
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Therapeutic potential of adenosine receptor modulators in cancer treatment
Prasenjit Maity
 ,
Swastika Ganguly
and
Pran Kishore Deb
*
,
Swastika Ganguly
and
Pran Kishore Deb
*
Department of Pharmaceutical Sciences and Technology, Birla Institute of Technology, Mesra, Ranchi, 835215, Jharkhand, India. E-mail: prankishore1@gmail.com; prankishoredeb@bitmesra.ac.in
First published on 17th June 2025
Abstract
All human cells contain the universal autocoid adenosine, which interacts with four types of G protein-coupled receptors (GPCRs), namely A1, A2A, A2B, and A3 adenosine receptors (ARs). Among these receptors, A2A and A2B ARs activate adenylate cyclase, while A1 and A3 ARs suppress the adenylate cyclase activity. Adenosine-receptor interactions play a crucial role in cancer biology by modulating the immune microenvironment, which tumors exploit to create immunosuppression that promotes their growth and metastasis. When the A2A AR is activated on natural killer (NK) cells and T cells, it reduces their ability to carry out cytotoxic functions. This activation also encourages the formation of immune-suppressing cell types, such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs), further weakening the immune response. Targeting adenosine receptors, particularly the A2A subtype, represents a promising therapeutic strategy. By antagonizing these receptors, it may be possible to restore T cell function, helping the body to recognize and attack cancer cells more effectively. Despite recent advancements in the discovery of novel, targeted anticancer agents, these treatments have shown limited effectiveness against metastatic tumours, complicating cancer management. Moreover, developing adenosine receptor agonists or antagonists with high target selectivity and potency remains a significant challenge, as the widespread distribution of adenosine receptors throughout the body raises concerns about off-target effects and reduced therapeutic efficacy. In order to improve outcomes for patients with advanced cancer, researchers are actively investigating safer and more efficient chemotherapy substitutes. However, drugs that activate A3 adenosine receptors and block A2A receptors are being explored as a novel approach for cancer treatment. Monoclonal antibodies and small-molecule inhibitors targeting the CD39/CD73/A2A AR axis are also being tested in clinical trials, both as standalone treatments and in combination with anti-PD-1/PD-L1 immunotherapies. This review primarily focuses on the signaling pathways and the therapeutic potential of various adenosine receptor agonists and antagonists across various cancer types, highlighting their ongoing evaluation in preclinical and clinical trials, both as monotherapies and in rational combination with immunotherapy, chemotherapy, or targeted therapies, potentially leading to the development of advanced treatments that could aid in tumor suppression.
 Prasenjit Maity | Mr Prasenjit Maity is a PhD research scholar in Pharmaceutical Medicinal Chemistry at the Department of Pharmaceutical Sciences and Technology, Birla Institute of Technology (BIT), Mesra, Ranchi, India. He is pursuing his doctoral studies under the supervision of Dr Pran Kishore Deb and the co-supervision of Dr Swastika Ganguly with the prestigious Institute Research Scholarship (IRS). Mr Maity earned his BPharm. and MPharm. degrees with first-class honours from the Bengal School of Technology, Chinsurah, Hooghly, West Bengal. His research interests focus on Computer-Aided Drug Design (CADD) and the discovery and development of novel chemical entities (NCEs) as adenosine receptor antagonists, as well as agents with anti-inflammatory and antibacterial potential. |
 Swastika Ganguly | Dr Swastika Ganguly is a Professor in the Department of Pharmaceutical Sciences and Technology at BIT Mesra, Ranchi, and served as Head of the Department from 2021 to 2023. She completed her BPharm., MPharm., and PhD (CSIR-SRF) at BIT Mesra. With over 27 years of experience in teaching and research, her expertise lies in Computer-Aided Drug Design and the synthesis of drugs targeting HIV and associated infections. She has guided more than 30 MPharm. dissertations and 14 PhD scholars, with five currently under her mentorship. Dr Ganguly has over 130 publications, more than 60 conference presentations, and has served as the APTI Women's Forum East Zone Coordinator. She is a Chartered Chemist recognized by the Indian Institution of Chemists and currently serves as the Chancellor's nominee at Jadavpur University, Kolkata. |
 Pran Kishore Deb | Dr Pran Kishore Deb is an Assistant Professor in the Department of Pharmaceutical Sciences and Technology at BIT Mesra, Ranchi, India. He earned his PhD in Pharmaceutical Medicinal Chemistry from the University Institute of Pharmaceutical Sciences (UIPS), Panjab University, Chandigarh, under the CSIR-SRF fellowship (New Delhi, India). His primary research interests include computer-aided drug design (CADD) and the development of novel chemical entities (NCEs) targeting adenosine receptors, COX-2, mosquito larvae, and tuberculosis. Dr Deb has published over 150 research articles and holds over 35 international patents. He has earned several national and international honors and has been consistently listed among the World's Top 2% Scientists by Stanford University, USA, for the past five years. |
1. Introduction
Adenosine is an endogenous purine nucleoside that comprises an adenine base linked to a sugar-containing ribose molecule via a β-N9-glycosidic bond, as depicted in Fig. 1.1 It serves as an essential element in human biology.2 Adenosine has been investigated for its potential role as a molecule with protective properties against cancer.3 Adenosine is well known for being an important local regulator of tissue function, particularly in situations where cellular energy demand exceeds the available energy supply.4 Adenosine is pivotal in maintaining cellular protection and modulating diverse physiological and pathological processes. In 1929, Drury and Szent-Gyorgyi elucidated adenosine's role as an extracellular signalling molecule, highlighting its broad impact on physiological functions.5 | ||
| Fig. 1 Structure of adenosine. | ||
Adenosine exerts an important function in inhibiting the immune response against tumours by encouraging the growth of blood vessels (angiogenesis) and boosting the growth, progression, and mobility of tumour cells.6,7 However, there is ongoing debate about how adenosine specifically affects the death of cancer cells through apoptosis.8 As a result, ARs are considered promising targets for treating various medical conditions.9–13 Adenosine plays a role in the body's immune suppression in tumors. Some studies have shown that blocking adenosine or its receptors might lead to enhanced immunity against tumours, but it could also reduce the body's anti-tumour responses under certain conditions.13
Adenosine is a type of molecule derived from adenosine triphosphate (ATP) which is crucial for cellular energy. It serves as a significant regulator within the tumour microenvironment (TME). In conditions where there is low oxygen, limited blood flow, or inflammation, the level of adenosine increases in the region surrounding the tumour. This rise in adenosine can influence both the immune system's reactions and the tumor's growth dynamics.14,15
All four types of adenosine receptors have been identified as contributing to the progression of cancer.10 Numerous drug candidates that affect adenosine receptors in the body, including agonists, antagonists, partial agonists, and allosteric modulators, have been recently discovered and patented, and many of them are currently undergoing clinical trials.14–23
Recently, a growing focus has been on understanding the role of adenosine receptors' impact on cancer, affecting tumour growth, spread, and the immune system's response. Investigating these roles is vital for developing new treatments to combat cancer effectively. This review is focused on possible treatment strategies emphasizing the role of adenosine and its receptors in cancer development.
2. Adenosine & adenosine receptors
Adenosine is ubiquitous, released by nearly all cells, and produced in the extracellular environment through the breakdown of ATP by a cascade of ectoenzymes, including apyrase (CD39) and 5′-nucleotidase (CD73).24 When adenosine levels become excessive, the body has mechanisms to reduce them. Adenosine kinase can convert adenosine back into adenosine monophosphate (AMP) through phosphorylation, and adenosine deaminase (ADA) can deaminate adenosine into inosine.16,17 Both processes require sufficient oxygen to function effectively. However, these enzymes may not work efficiently in areas with low oxygen levels, such as in tumours affected by hypoxia. This can lead to the accumulation of adenosine in these regions, which can affect inflammation and contribute to tumour growth. Thus, the oxygen-dependent regulation of adenosine metabolism plays a crucial role in the tumour microenvironment.17Elevated levels of CD73 have been noted in multiple cancer types, such as breast, colon, ovarian, melanoma, glioma, glioblastoma, leukemia, and bladder cancer.18
Adenosine, in turn, can act on immune cells and other cells in the tumour microenvironment, promoting immunosuppression and supporting tumour growth and metastasis. Therefore, CD73 expression in cancer cells is of interest as a potential target for therapeutic interventions to modulate the immune response against tumours.19,25,26
Adenosine is a potent compound that influences various cells and tissues, including platelets, coronary arteries, smooth muscle, cardiac muscle, and immune cells.27 As an extracellular messenger, it plays a role in conditions such as neurodegenerative diseases, psychiatric disorders, heart issues, lung injuries, cancers, and eye diseases.28
Adenosine receptors, encoded by separate genes, belong to the G protein-coupled receptor (GPCR) family. These receptors are categorized as A1, A2A, A2B, and A3 adenosine receptors (AR).29,30 The ARs are found throughout the human body in various organs and tissues, where they play crucial roles in regulating essential physiological functions, as shown in Table 1.22,31–35 Extracellular adenosine serves as a natural ligand for all these receptors. Each AR subtype exhibits unique binding affinities and is activated by adenosine in different ways, influencing diverse physiological processes.36,37 This interaction pattern allows adenosine to regulate neurotransmission, immune responses, inflammation, and vascular function across different tissues and organ systems in the body.38 The A1 AR, A2A AR, and A3 ARs have moderate to high affinities for adenosine, and they usually require 1–10 nM, 30 nM and 100 nM concentrations of adenosine, respectively, for their activation. The A2B AR, has the lowest affinity and requires a higher concentration of adenosine (approximately 1000 nM) for its activation.10,23,39
| Receptors | Gene | Chromosomal location | Molecular weight (by amino acid sequence)/length (residues) | Affinity for adenosine (nM) | G-protein coupling | Signalling system | Effects on ion channels | Distribution | ||
|---|---|---|---|---|---|---|---|---|---|---|
| High expression | Intermediate expression | Low expression | ||||||||
| a AC (adenyl cyclase), PLC (phospholipase C), Gi/Go (inhibitory G-proteins), DAG (diacylglycerol), IP3 (inositol triphosphate), Gs/Golf (stimulatory G-proteins), cAMP (cyclic adenosine monophosphate), K+ (potassium ion), Ca2+ (calcium ion), WBCs (white blood cells), CNS (central nervous system), PI3K (phosphoinositide 3-kinase), ADORA (adenosine receptor A), nM (nanomolar), MAPK (mitogen-activated protein kinase). | ||||||||||
| A1 AR | ADO RA1 | 1q32.1 | 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 512/326 512/326 |
1–10 | Gi/Go | Block AC (↓cAMP), stimulate PLC (↑IP3/DAG), ↑PI3 kinase, ↑MAPK, ↑K+, Ca2+ | ↑K+ & ↓Ca2+ | CNS, spinal cord, adrinal gland, atria | Adipose tissue, liver, renal tissue & skeletal muscle | In the higher bronchi & pancreas |
| A2A AR | ADO R A2A | 22q11.2 | 44707/412 |
30 | Gs/Golf | Stimulate AC (↑cAMP), ↑MAPK | Inhibit Ca2+ channels | Lymphoid tissue (spleen, thymus, WBC & platelets) | Blood vessels, cardiac tissue, lung & peripheral nerve | Other regions of CNS |
| A2B AR | ADOR A2B | 17p11.2-12 | 36333/332 |
1000 | Gs/Gq | Stimulate AC (↑cAMP), activate PLC (↑IP3/DAG), ↑MAPK | Inhibit Ca2+ channels | Intraperitoneal pouch (cecum), urinary bladder, and colon | Lungs, eye, must cell & blood vessel | Adipose tissue, CNS, kidney & adrenal gland |
| A3 AR | ADOR A3 | 1p13.2 | 36185/318 |
100 | Gi/Gq | Block AC (↓cAMP), activate PLC (↑IP3/DAG), ↑PI3 kinase, ↑MAPK | Testis & must cell | CNS (hippocampus and cerebellum) | Liver, lymphatic tissue, thyroid and adrenal gland | |
This similarity percentage gives us a rough estimate of how closely these receptors are related to each other; comparing the amino acid sequences of these receptors are roughly 49% of the residues in the A1 receptor are identical to those in the A3 receptor. Roughly 59% of the residues in the A2A receptor are identical to those in the A2B receptor.37,40
