
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparative study of the reaction of 2-mercaptobenzimidazole with 2-bromo-1,3-diketones under conventional and green conditions: regioselective access to N/S-difunctionalized benzimidazoles and benzimidazo[2,1-b]thiazoles†
Ranjana Aggarwal *ab,
Prince Kumara and
Suresh Kumara
*ab,
Prince Kumara and
Suresh Kumara
aDepartment of Chemistry, Kurukshetra University, Kurukshetra-136119, Haryana, India
bCouncil of Scientific and Industrial Research-National Institute of Science Communication and Policy Research, New Delhi 110012, India. E-mail: ranjanaaggarwal67@gmail.com; ranjana67in@yahoo.com; Tel: +91-9896740740
First published on 17th June 2025
Abstract
The development of eco-friendly and energy-efficient synthetic methods remains a priority in modern organic chemistry. In this study, we investigated the reaction of 2-mercaptobenzimidazole and 1,3-diketones in the presence of N-bromosuccinimide (NBS) under conventional and sustainable conditions. While the DCM-mediated and solvent-free approaches yielded 2-((1-acetyl-1H-benzo[4,5]imidazole-2-yl)thio)-1-arylethan-1-one derivatives, the visible-light irradiation strategy promoted a distinct regioselective [3 + 2] cyclo-condensation, affording corresponding cyclized products, i.e., 6-substituted-2-aroyl-3-methylbenzimidazo[2,1-b]thiazole derivatives in high yields. These one-step protocols feature an eco-friendly nature, short reaction times, operational simplicity, high efficiency, and excellent selectivity. Structures of the synthesized compounds were confirmed via comparative analysis of their Rf values, IR, NMR, and HRMS data, with further structural validation via detailed 2D NMR studies, and plausible mechanisms have been proposed for both reaction pathways. Overall, the work highlights condition-driven regioselectivity and the potential of energy-dependent strategies in benzimidazole-based heterocycle synthesis.
Introduction
Benzimidazo[2,1-b]thiazole is a fascinating fused heterocyclic scaffold that has gained significant attention across various scientific domains.1 Its unique structure and diverse reactivity make it appealing for synthetic chemistry, while its broad range of biological activities such as antimicrobial, anticancer, anti-inflammatory, and antiviral properties, positions it as a valuable target in medicinal chemistry.2 Compounds like Levamisole 1, with immunomodulatory effects, and Tilomisole (Wy-18,251) (2-(3-(4-chlorophenyl)benzo[4,5]imidazo-[2,1-b]thiazol-2-yl)acetic acid) 2, showing anticancer potential, highlight its therapeutic promise3,4 (Fig. 1). Recently, Alkhaldi et al. reported 3-methylthiazolo[3,2-a]benzimidazole–benzenesulfonamide conjugates as potent carbonic anhydrase inhibitors with anticancer activity, further demonstrating the medicinal relevance of benzimidazo[2,1-b]thiazole frameworks.5 This highlights the continuing interest in developing structurally diverse benzimidazothiazole derivatives for therapeutic applications. Additionally, benzimidazo[2,1-b]thiazoles have shown potential in material science, especially in organic electronics and sensors, due to their efficient charge transport properties.6 | ||
| Fig. 1 Structures of Levamisole and Tilomisole (Wy-18,251). | ||
Growing interest in benzimidazo[2,1-b]thiazole derivatives is evident from numerous synthetic reports detailing their synthesis and functionalization through diverse methodologies. 2-Mercaptobenzimidazole has been widely used as a synthon for synthesizing biologically active benzimidazo[2,1-b]thiazoles by reacting with various substrates,2,5,7–13 such as carbonyl compounds, propargyl bromides/tosylates, active alkynes and 1,1-dibromoalkenes. Additionally, copper-catalyzed coupling reactions,14,15 e.g. 1,2-aminothiolation of 1,1-dibromoalkenes and coupling of trans-1,2-diiodoalkenes with 2-mercaptobenzimidazole, 1,2-aminothiolation of terminal alkynes and thioamination of nitrogen compounds has been documented16,17 (Fig. 2a).
 | ||
| Fig. 2 (a) Synthetic strategies for benzimidazo[2,1-b]thiazole using 2-mercaptobenzimidazole as synthon. (b) Alternative synthetic strategies for benzimidazo[2,1-b]thiazole using 2-amino thiazoles as synthons. | ||
An alternative route is the construction of an imidazole ring by the reaction of 2-aminothiazole with 2-haloarylboronic acid or quinone under Cu(II)-catalyzed and acetic acid-catalyzed reactions,1,18 respectively (Fig. 2b).
Despite significant advancements in synthesizing benzimidazo[2,1-b]thiazole derivatives, several challenges persist, including reliance on metal catalysts, low atom economy, and harsh reaction conditions. These limitations underscore the need for one-step, environmentally sustainable method to enhance efficiency and versatility. Moreover, direct and selective synthesis of acyl-functionalized benzimidazo[2,1-b]thiazoles remains underexplored. Addressing these gaps could not only enhance the efficiency and versatility of existing methods but also pave the way for greener practices in drug discovery and material science applications.
Prompted by the aforementioned facts and as a continuation of our ongoing efforts toward the synthesis of biologically significant acylated heterocyclic scaffolds,19–21 we herein report a study on the reaction of 1,3-diketones, 2-mercaptobenzimidazoles, and N-bromosuccinimide (NBS) under various reaction conditions including solvent-mediated, solvent-free and visible-light-assisted protocols. These strategies enable regioselective access to two different products i.e. 2-((1-acetyl-1H-benzo[4,5]imidazole-2-yl)thio)-1-arylethan-1-one and acyl-functionalized benzimidazo[2,1-b]thiazole derivatives.
Result and discussion
Chemistry
As discussed, benzimidazo[2,1-b]thiazole derivatives have attracted interest for their biological and material applications, but existing methods often involve multistep procedures, metal catalysts, toxic reagents, and harsh conditions, limiting their sustainability and scalability.1,5,11–13 To overcome these challenges, we designed a one-pot protocol utilizing 1,3-diketones 3, 2-mercaptobenzimidazole 5, and NBS under varying reaction conditions. In principle, the reaction between trielectrophilic (α1, α2, α3) 2-bromo-1,3-diketones 4 and trinucleophilic (β1, β2, β3) 3-mercapto-1,2,4-triazoles 5 introduces regioselectivity in product formation. The more nucleophilic sulphur site (β2) of 2-mercaptobenzimidazole preferentially attacks the soft site (α2) of 2-bromo-1,3-diketone, initiating the reaction. However, competition between other sites (α1 and α3) and (β1 and β3) can lead to four possible regioisomers viz. 6-substituted-2-aroyl-3-methylbenzimidazo[2,1-b]thiazole 6, 6-substituted-2-acetyl-3-arylbenzimidazo-[2,1-b]thiazole 7, 7-substituted-2-aroyl-3-methylbenzimidazo-[2,1-b]thiazole 8 and 7-substituted-2-acetyl-3-arylbenzimidazo[2,1-b]thiazole 9 (Scheme 1). | ||
| Scheme 1 Possible regioisomeric products by the reaction of 2-bromo-1,3-diketones 4 with 2-mercaptobenzimidazole 5. | ||
Keeping in mind the formation of four possible regioisomers, firstly, the reaction of 1-phenylbutane-1,3-dione 3a with 1,3-dihydro-2H-benzimidazole-2-thione 5a was investigated in the presence of the brominating agent NBS using conventional approaches. The reaction was explored using a range of solvent systems, including DCM, CH3CN, THF, CH3OH, C2H5OH and H2O (Runs 1–6, Table 1), as well as under solvent-free conditions (Run 7, Table 1). Optimization experiments showed that the reaction faced challenges with solvents such as THF, CH3CN, CH3OH, C2H5OH, and H2O, resulting in low product yields, of up to 35%. However, using DCM solvent at room temperature furnished a single regioisomeric product with high yields of 82% with a retention factor (Rf) value of 0.82, indicated by Thin Layer Chromatography (TLC) (ethyl acetate-petroleum ether (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80, v/v)).
