complex.
5.5 Antimicrobial activity
Moreover, the zones of inhibition results revealed the compound's potential to inhibit the growth of bacteria and fungus strains. Thus, emphasizing that the synthesized compound 7a is a bioactive compound with significant antimicrobial potential leading to growth inhibition. The diameter of the zone of inhibition increases as the concentration of compound 7a increases (Table 3).
| MIC values (μg ml−1) | 7a | 7b | 7c | 7d | 7e | 7f | 7g | 7h | 7i | 7j |
|---|---|---|---|---|---|---|---|---|---|---|
| L. rhamnosus | 8 | 8 | 8 | 16 | 64 | 8 | 32 | 32 | 8 | 128 |
| B. subtilis | 16 | 16 | 16 | 32 | 16 | 128 | 128 | 16 | 16 | 256 |
| E. coli | 8 | 16 | 8 | 16 | 16 | 32 | 32 | 64 | 16 | 128 |
5.6 ct-DNA binding activity
 | ||
| Fig. 16 UV-Vis spectral changes were observed in 7a, 7b, and 7e at a concentration of 10 μM in 50 mM Tris–HCl/NaCl buffer (pH = 7.35) at 25 °C, as the ct-DNA concentration increased from (20–100 μM). The arrow indicates a decrease in absorbance as the ct-DNA concentration increases. | ||
The intrinsic binding constant (Kb) was evaluated from intercept by slope ratio from the equivalent double reciprocal graph of A0/A − A0 against 1/[DNA] mentioned in Table 5. The hypochromic effects of compounds with ct-DNA and the Kb values confirmed their stable interaction.
| Compounds | 7a | 7b | 7e |
|---|---|---|---|
| Binding constant (Kb) | 3.61 × 105 | 1.80 × 105 | 8.03 × 104 |
| Gibbs free energy (ΔG) | −31.70 | −29.98 | −27.98 |
The Gibbs free energy (ΔG) of the compounds (7a, 7b, 7e)/ctDNA complex can account for the complex's stability was assessed when ct-DNA was introduced into the solution. The equation below was used to calculate the free energy of the system utilizing the relevant parameters:
| ΔG = −RTln(Kb) |
The analysis of the Gibbs free energy cited in Table 5 for the complexes revealed that the binding of compound 7a with ct-DNA was thermodynamically more stable.
 | ||
| Fig. 17 Fluorescence emission spectra of 7a (10 μM) with an increasing concentration of ct-DNA (0–100 μM). The inset shows the Stern–Volmer constant (ksv). The arrow shows a decrease in intensity with an increase in ct-DNA concentration. | ||
Fluorescence quenching is a process where the fluorescence intensity of a substance diminishes, typically occurring due to various molecular interactions such as ground state complex formation, excited state reactions, and molecular rearrangement. The effectiveness of fluorescence quenching can be assessed using the Stern–Volmer equation to determine the Stern–Volmer quenching constant (Ksv):

In this context, F0 and F represent the fluorescence intensities in the absence and presence of the quencher, respectively while [Q] denotes the quencher concentration. The stern–Volmer quenching constant (Ksv) is obtained from the slope of the F0/F vs. [ct-DNA] plot. For compound 7a, the Ksv value was determined to be 2.0 × 103 M−1, suggesting a non-intercalative binding mode, as this value is lower than those typically associated with intercalators. The distinction between static and dynamic quenching can be determined by examining the linearity of the Stern–Volmer plot and applying the relevant equation.

In this equation, Kq represents the biomolecular quenching rate constant and τo is the biomolecule's fluorescence lifetime of the biomolecule in the quencher's absence. With τo at 10−8 s, Kq was calculated to be 2.0 × 1011 M−1 s−1. A high Kq value suggests that the quenching occurs via static quenching through complex formation rather than dynamic quenching. Hence, compound 7a can bind with ct-DNA at the groove binding site.