When adenosine binds to its receptors on the cell surface, it triggers a cascade of molecular events inside the cell. This includes activating MAPK (mitogen-activated protein kinase) proteins, which are enzymes that relay signals from the cell membrane to the nucleus. Once activated, MAPKs phosphorylate various target proteins involved in gene expression, cell cycle progression, and differentiation pathways.41 This pathway is essential for regulating fundamental cellular processes, including proliferation (cell division) and differentiation (maturation into specialized cell types). Various external signals can activate the MAPK pathway, with GPCRs playing a significant function in this process.42
AR, which is indeed a type of GPCR, regulates adenyl cyclase (AC) activity by either stimulating or inhibiting it. When AC is inhibited, it reduces the amount of cAMP in the cell. This affects the activity of PKA, which in turn phosphorylates proteins involved in the MAPK and AKT signalling pathways.43,44 The A2A and A2B receptors increase the activity of AC through the Gs protein. The A2A receptor activates the Gs protein, while the A2B receptor activates phospholipase C (PLC) through the Gq protein Which results in the formation of IP3 (Inositol trisphosphate) and DAG (Diacylglycerol).45,46
3. Purpose, mode of action, storage, release, and synthesis of adenosine
Adenosine is not stored in vesicles but is continuously released in response to metabolic changes. It modulates neuronal activity both presynaptically and postsynaptically. After acting, adenosine is either internalized for recycling or metabolized within the cell.1,47Adenosine is primarily produced extracellularly through the dephosphorylation of ATP, ADP, and AMP by two enzymes: CD39 (NTPDase 1), which converts ATP to ADP and AMP, and CD73 (5′-NT), which converts AMP to adenosine under stress (Fig. 2). Additionally, ecto-phosphodiesterase (ecto-PDE) enhances adenosine production by converting cAMP to AMP, further activating CD73.48,49
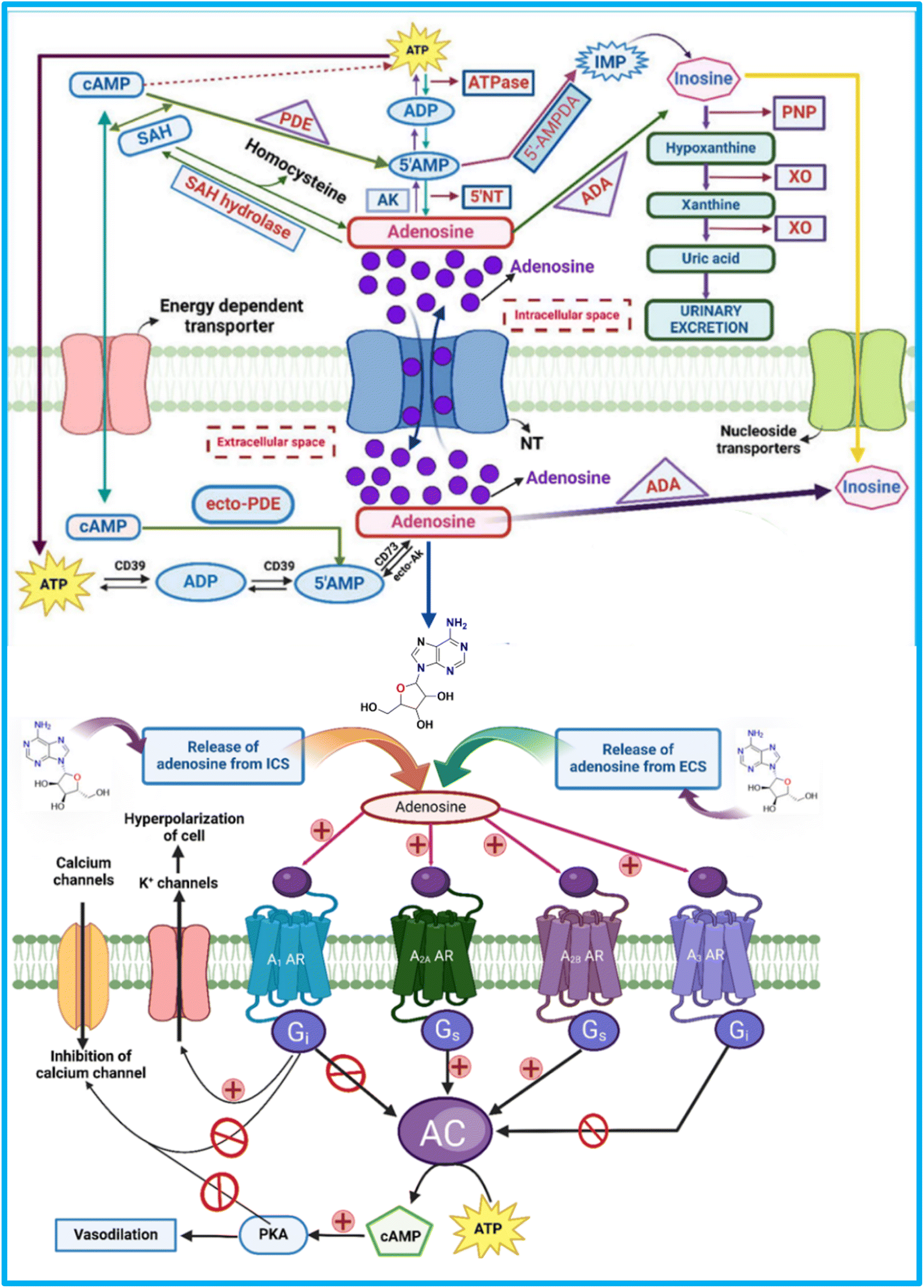 | ||
| Fig. 2 Synthesis, storage, release, and signalling pathways of adenosine through adenosine receptors. ATP – “Adenosine triphosphate”, CD73 – “Ecto-5′-nucleotidase” cAMP – “Cyclic adenosine monophosphate”, ADA – “Adenosine deaminase”, ADA – “Adenosine deaminase”, XO – “Xanthine oxidase”, CD39 – Ectonucleoside triphosphate diphosphohydrolase-1, PDE – “Phosphodiesterase”, SAH – “S-adenosyl-homocysteine”, 5-AMP – “5-Adenosine monophosphate”, PNP – “Purine nucleoside phosphorylase”, ADP – “Adenosine diphosphate”, 5NT – “5′-Nucleotidase”, ecto-AK – “Extracellular adenosine kinase”, A1 AR – “A1 adenosine receptor”, K+ channel – “Potassium channels”, A2B AR – “A2B adenosine receptor”, cAMP – “cyclic adenosine monophosphate”, Gi – “inhibitory G-proteins”, A2A AR – “A2A adenosine receptor”, AC – “Adenyl cyclase”, Gs – “Stimulatory G-proteins”, PKA – “Protein kinase A” A3 AR – “A3 adenosine receptor”. The figure was created in BioRender. Deb, P. (2025). | ||
Once produced, adenosine is transported across the cell membrane by concentrative nucleoside transporters (CNTs) and equilibrative nucleoside transporters (ENTs).50,51 Equilibrative nucleoside transporters (ENTs) are membrane proteins that regulate intracellular nucleoside levels by facilitating their passive, bidirectional transport across the cell membrane via facilitated diffusion. They allow adenosine and other nucleosides to move in response to concentration gradients, helping maintain extracellular adenosine levels without ATP or ionic gradients.52
Inside the cell, adenosine undergoes key enzymatic transformations, including hydrolysis to form SAH by SAHH, phosphorylation to AMP by AK, and deamination to inosine by ADA. These processes are essential for regulating adenosine levels.10,53,54 The ecto-ADA removes extracellular adenosine and transports it inside cells via ENTs.10,55–57 Fig. 2 represents the synthesis, storage, release, and signalling pathways of adenosine through adenosine receptors.
4. Adenosine's mode of action on its receptor subtypes
The affinity of each adenosine receptor for adenosine determines its activation, triggering G-proteins that regulate enzymes like adenylate cyclase (AC) to produce cAMP and modulate ion channels, affecting ion flow across the membrane. This regulation is crucial for intracellular signal transduction. Activating the A1 receptor by adenosine opens K+ channels, causing cell hyperpolarisation and inhibiting Ca2+ channels (Fig. 2), reducing calcium entry. This leads to vasodilation, lowering blood pressure, and increasing blood flow.58Adenosine receptor activation affects cellular signalling by modifying critical pathways like cAMP production, activating PKA, and MAPK signalling, which influence gene expression, cell proliferation, and survival.59–62 The activation of the A1 AR inhibits AC and reduces cAMP levels. This diminishes PKA activity and CREB-1 phosphorylation, both crucial for gene regulation.63,64 A1 AR also activates MAPKs, including JNK, ERK1/2, and p38, which influence tumor growth and gene expression.65–69
Activation of A2A AR triggers Gs protein signalling, which increases cAMP production via AC activation. PKA is activated by elevated cAMP, which also affects phosphodiesterases (PDES), CREB, and signalling pathways that control cell survival and proliferation. PKA also phosphorylates DARPP-32, modulating various cellular processes.70,71
A2B AR directly affects essential proteins like p38 MAPK, ERK1/2, and JNK through its involvement in the MAPK signalling cascade. A2B receptor activation plays a significant role in cancer cell growth and tumour progression by modulating stress-activated protein kinases (SAPK) and other MAPK-related processes.72,73
Through Gi protein signalling, A3 AR activation decreases cAMP levels and reduces PKA activity. As a result, increased activation of GSK-3β leads to decreased β-catenin, cyclin D1, and c-MYC expression, which inhibit cell growth and proliferation. Additionally, A3 AR suppresses NF-κB, which is involved in immune responses and inflammation.74
5. Role of adenosine and adenosine receptors on cancer
In cancer, the TME often exhibits higher levels of adenosine production, primarily due to reduced oxygen availability (hypoxia). Hypoxia promotes the breakdown of ATP, resulting in increased amounts of adenosine within the TME.75 The secretion of adenosine during hypoxic conditions stimulates angiogenesis, which in turn promotes tumor growth. In solid tumors, hypoxia enhances the production of CD39 and CD73, enzymes that generate adenosine, supporting tumor survival.76,77 Hypoxia activates transcription factors like HIF-1α, which regulate these enzymes, maintaining adenosine production under low-oxygen conditions.78 Elevated adenosine levels promote tumor growth, immune suppression, and angiogenesis by binding to A1, A2A, A2B, and A3 receptors.79Adenosine accumulation in tumor hypoxic regions impairs immune cells' ability to target and destroy cancer cells. While lymphokine-activated killer (LAK) cell therapy shows promise for cancers resistant to standard treatments, its effectiveness is limited in colon cancer due to the tumor's immune-suppressive environment.80,81 Colon adenocarcinoma cells release a substance, distinct from TGF-β or prostaglandins, that inhibits the activation of anti-CD3-activated killer cells, helping the tumor evade immune attacks.82 In hypoxic tumor regions, adenosine accumulates and interacts with specific receptors on cytotoxic T cells, reducing their adhesion to cancer cells and impairing their activation.82–84 This effect, mediated by the A3 AR, inhibits cytotoxic T-cell activation and function in mice,85 contributing to immunosuppression and reduced efficacy of immunotherapy. Additionally, adenosine interferes with integrin α4β7, further impairing T cell adhesion to tumor cells.86
While adenosine's impact on other angiogenic factors is debated, it is known to boost the synthesis of certain phospholipases and support endothelial cell migration, further facilitating tumor growth and spread.87,88 Adenosine's capacity to stimulate the creation of PLCs and increase endothelial cell motility contributes to its involvement in promoting angiogenesis.89,90
6. A1 adenosine receptors' role in cancer: molecular signaling pathways
The A1 adenosine receptor (A1 AR) plays several roles in cancer by influencing the TME. A1 AR signalling also affects cellular migration and invasion, potentially impacting metastasis and therapeutic resistance.91 Activation of the A1 AR stimulates phospholipase C-beta (PLC-β), which hydrolyzes PIP2 to produce IP3 and DAG. IP3 triggers calcium release from intracellular stores, activating calcium-dependent PKC and other calcium-binding proteins, impacting cellular functions.10,64,92 A1 AR activation also opens potassium channels, causing hyperpolarization and reducing cellular excitability, while inhibiting N-type and P/Q-type calcium channels (Fig. 3), further decreasing calcium influx and excitability in neurons and heart muscle.93,94 | ||
| Fig. 3 A1 adenosine receptors' role in cancer: molecular signaling pathways. GTP – “Guanosine triphosphate”, PKC – “Protein kinase C”, PKA – “Protein kinase A”, DAG – “Diacylglycerol”, GDP – “Guanosine diphosphate”, ERK1/2 – “Extracellular signal-regulated kinases”, ATP – “Adenosine triphosphate”, PIP2 – “Phosphatidylinositol 4,5-bisphosphate”, and IP3 – “Inositol trisphosphate”, cAMP – “Cyclic adenosine monophosphate” NK – “c-Jun N-terminal kinases”. The figure was created in BioRender. Deb, P. (2025). | ||
Overexpression of AC-3 increases molecules like matrix metalloproteinase 2 (MMP2), matrix metalloproteinase 9 (MMP9), and cAMP, promoting tumour growth.68,69 Inhibition of AC-3 slows tumor progression, while AC-2 is a marker for poor colon cancer outcomes. A1 receptor agonists reduce glioblastoma formation and microglial proliferation, and the absence of A1 AR increases microglial density around tumors.95,96
In brain tumors, hypoxic conditions within the TME cause adenosine levels to rise.97 This elevated adenosine activates theA1 AR in microglia, the brain's immune cells.98 Once A1 AR is activated, microglia become less effective at targeting and destroying cancer cells. This reduction in immune activity allows tumours to grow and spread more easily.99
The A1 AR and other adenosine receptors significantly aid tumor growth by promoting angiogenesis and forming new blood vessels. Tumors, especially in low-oxygen conditions, release high levels of adenosine which stimulates the production of vascular endothelial growth factor (VEGF) by interacting with A1 AR and other receptors on surrounding cells.100 VEGF then prompts endothelial cells to grow and form new blood vessels, which extend into the tumor, enhancing its blood supply, resulting in increased supply of nutrients and oxygen facilitating the tumor's growth and spread to other parts of the body.100,101
Blocking or modifying A1 AR function can counteract this suppression, enhancing the immune response and improving tumor control. This approach can be used alongside existing therapies, such as chemotherapy, radiation, or immunotherapy, to overcome resistance and improve patient outcomes. By addressing specific mechanisms of immune suppression, targeting A1 AR offers a promising strategy to enhance cancer treatment and survival rates.102–104
However, adenosine can also promote microglial proliferation through the combined action of A1 and A2ARs. While A1 ARs are often linked with suppressing immune responses, their interaction with A2 ARs can have different effects. Specifically, when adenosine activates both A1 and A2 ARs, it can stimulate the growth of microglia, which may alter the TME in ways that facilitate tumor growth and progression.103 A separate study found that blocking A1 AR prevented cell death caused by adenosine, while activating A1 AR led to the death of human colorectal carcinoma (CW2) cells. These findings imply that A1 AR contributes to the tumor-inhibitory properties of adenosine, suggesting that activation of this receptor may cause cancer cells to undergo apoptosis.105 The role of A1 AR in cancer was confirmed through studies where A1 AR levels were reduced in breast glands106 and kidney cancers.107 This was achieved using RNA interference and the A1 AR blocker DPCPX. Reducing A1 AR not only triggered cell death (apoptosis) in breast cancer cells but also slowed down tumour growth. Additionally, it caused cancer cells to stop progressing through the cell cycle at the G2/M phase and reduce the number of cells in the S phase, which is crucial for cell division.106,108
7. A2A adenosine receptors' role in cancer: molecular signaling pathways
A2A AR receptors play a key role in several cancer processes, including rapid cell proliferation, angiogenesis, immune escape, and metastasis.109,110 Thus, the increased presence of A2A ARs enhances the cancer's ability to grow and metastasize uncontrollably (Fig. 4).110 | ||
| Fig. 4 A2A adenosine receptors' role in cancer: molecular signaling pathways. GTP – “Guanosine triphosphate”, CREB – “cAMP-response element binding protein”, JNK – “c-Jun N-terminal kinases”, ERK1/2 – “Extracellular signal-regulated kinases”, ATP – “Adenosine triphosphate” AKT – “Protein kinase B”, GDP – “Guanosine diphosphate”, and cAMP – “Cyclic adenosine monophosphate”. The figure was created in BioRender. Deb, P. (2025). | ||