:80, v/v)).
| Run | Solvent | Energy source | Time | Yieldb (%) | |
|---|---|---|---|---|---|
| Product with Rf = 0.82 | Product with Rf = 0.56 | ||||
| a Reaction of 3a (1.0 mmol) and 5a (1.0 mmol) was reacted as per reaction conditions.b Isolated yield. | |||||
| 1 | DCM | Rt | 4 h | 82 | — |
| 2 | CH3CN | Rt/reflux | 5 h | Trace/20 | — |
| 3 | THF | Rt/reflux | 6 h | Trace/10 | — |
| 4 | CH3OH | Rt/reflux | 3 h | 10/30 | — |
| 5 | C2H5OH | Rt/reflux | 3 h | 10/35 | — |
| 6 | H2O | Rt/reflux | 2 h | Trace/10 | — |
| 7 | Solvent-free | Rt | 20 min | 86 | — |
| 8 | DCM | LED (9 W) | 2.5 h | — | 58 |
| 9 | CH3CN | LED (9 W) | 2.5 h | — | 56 |
| 10 | THF | LED (9 W) | 9 h | — | Trace |
| 11 | CH3OH | LED (9 W) | 2 h | — | 63 |
| 12 | C2H5OH | LED (9 W) | 30 min | - | 88 |
| 13 | H2O | LED (9 W) | 5 h | — | 25 |
The reaction was further conducted under solvent-free conditions to align the synthetic procedure with environmentally friendly approaches. The solvent-free approach delivered the same regioisomeric product (Rf = 0.82) with an improved yield of 86% in just 20 minutes, highlighting its advantages in terms of operational simplicity, shorter reaction time, and the elimination of hazardous solvents.
In recent years, several studies have highlighted the significance of visible-light-mediated synthesis of benzimidazo[2,1-b]thiazoles, emphasizing the advantages of green synthetic methods.22,23 Moreover, we have carried out the above reaction under visible-light irradiations, specifically, the reaction was performed under the illumination of a 9 W light-emitting diode (LED) lamp, placed 5 cm above the reaction mixture, across a series of experiments (Runs 8–13, Table 1). Interestingly, the product obtained under visible-light conditions displayed a distinct Rf value of 0.56 on TLC, contrasting with the products formed under conventional and solvent-free conditions. Various solvent systems have been tested systematically to identify the most effective conditions to further optimize the LED reaction conditions. Notably, the best results were achieved with ethanol, yielding an impressive 88% in just 30 minutes (Run 12, Table 1). This successful application of visible light and ethanol not only resulted in high yields but also demonstrated the potential for environmentally sustainable chemical transformations.
The attention-grabbing products were comprehensively characterized using various spectral techniques including IR, 1D & 2D NMR ((1H–13C) HSQC and (1H–13C) HMBC) and mass spectrometric analysis. Based on these extensive spectral analyses, the compound with an Rf value of 0.82 was identified as a reported N/S-difunctionalized product,11 namely 2-((1-acetyl-1H-benzimidazol-2-yl)thio)-1-phenylethan-1-one 10a. In contrast, the compound obtained under visible-light conditions, exhibiting an Rf value of 0.56, was characterized and found as the corresponding cyclized product i.e. (3-methylbenzo[4,5]imidazo[2,1-b]thiazol-2-yl)(phenyl)methanone 6a (Scheme 2). Notably, when compound 10a was refluxed with acetic anhydride the corresponding cyclized compound 6a was obtained.11 The current synthetic protocol offers several advantages, including a one-step synthesis significantly reduced time, environmentally benign reaction media and reaction conditions, etc.
 | ||
| Scheme 2 Synthesis of benzo[4,5]imidazo[2,1-b]thiazoles. | ||
IR spectrum of product 10a exhibited two strong bands at 1708 cm−1 and 1670 cm−1, indicating the presence of two carbonyl groups instead of the expected single band for possible regioisomers (6–9). Conversely, the IR spectrum of product 6a exhibited a single C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O absorption band at 1680 cm−1, suggesting intramolecular cyclization followed by dehydration in the product.
O absorption band at 1680 cm−1, suggesting intramolecular cyclization followed by dehydration in the product.
In the 1H NMR (CDCl3) analysis of product 10a, an interesting observation emerged in the aliphatic region, two distinct peaks appeared at δ 2.81 ppm and δ 4.85 ppm, deviating from the expected single peak corresponding to the methyl group for each envisaged regioisomer (6–9). However, in the 1H NMR (DMSO-d6) analysis of compound 6a, a single peak in the aliphatic region at δ 2.73 ppm was observed, contrasting with the spectrum of the N/S-difunctionalized product 10a, which displayed two peaks at δ 2.81 and 4.85 ppm.
Additionally, the 13C NMR (CDCl3) spectrum of 10a provided further validation of the findings from the proton NMR and IR analyses. Notable, the spectrum revealed two distinct peaks in the aliphatic region at δ 26.2 and 40.5 ppm, supporting the formation of interesting product 10a rather than one of the four probable regioisomers. Consequently, through the comprehensive spectral studies conducted, the product was definitively identified as an N/S-difunctionalized compound, specifically 2-((1-acetyl-1H-benzimidazol-2-yl)thio)-1-phenylethan-1-one 10a. The structural determination of the compound can be rationalized by considering an in situ cleavage of the 1,3-diketone moiety. In contrast, the 13C NMR (DMSO-d6) spectrum of compound 6a revealed a sole distinctive peak in the aliphatic region at δ 15.7 ppm, indicating the successful formation of the cyclized product, differing from the N,S-difunctionalized product.
Furthermore, the structural characterization of the synthesized compounds was reinforced through meticulous mass spectrometric analysis. For compound 10a, the mass spectrum revealed a prominent peak at an m/z value of 311.0786 for [M + H]+, further supported the formation of the N/S-difunctionalized product. Conversely, the mass spectrum of compound 6a exhibited a distinct peak at m/z value of 293.0656 for [M + H]+, consisting with the expected cyclized regioisomer via dehydration (loss of H2O, m/z = 18), confirming the successful formation of the desired compound.