Conclusion
Ten novel derivatives of dibenzoxepine-11-one were synthesized and demonstrated the ability to bind effectively with key biological targets, including bacterial DNA gyrase (PDB ID:1KZN, E. coli), N-myristoyltransferase (PDB ID:1IYL, C. albicans), and the DNA dodecamer (PDB ID:1BNA, ct-DNA). Molecular docking studies revealed favorable binding energies across all targets, with compound 7a exhibiting the highest binding affinities of −9.2 kcal mol−1 with E. coli, −11.0 kcal mol−1 with C. albicans, and −8.1 kcal mol−1 with ct-DNA. The in vitro antimicrobial activity was done compound 7a against B. subtilis, C. albicans, and E. coli. At a concentration of 50 μg, compound 7a showed the highest zone of inhibition, with diameters of 17 mm against the bacterial strains and 18 mm against the fungal strain comparable to the standard antimicrobial agents Ampicillin and Fluconazole. These results were further validated for all the compounds by RMDA method to find MIC values against Gram-positive strains (B. subtilis, L. rhamnosus) and a Gram-negative strain (E. coli). Compounds 7a, 7b, 7c, and 7i demonstrated notable antibacterial potency, exhibiting lowest minimum inhibitory concentration (MIC) values for the Gram-positive strains, however, 7a & 7c have lowest MIC values for both Gram-positive and negative strains. Additionally, DNA-binding studies with calf thymus DNA on compounds 7a, 7b, 7e, again confirmed that compound 7a was the most potent groove binder, with a binding constant (Kb) of 3.61 × 105 M−1 and a Gibbs free energy of −31.70 kJ mol−1. Overall, compound 7a emerged as the most promising candidate, showing superior performance across all in vitro evaluations, which was further validated by in silico approaches, including DFT calculations and molecular dynamics simulations.Abbreviations
| DFT | Density functional theory |
| ct-DNA | Calf thymus seoxyribose nucleic acid |
| PSA | Polar surface area |
| UV | Ultra-violet |
| BBB | Blood–brain barrier |
Data availability
The authors confirm that the supporting findings of this study are available within the article and are ESI.†Author contributions
Dr Leena Khanna and Prof Pankaj Khanna: conceptualization of idea, supervision. Shilpa Yadav, Asmita Singh: literature search and primary draft and drawing of figures and tables. Shilpa Yadav, Priyanshu, Pratibha Chanana: performed in vitro experiments. Shilpa Yadav & Mansi: DFT study, execution of molecular docking and simulation of ligands. All authors reviewed the manuscript.Conflicts of interest
The authors state no conflict of interest.Acknowledgements
The authors would like to thank the Faculty Research Grant Scheme (FRGS 2024-25), Guru Gobind Singh Indraprastha University, Dwarka, New Delhi, for the financial support of this project. Shilpa Yadav, Asmita Singh and Mansi are thankful to Guru Gobind Singh Indraprastha University, Dwarka, New Delhi, for providing STRF.References
- K. Zimmermann, P. C. Waldmeier and W. G. Tatton, Dibenzoxepines as Treatments for Neurodegenerative Diseases, Pure Appl. Chem., 1999, 71(11), 2039–2046, DOI:10.1351/pac199971112039.
- C. Limban and M. C. Chifiriuc, Antibacterial Activity of New Dibenzoxepinone Oximes with Fluorine and Trifluoromethyl Group Substituents, Int. J. Mol. Sci., 2011, 12(10), 6432–6444, DOI:10.3390/ijms12106432.
- F. Naporra, S. Gobleder, H. J. Wittmann, J. Spindler, M. Bodensteiner, G. Bernhardt, H. Hübner, P. Gmeiner, S. Elz and A. Strasser, Dibenzo[b,f][1,4]Oxazepines and Dibenzo[b,e]Oxepines: Influence of the Chlorine Substitution Pattern on the Pharmacology at the H1R, H4R, 5-HT2AR and Other Selected GPCRs, Pharmacol. Res., 2016, 113, 610–625, DOI:10.1016/j.phrs.2016.09.042.
- W. F. Yeung, K. F. Chung, K. P. Yung and T. H. Y. Ng, Doxepin for Insomnia: A Systematic Review of Randomized Placebo-Controlled Trials, Sleep Med. Rev., 2015, 19, 75–83, DOI:10.1016/j.smrv.2014.06.001.
- J. Katwala, A. K. Kumar, J. J. Sejpal, M. Terrence and M. Mishra, Therapeutic Rationale for Low Dose Doxepin in Insomnia Patients, Asian Pacific J. Trop. Dis., 2013, 3(4), 331–336, DOI:10.1016/S2222-1808(13)60080-8.