Alternatively, the overexpression of A2A ARs can also affect how immune cells, such as T cells, recognize and target cancer cells. Elevated levels of A2A ARs can impair T cells' ability to effectively identify and attack tumour cells, as adenosine signalling through these receptors inhibits T cell activation and functionality,111–113 leading to suppression of immune cells, which in turn causes increased hypoxia within the tumour cells.114 Under hypoxic conditions, adenosine levels increase in the tumor microenvironment (TME), leading to immune suppression. When adenosine accumulates in the TME, it binds to the A2A AR, and inhibits the immune system. This binding particularly impairs the function of tumor-reactive immune cells, such as T cells and natural killer (NK) cells115,116
8. A2B adenosine receptors' role in cancer: molecular signalling pathways
Chinese hamster ovary (CHO) cells that produce recombinant human A2B AR have been the primary and original demonstration source for classical A2B AR signalling.117–119 Like the A2A receptor, the A2B AR is coupled with the Gs protein, which is crucial for intracellular signalling.120 Adenosine activates PKA (Fig. 5) by binding to A2B AR, which phosphorylates target proteins and also recruits effectors like Epac (exchange protein directly activated by cAMP).120–122 This Epac impacts the proliferation of umbilical vein endothelial cells and triggers the expression of early genes, ultimately reducing smooth muscle cell growth in human coronary arteries.123–125 Additionally, A2B AR is coupled with Gq proteins, which activate PLC that converts a molecule called PI2 into two important products: DAG and IP3. IP3 releases calcium ions within the cell, while DAG activates PKC.126,127 According to recent studies, activating A2B AR reduces the activity of signalling molecules that are usually activated by the RANKL protein, such as p38, NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), ERK1/2 (extracellular signal-regulated kinases 1 and 2), and p38. These signalling pathways collectively support tumour development and growth.128 Many studies have highlighted the importance of A2B AR signalling in various disorders, including atherosclerosis,129,130 neurological inflammation,131,132 inflammatory bowel disease,133 ischemic heart preconditioning134 and the prevention of cardiovascular fibrosis. Overexpression of A2B receptors is also linked to renal disorders.135 | ||
| Fig. 5 A2B adenosine receptors' role in cancer: molecular signalling pathways. ATP – “Adenosine triphosphate”, CREB – “cAMP-response element binding protein”, PKC – “Protein kinase C”, IP3 – “Inositol trisphosphate”, GDP – “Guanosine diphosphate”, PLC-β – “phospholipase C-β”, IL-10 – “Interleukin 10”, AKT – “Protein kinase B”, ERK1/2 – “Extracellular signal-regulated kinases”, cAMP – “Cyclic adenosine monophosphate”, FoxP3 – “Forkhead box P3”, DAG – “Diacylglycerol”, PKA-“Protein kinase A”, GTP – “Guanosine trihosphate”, JNK – “c-Jun N-terminal kinases”, PIP2 – “Phosphatidylinositol 4,5-bisphosphate” and TGF-β “Transforming growth factor β”. The figure was created in BioRender. Deb, P. (2025). | ||
Wei et al. discovered that the A2B AR is the most highly expressed adenosine receptor in human prostate cancer cell lines. According to their findings, TNFα and chemotherapy-induced cell death are lessened when the A2B receptor is activated.136 J. Linden and colleagues showed the selective antagonist ATL801 to block the A2B receptor, which prevents the development of 4T1 mammary carcinoma and MB49 urinary bladder tumours in vivo.137
9. A3 adenosine receptors' role in cancer: molecular signalling pathways
The A3 AR is linked to Gq proteins that activate phospholipase C-β (PLC-β), producing IP3, which further increases intracellular calcium and activates PKC (Fig. 6).10,138,139 | ||
| Fig. 6 A3 adenosine receptors' role in cancer: molecular signaling pathways. ERK1/2 – “Extracellular signal-regulated kinases”, ATP – “Adenosine triphosphate”, PKA – “protein kinase A”, PIP2 – “Phosphatidylinositol 4,5-bisphosphate”, FoxP3 – “Forkhead box P3”, GSK-3β – “Glycogen synthase kinase-3β”, IP3 – “Inositol trisphosphate”, cAMP – “Cyclic adenosine monophosphate”, JNK – “c-Jun N-terminal kinases”, DAG “Diacylglycerol”, PKC – “Protein kinase C”, PKB – “Protein kinase B”, RhoA – “Ras homolog family member A” and NFκB – “Nuclear factor kappa-light-chain-enhancer of activated B cells”. The figure was created in BioRender. Deb, P. (2025). | ||
In normal tissue, A3 AR expression is minimal, but in tumour cells, it is markedly elevated;140 as a result, it can be regarded as a possible tumour marker. A3 AR levels are elevated in colorectal cancer and other malignant tumours, including pancreatic carcinoma, small-cell lung cancer, breast cancer, and melanoma, and are associated with disease progression.141–144 According to reports, human melanoma cells that express A3 AR signalling exhibit pro-survival effects.145 Glycogen synthase kinase (GSK)-3β, an enzyme essential for β-catenin phosphorylation, regulates this pathway, increasing tumour cell development.146 The expression of genes crucial for advancing the cell cycle, such as cyclin D1 and c-myc, is encouraged by phosphorylated β-catenin.147
Chromosome 1 at location p13.3 in humans contains the gene that codes for the A3 AR.148 A3 AR's overexpression in various tumour cells and tissues suggests that it may be useful as a therapeutic target and a cancer marker.38,143
10. Potential use of adenosine receptor ligands in cancer treatment
Selective modulators of adenosine receptors provide promising strategies for cancer treatment by directly inhibiting tumour growth or modifying the tumour microenvironment to enhance anti-tumour immune responses. These modulators target specific adenosine receptors: A1, A2A, A2B, and A3, each playing distinct roles in cancer progression (Table 2). Their effects and mechanisms in cancer treatment are actively being explored, and future research will likely continue to refine these pathways for more targeted and effective therapies. Below is an elaboration on the selective modulators for each receptor type, their effects, and mechanisms in cancer treatment.| S. No. | Name of the compounds | Agonist/antagonist | Target receptor subtype | Transduction mechanism | Description role |
|---|---|---|---|---|---|
| 1 | CHA | Agonist | A1 receptor | AC ↓ PLC ↑ | Binds to A1AR and activates it; this activation leads to decreased AC and decreased cAMP in the cell |
| 2 | CPA | Agonist | A1 receptor | AC ↓ PLC ↑ | Activated Gi proteins inhibit AC activity, decreasing the production of cAMP from ATP. Lower cAMP levels result in reduced activation of PKA and downstream signaling pathways |
| 3 | CCPA | Agonist | A1 receptor | AC ↓ PLC ↑ | cAMP levels can lead to decreased activation of protein kinase A (PKA), which promotes cell growth and proliferation |
| 4 | S-ENBA | Agonist | A1 receptor | AC ↓ PLC ↑ | cAMP levels due to A1AR activation can lead to decreased activation of protein kinase A (PKA) |
| 5 | 5′-Cl-ENBA | Agonist | A1 receptor | AC ↓ PLC ↑ | Can reduce PKA activity, potentially inhibiting the proliferation of cancer cells |
| 6 | Sele-denoson or DTI 0009 | Agonist | A1 receptor | AC ↓ PLC ↑ | Lowering AC level |
| 7 | GR79236X | Agonist | A1 receptor | AC ↓ PLC ↑ | Decrease the cAMP level |
| 8 | Tiazofurin | Agonist | A1 receptor | AC ↓ PLC ↑ | Inhibit inosine monophosphate dehydrogenase (IMP dehydrogenase) activity so, it can decrease the levels of downstream purine nucleotides, which can impact cell proliferation, especially in rapidly dividing cells such as cancer cells |
| 9 | Cyclosaligenyl-tiazofurin monophosphate | Agonist | A1 receptor | AC ↓ PLC ↑ | Reduction in cAMP levels may affect downstream effectors, such as protein kinase A (PKA), which can influence cell growth |
| 10 | 1,3-Dipropyl-8-cyclopentylxanthine | Antagonist | A1 receptor | AC ↑ | The inhibitory effect that adenosine has on adenylate cyclase, increased adenylate cyclase activity and higher levels of cAMP within the cell |
| 11 | HENECA | Agonist | A2A receptor | AC ↑ | Activating A2A receptors may influence the immune response within the tumor microenvironment |
| 12 | CGS15943 | Antagonist | A2A receptor | AC ↓ | Binding to the A1 receptor usually results in reduced cyclic AMP (cAMP) levels due to inhibition of adenylate cyclase |
| 13 | ZM241385 | Antagonist | A2A receptor | AC ↓ | Antagonising A2A adenosine receptors, leading to decreased cAMP production |
| 14 | SCH58261 | Antagonist | A2A receptor | AC ↓ | Promote T cell function and enhance anti-tumour immune responses |
| 15 | SCH-442416 | Antagonist | A2A receptor | AC ↓ | Blocking the A2A receptor |
| 16 | SYN115 (tozadenant) | Antagonist | A2A receptor | AC ↓ | Inhibition of the A2A receptor can disrupt pathways that allow cancer cells to evade immune surveillance, potentially leading to reduced tumor growth |
| 17 | TP455 | Antagonist | A2A receptor | AC ↓ | Inhibition of A2A receptors can lead to increased activation and proliferation of T cells |
| 18 | CPI-444 | Antagonist | A2A receptor | AC ↓ | Inhibits the A2A adenosine receptor |
| 19 | PBF-509 | Antagonist | A2A receptor | AC ↓ | The antagonism of A2A receptors may also help inhibit pathways that contribute to tumor cell survival and proliferation |
| 20 | AZD4635 | Antagonist | A2A receptor | AC ↓ | Blocking the A2A receptor |
| 21 | TT-10 | Antagonist | A2A receptor | AC ↓ | Inhibits the A2A adenosine receptor |
| 22 | Preladenant | Antagonist | A2A receptor | AC ↓ | Inhibits the A2A receptor |
| 23 | Etrumadenant (AB928) | Antagonist | A2A receptor | AC ↓ | Blocking A2A receptors helps to counteract the immunosuppressive effects of adenosine |
| 24 | Inupadenant | Antagonist | A2A receptor | AC ↓ | Inhibits the A2A AR |
| 25 | ANR 94 | Antagonist | A2A receptor | AC ↓ | Selectively inhibits the A2A receptor |
| 26 | CPI-444 analog | Antagonist | A2A receptor | AC ↓ | Inhibition of A2A receptors |
| 27 | Istradefylline | Antagonist | A2A receptor | AC ↓ | Inhibition of A2A receptors |
| 28 | PSB1115 | Antagonist | A2B receptor | AC ↑ | Inhibit A2B receptor |
| 29 | PSB603 | Antagonist | A2B receptor | AC ↑ | Blocked A2B receptor |
| 30 | ATL801 (30) | Antagonist | A2B receptor | AC ↑ | A2B receptor blocked |
| 31 | AB928 | Antagonist | A2B receptor | AC ↑ | A2B receptor inhibitor |
| 32 | Piclidenoson (IB-MECA) | Agonist | A3 receptor | AC ↓ PLC ↑ | Boost the activity of regulatory T cells (Tregs), thereby increasing immunosuppression within the TME |
| 33 | Namodenoson (Cl-IB-MECA) | Agonist | A3 receptor | AC ↓ PLC ↑ | Promoting anti-tumor immunity by reducing inflammation while also potentially inducing apoptosis in tumor cells |
| 34 | Thio-Cl-IB-MECA | Agonist | A3 receptor | AC ↓ PLC ↑ | Thio-Cl-IB-MECA binds to the A3 adenosine receptor. Upon activation, the A3 receptor inhibits adenylate cyclase, leading to decreased levels of cAMP in the cell |
| 35 | Cordycepin | Agonist | A3 receptor | AC ↓ PLC ↑ | A3 receptor activation |
| 36 | N6-(2-Isopentenyl) adenosine | Agonist | A3 receptor | AC ↓ PLC ↑ | Activation of A3 receptors lead to the inhibition of cell proliferation |
| 37 | Resveratrol-3-O-D-glucuronide | Agonist | A3 receptor | AC ↓ PLC ↑ | A3 receptor activation |
| 38 | Resveratrol 4′-O-D-glucuronide | Agonist | A3 receptor | AC ↓ PLC ↑ | A3 receptor activation |
| 39 | Linagliptin | Agonist | A3 receptor | AC ↓ PLC ↑ | A3 receptor activation |
| 40 | Oxidative degradation product of linagliptin | Agonist | A3 receptor | AC ↓ PLC ↑ | A3 receptor activation |
| 41 | MRS 1523 | Antagonist | A3 receptor | AC ↑ | Inhibit A3 receptor |
| 42 | Truncated thio-Cl-IB-MECA | Antagonist | A3 receptor | AC ↑ | Inhibit A3 receptor |
| 43 | N6-(2,2-Diphenylethyl)-2-phenylethynylAdo | Antagonist | A3 receptor | AC ↑ | Inhibit A3 receptor |
| 44 | [1,2,4]-Triazolo[1,5-c]pyrimidines | Antagonist | A3 receptor | AC ↑ | Inhibit A3 receptor |
| 45 | MRS 1097 | Antagonist | A3 receptor | AC ↑ | Inhibit A3 receptor |
| 46 | MRS 1067 | Antagonist | A3 receptor | AC ↑ | Inhibit A3 receptor |
| 47 | MRS 1220 | Antagonist | A3 receptor | AC ↑ | Inhibit A3 receptor |
| 48 | MRS 1191 | Antagonist | A3 receptor | AC ↑ | Inhibit A3 receptor |
Many studies have been conducted on the structure–activity relationship (SAR) of adenosine derivatives as agonists of the AR. Adenosine and xanthosine, two purine nucleoside derivatives, constitute the basis for the majority of known AR agonists. Many agonists that target distinct AR subtypes have been developed due to modifications to the physiological agonist adenosine. Adenosine's affinity for adenosine receptors is increased by adding nitrogen atoms at positions 3 and 7.149 Mono-substitution of the N6 position (exocyclic amino group) of adenosine with a large, non-polar (hydrophobic) group enhances the molecule's ability to preferentially bind to the A1 and A3 AR subtypes.33,149 The existence of a hydrogen atom at position N6 is critical for agonist activation because it creates hydrogen bonds with the receptor.150 For tiny halogen atoms or bigger substituents, modifications to adenine at the C2 position are generally well tolerated.151 Strong and focused agonistic action at ARs is produced by substitutions at the 5′-position, which are often well tolerated and require the presence of a ribose sugar moiety. In the adenosine structure, the 2′- and 3′-hydroxyl (OH) groups of the ribose moiety are essential for agonist activity at adenosine receptors (Fig. 7).