The conclusive evidence for the formation of 2-((1-acetyl-1H-benzimidazol-2-yl)thio)-1-phenylethan-1-one 10a was obtained through a comprehensive analysis involving heteronuclear 2D NMR experiments. The correlations of protons and their corresponding carbon atoms within compound 10a were meticulously examined (Fig. 3a). The (1H–13C) HMBC results revealed significant cross-peaks, including the carbonyl carbon (δ 193.9 ppm) exhibiting correlations with the 2′/6′-H proton (δ 8.09–8.11 ppm) of the aryl ring and CH2 (δ 4.85 ppm), indicating the presence of –CH2COAr fragment, ruled out the possibility of the formation of regioisomer 11a. The cross-peak between C-2 (δ 154.3 ppm) of the benzimidazole core and methylene protons (δ 4.85 ppm) indicated the presence of a 2-oxo-2-arylethylthio group at the 2nd position of the benzimidazole ring. Similarly, the cross-peak of carbonyl carbon (δ 168.8 ppm) with methyl protons (δ 26.2 ppm), indicated the presence of the –COCH3 group. All other expected correlations were evident in (1H–13C) HMBC and (1H–13C) HSQC spectra, supported the substituents distribution around the benzimidazole nucleus.
 | ||
| Fig. 3 (a) 1H, 13C and 2D NMR correlations of 2-((1-acetyl-1H-benzimidazol-2-yl)thio)-1-phenylethan-1-one 10a. (b)1H, 13C and 2D NMR correlations data of (3-methylbenzo[4,5]imidazo[2,1-b]thiazol-2-yl)(phenyl)methanone 6a. | ||
Additionally, a thorough characterization of regioisomer 6a was also achieved through extensive 2D NMR spectral studies, conclusively identified the product as the regioisomer (3-methylbenzo[4,5]imidazo[2,1-b]thiazol-2-yl)(phenyl)methanone 6a. The (1H–13C) HMBC spectrum of the compound showed correlation peaks of methyl protons (δ 2.73 ppm) with C-2 (δ 130.8 ppm) and C-3 (δ 154.8 ppm), confirming the presence of the methyl group at the 3rd position of benzo[4,5]imidazo[2,1-b]thiazole core (Fig. 3b). Similarly, a cross peak between carbonyl carbon at δ 189.3 ppm and 2′/6′-H protons (δ 7.84–7.87 ppm) of the aryl ring indicated the presence of carbonyl carbon adjacent to the aryl ring, thereby, eliminating the possibility of regioisomers with acetyl group 7 and 9. The formation of 7-substituted-2-aroyl-3-methylbenzimidazo[2,1-b]thiazoles 8 was also ruled out, as the HMBC spectrum of the 7-methyl-substituted product did not indicate any correlation between the methyl group protons and the C-12 carbon (δ 125.0 ppm). Therefore, the regioisomeric structure of the final compounds was assigned as 6-substituted-2-aroyl-3-methylbenzimidazo[2,1-b]thiazoles 6.
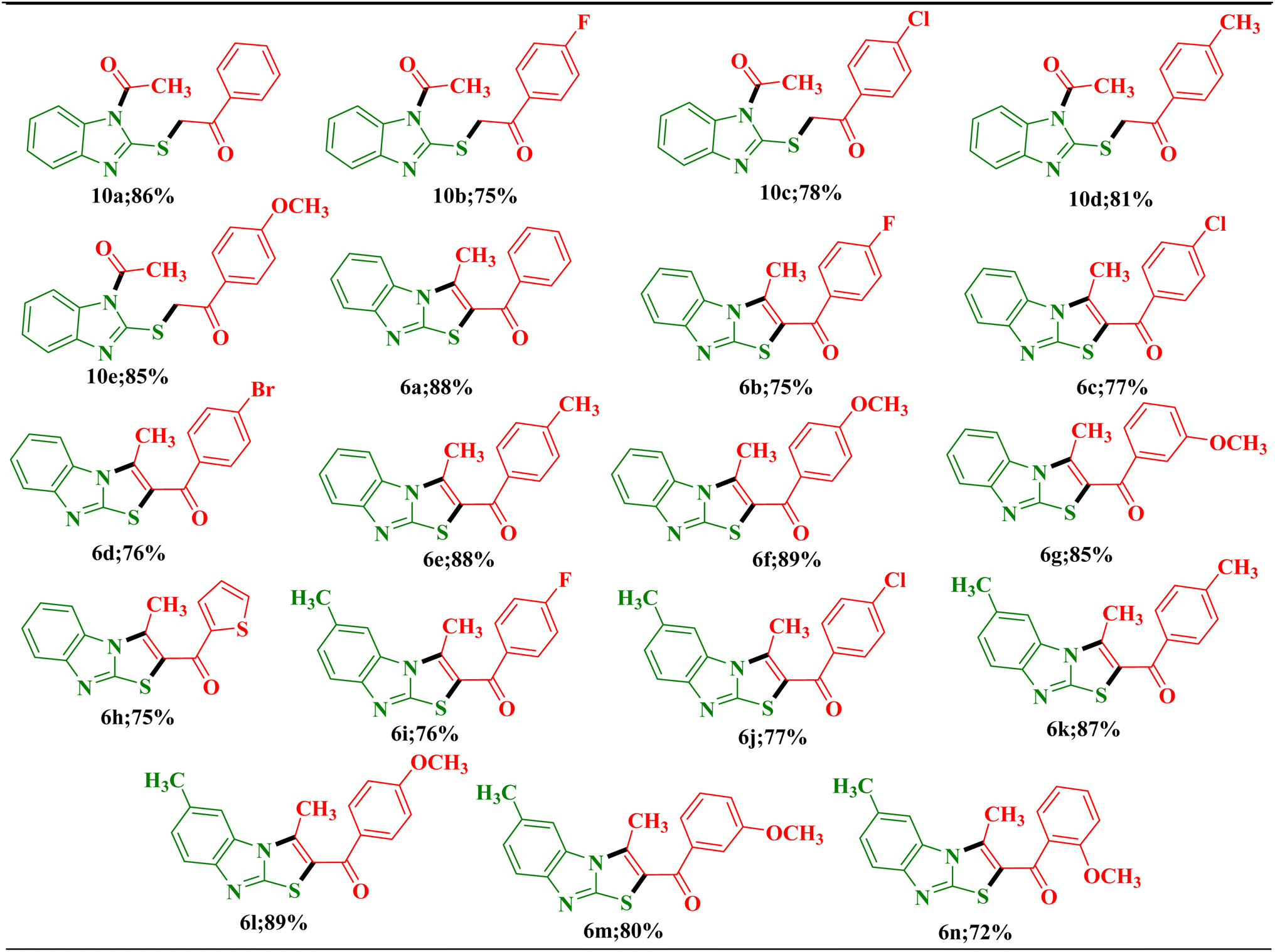
After optimizing reaction conditions and identifying specific regioisomeric structures, an extensive investigation of substrate tolerance was undertaken. This investigation utilized optimized protocols: a solvent-free protocol for synthesizing N/S-difunctionalized product, 2-((1-acetyl-1H-benzimidazol-2-yl)thio)-1-arylethan-1-one 10(a–e) and a visible-light-mediated synthesis for (3-methylbenzo[4,5]imidazo[2,1-b]thiazol-2-yl)(aryl)methanone 6(a–n). The substrate scope of the reaction protocols was explored by taking a diverse array of unsymmetrical 1,3-diketones, featuring aryl moieties with electron-donating or electron-withdrawing substituents, which were subjected to the reaction (Scheme 3). Remarkably, the results highlighted the versatility of the reaction procedure, demonstrating its smooth progression with various substituents. Considering the electron-donating and electron-withdrawing groups associated with the diketones, it was observed that the reaction exhibited enhanced yields when the aryl moiety featured an electron-donating group (Table 2). The present one-step synthetic protocol for N/S-difunctionalized product 10 represents significant advantages over previously reported methods,24 including eco-friendliness, short reaction period, high yields, easy workup, and high regioselectivity.