- H. Krawczyk, Dibenzo[b,f]Oxepine Molecules Used in Biological Systems and Medicine, Int. J. Mol. Sci., 2023, 24(15), 12066, DOI:10.3390/ijms241512066.
- B. Sadek, C. Limban, C. E. Stecoza and S. Elz, Synthesis and Antimicrobial Evaluation of Dibenzo[b,e]Oxepin-11(6H)-One O-Benzoyloxime Derivatives, Sci. Pharm., 2011, 79(4), 749–761, DOI:10.3797/scipharm.1107-02.
- C. X. Liu, Z. X. Lin and A. M. Zhou, Design, Synthesis, Cytotoxicities and DNA Cleavage Activities of Dibenzoxepine and Isoquinoline Derivatives Starting from Dehydroabietylamine, J. Asian Nat. Prod. Res., 2016, 18(12), 1169–1177, DOI:10.1080/10286020.2016.1232251.
- D. Garbicz, P. Tobiasz, F. Borys, T. Pilżys, M. Marcinkowski, M. Poterała, E. Grzesiuk and H. Krawczyk, The Stilbene and Dibenzo[b,f]Oxepine Derivatives as Anticancer Compounds, Biomed. Pharmacother., 2020, 123(20), 109781, DOI:10.1016/j.biopha.2019.109781.
- P. Khanna, D. Gupta, S. Yadav and L. Khanna, Hydrotrope Assisted Green Synthesis of Dicoumarols and in Silico and in Vitro Antibacterial, Antioxidant and Xanthine Oxidase Inhibition Studies, J. Biomol. Struct. Dyn., 2023, 41(19), 9651–9665, DOI:10.1080/07391102.2022.2145368.
- S. Yadav, N. Misra, P. Khanna and L. Khanna, Novel 10,11-Dihydro-5H-Dibenzo[b,f]Azepine Triazoles Hybrids: Synthesis, in Vitro Antioxidant Activity and Xanthine Oxidase Inhibition and Computational Study, J. Mol. Struct., 2024, 1312(P1), 138639, DOI:10.1016/j.molstruc.2024.138639.
- L. Khanna, S. Singhal, S. C. Jain and P. Khanna, Spiro-Indole-Coumarin Hybrids: Synthesis, ADME, DFT, NBO Studies and In Silico Screening through Molecular Docking on DNA G-Quadruplex, ChemistrySelect, 2020, 5(11), 3420–3433, DOI:10.1002/slct.201904783.
- S. Yadav, N. Misra, P. Khanna and K. Batra, A DFT Study on Diels-Alder Reaction of Dibenzazepine and 2,5-Dimethylfuran Using Different Solvents and Temperature Conditions A DFT Study on Diels-Alder Reaction of Dibenzazepine And, Polycycl. Aromat. Compd., 2022, 1–12, DOI:10.1080/10406638.2022.2056622.
- P. Khanna, S. Yadav, A. Singh and L. Khanna, Inclusion Complexes of Novel Formyl Chromone Schiff Bases with β-Cyclodextrin: Synthesis, Characterization, DNA Binding Studies and in-Vitro Release Study, Carbohydr. Polym., 2025, 347(2024), 122667, DOI:10.1016/j.carbpol.2024.122667.
- S. Sevvanthi, S. Muthu, M. Raja, S. Aayisha and S. P. E. S. Janani, Molecular Structure, Spectroscopic (FT-IR, FT-Raman), Electronic (UV-Vis, HOMO-LUMO), Quantum Chemical and Biological (Docking) Studies on a Potent Membrane Permeable Inhibitor: Dibenzoxepine Derivative, Heliyon, 2020, 6(8), e04724, DOI:10.1016/j.heliyon.2020.e04724.
- A. Manwal, P. Mekoung, A. Malloum, M. Govindarajan, R. N. Mballa, I. Patouossa, A. Abouem A Zintchem, C. P. N. Nanseu and I. N. Mbouombouo, Spectroscopic Properties (FT-IR, NMR and UV) and DFT Studies of Amodiaquine, Heliyon, 2023, 9(12), e30645, DOI:10.1016/j.heliyon.2023.e22187.