 | ||
| Fig. 7 Impact of structural modifications of adenosine and xanthine derivatives on adenosine receptor binding. | ||
Structure–activity relationship (SAR) studies at adenosine receptors show that xanthines with strong inhibitory activity were consistently alkylated at the 1-position, where substituting hydrogen with a methyl group enhanced potency by approximately 20-fold. Modifications at the 3-position were not essential for high activity. Substitution at the 7- and 8-position might slightly increase or decrease the potency, while substitutions at the 7-position are typically considered unfavourable. However, certain alkyl modifications at these positions can selectively boost affinity for the A2 receptor subtype.152
10.1 The A1 receptor agonists
The selectivity towards A1 AR was found to be increased mainly upon the introduction of a larger cycloalkyl group at the N6-position of the agonists like CHA (N6-cyclohexyladenosine) (1) and CPA (N6-cyclopentyladenosine) (2), being 400- and 800-fold selective.153 Combined substitutions at the N6- and 2-positions have yielded 2-chloro-CPA (CCPA) (3), which is 1500-fold A1-selective, and helpful in wide pharmaceutical applications.33,149–151,154,155Mice deficient in A1 AR and their wild-type littermates responded differently to injections of Gl261 glioblastoma tumour cells and treatment with adenosine and N6-cyclopentyladenosine (CPA), where CPA significantly reduced the growth of the tumour. Remarkably, research revealed that in brain slices taken from A1 AR-deficient mice, neither adenosine nor CPA (N6-cyclopentyladenosine) had any impact on tumour formation. These results suggest that CPA and adenosine influence tumour size reduction by selectively interacting with A1 ARs on microglial cells.96
Like numerous other N6-substituted adenosine analogues, both CPA and CCPA derivatives show notable affinity for the A3 AR as well. In contrast to A1 AR, CCPA has been identified as an antagonist at the human A3 AR, with a Ki value of 35 nM.156 N6-Bicycloalkyladenosine is even more A1-selective, with S-ENBA (4), showing 4700-fold selectivity for the A1 receptor.157–159 Among the most selective A1 AR agonists, 5′-Cl-ENBA (5) demonstrates exceptionally high affinity and selectivity for the A1 AR compared to other AR subtypes.160,161 Through the activation of A1 AR, N6-cyclopentyl-NECA, often referred to as sele-denoson or DTI 0009 (6), exhibited the strongest negative dromotropic impact (A1 AR).162 Seledenoson has been tested in phase 2 clinical trials to determine its efficacy in treating atrial fibrillation via oral and intravenous (IV) routes.163 In addition the other A1-selective adenosine derivatives, including GR79236, clinical investigations have examined the possible use of N6-[(1S, trans)-2-hydroxy-cyclopentyl] adenosine (GR79236X) (7), a hydroxylated derivative of CPA, in the treatment of myocardial ischemia, diabetes, and pain.33
Tiazofurin (8) is a C-nucleoside that inhibits inosine monophosphate dehydrogenase (IMPDH) and has demonstrated anticancer efficacy in clinical settings. Tiazofurin adenine dinucleotide, produced when metabolized, inhibits the IMPDH enzyme, stopping nucleotide synthesis and inhibiting the growth of cancerous cells.108 A new tiazofurin pronucleotide called cyclosaligenyl-tiazofurin monophosphate (9) is a selective agonist of the A1 AR. Its binding affinity is comparable to that of tiazofurin, suggesting a similar mechanism of action at this receptor subtype. Additionally, it exhibits significant efficacy against the K-562 cell line, which is a model for human chronic myelogenous leukaemia (in vitro). Structural representations of interesting A1 AR agonists with anti-cancer activity are shown in Fig. 8.
 | ||
| Fig. 8 Potential A1 adenosine receptor agonists exhibiting promising anticancer properties. | ||
10.2 The A1 receptor antagonists
The A1 AR antagonist has diverse effects on different forms of cancer and may help in preventing the development of specific cancer types. Recent investigations have employed quantitative real-time PCR and western blotting analysis to ascertain the function of A1 AR in kidney cancer in 786-O & ACHN cell lines. In addition, investigations on anticancer research have shown that 1,3-dipropyl-8-cyclopentylxanthine (10), an A1 AR antagonist (Fig. 9), efficiently decreases the formation of tumours in vivo and the proliferation of RCC cells in vitro. Compound 10 also inhibited the migration of RCC cells; however, the selective A1 agonist N6-cyclopentyladenosine (CPA) increased the migration of RCC cells. Furthermore, the xanthine derivative 10 produced an arrest in the S-phase of the cell cycle and increased apoptosis in 786-O and ACHN cells.108 | ||
| Fig. 9 Potential A1 adenosine receptor antagonist exhibiting promising anticancer properties. | ||
10.3 The A2A receptor selective agonists
With A-375 cells, the anticancer effects were studied using a particular A2A agonist, (2S,3R,5R)-HENECA (2-hexynyl-NECA) (11) as shown in Fig. 10, which demonstrated consistent, modest effects in inhibiting cell proliferation and lowering cytotoxicity in A-375 cell lines.106,164 Compound 11 causes concentration-dependent cell death, with peak effects being shown at 100 nM. Higher concentrations, however, resulted in a decrease in its effectiveness. | ||
| Fig. 10 Potential A2A adenosine receptor agonist exhibiting promising anticancer properties. | ||
10.4 The A2A receptor-selective antagonists
Hypoxic solid tumours are known to include an increased amount of adenosine, which hinders the capacity of cytolytic T lymphocytes that are essential to identify cancer cells as targets.112,165 It has been discovered that the activation of A2A AR stimulates angiogenesis and boosts the growth of melanoma and mammary cells. These findings emphasise the significance of developing A2A AR antagonists for the treatment of cancer.166,167 Examples of A2A AR antagonists with anticancer effects are shown in Fig. 11. | ||
| Fig. 11 Potential A2A adenosine receptor antagonists exhibiting promising anticancer properties. | ||
The effects of adding styryl groups to xanthines at the 8-position have been brought to light by recent developments in search of A2A AR antagonists. This has increased the A2A AR selectivity.168–170
However, replacing the xanthine core with various heterocyclic ring systems has resulted in exceptionally high affinity and selectivity for the A2A AR. An early example of a heterocyclic structure proposed as an A2A AR antagonist was triazoloquinazoline (CGS15943) (12), which was later found to exhibit only modest selectivity. Subsequent modifications, such as adding a third ring or altering the nitrogen arrangement within the heterocyclic system, significantly enhanced A2A AR selectivity.5,171 Triazoloquinazoline CGS15943 (12), an early example of a heterocyclic antagonist, was discovered to have only moderate selectivity, contrary to initial predictions.
Selectivity for the A2A AR was markedly increased by further modifications to the triazoloquinazoline, with adjustments to the nitrogen arrangement within the heterocyclic structure leading to the development of a compound with improved potency, triazolotriazine ZM241385 (13).172
ZM241385 “(4-(2-((7-amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-yl)amino)ethyl)phenol)” is an A2A AR antagonist that enhances the ability of antitumor T lymphocytes to suppress tumour growth, eradicate cancer, and prevent the production of new blood vessels. Compound 13 stimulated the development of anti-CL8-1 CD8+ T cells and markedly slowed the growth of CL8-1 tumours in wild-type mice. When this antagonist (compound 13) is used in conjunction with anti-CD8+ T cells in adoptive immunotherapy models, tumour growth is successfully prevented; however, the antagonist by itself does not have the same impact.170 In melanoma-bearing mice, the efficacy of compound 13, was investigated in combination with an anti-CTLA4 monoclonal antibody.173 Tumor growth was significantly inhibited in mice receiving just compound 13 treatment. In contrast to either the control group or treatment with compound 13 alone, the combination therapy led to a more noticeable delay in the progression of the tumor. There is a theory that this enhanced effect is linked to a decrease in regulatory T cells (TREGs) and an increase in CD8-positive (CD8+) T lymphocyte infiltration within the tumor tissue. In cutaneous melanoma tissue, treatment with the compound 13 alone resulted in increased infiltration of CD8+ T cells and a decrease in regulatory T cells (TREGs).168
Interestingly, the pyrazolotriazolopyrimidine group plays a significant role in improving A2A AR selectivity. In SCH58261 (14), the xanthine core is also modified, but it includes additional aromatic and heteroatom-containing groups. SCH58261 (14) “2-(furan-2-yl)-7-phenethyl-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine” is an A2A receptor blocker that has been studied for its potential to prevent metastasis using the 4T1.2 cancer model, a highly aggressive metastatic mammary gland tumor cell line.174 Research has demonstrated that by blocking the A2A AR, the triazolo-pyrimidine derivative 14 dramatically reduces cancer cell proliferation in B16F10 CD73+ malignancies. Additionally, compared to the control group, therapy with the A2A antagonist 14 resulted in a significant reduction in the metastasis of 4T1.2 tumors. Moreover, A2A−/– animals showed significant defence against B16-F10 CD73+ tumor cell metastasis.175 In non-A2A−/– animals, the A2A antagonist 14 likewise showed decreased efficacy, demonstrating the drug's specificity for the A2A receptor.175,176 In animal models of skin and breast tumor, compound 14 was also demonstrated to improve survival and lessen the stress caused by metastatic cancer when given in combination with an anti-PD-1 monoclonal antibody.176
Incorporating pyrazole and triazole rings into the structure of SCH442416 (15) enhances its binding affinity by altering the electronic properties of the xanthine core. These modifications improve interactions with the A2A receptor.29 Specifically, the addition of an N′-substituted pyrazolotriazolopyrimidine group refines the molecule's conformation, increasing A2A receptor selectivity while reducing affinity for other adenosine receptor subtypes. As a result, SCH442416 exhibits high affinity for the A2A receptor (hA2A Ki = 1.1 nM) and significantly lower affinity for the A1 receptor (hA1 Ki = 549 nM), establishing it as a widely used reference antagonist for A2A receptor studies.177
In a follow-up investigation, researchers evaluated the benzothiazole derivative SYN115 (tozadenant) (16) for its potential to act as an antagonist of the A2A AR. The study focused on how this property could enhance the anticancer effects of an anti-PD-1 monoclonal antibody, which is used in cancer immunotherapy. By blocking the A2A receptor, SYN115 may help improve immunological reaction to malignancies, thereby possibly increasing the effectiveness of the anti-PD-1 treatment in combating cancer. It was demonstrated that the A2A receptor blocker 16 significantly increased the anticancer efficacy of the anti-PD-1 antibody, with results comparable to compound 15.70
The role of A2A AR in a number of human cancers, such as rat MRMT-1 breast cancer, A-375 melanoma, and A-549 lung cancer, has been extensively studied. Special attention has been paid to the signaling pathways involved and the outcomes of a novel A2A receptor antagonist, TP455 (17) (2-(furan-2-yl)-N5-(2-methoxybenzyl)thiazolo[5,4-d]pyrimidine-5,7-diamine), as well as the actions of this antagonist.178
The immune-activating and anti-tumor properties of (S)-7-(5-methylfuran-2-yl)-3-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-5-amine, CPI-444 (18), either alone or in combination with anti-PD-1/PD-L1 monoclonal antibodies, have been evaluated in vitro using activated primary human T cells. The results showed that pyrimidine derivative 18 completely inhibited the production of intracellular cAMP when the cells were incubated with 5′-N-ethylcarboxamidoadenosine (NECA), a stable adenosine analog. Activated T lymphocytes produce less IL-2 and IFN-γ when the A2A AR agonist prevents fast TCR-mediated ERK phosphorylation. On the other hand, T cell signalling and function were restored when antagonist 17 blocked the A2A AR. Efficacy of the compound 18 has been examined using CT26 and MC38 syngeneic animal tumor models. The absence of fresh tumour development in cured animals when re-challenged with MC38 cells indicates that antagonist 18 produced systemic anti-cancer immunological memory. Molecule 17, in conjunction with an anti-PD-L1 monoclonal antibody in the MC38 model, led to a synergistic decrease in tumour development and total tumour eradication in 9 out of 10 treated mice. In the CT26 model, antagonist 17 and anti-PD-1 monoclonal antibody also showed synergistic effects; the combination dramatically reduced tumour development and increased survival over antagonist 17 treatment alone.179,180
The A2A AR-mediated immunological checkpoint is activated when adenosine levels rise in the tumour microenvironment, suppressing anti-tumour responses. Enhancing anti-tumour T cell function may be possible by focusing on this immunological checkpoint. PBF-509 (5-bromo-2,6-di(1H-pyrazol-1-yl)pyrimidin-4-amine) (19) is a novel A2A AR antagonist that has been developed as a possible treatment for non-small cell pulmonary carcinoma in this regard.181 According to research, PBF-509 has strong selectivity for the A2A AR. PBF-509 therapy decreased lung metastases in a mouse model compared to the control group. Additionally, among recently extracted tumor-infiltrating lymphocytes from lung cancer patients, investigations showed varied expression of the A2A receptor in CD8+ cells and enhanced expression in CD4+ cells. Additionally, when PBF-509 was administered in conjunction with anti-PD-1 or anti-PD-L1 therapy, human tumor-infiltrating lymphocytes showed enhanced reactivity, according to in vitro investigations.182 These results imply that blocking the A2A AR may open up new avenues for the development of creative immunotherapeutic approaches to treat non-small-cell lung cancer.183
The use of A2A AR antagonists in the treatment of cancer, either as monotherapy or in addition to different immunotherapies, is now being investigated in a number of clinical trials, which is relevant.91,184 A number of A2A AR antagonists are currently being evaluated in clinical settings including AZD4635 (20). In TT-10 (21), [4-amino-2-(prop-2-enylamino)-1,3-thiazol-5-yl]-(5-fluorothiophen-2-yl)methanone is an immunomodulatory drug that is being developed by Portage Biotech for oral administration. TT-10 exhibited a higher level of tumor growth suppression in preclinical investigations employing the 4T1 syngeneic mouse model of breast cancer than both the vehicle control and anti-PD-1 treatment groups. TT-10 therapy also resulted in a notable decrease in myeloid-derived suppressor cell (MDSC) numbers.185,186 The adenosine pathway is a target for cancer immunotherapy, and 2020 saw the publication of the first clinical data supporting this theory. In this trial, 68 patients with renal cell carcinoma were treated with either ciforadenant alone or with atezolizumab (PD-L1 inhibitor). Many of these patients had tumors that were primarily PD-L1-negative and were resistant to or refractory to anti-PD-1/PD-L1 antibodies. The research emphasized that in patients with resistant renal cell carcinoma, anti-PD-L1 combination therapy, and monotherapy both had antitumor efficaciousness. Compared to combination therapy, which had a median progression-free survival of 5.8 months, monotherapy had a median survival of 4.1 months. Furthermore, overall survival rates for monotherapy at 16 months and combination therapy at 25 months were higher than 69% and 90%, respectively. A2A AR antagonists' efficacy in immunotherapy for various cancer types has also been shown in additional trials. The research emphasized that in patients with resistant renal cell carcinoma, anti-PD-L1 combination therapy and monotherapy both had antitumor efficaciousness. In comparison to combination therapy, which had a median progression-free survival of 5.8 months, monotherapy had a median survival of 4.1 months. A2A AR antagonists' efficacy in immunotherapy for a range of cancer types has also been shown in additional trials.187–189
A recent Phase Ib/II trial (NCT03099161) evaluated the safety of Preladenant (22), both standalone and in combination with the anti-PD-1 drug Pembrolizumab, in patients with advanced cancers.190 However, the results have not yet been released.