 | ||
| Scheme 3 Synthesis of benzimidazole analogous. | ||
| a Reaction conditions: a mixture of 5(a–b) (1.0 mmol) and 3(a–i) (1.0 mmol) in the presence of NBS in 10.0 mL ethanol was reacted. |
|---|
 |
Proposed mechanism
(a) The proposed potential mechanism for the unexpected synthesis of 2-((1-acetyl-1H-benzo[4,5]imidazole-2-yl)thio)-1-arylethan-1-one 10(a–e) is illustrated in Scheme 4a. Initially, 2-mercaptobenzimidazole undergoes S-alkylation by replacing the bromine in 2-bromo-1,3-diketones with sulphur, leading to the formation of intermediate A. Subsequently, the nucleophilic attack of nitrogen atom on less sterically hindered and more electrophilic carbonyl carbon adjacent to the CH3 group provides favourable sites for the intramolecular cyclization of A. However, instead of intramolecular cyclization, the reaction leads to the N-acylation of intermediate A via C–C bond cleavage,24,25 thus generating an unexpected N/S-difunctionalized product; 2-((1-acetyl-1H-benzo[4,5]imidazole-2-yl)thio)-1-arylethan-1-one 10. Upon refluxing with acetic anhydrid, the carbonyl group adjacent to the thioether moiety in compound 10 undergoes acetylation to form intermediate B. Intermediate B undergoes intramolecular cyclization yielding C, which subsequently undergoes dehydration and deacylation to furnish the desired cyclized product 6. | ||
| Scheme 4 (a) Proposed mechanism for the N/S-difunctionalized product: 2-((1-acetyl-1H-benzo[4,5]imidazole-2-yl)thio)-1-arylethan-1-one 10(a–e). (b) Proposed mechanism for the cyclized product: (3-methylbenzo[4,5]-imidazo[2,1-b]thiazol-2-yl)(aryl)methanone 6(a–n). | ||
(b) The possible mechanistic pathway for the regioselective synthesis of (3-methylbenzo[4,5]imidazo[2,1-b]thiazol-2-yl)(aryl)methanone 6(a–n) is depicted in Scheme 4b. Initially, under the influence of visible light, homonuclear fission occurs in both the S–H bond of 2-mercaptobenzimidazole 5 and the C–Br bond of 2-bromo-1,3-diketones 4, leading to the formation of free radicals E and F, respectively, which mutually share their electrons to form the S-alkylated intermediate A. Subsequently, the bromine free radical initiates the homolytic cleavage of the N–H bond, leading to the formation of a new N-benzimidazole radical. This radical, in conjunction with the less sterically hindered carbonyl carbon and an oxygen atom, forms a stable complex, 3-hydroxy-3-methyl-2,3-dihydrobenzo[4,5]imidazo-[2,1-b]thiazol-2-yl)(aryl)methanone D, which finally undergoes dehydration to yield exclusive final cyclized product 6.
To support the proposed reaction mechanism, controlled experiments with radical initiation and trapping were systematically conducted. Under standard conditions, the addition of free radical initiator benzoyl peroxide26,27 significantly enhanced the reaction yield (6a; 92%) and a better reaction rate. In contrast, the introduction of radical scavenger TEMPO28,29 to the reaction mixture resulted in notable inhibition, yielding only 18% of product 6a. These results strongly suggest a free radical pathway involvement, with benzoyl peroxide improving and TEMPO inhibiting the reaction. However, solvent-free controlled experiments showed no significant impact on reaction yield and rate, supporting the proposed ionic mechanism for product formation.
Conclusion
In conclusion, a comparative study has been carried out for the regioselective synthesis of benzimidazole-based heterocycles via the reaction of 2-mercaptobenzimidazoles 5(a–b) with 1,3-diketones 3(a–i) in the presence of NBS under both conventional and sustainable conditions. Under DCM-mediated and solvent-free conditions, 2-((1-acetyl-1H-benzo[4,5]-imidazole-2-yl)thio)-1-arylethan-1-ones 10(a–e) were obtained, whereas visible-light-mediated conditions furnished 6-substituted-2-aroyl-3-methylbenzimidazo[2,1-b]thiazoles 6(a–n). The formation of these structurally distinct scaffolds was confirmed through comparative analysis of their Rf values, IR, NMR, and HRMS data, with further structural validation via detailed 2D NMR studies. Proposed reaction mechanisms suggest that the formation of N/S-difunctionalized products proceeds via a C–C bond cleavage through an ionic pathway, while the visible-light-induced products are formed through a free-radical mechanism, supported by radical initiation and trapping experiments. Both developed protocols are metal-free, environmentally benign, and operationally simple, offering excellent yields within short reaction times. These green methodologies pave the way for the efficient synthesis of structurally diverse and potentially bioactive thiazole-fused benzimidazole derivatives. Further investigations, including exploration of a broader substrate scope and biological evaluation coupled with structure–activity relationship (SAR) studies of these compounds, are currently underway in our laboratory.Experimental
Melting points were taken with an electric digital Melting Point Apparatus (MEPA) in open capillaries and may be uncorrected. Thin-layer chromatography (TLC) utilized Merck Kieselgel 60 F254 silica gel plates, visualized under UV light (254 nm). LED of 9 W power positioned 5 cm from the reaction mixture in an Erlenmeyer borosilicate flask. Spectroscopic analyses included IR spectra recorded on a Buck Scientific IR M-500 spectrophotometer with KBr pellets (υmax in cm−1). Proton (1H) and carbon-13 (13C) NMR spectra were acquired on a Bruker instrument at frequencies of 400 MHz and 101 MHz, respectively, using CDCl3 and DMSO-d6 as solvents. Chemical shifts were expressed in parts per million (ppm) and coupling constants (J) in Hz, with TMS as the internal standard. High-resolution mass spectra (HRMS) were obtained in ESI+ mode at SAIF, Panjab University, Chandigarh. The reaction was carried out under visible-light irradiation using a 9 W white LED lamp (Havells India Ltd), positioned 5 cm above the reaction mixture. The reaction setup ensured uniform exposure to light throughout the process.1,3-Diketones were synthesized using methods described in the literature.20 Commercially available 2-mercaptobenzimidazole (Avra Chemicals, India) and NBS (Avra Chemicals, India) were used without any purification.
General method for preparation of 6-substituted-2-aroyl-3-methylbenzimidazo[2,1-b]thiazoles 6(a–n)
1,3-Diketones 3 (1.0 mmol) were ground with N-bromosuccinimide (1.0 mmol) in a dry mortar for 15–30 min, forming a thick paste. The mixture was then transferred to a conical flask and stirred with 15 mL of absolute ethanol under visible-light irradiation. Next, 2-mercaptobenzimidazoles 5 (1.0 mmol) were added and stirred for 30–40 min until completion, monitored by TLC using ethyl acetate–petroleum ether (20:80, v/v). Excess ethanol was removed under reduced pressure and the reaction mixture was neutralized with aqueous sodium bicarbonate. The obtained solid products 6(a–n) were recrystallized using ethanol and dried, yielding high-purity products.