- S. Murugavel, C. S. Jacob Prasanna Stephen, R. Subashini and D. AnanthaKrishnan, Synthesis, Structural Elucidation, Antioxidant, CT-DNA Binding and Molecular Docking Studies of Novel Chloroquinoline Derivatives: Promising Antioxidant and Anti-Diabetic Agents, J. Photochem. Photobiol. B Biol., 2017, 173, 216–230, DOI:10.1016/j.jphotobiol.2017.05.043.
- S. Murugavel, S. Deepa, C. Ravikumar, R. Ranganathan and P. Alagusundaram, Synthesis, Structural, Spectral and Antibacterial Activity of 3,3a,4,5-Tetrahydro-2H-Benzo[g]Indazole Fused Carbothioamide Derivatives as Antibacterial Agents, J. Mol. Struct., 2020, 1222, 128961, DOI:10.1016/j.molstruc.2020.128961.
- W. Liu, E. Li, L. Liu, F. Tian, X. Luo, Y. Cai, J. Wang and X. Jin, Antifungal Activity of Compounds from Gordonia Sp. WA8-44 Isolated from the Gut of Periplaneta Americana and Molecular Docking Studies, Heliyon, 2023, 9(7), e17777, DOI:10.1016/j.heliyon.2023.e17777.
- M. I. Sulistyowaty, G. S. Putra, T. Budiati, A. W. Indrianingsih, F. Anwari, D. Kesuma, K. Matsunami and T. Yamauchi, Synthesis, In Silico Study, Antibacterial and Antifungal Activities of N-Phenylbenzamides, Int. J. Mol. Sci., 2023, 24(3), 2745, DOI:10.3390/ijms24032745.
- F. Wang, W. Yang and B. Zhou, Studies on the Antibacterial Activities and Molecular Mechanism of GyrB Inhibitors by 3D-QSAR, Molecular Docking and Molecular Dynamics Simulation, Arab. J. Chem., 2022, 15(6), 103872, DOI:10.1016/j.arabjc.2022.103872.
- K. M. Prabhu Kumar, B. C. Vasantha Kumar, P. R. Kumar, R. J. Butcher, H. K. Vivek, P. A. Suchetan, H. D. Revanasiddappa and S. Foro, Synthesis, Characterization, CT-DNA Binding and Docking Studies of Novel Selenated Ligands and Their Palladium Complexes, Appl. Organomet. Chem., 2020, 34(6), 1–17, DOI:10.1002/aoc.5634.
- K. K. Chaudhary and N. Mishra, Investigation of the Interaction Between Chalcones with CT-DNA by Molecular Docking, ADMET and Fluorescence Spectroscopy, Proc. Natl. Acad. Sci., India, Sect. A, 2017, 87(2), 195–206, DOI:10.1007/s40010-017-0346-9.
- A. Diamanti, Z. Ganase, E. Grant, A. Armstrong, P. M. Piccione, A. M. Rea, J. Richardson, A. Galindo and C. S. Adjiman, Mechanism, Kinetics and Selectivity of a Williamson Ether Synthesis: Elucidation under Different Reaction Conditions, React. Chem. Eng., 2021, 6(7), 1195–1211, 10.1039/d0re00437e.
- M. Rueping and B. J. Nachtsheim, A Review of New Developments in the Friedel-Crafts Alkylation - From Green Chemistry to Asymmetric Catalysis, Beilstein J. Org. Chem., 2010, 6, 1–24, DOI:10.3762/bjoc.6.6.
- Y. Otake, J. D. Williams, J. A. Rincón, O. De Frutos, C. Mateos and C. O. Kappe, Photochemical Benzylic Bromination in Continuous Flow Using BrCCl 3 and Its Application to Telescoped P-Methoxybenzyl Protection, Org. Biomol. Chem., 2019, 17(6), 1384–1388, 10.1039/c9ob00044e.
- Y. Jiang, W. Zhu, J. Huang, F. Luo, X. Chen, C. Fang, X. Chen, S. Liu, Y. Hu and S. A. Zhang, Simple Method for N-Arylation of Secondary Amides/Amines through a NaH-Initiated Aryne Generation Strategy, Org. Chem. Front., 2023, 11(1), 12–20, 10.1039/d3qo01109g.