In a Phase 1 open-label, multicenter trial, patients with advanced solid tumors will receive continuous oral administration of AZD4635 (20) for evaluation.103,104 Determining the maximal safe dosage of AZD4635 in conjunction with the anti-PD-L1 medication Durvalumab (NCT02740985) is the main goal of the trial.191 In patients with non-small cell lung cancer (NSCLC), this A2A antagonist will also be evaluated for safety, tolerability, and anticancer effectiveness in combination with the anti-CD73 medication MEDI9497 and the EGFR inhibitor osimertinib (NCT03381274).183
There are several other high-affinity and selective adenosine A2A receptor inhibitors that have been discovered with potential clinical applications. These include Etrumadenant (AB928) (23), Inupadenant (24), ANR 94 (25), CPI-444 analogue (26). A2A AR selectivity has been achieved by altering xanthines at the 8-position with alkenes, especially styryl groups. One of the earliest recognised A2A AR antagonists was 8-styryl-xanthine, istradefylline (27) (KW6002).183,192,193 The US-FDA has approved istradefylline for the treatment of Parkinson's disease.194 This milestone, coupled with advancing insights into the role of adenosine (ADO) in cancer biology, is anticipated to accelerate the development of ADO receptor ligands as promising anticancer agents or as adjunct therapy to existing treatments.
10.5 The A2B receptor selective antagonists
The release of angiogenic factors from vascular smooth muscle, endothelial cells, and host immune cells is thought to be facilitated by the stimulation of the A2B AR, which in turn aids in the formation of tumors.145,195 Conversely, blocking the A2B AR increases the activation of dendritic cells (DC) and thus increases the synthesis of CXCL10 (C-X-C pattern chemokine 10) which is induced by IFN-γ. This chemokine contributes to the activation of lymphocytes and the induction of an angiostatic response in malignancies.196 Antitumor actions of several synthetic A2B adenosine receptor antagonists have shown promise, as illustrated in Fig. 12. | ||
| Fig. 12 Potential A2B adenosine receptor antagonists exhibiting promising anticancer properties. | ||
Specific modifications of the xanthine core at the 8-position with aryl groups have been shown to confer selectivity for the A2B AR. In PSB-1115 (28), modification of the xanthine core with an aryl group at the 8-position enhances receptor selectivity. Studies have demonstrated that melanoma growth can be effectively slowed by selectively inhibiting the A2B AR with the antagonist “PSB1115 (4-(2,6-dioxo-1-propyl-3,7-dihydropurin-8-yl)benzenesulfonic acid)” (28). To do this, antitumor immune responses are reactivated, and the growth of myeloid-derived suppressor cells (MDSCs) within tumors is suppressed.197
Furthermore, studies have shown that employing the antagonist 28 targets the A2B AR, which is known to play a role in suppressing the immune response within tumors. By inhibiting this receptor, the immune system can become more effective against the cancer, leading to a significant reduction in tumor growth. Compound 28, derived from xanthine, is effective in diminishing specific immune cell populations associated with tumors (Gr-1+CD11b+ cells) in melanoma. Additionally, it decreases the production of key regulatory molecules, such as interleukin-10 (IL-10) and monocyte chemoattractant protein 1 (MCP-1) that can suppress immune responses, potentially enhancing the body's ability to fight the tumor. The T cells that release cytokines like those produced by T helper 1 (Th1) cells are linked to these effects, and there are higher numbers of CD8+ T cells and natural killer T (NKT) cells within tumours. These outcomes imply that the ability of the A2B AR antagonist 28 to minimise the infiltration of MDSCS (myeloid-derived suppressor cells) in tumours and to strengthen the anti-cancer T cell response is related to its efficacy. Furthermore, the xanthine derivative 28 has been found to drastically reduce the metastasis of B16F10 CD73+ malignancies by blocking the A2B AR.198
“PSB603 (8-(4-(4-chlorophenyl)piperazide-1-sulfonyl)phenyl)-1-propylxanthine” (29), a recognized A2B receptor antagonist, exhibits particularly high affinity and selectivity for the A2B receptor, not only in humans but also in rodents. PSB-1115 also offers high water solubility, making it suitable for in vivo studies; however, its A2B receptor affinity and selectivity are lower than other A2B antagonists.199 PSB603 is being investigated in C57BL/6 mice bearing B16 melanoma to see how it affects tumour progression. The in vivo observations revealed a significant decrease in tumour volume, postponed tumour growth, and decreased metastasis, associated with a decrease in the regulatory T-cell population in mice. This antagonist increases CD4+ helper T cell and CD8+ cytotoxic T cell populations, which improves anti-tumor immunity in tumor-bearing animals. As a result, inhibiting the A2B receptor increases the quantity of helper and cytotoxic T cells, that are necessary for the development of cancer immunity.137,176
ATL801 (30), an A2B receptor antagonist, has been demonstrated to successfully block the growth of 4T1 mammary carcinoma and MB49 urinary bladder cancer in syngeneic animal models. It also stops breast cancer cells from metastasizing. When administered intravenously, compound 30 can elicit adaptive immunological responses in a way that is dependent on CXCR3, most likely via indirectly increasing the activity of dendritic cells (DCs). This process prevents the growth of tumors and helps offset the immunosuppressive effects of adenosine. These results imply that stimulating T cell activation and inhibiting formation of new blood vessel in solid tumors may be accomplished by selectively blocking the A2B adenosine receptor.200
Lastly, it is noteworthy that a number of antagonists are presently being studied in clinical studies for different types of cancer. Notably, AB928 (31), a dual A2A/A2B antagonist, is currently being investigated in patients with breast tumor, non-small-cell pulmonary carcinoma, and ovarian carcinoma after demonstrating encouraging findings in a Phase I clinical trial.137,201
10.6 The A3 receptor selective agonists
The possibility of A3 receptors as a therapeutic target for the treatment of cancer is highlighted by their overexpression in malignant and inflammatory cells.202 Various potential A3 receptor agonists are illustrated in Fig. 13. Interestingly, the effects of these agonists on tumor cell proliferation differ from those on normal cell growth. Most of the A3 AR agonists developed to date are based on the nucleoside structure of the endogenous ligand, adenosine. The most effective enhancements in A3 AR potency and selectivity have been achieved through substitutions at the N6-, C2-, and 5′-positions, or through strategic combinations of these modifications. | ||
| Fig. 13 Potential A3 adenosine receptor agonists exhibiting promising anticancer properties. | ||
Potent and selective A3 AR agonists were developed by combining N6-substitution with a 5′-uronamide group. The first A3 AR-selective compounds featured a 5′-N-alkyluronamide moiety paired with an N6-benzyl group. A key representative of this series, N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide (IB-MECA; 32), also known as piclidenoson was discovered in 1994.203,204
Extensive studies have explored the effects of various substitution patterns at the N6- and C2-positions of the adenine core in 5′-N-alkylcarbamoyladenosine derivatives. Introducing small functional groups, such as halogens, methylamino, or thiomethyl, at the C2-position of IB-MECA has enhanced both affinity and selectivity for the A3 adenosine receptor (A3AR). This led to the development of C2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine (Cl-IB-MECA), a highly selective A3 AR agonist now considered the prototypical ligand for this receptor subtype. Cl-IB-MECA exhibits very high affinity towards rat A3 AR (Ki = 0.33 nM), compared to the rat A1 and A2A receptors with Ki 820 nM and 470 nM, respectively.205 Both the A3 receptor agonists piclidenoson (IB-MECA; 32) and namodenoson (Cl-IB-MECA; 33), have a noteworthy effect even at low doses on the proliferation of tumor cells.139,206
When combined with 5-fluorouracil, compound 32 demonstrates more effective growth inhibition of HCT-116 human colon carcinoma cells compared to the use of 5-fluorouracil alone.207 Studies looking into the effects of this ligand on cells expressing estrogen receptor α (ERα) have discovered that derivative 33 quickly lowers the levels of ERα at both the mRNA and protein levels. Different kinds of breast cancer cells may experience apoptosis as a result of this action, which hinders cell proliferation.208,209
The synthetic A3 AR agonist 32, even at low nanomolar concentrations, can inhibit the growth of HCT-116 human colon carcinoma cells. Moreover, the agonist 32 has a synergistic anticancer action when coupled with 5-FU. Additionally, it reduces the myelotoxicity brought on by 5-FU, preserving normal neutrophil and white blood cell numbers. These results show that in mice with colon cancer, the A3 AR agonist 32 provides systemic anticancer, antimetastatic, and myeloprotective effects. It may also function as an adjuvant therapy to increase the efficiency of chemotherapy and lessen myelotoxicity.210
The chloro-substituted adenosine derivative 33 showed a dose-dependent reduction of Hep-3B cell proliferation at dosages of 1 and 10 nM in a xenograft animal model employing Hep-3B hepatocellular carcinoma (HCC) cells. Compound 33 effectively reduces HCC tumor growth in vivo, as evidenced by the differences in tumor size between the compound-treated and vehicle-treated groups after 45 days of tumor inoculation. The leading cause of this effect is the elevated production of apoptotic proteins, which are activated by the compound 33, including FasR, caspase-8, Bax, Bad, cytochrome-c, and caspase-3.211
It has been demonstrated that micromolar doses of the A3 AR agonist 2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarbamoyl-4′-thioadenosine, a sulfur-containing analogue of Cl-IB-MECA,212 often referred to as LJ-529 or thio-Cl-IB-MECA (34), cause anti-leukemic actions in HL-60 human leukemia cell cultures. Studies on poly(ADP-ribose) polymerase (PARP) cleavage and DNA fragmentation support the apoptotic explanation for this impact.213 In a different investigation, it was discovered that agonist 34 inhibited the growth of breast cancer cells' tumors in vivo and their in vitro proliferation by causing apoptosis and interfering with the Wnt signaling pathway. The thioadenosine derivative 34 has been connected to the down-regulation of c-ErbB2, a critical marker for the prognosis and treatment of breast cancer, as well as the molecular mechanisms behind these effects. In both in vitro and in vivo experiments, this down-regulation is seen in SK-BR-3 breast cancer cells, suggesting that the molecule may be useful in the treatment of breast cancer.214 Interestingly, by acting on A3 AR, the herbal compound cordycepin (3′-deoxyadenosine) (35) that is extracted from the parasitic fungus Cordyceps sinensis, which is utilized in traditional Chinese medicine, inhibits the growth of tumor cells.215 At micromolar concentrations, this molecule significantly inhibited the growth of murine B16-BL6 melanoma and Lewis lung carcinoma (LCC) tumor cells. Aqueous extracts from Cordyceps sinensis have been investigated recently for their potential anticancer and antimetastatic effects in vitro using mouse melanoma B16 and LLC cells as models. These extracts showed direct cytotoxic effects at 10 and 30 μg mL−1, respectively. Furthermore, an in vivo investigation employing oral cordycepin (35) in B16-BL6 cell tumor-bearing C57BL/6Cr mice demonstrated a decrease in the main tumor weight without resulting in a reduction in body weight or systemic toxicity. With little side effects, cordycepin seems to be a potential treatment for melanoma.216