O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.04–8.02 (d, 1H, 3J = 8.0 Hz, 4H), 7.86–7.84 (m, 2H, 2′,6′-H), 7.75–7.70 (m, 2H, 4′,7H), 7.62–7.58 (m, 2H, 3′,5′-H), 7.45–7.41 (m, 1H, 6H), 7.34–7.31 (m, 1H, 5H), 2.73 (s, 3H, 3-CH3); 13C NMR (101 MHz) δ (ppm) 189.3, 154.8, 148.8, 139.9, 138.7, 133.4, 130.8, 129.3, 129.2, 125.0, 122.0, 121.7, 119.2, 113.4, 15.7; anal. calcd. for C17H12N2OS: C, 69.84; H, 4.14; N, 9.58% found: C, 69.80; H, 4.11; N, 9.53%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.04–8.02 (d, 1H, 3J = 8.0 Hz, 4H), 7.98–7.94 (m, 2H, 2′,6′-H), 7.75–7.73 (d, 1H, 3J = 8.0 Hz, 7H), 7.46–7.40 (m, 3H, 3′,5′,6H), 7.35–7.30 (m, 1H, 5H), 2.74 (s, 3H, 3-CH3); 13C NMR (101 MHz) δ (ppm) 187.8, 166.5, 164.0, 154.8, 148.8, 139.8, 135.2 (d, 3JCF = 12 Hz), 132.4 (d, 2JCF = 36 Hz), 132.3, 130.8, 125.0, 122.0, 121.4, 119.2, 116.5, 116.3, 113.3, 15.8; 19F NMR (376 MHz) δ (ppm) −105.5.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.04–8.02 (d, 1H, 3J = 8.0 Hz, 4H), 7.88–7.86 (m, 2H, 2′,6′-H), 7.78–7.76 (d, 1H, 3J = 8.0 Hz, 7H), 7.48–7.46 (m, 2H, 3′,5′-H), 7.44–7.42 (m, 1H, 6H), 7.35–7.32 (m, 1H, 5H), 2.80 (s, 3H, 3-CH3); 13C NMR (101 MHz) δ (ppm) 187.5, 153.8, 147.6, 141.2, 138.6, 131.5, 131.2, 130.4, 128.2, 124.9, 121.9, 120.9, 119.1, 112.9, 15.4; anal. calcd. for C17H11ClN2OS: C, 62.48; H, 3.39; N, 8.57% found: C, 62.40; H, 3.33; N, 8.48%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.04–8.02 (d, 1H, 3J = 8.0 Hz, 4H), 7.84–7.78 (m, 4H, 2′,3′,5′,6′-H), 7.75–7.73 (d, 1H, 3J = 8.0 Hz, 7H), 7.46–7.42 (m, 1H, 6H), 7.35–7.31 (m, 1H, 5H), 2.74 (s, 3H, 3-CH3); 13C NMR (101 MHz) δ (ppm) 188.2, 154.8, 148.8, 140.1, 137.7, 132.4, 131.3, 130.8, 127.4, 125.0, 122.0, 121.3, 119.3, 113.4, 15.8; HRMS (ESI) m/z for C17H11BrN2OS: 370.9831 [M + H]+, 372.9811 [M + H + 2]+ (1:1); anal. calcd. for C17H11BrN2OS: C, 55.00; H, 2.99; N, 7.55% found: C, 54.93; H, 2.91; N, 7.50%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.01–7.99 (d, 1H, 3J = 8.1 Hz, 4H), 7.77–7.74 (m, 2H, 2′,6′-H), 7.74–7.72 (d, 1H, 3J = 8.0 Hz, 7H), 7.44–7.39 (m, 3H, 3′,5′,6H), 7.33–7.29 (m, 1H, 5H), 2.73 (s, 3H, 4′-CH3), 2.43 (s, 3H, 3-CH3); 13C NMR (101 MHz) δ (ppm) 188.7, 154.8, 148.8, 144.0, 139.3, 135.9, 130.8, 129.8, 129.5, 124.9, 121.9, 121.6, 119.2, 113.3, 21.7, 15.7; anal. calcd. for C18H14N2OS: C, 70.56; H, 4.61; N, 9.14% found: C, 70.51; H, 4.55; N, 9.10%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.05–8.03 (d, 1H, 3J = 8.0 Hz, 4H), 7.90–7.87 (d, 2H, 3J = 8.7 Hz, 2′,6′-H), 7.75–7.73 (d, 1H, 3J = 8.0 Hz, 7H), 7.45–7.41 (t, 1H, 3J = 8.0 Hz, 6H), 7.34–7.30 (t, 1H, 3J = 8.0 Hz, 5H), 7.13–7.11 (d, 2H, 3J = 8.7 Hz, 3′,5′-H), 3.89 (s, 3H, 4′-OCH3), 2.77 (s, 3H, 3-CH3); 13C NMR (101 MHz) δ (ppm) 187.6, 163.8, 154.8, 148.8, 138.6, 132.1, 130.8, 124.8, 121.9, 121.5, 119.2, 114.6, 113.3, 56.2, 15.8; anal. calcd. for C18H14N2O2S: C, 67.06; H, 4.38; N, 8.69% found: C, 67.01; H, 4.33; N, 8.61%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.04–8.02 (d, 1H, 3J = 8.0 Hz, 4H), 7.75–7.73 (d, 1H, 3J = 8.0 Hz, 7H), 7.53–7.49 (m, 1H, 5′-H), 7.45–7.40 (m, 2H, 6,6′-H), 7.36–7.30 (m, 2H, 5,2′-H), 7.28–7.26 (dd, 1H, J = 8.0 Hz, J = 2.0 Hz, 4′-H), 3.84 (s, 3H, 3′-OCH3), 2.73 (s, 3H, 3-CH3); 13C NMR (101 MHz) δ (ppm) 189.0, 159.8, 154.9, 148.8, 140.1, 130.8, 130.6, 125.0, 121.9, 121.7, 121.4, 119.6, 119.2, 113.7, 113.4, 56.0, 15.8; anal. calcd. for C18H14N2O2S: C, 67.06; H, 4.38; N, 8.69% found: C, 66.98; H, 4.30; N, 8.58%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.19–8.17 (dd, 1H, J = 5.2 Hz, J = 1.1 Hz, 3′-H), 8.09–8.07 (m, 2H, 4,5′-H), 7.76–7.74 (d, 1H, 3J = 8.0 Hz, 7H), 7.47–7.43 (m, 1H, 6H), 7.37–7.33 (m, 2H, 5,4′-H), 2.98 (s, 3H, 6-CH3); 13C NMR (101 MHz) δ (ppm) 179.6, 154.5, 148.7, 143.2, 139.3, 136.7, 135.7, 130.7, 129.4, 125.0, 122.0, 119.2, 118.8, 113.4, 15.5; anal. calcd. for C15H10N2OS2: C, 60.38; H, 3.38; N, 9.39% found: C, 60.30; H, 3.33; N, 9.35%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 7.96–7.92 (m, 2H, 2′,6′-H), 7.87–7.84 (m, 1H, 4H), 7.60–7.58 (d, 1H, 3J = 8.0 Hz, 7H), 7.44–7.40 (m, 2H, 3′,5′-H), 7.12–7.10 (d, 1H, 3J = 8.0 Hz, 6H), 2.71 (s, 3H, 3-CH3), 2.47 (s, 3H, 5-CH3); 13C NMR (101 MHz) δ (ppm) 187.8, 164.0, 154.1, 149.1, 146.9, 139.8, 135.3–135.2 (d, 3JCF = 12 Hz), 132.4–132.3 (d, 2JCF = 36 Hz), 131.5, 130.9, 126.3, 121.0, 118.8, 116.5, 116.3, 113.2, 21.7, 15.1; 19F NMR (376 MHz) δ (ppm) −103.9.