- J. Scoccia, M. J. Castro, M. B. Faraoni, C. Bouzat, V. S. Martín and D. C. Gerbino, Iron(II) Promoted Direct Synthesis of Dibenzo[b,e]Oxepin-11(6H)-One Derivatives with Biological Activity. A Short Synthesis of Doxepin, Tetrahedron, 2017, 73(20), 2913–2922, DOI:10.1016/j.tet.2017.03.085.
- C. C. Silveira, S. R. Mendes, L. Wolf and G. M. Martins, The Use of Anhydrous CeCl3 as a Catalyst for the Synthesis of 3-Sulfenyl Indoles, Tetrahedron Lett., 2010, 51(15), 2014–2016, DOI:10.1016/j.tetlet.2010.02.038.
- T. Warashina, D. Matsuura and Y. Kimura, Efficient Preparation of Dichloromethyl Alkyl Ethers and Their Application in the Formylation of Aromatic Compounds: Scope and Limitations, Tetrahedron, 2019, 75(5), 608–616, DOI:10.1016/j.tet.2018.12.030.
- J. Ohata, Friedel-Crafts Reactions for Biomolecular Chemistry, Org. Biomol. Chem., 2024, 22(18), 3544–3558, 10.1039/d4ob00406j.
- D. Cantillo, O. De Frutos, J. A. Rincon, C. Mateos and C. A. Oliver Kappe, Scalable Procedure for Light-Induced Benzylic Brominations in Continuous Flow, J. Org. Chem., 2014, 79(1), 223–229, DOI:10.1021/jo402409k.
- S. B. Radder, R. Melavanki, S. M. Hiremath, R. Kusanur, S. S. Khemalapure and S. C. Jeyaseelan, Synthesis, Spectroscopic (FT-IR, FT-Raman, NMR & UV-Vis), Reactive (ELF, LOL, Fukui), Drug Likeness and Molecular Docking Insights on Novel 4-[3-(3-Methoxy-Phenyl)-3-Oxo-Propenyl]-Benzonitrile by Experimental and Computational Methods, Heliyon, 2021, 7(11), e08429, DOI:10.1016/j.heliyon.2021.e08429.
- R. S. Saji, J. C. Prasana, S. Muthu and J. George, Experimental and Theoretical Spectroscopic (FT-IR, FT-Raman, UV-VIS) Analysis, Natural Bonding Orbitals and Molecular Docking Studies on 2-Bromo-6-Methoxynaphthalene: A Potential Anti-Cancer Drug, Heliyon, 2021, 7(6), e07213, DOI:10.1016/j.heliyon.2021.e07213.
- M. Sima Abdollahi, E. Nemati-Kande and A. Poursattar Marjani, Experimental and DFT Studies on the FT-IR, NMR and UV/Vis Spectra of a Xanthene Derivative: The Case of 9-Benzoyl-3,4,5,6,7,9-Hexahydro-1h-Xanthene-1,8(2h)-Dione, ChemistrySelect, 2020, 5(13), 3971–3980, DOI:10.1002/slct.201904165.
- K. Gullapelli, G. Brahmeshwari, M. Ravichander and U. Kusuma, Synthesis, Antibacterial and Molecular Docking Studies of New Benzimidazole Derivatives, Egypt. J. Basic Appl. Sci., 2017, 4(4), 303–309, DOI:10.1016/j.ejbas.2017.09.002.
- D. Nahar, P. Mohite, A. Lonkar, V. R. Chidrawar, R. Dodiya, M. J. Uddin, S. Singh and B. G. Prajapati, An Insight into New Strategies and Targets to Combat Antifungal Resistance: A Comprehensive Review, Eur. J. Med. Chem. Reports, 2024, 10, 100120, DOI:10.1016/j.ejmcr.2023.100120.
- J. D. Dimitrijević, N. Solovjova, A. M. Bukonjić, D. L. Tomović, M. Milinkovic, A. Caković, J. Bogojeski, Z. R. Ratković, G. V. Janjić, A. A. Rakić, N. N. Arsenijevic, M. Z. Milovanovic, J. Z. Milovanovic, G. P. Radić and V. V. Jevtić, Docking Studies, Cytotoxicity Evaluation and Interactions of Binuclear Copper(II) Complexes with S-Isoalkyl Derivatives of Thiosalicylic Acid with Some Relevant Biomolecules, Int. J. Mol. Sci., 2023, 24(15), 12504, DOI:10.3390/ijms241512504.