Comparable antiproliferative effects to Cl-IB-MECA (33) were shown by the naturally occurring anticancer nucleoside N6-(2-isopentenyl) adenosine (36), which shows strong affinity and selectivity for A3 AR in human and rat tumor cell lines LNCaP and N1S1.217
It has been demonstrated that the resveratrol glucuronides like resveratrol-3-O-D-glucuronide (37) and resveratrol 4′-O-D-glucuronide (38), with IC50 values ranging from 9.8 to 31 μM, suppress the development of colon cancer cells Caco-2, HCT-116, and CCL-228. In CCL-228 and Caco-2 cells, these glucuronides similarly caused a G1 phase arrest.218 However, the adenosine A3 receptor antagonist MRS1191 (48), which reversed the growth inhibition caused by these two compounds, provided direct proof that the biological activity of these drugs is mediated through A3 AR. Further evidence that A3 AR is involved in this process comes from the G1 phase arrest and cyclin D1 depletion mechanisms used to suppress cell proliferation.170,219
The compound 3,7-dihydro-1H-purine-2,6-dione, known as linagliptin (39), is an FDA-certified anti-hyperglycemic medication used primarily to manage type 2 diabetes. The principal oxidative degradation product of linagliptin (40) is a pyrimidine derivative. Both compounds, 39 and 40, have been assessed due to their capacity to harm cells, A3 AR binding compatibility, cAMP levels, and apoptosis-inducing capabilities. They demonstrated inhibitory effects against hepatocellular carcinoma cell lines, inducing apoptosis at the G2/M phase, increasing caspase-3 levels, and causing suppression of the A3 AR gene and protein expression, which was followed by an enhancement in cAMP levels. Linagliptin's quantitative in vitro binding affinity for A3 AR displays a blocking characteristic with a 37.7 nM Ki value.220
10.7 The A3 receptor selective antagonists
A3 AR antagonists have garnered attention due to their potential as cancer therapeutics and are frequently linked to anti-inflammatory properties.221,222MRS1523 (41), a pyridine derivative, is currently the most commonly used selective antagonist for the rat A3 AR. However, its reported affinity and selectivity show some variability across different studies.223,224 The reported Ki values of MRS1523 for the A3 adenosine receptor are 43.9 nM in humans, 349 nM in mice, and 216 nM in rats.225
The antagonistic action of truncated thio-Cl-IB-MECA (42) (Fig. 14) on A3 AR is responsible for the inhibition of T24 human urinary bladder carcinoma cells, resulting in sub-G1 cell cycle arrest and early and late-stage cell death.203,226
 | ||
| Fig. 14 Potential A3 adenosine receptor antagonists exhibiting promising anticancer properties. | ||
N6-(2,2-Diphenylethyl)-2-phenylethynylAdo (43) in particular showed the most efficacy, indicating that it has the potential to be a strong anticancer drug. The cytostatic effects seen with the A3 receptor blocker Cl-IB-MECA and antagonists 46 and other related compounds highlight the fact that various cellular pathways contribute to the impact of these ligands against tumors, in addition to A3 AR activity, which is not the only factor responsible for the anticancer consequences.226
Several “[1,2,4]-triazolo[1,5-c]pyrimidines” (44) demonstrated a strong tumor-suppressive effect on human cancer cell lines HCT16 and THP1. Compound 44 (Fig. 14), in particular, showed notable efficacy against both cell lines. To further investigate its anticancer properties, the researchers assessed its ability to penetrate the phospholipid bilayer of the cell membrane. The compound can enter cells and interact with possible target molecules, according to the Parallel Artificial Membrane Permeability Assay (PAMPA) results, which supports the theory of an as-yet-undefined mechanism.227
Adenosine compounds that jointly oppose A3 & A2A ARs and have cancer immunotherapeutic efficacy are highlighted in a recent study. As mentioned earlier, these compounds suppress the immunological checkpoint activity mediated by A2A AR in addition to targeting the anticancer signaling pathway triggered by A3 AR inhibition. This reduces aberrant immune responses.228
MRS 1097 (45), MRS 1067 (46), MRS 1220 (47), & MRS 1191 (48), in contrast to focusing on a specific second messenger pathway, act as antagonists of the stimulation of [35S]GTPγS binding produced by agonists, according to a separate study. MRS 1220 and MRS 1191, with KB values of 1.7 nM and 92 nM, respectively, demonstrated strong selectivity for the human A3 AR over the human A1 AR in mediating effects on adenylate cyclase.229 These agents demonstrated high selectivity in blocking the inhibitory effects of AC mediated by human A3 receptors compared to those mediated by human A1 receptors.
Saturation binding experiments with the radiolabeled agonist [125I]AB-MECA (N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide) at cloned human brain A3 AR produced in HEK-293 cells demonstrated the competitiveness of MRS 1220, MRS 1191, and MRS 1067. Functional tests, such as agonist-induced suppression of AC and activation of [35S]GTPγS binding to related G-proteins, were used to validate their antagonistic characteristics. When it came to their actions on AC, MRS 1220 and MRS 1191, whose KB values were 1.7 nM and 92 nM, respectively, showed strong selectivity for human A3 receptors as opposed to human A1 receptors. Additionally, MRS 1220 has shown effectiveness at reversing the A3 agonist-induced reduction of tumour necrosis factor-α (TNF-α) production in the human macrophage U-937 cell line.193
Research has explored the pre- or co-administration of pharmaceutical formulations containing high-affinity adenosine A3 AR antagonists like the triazine derivative MRE-3008-F20 (49), to enhance the effectiveness of chemotherapy. This includes treatments in combination with taxanes (paclitaxel), vinca alkaloids (vincristine), camptothecins (irinotecan), or antibacterial agents (doxorubicin).230
11. Clinical trials updates on adenosine receptors' modulators
Adenosine receptor antagonists are in clinical trials to investigate the treatment of various cancers. These agents block adenosine signalling to overcome tumour-induced immune suppression and boost anti-tumour immunity. Table 3 summarises key modulators under investigation, including their targets, cancer types, and combination strategies.185,231| Compounds | Clinical trial identifier | Phase | Pharmaceutical sponsor | Patients | Description |
|---|---|---|---|---|---|
| a Study terminated. | |||||
| A2Areceptor antagonists | |||||
| Ciforadenant (CPI-444) | NCT02655822 | I/Ib | Corvus pharmaceuticals, Inc | Carcinoma of the renal cells and mCRPC | A selective A2A AR antagonist that binds to the A2A AR with a Ki value of 3.54 nM and exhibits over 50 times greater selectivity for A2A receptors compared to other adenosine receptor subtypes232 |
| NCT04280328 | Ib | Relapsed multiple myeloma | |||
| NCT03454451 | I | ||||
| NCT03237988 | I | Healthy subjects | |||
| NCT03337698 | Ib/II | Hoffmann-La Roche | Non-small cell lung cancer | ||
| NCT05501054 | Ib/II | MD Anderson Cancer Center | Advanced renal cell carcinoma | ||
| Taminadenant (PBF-509/NIR 178) | NCT03207867 | IIa | Novartis Pharmaceuticals II | NSCLC, carcinoma of the renal cells, pancreatic cancer, head and neck cancer, urothelial cancer, diffused large B cell lymphoma, TNBC, microsatellite stable colon cancer, melanoma, and mCRPC | A2A AR antagonist185 |
| NCT03549000 | I/Iba | NSCLC, TNBC, pancreatic ductal adenocarcinoma, colorectal cancer microsatellite stable, ovarian cancer, carcinoma of the renal cells, and mCRPC | |||
| NCT04895748 | I/Ib | Renal cell cancer | |||
| NCT02403193 | I/II | Palobiofarma SL/Novartis Pharmaceuticals | Advanced NSCLC | ||
| Preladenant | NCT03099161 | Ib/IIa | Merck Sharp & Dohme LLC | Advanced solid tumours | Strong and competitive adenosine A2A AR antagonist233 |
| ILB-2109 | NCT05278546 | Ia | Innolake Biopharm | Advanced solid tumours | A2A AR antagonist |
| NCT05955105 | Advanced solid tumours | ||||
| AZD | NCT02740985 | Ia/Ib | AstraZeneca | Advanced solid malignancies, colorectal carcinoma, mCRPC, and NSCLC | Oral A2A AR antagonist (Ki value of 1.7 nM) binds to the human A2A receptor and has about 30 times the selectivity for A2A receptors over other adenosine receptor subtypes234 |
| NCT04089553 | II | Prostate cancer and mCRPC | |||
| NCT03980821 | I | Advanced solid malignancies | |||
| NCT04495179 | II | Progressive mCRPC | |||
| NCT03381274 | Ib/II | MedImmune LLC | Advanced epidermal growth factor receptor mutant NSCLC | ||
| Inupadenant (EOS-100850/EOS-850) | NCT05060432 | I/II | iTeosTherapeutics | Advanced solid tumors | Inupadenant is a highly selective oral A2A AR antagonist that is blood–brain barrier insensitive235 |
| NCT05403385 | II | Advanced or metastatic non-small cell lung cancer | |||
| NCT05117177 | I | Advanced lung non-small cell carcinoma | |||
| TT-10 | NCT04969315 | I/II | Portage Biotech | NSCLC, mCRPC, renal cell cancer, and head and neck cancer | Potent and selective antagonists of A2A AR185,236 |
|
|||||
| Antagonists of both A2Aand A2Breceptors | |||||
| Etrumadenant (AB928) | NCT03720678 | I/Ib | Arcus Biosciences | Gastroesophageal cancer and colorectal cancer | A2A AR and A2B AR have Kd values of 1.4 nM and 2 nM, respectively, for this new dual-active A2A/A2B AR antagonist201 |
| NCT04381832 | Ib/II | mCRPC | |||
| NCT04892875 | Ib | Vanderbilt-Ingram Cancer Center | Locally advanced head and neck cancers | ||
| NCT05024097 | I/II | Weill Medical College of Cornell University | Rectal cancer | ||
| NCT05886634 | II | Memorial Sloan Kettering Cancer Center | Advanced dedifferentiated liposarcoma | ||
| NCT04660812 | Ib/II | Arcus Biosciences | Metastatic colorectal cancer | ||
| NCT05177770 | II | Surface oncology | mCRPC | ||
| M1069 | NCT05198349 | Ia | EMD Serono Research & Development Institute | Locally advanced unresectable solid tumors | A2A/A2B AR antagonist |
|
|||||
| A3receptor agonists | |||||
| Namodenoson/Cl-IB-MECA | NCT00790218 | I-II | Can-Fite BioPharma Ltd | Hepatocellular carcinoma | Only an A3 AR agonist with such high affinity has been found to date |
| NCT02128958 | II | Hepatocellular carcinoma | |||
12 Summary and future perspectives