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 7.88–7.84 (m, 3H, 4,2′,6′-H), 7.66–7.64 (d, 2H, 3J = 8.0 Hz, 3′,5′-H), 7.52–7.51 (m, 1H, 7H), 7.13–7.11 (d, 1H, 3J = 8.0 Hz, 6H), 2.69 (s, 3H, 3-CH3), 2.45 (s, 3H, 5-CH3); 13C NMR (101 MHz) δ (ppm) 188.0, 154.7, 149.3, 140.0, 138.3, 137.4, 134.6, 131.2, 129.5, 128.8, 123.3, 121.1, 119.1, 112.8, 21.7, 15.7; HRMS (ESI) m/z for C18H13ClN2OS: 341.0503 [M + H]+, 343.0514 [M + H + 2]+ (3:1); anal. calcd. for C18H13ClN2OS: C, 63.43; H, 3.84; N, 8.22% found: C, 63.39; H, 3.75; N, 8.16%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 7.87–7.85 (m, 1H, 4H), 7.76–7.73 (m, 2H, 2′,6′-H), 7.60–7.58 (d, 1H, 3J = 8.0 Hz, 7H), 7.40–7.38 (d, 2H, 3J = 8.0 Hz, 3′,5′-H), 7.13–7.11 (d, 1H, 3J = 8.0 Hz, 6H), 2.70 (s, 3H, 3-CH3), 2.45 (s, 3H, 5-CH3), 2.43 (s, 3H, 4′-CH3); 13C NMR (101 MHz) δ (ppm) 189.3, 154.8, 148.8, 139.9, 138.7, 133.8, 130.8, 129.2, 125.6, 122.5, 120.7, 118.5, 112.5, 21.1, 15.1, 15.0; anal. calcd. for C19H16N2OS: C, 71.22; H, 5.03; N, 8.74% found: C, 71.16; H, 4.99; N, 8.69%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 7.88–7.86 (d, 2H, 3J = 8.0 Hz, 2′,6′-H), 7.84–7.83 (m, 1H, 4H), 7.61–7.59 (d, 1H, 3J = 8.0 Hz, 7H), 7.25–7.23 (d, 1H, 3J = 8.0 Hz, 6H), 7.12–7.10 (d, 2H, 3J = 8.0 Hz, 3′,5′-H), 3.88 (s, 3H, 4′-OCH3), 2.75 (s, 3H, 3-CH3), 2.48 (s, 3H, 5-CH3); 13C NMR (101 MHz) δ (ppm) 187.0, 163.2, 153.6, 146.3, 138.0, 131.5, 130.8, 130.4, 130.3, 125.6, 120.5, 118.2, 114.0, 112.5, 55.6, 21.2, 15.2; anal. calcd. for C19H16N2O2S: C, 67.84; H, 4.79; N, 8.33% found: C, 67.76; H, 4.77; N, 8.28%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 7.90–7.88 (m, 1H, 5′-H), 7.53–7.48 (m, 2H, 6′,6H), 7.40–7.39 (m, 1H, 4H), 7.35–7.34 (m, 1H, 2′-H), 7.27–7.25 (m, 1H, 4′-H), 7.15–7.13 (m, 1H, 7H), 3.84 (s, 3H, 3′-OCH3), 2.71 (s, 3H, 3-CH3), 2.46 (s, 3H, 5-CH3); 13C NMR (101 MHz) δ (ppm) 188.4, 159.2, 154.2, 148.6, 139.6, 139.5, 134.0, 130.0, 128.3, 122.6, 120.8, 118.9, 118.5, 113.1, 112.3, 55.4, 21.2, 15.1; anal. calcd. for C19H16N2O2S: C, 67.84; H, 4.79; N, 8.33% found: C, 67.77; H, 4.79; N, 8.30%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm) 7.86–7.84 (m, 1H, 6′-H), 7.63–7.58 (m, 2H, 4′,6H), 7.44–7.41 (m, 1H, 4H), 7.25–7.23 (m, 2H, 3′,5′-H), 7.15–7.11 (m, 1H, 7H), 3.80 (s, 3H, 2′-OCH3), 2.63 (s, 3H, 3-CH3), 2.46 (s, 3H, 5-CH3); 13C NMR (101 MHz) δ (ppm) 188.4, 156.8, 140.1, 134.6, 133.3, 133.2, 131.5, 128.8, 126.4, 123.7, 121.4, 118.8, 113.3, 112.5, 56.3, 21.7, 14.1; anal. calcd. for C19H16N2O2S: C, 67.84; H, 4.79; N, 8.33% found: C, 67.74; H, 4.75; N, 8.25%.General method for preparation of 2-((1-acetyl-1H-benzimidazol-2-yl)thio)-1-arylethan-1-one 10(a–e)
1,3-Diketones 3 (1.0 mmol) were grounded with NBS (1.0 mmol) in a dry mortar for 15–30 min to form a thick paste. Subsequently, 2-mercaptobenzimidazole 5 (1.0 mmol) was added and the reaction mixture was further grounded thoroughly at room temperature for 15–20 min. Reaction progress was monitored by TLC using ethyl acetate–petroleum ether (20:80, v/v). After completion, 30 mL of saturated sodium bicarbonate solution was added and the mixture was filtered to obtain the crude solid. N/S-difunctionalized benzimidazole derivatives 10(a–e), were recrystallized from ethanol and dried, yielding high-purity products.
O), 1670 cm−1 (CO); 1H NMR (500 MHz, CDCl3) δ (ppm) 8.11–8.09 (m, 2H, 2′,6′-H), 7.63–7.60 (m, 2H, 4,7H), 7.54–7.50 (m, 3H, 3′,4′,5′-H), 7.30–7.25 (m, 2H, 5,6H), 4.85 (s, 2H, CH2), 2.81 (s, 3H, CH3); 13C NMR (126 MHz) δ (ppm) 193.9, 168.8, 154.3, 144.0, 136.1, 133.6, 133.2, 128.8, 128.6, 124.6, 123.6, 119.1, 113.2, 40.5, 26.2; anal. calcd. for C17H14N2O2S: C, 65.79; H, 4.55; N, 9.03% found: C, 65.73; H, 4.50; N, 9.00%.O), 1690 cm−1 (CO); 1H NMR (500 MHz, DMSO-d6) δ (ppm) 8.06–8.04 (d, 1H, 3J = 8.0 Hz, 7H), 7.98–7.93 (m, 2H, 2′,6′-H), 7.77–7.75 (d, 1H, 3J = 8.0 Hz, 4H), 7.47–7.41 (m, 3H, 3′,5′,6H), 7.36–7.32 (m, 1H, 5H), 5.65 (s, 2H, CH2), 2.74 (s, 3H, CH3); 13C NMR (126 MHz) δ (ppm) 187.3, 166.0, 154.2, 147.7, 139.2, 134.6 (d, 3JCF = 12 Hz), 131.9–131.8 (d, 2JCF = 40 Hz), 130.2, 124.6, 121.6, 118.5, 116.0, 115.8, 112.9, 38.8, 15.3; 19F NMR (376 MHz) δ (ppm) −110.1.O), 1692 cm−1 (CO); 1H NMR (500 MHz, CDCl3) δ (ppm) 8.07–8.05 (d, 1H, 3J = 7.8 Hz, 7H), 7.98–7.95 (m, 2H, 2′,6′-H), 7.77–7.75 (d, 1H, 3J = 7.8 Hz, 4H), 7.44–7.38 (m, 3H, 3′,5′,6H), 7.34–7.32 (m, 1H, 5H), 5.51 (s, 2H, CH2), 2.75 (s, 3H, CH3); 13C NMR (126 MHz) δ (ppm) 187.8, 154.7, 148.2, 139.8, 132.4, 132.2, 130.7, 125.1, 122.1, 119.1, 116.6, 116.3, 113.4, 39.4, 15.8; anal. calcd. for C17H13ClN2O2S: C, 59.22; H, 3.80; N, 8.12% found: C, 59.18; H, 3.08; N, 8.05%.O), 1680 cm−1 (CO); 1H NMR (500 MHz, CDCl3) δ (ppm) 8.04–8.02 (d, 1H, 3J = 8.0 