- S. Sharma, A. Sharma and U. Gupta, Molecular Docking studies on the Anti-fungal activity of Allium sativum (Garlic) against Mucormycosis (black fungus) by BIOVIA discovery studio visualizer 21.1.0.0, Ann. Antivir. Antiretrovir., 2021, 5(1), 028–032, DOI:10.17352/aaa.000013.
- C. A. Lipinski, Rule of Five in 2015 and Beyond: Target and Ligand Structural Limitations, Ligand Chemistry Structure and Drug Discovery Project Decisions, Adv. Drug Deliv. Rev., 2016, 101, 34–41, DOI:10.1016/J.ADDR.2016.04.029.
- M. J. Waring, Lipophilicity in Drug Discovery, 2010, 5(3), 235–248, DOI:10.1517/17460441003605098.
- D. E. Clark, In Silico Prediction of Blood–Brain Barrier Permeation, Drug Discov. Today, 2003, 8(20), 927–933, DOI:10.1016/S1359-6446(03)02827-7.
- D. Osmaniye, N. Baltacı Bozkurt, S. Levent, G. Benli Yardımcı, B. N. Sağlık, Y. Ozkay and Z. A. Kaplancıklı, Synthesis, Antifungal Activities, Molecular Docking and Molecular Dynamic Studies of Novel Quinoxaline-Triazole Compounds, ACS Omega, 2023, 8(27), 24573–24585, DOI:10.1021/acsomega.3c02797.
- D. Zaeifi, A. Najafi and R. Mirnejad, Molecular Dynamics Simulation of Antimicrobial Peptide CM15 in Staphylococcus Aureus and Escherichia Coli Model Bilayer Lipid, Iran. J. Biotechnol., 2023, 21(2), 15–26, DOI:10.30498/ijb.2023.337246.3344.
- Clinical and Laboratory Standards Institute, Performance Standards for Antimicrobial Disk Susceptibility Tests: Approved Standard - Eleventh Edition, 2012, vol. 32, M02-A11 Search PubMed.
- X. Wang, H. Y. Fu, W. He, Y. T. Xiang, Z. C. Yang, Y. Kuang and S. X. Yang, Synthesis and Antibacterial Activity Evaluation of Biphenyl and Dibenzofuran Derivatives as Potential Antimicrobial Agents against Antibiotic-Resistant Bacteria., Curr. Issues Mol. Biol., 2022, 44(9), 4087–4099, DOI:10.3390/cimb44090280.
- V. L. Barnes, D. M. Heithoff, S. P. Mahan, J. K. House and M. J. Mahan, Antimicrobial Susceptibility Testing to Evaluate Minimum Inhibitory Concentration Values of Clinically Relevant Antibiotics, STAR Protoc., 2023, 4(3), 102512, DOI:10.1016/j.xpro.2023.102512.
- B. GKowalska-Krochmal and R. Dudek-Wicher, The Minimum Inhibitory Concentration of Antibiotics: Methods, Interpretation, Clinical Relevance, Pathogens, 2021, 10(2), 1–21, DOI:10.3390/pathogens10020165.
- M. Sunita, B. Anupama, B. Ushaiah and C. Gyana Kumari, Synthesis, Characterization, DNA Binding and Cleavage Studies of Mixed-Ligand Copper (II) Complexes, Arab. J. Chem., 2017, 10, S3367–S3374, DOI:10.1016/j.arabjc.2014.01.017.
- K. Yi, X. Wang, S. K. Filippov and H. Zhang, Emerging CtDNA Detection Strategies in Clinical Cancer Theranostics, Smart Med., 2023, 2(4), 1–17, DOI:10.1002/smmd.20230031.
- C. Levine, H. Hiasa and K. J. Marians, DNA Gyrase and Topoisomerase IV: Biochemical Activities, Physiological Roles during Chromosome Replication, and Drug Sensitivities, Biochim. Biophys. Acta, 1998, 1400(1–3), 29–43, DOI:10.1016/S0167-4781(98)00126-2.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopy spectrums, molecular docking binding energy tables, ADME, and physiochemical properties and DFT calculations of all the derivatives can be accessed here. See DOI: https://doi.org/10.1039/d5ra01068c |
| This journal is © The Royal Society of Chemistry 2025 |