AR activation affects immune suppression, angiogenesis, development of tumours, cell proliferation, and metastasis, all of which are essential to the progression of cancer. In the TME, extracellular adenosine concentration is much higher than normal. This elevated level stimulates adenosine receptors, which then trigger various biological responses that can inhibit cancer cell growth and enhance the immune system's ability to target tumors. The existing literature on AR subtypes indicates that each subtype is significantly involved in cancer, supported by findings from both in vitro and in vivo research. Because of this, all four AR subtypes are thought to be viable targets for the development of innovative therapeutic approaches in the treatment of cancer. Clinical trials focusing on Parkinson's disease have assessed the safety of A2A AR antagonists. The role of A2B ARs in tumors is not fully understood. While these receptors may aid in tumour growth by releasing substances that encourage the formation of angiogenesis, they might also send signaling that blocks the growth of cancer cells. Their feasibility as therapeutic targets in the therapy of cancer is complicated by this contradiction. Medications that target A2B adenosine receptors (ARs) by either activating or inhibiting them have proven to be more effective in treating cancer. Meanwhile, synthetic drugs that activate A3 ARs have been effective in slowing down cancer cell growth and promoting programmed apoptosis on various types of cancer. This effectiveness has been validated through laboratory studies and experiments on animals, highlighting the potential of these receptors as therapeutic targets in cancer treatment. Specifically, activating the A3 receptor or blocking the A1, A2A, and A2B receptors could help shift the tumor environment from one that supports cancer growth to one that inhibits it, enhancing the body's ability to fight cancer. In this regard, numerous modulators targeting adenosine receptors (A1, A2A, A2B, and A3), including preladenant, tozadenant, CPI444, NIR 178, PBF-509, M1069, Inupadenant, and Cl-IB-MECA, are in advanced clinical stages. However, many have failed for various reasons,185 including unsatisfactory pharmacokinetic properties. Moreover, developing adenosine receptor agonists or antagonists with high target selectivity and potency remains a significant challenge due to the body's widespread presence of adenosine receptors,31 raising concerns about off-target effects and limited efficacy. Moreover, data on the long-term impact of AR blockade, including tumour resistance and compensatory GPCR signalling, remain unclear. Even clinical-stage compounds often lack complete optimisation for durable and selective anti-tumour activity. These gaps emphasise the need for improved mechanistic understanding and more targeted drug development strategies.22Synthetic agonist activation of the A3 AR decreases cell proliferation and enhances apoptosis in a range of cancer cells, as demonstrated by both in vitro and in vivo models. These agonists have already been shown in preclinical and Phase I/II trials to be safe and generally well-tolerated in human patients. However, a couple of studies revealed that A3 AR antagonists may also be a viable therapeutic approach for cancer by preventing hypoxia-induced rises in HIF-1α and decreasing angiogenesis and cell invasion in the tumor microenvironment. The A2A receptor activation contributes to immune system suppression in the tumour microenvironment (TME) via cAMP signalling, making it easier for cancer to evade the immune system. Interestingly, the enzyme that changes AMP into adenosine, CD39 and CD73 are highly expressed in various TME cell types and contribute to this immune suppressive effect. Recent studies have identified CD39, CD73, and A2A AR as promising targets for boosting antitumor immunity. Monoclonal antibodies and small-molecule inhibitors targeting the CD39/CD73/A2A AR axis are currently being tested in clinical trials, both as standalone treatments and in combination with anti-PD-1/PD-L1 immunotherapies.237
Results indicate that the antitumor immune response is enhanced when these antibodies are combined with A2A AR antagonists, potentially providing an impactful therapeutic strategy for cancer treatment.170,185
Clinical trials involving A2A antagonists and A3 agonists will also take some time to conclude which therapy approach is more promising for cancer. The two receptors do have different biological effects: A2A receptor signalling decreases immune cell antitumor activity, whereas A3 receptor signalling enhances immune function while inhibiting tumour growth. Thus, drugs that activate A3 adenosine receptors and block A2A receptors are being explored as a novel approach for cancer treatment. This strategy aims to enhance the body's anti-tumour response by leveraging the beneficial effects of A3 receptor activation while reducing the tumour-supportive signals associated with A2A receptor activation. As a result, this combination could potentially improve therapeutic outcomes for cancer patients. However, they are still in the early stages of development and must overcome several critical challenges, including stability, bioavailability, toxicity, and safety profile studies.238
Abbreviations
| 5-AMP | 5-Adenosine monophosphate |
| A1 AR | A1 adenosine receptor |
| A2A AR | A2A adenosine receptor |
| A2B AR | A2B adenosine receptor |
| A3 AR | A3 adenosine receptor |
| AC | Adenylate cyclase |
| ADA | Adenosine deaminase |
| ADP | Adenosine diphosphate |
| AK | Adenosine kinase |
| AK-T cell | Activated T cell |
| AMP | Adenosine monophosphate |
| ARs | Adenosine receptors |
| ATP | Adenosine triphosphate |
| Ca2+ | Calcium channels |
| cAMP | Cyclic adenosine monophosphate |
| CNS | Central nervous system |
| CNTs | Concentrative nucleoside transporters |
| CPA | Cyclopentyl adenosine |
| CREB | Camp-response element binding protein |
| CTLs | Cytotoxic T lymphocytes |
| DAG | Diacylglycerol |
| ecto-PDE | Ecto-phosphodiesterase |
| ENTs | Equilibrative nucleoside transporters |
| ERK1/2 | Extracellular signal-regulated kinases |
| FoxP3 | Forkhead box P3 |
| GDP | Guanosine diphosphate |
| GFR | Glomerular filtration rate |
| Gi/Go | Inhibitory G-proteins |
| GPCRs | G protein-coupled receptors |
| Gs | Stimulatory G-proteins |
| Gs/Golf | Stimulatory G-proteins |
| GSK-3β | Glycogen synthase kinase -3β |
| GTP | Guanosine triphosphate |
| IMPDH | Inosine monophosphate dehydrogenase |
| IP3 | Inositol triphosphate |
| JNK | C-Jun N-terminal kinases |
| K+ | Potassium channels |
| LAK Cell | Lymphokine-activated killer cells |
| MAPKs | Mitogen-activated protein kinase |
| MMP2 | Matrix metalloproteinase 2 |
| NK | Natural killer |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death-ligand 1 |
| PGs | Prostaglandins |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PKA | Protein kinase A |
| PLC | Phospholipase C |
| PLC-β | Phospholipase C-beta |
| PNP | Purine nucleoside phosphorylase |
| RhoA | Ras homolog family member A |
| ROS | Reactive oxygen species |
| SAH | S-Adenosyl-homocysteine |
| SAHH | S-Adenosyl-homocysteine hydrolase |
| SAMe | S-Adenosylmethionine |
| TGF-β | Transforming growth factor-beta |
| TME | Tumor microenvironment |
| VEGFs | Vascular endothelial growth factors |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Author, Prasenjit Maity, gratefully acknowledges the Department of Pharmaceutical Sciences and Technology, BIT Mesra, Ranchi, for awarding the Institute Research Scholarship (IRS) (Application Number PHDSP 24-43). Author Pran Kishore Deb thanks the Department of Pharmaceutical Sciences and Technology, BIT Mesra, Ranchi, for providing the research fund under the Seed Money Scheme (Grant Number BIT/DRIE/SMS/2024-25/1877) and the necessary research facilities.References
- S. Sachdeva and M. Gupta, Saudi Pharm. J., 2013, 21, 245–253 CrossRef PubMed.
- J. Layland, D. Carrick, M. Lee, K. Oldroyd and C. Berry, JACC Cardiovasc. Interv., 2014, 7, 581–591 CrossRef PubMed.
- S. Bar-Yehuda, F. Barer, L. Volisson and P. Fishman, Neoplasia, 2001, 3, 125–131 CrossRef CAS PubMed.
- A. C. Newby, Trends Biochem. Sci., 1984, 9, 42–44 CrossRef CAS.
- M. de Lera Ruiz, Y.-H. Lim and J. Zheng, J. Med. Chem., 2014, 57, 3623–3650 CrossRef CAS PubMed.
- A. Bahreyni, M. Khazaei, M. Rajabian, M. Ryzhikov, A. Avan and S. M. Hassanian, J. Pharm. Pharmacol., 2018, 70, 191–196 CrossRef CAS PubMed.
- S. Gessi, S. Merighi, V. Sacchetto, C. Simioni and P. A. Borea, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 1400–1412 CrossRef CAS PubMed.
- A. Bahreyni, S. S. Samani, F. Rahmani, R. Behnam-Rassouli, M. Khazaei, M. Ryzhikov, M. R. Parizadeh, A. Avan and S. M. Hassanian, J. Cell. Physiol., 2018, 233(3), 1836–1843 CrossRef CAS PubMed.
- P. A. Borea, S. Gessi, S. Merighi and K. Varani, Trends Pharmacol. Sci., 2016, 37, 419–434 CrossRef CAS PubMed.
- P. A. Borea, S. Gessi, S. Merighi, F. Vincenzi and K. Varani, Physiol. Rev., 2018, 98, 1591–1625 CrossRef CAS PubMed.
- P. Kumar and P. K. Deb, Frontiers in Pharmacology of Neurotransmitters, Springer, 2020 Search PubMed.
- P. Borah, S. Deka, R. P. Mailavaram and P. K. Deb, Curr. Pharm. Des., 2019, 25, 2792–2807 CrossRef CAS PubMed.
- S. Gessi, K. Varani, S. Merighi, E. Cattabriga, V. Iannotta, E. Leung, P. G. Baraldi and P. A. Borea, Mol. Pharmacol., 2002, 61, 415–424 CrossRef CAS PubMed.
- L. Antonioli, C. Blandizzi, P. Pacher and G. Haskó, Nat. Rev. Cancer, 2013, 13, 842–857 CrossRef CAS PubMed.
- F. Jadidi-Niaragh, Immunotherapy, 2019, 11(16), 1353–1355 CrossRef CAS PubMed.
- P. A. Beavis, J. Stagg, P. K. Darcy and M. J. Smyth, Trends Immunol., 2012, 33, 231–237 CrossRef CAS PubMed.
- U. K. M. Decking, G. Schlieper, K. Kroll and J. Schrader, Circ. Res., 1997, 81, 154–164 CrossRef CAS PubMed.
- L. Antonioli, R. Colucci, C. La Motta, M. Tuccori, O. Awwad, F. Da Settimo, C. Blandizzi and M. Fornai, Curr. Drug Targets, 2012, 13, 842–862 CrossRef CAS PubMed.
- B. Allard, S. Pommey, M. J. Smyth and J. Stagg, Clin. Cancer Res., 2013, 19, 5626–5635 CrossRef CAS PubMed.
- P. K. Deb, Curr. Pharm. Des., 2019, 25, 2789–2791 CrossRef CAS PubMed.
- P. K. Deb, Curr. Pharm. Des., 2019, 25, 2695–2696 CrossRef CAS PubMed.
- B. Chandrasekaran, S. Samarneh, A. M. Y. Jaber, G. Kassab and N. Agrawal, Curr. Pharm. Des., 2019, 25, 2741–2771 CrossRef CAS PubMed.
- P. K. Deb, S. F. Kokaz, S. N. Abed, B. Chandrasekaran, W. Hourani, A. Y. Jaber, R. P. Mailavaram, P. Kumar and K. N. Venugopala, Front. Pharmacol., 2020, 325–359 Search PubMed.
- H. Zimmermann, Naunyn-Schmiedeberg’s Arch. Pharmacol., 2000, 362, 299–309 CrossRef CAS PubMed.
- C. M. Hay, E. Sult, Q. Huang, K. Mulgrew, S. R. Fuhrmann, K. A. McGlinchey, S. A. Hammond, R. Rothstein, J. Rios-Doria and E. Poon, Oncoimmunology, 2016, 5, e1208875 CrossRef PubMed.
- F. Jadidi-Niaragh, F. Atyabi, A. Rastegari, N. Kheshtchin, S. Arab, H. Hassannia, M. Ajami, Z. Mirsanei, S. Habibi and F. Masoumi, J. Controlled Release, 2017, 246, 46–59 CrossRef CAS PubMed.
- K. A. Jacobson and Z.-G. Gao, Nat. Rev. Drug Discovery, 2006, 5, 247–264 CrossRef CAS PubMed.
- G. Burnstock, Front. Pharmacol., 2017, 8, 661 CrossRef PubMed.
- C. E. Müller and K. A. Jacobson, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 1290–1308 CrossRef PubMed.
- B. B. Fredholm, Exp. Cell Res., 2010, 316, 1284–1288 CrossRef CAS PubMed.
- J.-F. Chen, H. K. Eltzschig and B. B. Fredholm, Nat. Rev. Drug Discovery, 2013, 12, 265–286 CrossRef CAS PubMed.
- K. Varani, F. Vincenzi, S. Merighi, S. Gessi and P. A. Borea, Protein Rev., 2017, 19, 193–232 Search PubMed.
- P. K. Deb, S. Deka, P. Borah, S. N. Abed and K.-N. Klotz, Curr. Pharm. Des., 2019, 25, 2697–2715 CrossRef CAS PubMed.
- A. K. Dhalla, J. W. Chisholm, G. M. Reaven and L. Belardinelli, A1 Adenosine Receptor: Role in Diabetes and Obesity, Adenosine Receptors in Health and Disease, 2009, pp. 271–295 Search PubMed.
- S. Merighi, P. A. Borea and S. Gessi, Pharmacol. Res., 2015, 99, 229–236 CrossRef CAS PubMed.
- F. Ciruela, C. Albergaria, A. Soriano, L. Cuffí, L. Carbonell, S. Sánchez, J. Gandía and V. Fernández-Dueñas, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 9–20 CrossRef CAS PubMed.
- F. BB, Biochem. Pharmacol., 2001, 61, 443–444 CrossRef PubMed.
- P. A. Borea, K. Varani, F. Vincenzi, P. G. Baraldi, M. A. Tabrizi, S. Merighi and S. Gessi, Pharmacol. Rev., 2015, 67, 74–102 CrossRef PubMed.
- J.-F. Chen and A. Mori, Adenosine A2A Receptor Antagonists, Elsevier, 2023, vol. 170 Search PubMed.
- B. B. Fredholm, E. Irenius, B. Kull and G. Schulte, Biochem. Pharmacol., 2001, 61, 443–448 CrossRef CAS PubMed.
- M. Raman, W. Chen and M. H. Cobb, Oncogene, 2007, 26, 3100–3112 CrossRef CAS PubMed.
- Z. G. Goldsmith and D. N. Dhanasekaran, Oncogene, 2007, 26, 3122–3142 CrossRef CAS PubMed.
- S. Seino and T. Shibasaki, Physiol. Rev., 2005, 85, 1303–1342 CrossRef CAS PubMed.
- S.-A. Poulsen and R. J. Quinn, Bioorg. Med. Chem., 1998, 6, 619–641 CrossRef CAS PubMed.
- S. Offermanns and M. I. Simon, J. Biol. Chem., 1995, 270, 15175–15180 CrossRef CAS PubMed.
- P. Fresco, C. Diniz and J. Gonçalves, Cardiovasc. Res., 2004, 63, 739–746 CrossRef CAS PubMed.
- B. Sperlágh and E. Sylvester Vizi, Curr. Top. Med. Chem., 2011, 11, 1034–1046 CrossRef PubMed.
- R. O. Godinho, T. Duarte and E. S. A. Pacini, Front. Pharmacol, 2015, 6, 58 Search PubMed.
- M. Peleli, B. B. Fredholm, L. Sobrevia and M. Carlström, Mol. Aspects Med., 2017, 55, 4–8 CrossRef CAS PubMed.
- N. T. Fried, M. B. Elliott and M. L. Oshinsky, Brain Sci., 2017, 7, 30 CrossRef PubMed.