Hz, 7H), 7.78–7.76 (m, 2H, 2′,6′-H), 7.75–7.74 (m, 1H, 4H), 7.43–7.40 (m, 2H, 3′,5′-H), 7.34–7.32 (m, 1H, 6H), 6.91–6.90 (m, 1H, 5H), 5.65 (s, 2H, CH2), 2.75 (s, 3H, CH3), 2.44 (s, 3H, 4′-CH3); 13C NMR (126 MHz) δ (ppm) 188.8, 154.8, 148.8, 144.0, 139.3, 135.9, 130.8, 129.8, 129.5, 124.9, 121.9, 119.2, 113.3, 39.4, 21.7, 15.7; HRMS (ESI) m/z for C18H13FN2OS: 325.0997 [M + H]+; anal. calcd. for C18H16N2O2S: C, 66.65; H, 4.97; N, 8.64% found: C, 66.62; H, 4.91; N, 8.60%.O), 1680 cm−1 (CO); 1H NMR (500 MHz, CDCl3) δ (ppm) 8.10–8.08 (d, 2H, 3J = 8.0 Hz, 2′,6′-H), 7.64–7.63 (m, 1H, 7H), 7.56–7.54 (m, 1H, 4 H), 7.31–7.24 (m, 2H, 5,6H), 6.98–6.96 (d, 2H, 3J = 8.0 Hz, 3′,5′-H), 4.83 (s, 2H, CH2), 3.89 (s, 3H, 4′-OCH3), 2.82 (s, 3H, CH3); 13C NMR (126 MHz) δ (ppm) 192.2, 168.7, 163.9, 154.3, 143.9, 133.2, 130.9, 128.9, 124.5, 123.5, 119.0, 113.8, 113.2, 55.5, 40.3, 26.2; anal. calcd. for C18H16N2O3S: C, 63.51; H, 4.74; N, 8.23% found: C, 63.48; H, 4.71; N, 8.20%.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interest.Acknowledgements
We are highly thankful to the Council of Scientific and Industrial Research (CSIR), New Delhi, India, for their generous financial support for the JRF & SRF awarded to Prince Kumar (Grant 09/105(0302)/2020-EMR-I).References
- K. A. Al-Rashood and H. A. Abdel-Aziz, Thiazolo[3,2-a]Benzimidazoles: Synthetic Strategies, Chemical Transformations and Biological Activities, Molecules, 2010, 15(6), 3775–3815, DOI:10.3390/molecules15063775.
- X. Deng, X. Tan, T. An, Q. Ma, Z. Jin, C. Wang, Q. Meng and C. Hu, Synthesis, Characterization, and Biological Activity of a Novel Series of Benzo[4,5]Imidazo[2,1-b]Thiazole Derivatives as Potential Epidermal Growth Factor Receptor Inhibitors, Molecules, 2019, 24(4), 682, DOI:10.3390/molecules24040682.
- S. C. Gilman and H. G. Bluestein, Effects of Tilomisole, Indomethacin and Levamisole on Regulation of Epstein Barr Virus-Induced B Cell Proliferation by Peripheral Blood Mononuclear Cells from Normal Individuals and Patients with Rheumatoid Arthritis, Agents Actions, 1987, 21(3–4), 266–268, DOI:10.1007/BF01966486.
- S. A. Darwish, M. M. El-Kerdawy, A. R. Elsheakh, R. S. Abdelrahman, M. A. Shaldam, H. A. Abdel-Aziz, G. S. Hassan and M. A. Ghaly, New Tilomisole-Based Benzimidazothiazole Derivatives as Anti-Inflammatory Agents: Synthesis, in Vivo, in Vitro Evaluation, and in Silico Studies, Bioorg. Chem., 2022, 120(March), 105644, DOI:10.1016/j.bioorg.2022.105644.
- A. A. M. Alkhaldi, M. M. Al-Sanea, A. Nocentini, W. M. Eldehna, Z. M. Elsayed, A. Bonardi, M. F. Abo-Ashour, A. K. El-Damasy, M. S. Abdel-Maksoud, T. Al-Warhi, P. Gratteri, H. A. Abdel-Aziz, C. T. Supuran and R. El-Haggar, 3-Methylthiazolo[3,2-a]Benzimidazole-Benzenesulfonamide Conjugates as Novel Carbonic Anhydrase Inhibitors Endowed with Anticancer Activity: Design, Synthesis, Biological and Molecular Modeling Studies, Eur. J. Med. Chem., 2020, 207, 112745, DOI:10.1016/j.ejmech.2020.112745.
- L. Cui, J. U. Kim, H. Nomura, H. Nakanotani and C. Adachi, Benzimidazobenzothiazole-Based Bipolar Hosts to Harvest Nearly All of the Excitons from Blue Delayed Fluorescence and Phosphorescent Organic Light-Emitting Diodes, Angew. Chem., 2016, 128(24), 6978–6982, DOI:10.1002/ange.201601136.
- X. Zhang, J. Jia and C. A. Ma, One-Pot Regioselective Synthesis of Benzo[d]Imidazo[2,1-b]Thiazoles, Org. Biomol. Chem., 2012, 10(39), 7944, 10.1039/c2ob26211h.
- S. Ambethkar, M. Vellimalai, V. Padmini and N. Bhuvanesh, Iodine-Mediated C–N and C–S Bond Formation: Regioselective Synthesis of Benzo[4,5]Imidazo[2,1-b]Thiazoles, New J. Chem., 2017, 41(1), 75–80, 10.1039/C6NJ02102F.
- M. Talebizadeh, M. Anary-Abbasinejad and A. A. Darehkordi, Simple One-pot Three-component Synthesis of Dihydrobenzo[4,5]Imidazo[2,1- b ]Thiazol-3-ols by Reaction of Acyl Chlorides, Isocyanides, and 2-Mercaptobenzimidazoles, J. Heterocycl. Chem., 2018, 55(12), 2737–2743, DOI:10.1002/jhet.3335.
- T. Beresneva, S. Belyakov, E. Abele and E. Lukevics, Mechanism of Formation of 3-Methyl Derivatives of Imidazo[2,1-b]Thiazoles and Their Benzo Analogs in the Reactions of 2-Mercaptoimidazole and 2-Mercaptobenzimidazole with 1,3-Dichloroacetone under Phase-Transfer Catalysis Conditions, Chem. Heterocycl. Compd., 2011, 46(11), 1400–1404, DOI:10.1007/s10593-011-0678-3.
- A. E.-W. A. O. Sarhan, H. A. H. El-Sherief and A. M. Mahmoud, A Convenient One-Pot Synthesis of 2-Benzimidazolyl-Thioacetophenones and Thiazolo[3,2-a]Benzimidazoles, Tetrahedron, 1996, 52(31), 10485–10496, DOI:10.1016/0040-4020(96)00569-8.
- W. M. Eldehna, M. A. El Hassab, M. F. Abo-Ashour, T. Al-Warhi, M. M. Elaasser, N. A. Safwat, H. Suliman, M. F. Ahmed, S. T. Al-Rashood, H. A. Abdel-Aziz and R. El-Haggar, Development of Isatin-Thiazolo[3,2-a]Benzimidazole Hybrids as Novel CDK2 Inhibitors with Potent in Vitro Apoptotic Anti-Proliferative Activity: Synthesis, Biological and Molecular Dynamics Investigations, Bioorg. Chem., 2021, 110, 104748, DOI:10.1016/j.bioorg.2021.104748.
- K. Anichina, A. Mavrova, D. Yancheva, J. Tsenov and R. Dimitrov, Tautomerism and Isomerism in Some Antitrichinellosis Active Benzimidazoles: Morphological Study in Polarized Light, Quantum Chemical Computations, J. Mol. Struct., 2017, 1150, 179–187, DOI:10.1016/j.molstruc.2017.08.073.
- G. Shen, B. Yang, X. Huang, Y. Hou, H. Gao, J. Cui, C. Cui and T. Zhang, Copper- and Palladium-Catalyzed Cross-Coupling Reactions for the Synthesis of N -Fused Benzo[4,5]Imidazo[2,1- b ]Thiazole Derivatives via Substituted Trans -1,2-Diiodoalkenes, 1 H -Benzo[ d ]Imidazole-2-Thiols, and Halobenzenes, J. Org. Chem., 2017, 82(7), 3798–3805, DOI:10.1021/acs.joc.7b00162.
- J. Gao, J. Zhu, L. Chen, Y. Shao, J. Zhu, Y. Huang, X. Wang and X. Lv, Synthesis of Benzimidazo[2,1-b]Benzothiazole Derivatives through Sequential Cu-Catalyzed Domino Coupling and Pd-Catalyzed Suzuki Reaction, Tetrahedron Lett., 2014, 55(22), 3367–3373, DOI:10.1016/j.tetlet.2014.04.070.
- S. Jana, A. Chakraborty, V. Z. Shirinian and A. Hajra, Synthesis of Benzo[4,5]Imidazo[2,1- b ]Thiazole by Copper(II)-Catalyzed Thioamination of Nitroalkene with 1 H -Benzo[ d ]Imidazole-2-thiol, Adv. Synth. Catal., 2018, 360(12), 2402–2408, DOI:10.1002/adsc.201800393.
- J. Kuang, Y. Xia, A. Yang, H. Zhang, C. Su and D. Lee, Copper-Catalyzed Aminothiolation of Terminal Alkynes with Tunable Regioselectivity, Chem. Commun., 2019, 55(12), 1813–1816, 10.1039/C8CC09122F.
- S. Rasheed, D. N. Rao and P. Das, Copper-Catalyzed Inter- and Intramolecular C–N Bond Formation: Synthesis of Benzimidazole-Fused Heterocycles, J. Org. Chem., 2015, 80(18), 9321–9327, DOI:10.1021/acs.joc.5b01396.
- R. Aggarwal, M. Hooda, P. Kumar, S. Kumar, S. Singh and R. Chandra, An Expeditious On-Water Regioselective Synthesis of Novel Arylidene-Hydrazinyl-Thiazoles as DNA Targeting Agents, Bioorg. Chem., 2023, 136, 106524, DOI:10.1016/j.bioorg.2023.106524.
- R. Aggarwal, M. Hooda, P. Kumar and M. C. Torralba, Visible-Light-Mediated Regioselective Synthesis of Novel Thiazolo[3,2-b][1,2,4]Triazoles: Advantageous Synthetic Application of Aqueous Conditions, Org. Biomol. Chem., 2022, 20(3), 584–595, 10.1039/d1ob02194j.
- R. Aggarwal, P. Kumar, M. Hooda, R. Singh and P. Kumar, Efficient Synthesis of Promising Antidiabetic Triazinoindole Analogues via a Solvent-Free Method: Investigating the Reaction of 1,3-Diketones and 2,5-Dihydro-3 H -[1,2,4]Triazino[5,6- b ]Indole-3-Thione, Org. Biomol. Chem., 2025, 23(1), 213–225, 10.1039/D4OB01487A.
- M. Nazeef, K. N. Shivhare, S. Ali, S. Ansari and I. R. Siddiqui, Visible-Light-Mediated One-Pot Efficient Synthesis of 1-Aryl-1H,3H-Thiazolo[3,4-a]Benzimidazoles: A Metal-Free Photochemical Approach in Aqueous Ethanol, Mol. Diversity, 2021, 25(4), 2479–2486, DOI:10.1007/s11030-020-10145-8.
- Z. Chen, F. Xue, Y. Zhang, W. Jin, B. Wang, Y. Xia, M. Xie, A. Abdukader and C. Liu, Visible-Light-Promoted [3 + 2] Cyclization of Chalcones with 2-Mercaptobenzimidazoles: A Protocol for the Synthesis of Imidazo[2,1- b ]Thiazoles, Org. Lett., 2022, 24(17), 3149–3154, DOI:10.1021/acs.orglett.2c00867.
- A. Ohno, T. Morishita and S. Oka, Acyl Migrations in Diacyl Derivatives of 2-Methylmercaptobenzimidazole, Bioorg. Chem., 1976, 5(4), 383–391, DOI:10.1016/0045-2068(76)90023-7.
- R. Aggarwal, P. Kumar, M. Hooda, S. Kumar and N. Serendipitous, S -Difunctionalization of Triazoles with Trifluoromethyl-β-Diketones: Access to Regioisomeric 1-Trifluoroacetyl-3-Aryl-5-(2-Oxo-2-Arylethylthio)-1,2,4-Triazoles as DNA-Groove Binders, RSC Adv., 2024, 14(10), 6738–6751, 10.1039/D4RA00083H.
- P. Garra, F. Morlet-Savary, C. Dietlin, J. P. Fouassier and J. Lalevée, On-Demand Visible Light Activated Amine/Benzoyl Peroxide Redox Initiating Systems: A Unique Tool To Overcome the Shadow Areas in Photopolymerization Processes, Macromolecules, 2016, 49(24), 9371–9381, DOI:10.1021/acs.macromol.6b02167.
- M. Bhardwaj, P. Grover, B. Rasool and D. Mukherjee, Recent Advances in N-Hyrdoxypthalimide: As a Free Radical Initiator and Its Applications, Asian J. Org. Chem., 2022, 11(11), e202200442, DOI:10.1002/ajoc.202200442.
- C. R. DeJulius, B. R. Dollinger, T. E. Kavanaugh, E. Dailing, F. Yu, S. Gulati, A. Miskalis, C. Zhang, J. Uddin, S. Dikalov and C. L. Duvall, Optimizing an Antioxidant TEMPO Copolymer for Reactive Oxygen Species Scavenging and Anti-Inflammatory Effects in Vivo, Bioconjugate Chem., 2021, 32(5), 928–941, DOI:10.1021/acs.bioconjchem.1c00081.
- A. Konopko and G. Litwinienko, Mutual Activation of Two Radical Trapping Agents: Unusual “Win–Win Synergy” of Resveratrol and TEMPO during Scavenging of Dpph • Radical in Methanol, J. Org. Chem., 2022, 87(22), 15530–15538, DOI:10.1021/acs.joc.2c02080.
Footnote |
| † Electronic supplementary information (ESI) available: The supporting information consists of additional experimental data (1H, 13C, HMBC, HSQC NMR and HRMS spectra) for final compounds. See DOI: https://doi.org/10.1039/d5ra02681d |
| This journal is © The Royal Society of Chemistry 2025 |