- A. Deussen and N. Schmiedebergs, Arch. Pharmacol., 2000, 362, 351–363 CrossRef CAS PubMed.
- K. A. Jacobson, B. K. Trivedi, P. C. Churchill and M. Williams, Biochem. Pharmacol., 1991, 41, 1399–1410 CrossRef CAS PubMed.
- S. Sheth, R. Brito, D. Mukherjea, L. P. Rybak and V. Ramkumar, Int. J. Mol. Sci., 2014, 15, 2024–2052 CrossRef PubMed.
- T. Stumpe and J. Schrader, Am. J. Physiol.: Heart Circ. Physiol., 1997, 273, H756–H766 CrossRef CAS PubMed.
- D. Boison, Pharmacol. Rev., 2013, 65, 906–943 CrossRef CAS PubMed.
- E. Gracia, D. Farré, A. Cortés, C. Ferrer-Costa, M. Orozco, J. Mallol, C. Lluís, E. I. Canela, P. J. McCormick and R. Franco, FASEB J., 2013, 27, 1048–1061 CrossRef CAS PubMed.
- R. Pacheco, J. M. Martinez-Navio, M. Lejeune, N. Climent, H. Oliva, J. M. Gatell, T. Gallart, J. Mallol, C. Lluis and R. Franco, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9583–9588 CrossRef CAS PubMed.
- B. Cacciari, G. Spalluto and S. Federico, Mini-Rev. Med. Chem., 2018, 18, 1168–1174 CrossRef CAS PubMed.
- L. López-Cruz, J. D. Salamone and M. Correa, Front. Pharmacol, 2018, 9, 353416 Search PubMed.
- S. Ferre, C. Quiroz, A. S. Woods, R. Cunha, P. Popoli, F. Ciruela, C. Lluis, R. Franco, K. Azdad and S. N. Schiffmann, Curr. Pharm. Des., 2008, 14, 1468–1474 CrossRef CAS PubMed.
- S. C. Holst and H.-P. Landolt, Curr. Sleep Med. Rep., 2015, 1, 27–37 CrossRef.
- I. Ballesteros-Yáñez, C. A. Castillo, S. Merighi and S. Gessi, Front. Pharmacol, 2018, 8, 320873 Search PubMed.
- B. B. Fredholm, G. Arslan, L. Halldner, B. Kull, G. Schulte, W. Wasserman and N. Schmiedebergs, Arch. Pharmacol., 2000, 362, 364–374 CrossRef CAS PubMed.
- M. J. Ellis, A. C. Lindon, K. J. Flint, N. C. Jones and S. Goodbourn, Mol. Endocrinol., 1995, 9, 255–265 CAS.
- I. Löffler, M. Grün, F. D. Böhmer and I. Rubio, BMC Cancer, 2008, 8, 1–18 CrossRef PubMed.
- K. M. Sakamoto and D. A. Frank, Clin. Cancer Res., 2009, 15, 2583–2587 CrossRef CAS PubMed.
- M. Boettcher, A. Lawson, V. Ladenburger, J. Fredebohm, J. Wolf, J. D. Hoheisel, C. Frezza and T. Shlomi, BMC Genomics, 2014, 15, 1–11 CrossRef PubMed.
- S.-J. Yu, J.-K. Yu, W.-T. Ge, H.-G. Hu, Y. Yuan and S. Zheng, World J. Gastroenterol., 2011, 17, 2028 CrossRef PubMed.
- S.-H. Hong, S.-H. Goh, S. J. Lee, J.-A. Hwang, J. Lee, I.-J. Choi, H. Seo, J.-H. Park, H. Suzuki and E. Yamamoto, Oncotarget, 2013, 4, 1791 CrossRef PubMed.
- R. A. Mohamed, A. M. Agha, A. A. Abdel-Rahman and N. N. Nassar, Neuroscience, 2016, 314, 145–159 CrossRef CAS PubMed.
- D. Preti, P. G. Baraldi, A. R. Moorman, P. A. Borea and K. Varani, Med. Res. Rev., 2015, 35, 790–848 CrossRef CAS PubMed.
- G. Schulte and B. B. Fredholm, Cell Signalling, 2003, 15, 813–827 CrossRef CAS PubMed.
- S. Loi, S. Dushyanthen, P. A. Beavis, R. Salgado, C. Denkert, P. Savas, S. Combs, D. L. Rimm, J. M. Giltnane and M. V Estrada, Clin. Cancer Res., 2016, 22, 1499–1509 CrossRef CAS PubMed.
- P. Fishman, S. Bar-Yehuda, L. Madi and I. Cohn, Anticancer Drugs, 2002, 13, 437–443 CrossRef CAS PubMed.
- P. Vaupel, F. Kallinowski and P. Okunieff, Cancer Res., 1989, 49, 6449–6465 CAS.
- G. Multhoff and P. Vaupel, Oxyg. Transp. to Tissue XLI, 2020, pp. 131–143 Search PubMed.
- M. V Sitkovsky, D. Lukashev, S. Apasov, H. Kojima, M. Koshiba, C. Caldwell, A. Ohta and M. Thiel, Annu. Rev. Immunol., 2004, 22, 657–682 CrossRef PubMed.
- M. Sitkovsky and D. Lukashev, Nat. Rev. Immunol., 2005, 5, 712–721 CrossRef CAS PubMed.
- M. H. Kazemi, S. Raoofi Mohseni, M. Hojjat-Farsangi, E. Anvari, G. Ghalamfarsa, H. Mohammadi and F. Jadidi-Niaragh, J. Cell. Physiol., 2018, 233, 2032–2057 CrossRef CAS PubMed.
- J. Blay, T. D. White and D. W. Hoskin, Cancer Res., 1997, 57, 2602–2605 CAS.
- A. Deussen, M. Stappert, S. Schäfer and M. Kelm, Circulation, 1999, 99, 2041–2047 CrossRef CAS PubMed.
- D. W. Hoskin, T. Reynolds and J. Blay, Cancer Immunol., Immunother., 1994, 38, 201–207 CAS.
- D. W. Hoskin, T. Reynolds and J. Blay, Int. J. Cancer, 1994, 59, 854–855 CrossRef CAS PubMed.
- D. W. Hoskin, T. Reynolds and J. Blay, Cell. Immunol., 1994, 159, 85–93 CrossRef CAS PubMed.
- D. W. Hoskin, J. J. Butler, D. Drapeau, S. M. M. Haeryfar and J. Blay, Int. J. Cancer, 2002, 99, 386–395 CrossRef CAS PubMed.
- W. M. MacKenzie, D. W. Hoskin and J. Blay, Exp. Cell Res., 2002, 276, 90–100 CrossRef CAS PubMed.
- G. Burnstock, Arterioscler., Thromb., Vasc. Biol., 2002, 22, 364–373 CrossRef PubMed.
- P. Van Daele, A. Van Coevorden, P. P. Roger and J.-M. Boeynaems, Circ. Res., 1992, 70, 82–90 CrossRef CAS PubMed.
- M. F. Ethier, V. Chander and J. G. Dobson Jr, Am. J. Physiol.: Heart Circ. Physiol., 1993, 265, H131–H138 CrossRef CAS PubMed.
- G. A. Lutty, M. K. Mathews, C. Merges and D. S. McLeod, Curr. Eye Res., 1998, 17, 594–607 CrossRef CAS PubMed.
- U. Testa, G. Castelli and E. Pelosi, Medicines, 2019, 6, 82 CrossRef CAS PubMed.
- R. Basheer, E. Arrigoni, H. S. Thatte, R. W. Greene, I. S. Ambudkar and R. W. McCarley, J. Neurosci., 2002, 22, 7680–7686 CrossRef CAS PubMed.
- G. E. Kirsch, J. Codina, L. Birnbaumer and A. M. Brown, Am. J. Physiol.: Heart Circ. Physiol., 1990, 259, H820–H826 CrossRef CAS PubMed.
- S. S. Kunduri, G. M. Dick, M. A. Nayeem and S. J. Mustafa, Physiol. Rep. Search PubMed.
- M. Shaban, R. A. Smith and T. W. Stone, Cell Proliferation, 1995, 28, 673–682 CrossRef CAS PubMed.
- M. Synowitz, R. Glass, K. Färber, D. Markovic, G. Kronenberg, K. Herrmann, J. Schnermann, C. Nolte, N. van Rooijen and J. Kiwit, Cancer Res., 2006, 66, 8550–8557 CrossRef CAS PubMed.
- Z. Chen, F. Han, Y. Du, H. Shi and W. Zhou, Signal Transduction Targeted Ther., 2023, 8, 70 CrossRef PubMed.
- G. Haskó, P. Pacher, E. S. Vizi and P. Illes, Trends Pharmacol. Sci., 2005, 26, 511–516 CrossRef PubMed.
- B. Lv, Y. Wang, D. Ma, W. Cheng, J. Liu, T. Yong, H. Chen and C. Wang, Front. Immunol., 2022, 13, 844142 CrossRef PubMed.
- H. Takagi, G. L. King, G. S. Robinson, N. Ferrara and L. P. Aiello, Invest. Ophthalmol. Visual Sci., 1996, 37, 2165–2176 CAS.
- K. N. Venugopala and M. Buccioni, Molecules, 2024, 29, 3501 CrossRef CAS PubMed.
- K. Sek, C. Mølck, G. D. Stewart, L. Kats, P. K. Darcy and P. A. Beavis, Int. J. Mol. Sci., 2018, 19, 3837 CrossRef PubMed.
- P. J. Gebicke-Haerter, F. Christoffel, J. Timmer, H. Northoff, M. Berger and D. Van Calker, Neurochem. Int., 1996, 29, 37–42 CrossRef CAS PubMed.
- B. Gorain, H. Choudhury, G. S. Yee and S. K. Bhattamisra, Curr. Pharm. Des., 2019, 25, 2828–2841 CrossRef CAS PubMed.
- T. Kaur, V. Borse, S. Sheth, K. Sheehan, S. Ghosh, S. Tupal, S. Jajoo, D. Mukherjea, L. P. Rybak and V. Ramkumar, J. Neurosci., 2016, 36, 3962–3977 CrossRef CAS PubMed.
- F. Ghiringhelli, M. Bruchard, F. Chalmin and C. Rébé, BioMed Res. Int., 2012, 2012, 473712 Search PubMed.
- A. Mirza, A. Basso, S. Black, M. Malkowski, L. Kwee, J. A. Patcher, J. E. Lachowicz, Y. Wang and S. Liu, Cancer Biol. Ther., 2005, 4, 1355–1360 CrossRef CAS PubMed.
- Y. Zhou, L. Tong, X. Chu, F. Deng, J. Tang, Y. Tang and Y. Dai, Cell. Physiol. Biochem., 2017, 43, 733–742 CrossRef CAS PubMed.
- A. Young, D. Mittal, J. Stagg and M. J. Smyth, Cancer Discovery, 2014, 4, 879–888 CrossRef CAS PubMed.
- F. Yu, C. Zhu, Q. Xie and Y. Wang, J. Med. Chem., 2020, 63, 12196–12212 CrossRef CAS PubMed.
- S. Merighi, P. Mirandola, K. Varani, S. Gessi, E. Leung, P. G. Baraldi, M. A. Tabrizi and P. A. Borea, Pharmacol. Ther., 2003, 100, 31–48 CrossRef CAS PubMed.
- S. Muller-Haegele, L. Muller and T. L. Whiteside, Expert Rev. Clin. Immunol., 2014, 10, 897–914 CrossRef CAS PubMed.
- S. Gessi, S. Bencivenni, E. Battistello, F. Vincenzi, V. Colotta, D. Catarzi, F. Varano, S. Merighi, P. A. Borea and K. Varani, Front. Pharmacol., 2017, 8, 888 CrossRef PubMed.
- V. Bova, A. Filippone, G. Casili, M. Lanza, M. Campolo, A. P. Capra, A. Repici, L. Crupi, G. Motta and C. Colarossi, Cancers, 2022, 14, 4032 CrossRef CAS PubMed.
- J. Xing, J. Zhang and J. Wang, Int. J. Mol. Sci., 2023, 24, 14928 CrossRef CAS PubMed.
- S. M. Hatfield and M. Sitkovsky, Curr. Opin. Pharmacol., 2016, 29, 90–96 CrossRef CAS PubMed.
- S. A. Rivkees and S. M. Reppert, Mol. Endocrinol., 1992, 6, 1598–1604 CAS.
- K. D. Pierce, T. J. Furlong, L. A. Selbie and J. Shine, Biochem. Biophys. Res. Commun., 1992, 187, 86–93 CrossRef CAS PubMed.
- G. Schulte and B. B. Fredholm, Exp. Cell Res., 2003, 290, 168–176 CrossRef CAS PubMed.
- B. B. Fredholm, A. P. IJzerman, K. A. Jacobson, J. Linden and C. E. Müller, Pharmacol. Rev., 2011, 63, 1–34 CrossRef CAS PubMed.
- Z.-G. Gao and K. A. Jacobson, Int. J. Mol. Sci., 2019, 20, 5139 CrossRef CAS PubMed.
- W. G. Robichaux III and X. Cheng, Physiol. Rev., 2018, 98, 919–1053 CrossRef PubMed.
- Y. Fang and M. E. Olah, J. Pharmacol. Exp. Ther., 2007, 322, 1189–1200 CrossRef CAS PubMed.
- P. Mayer, A. V. Hinze, A. Harst and I. von Kügelgen, Cardiovasc. Res., 2011, 90, 148–156 CrossRef CAS PubMed.
- S. Merighi, A. Benini, P. Mirandola, S. Gessi, K. Varani, E. Leung, S. Maclennan, P. G. Baraldi and P. A. Borea, Mol. Pharmacol., 2007, 72, 162–172 CrossRef CAS PubMed.
- E. A. Vecchio, P. J. White and L. T. May, Pharmacol. Ther., 2019, 198, 20–33 CrossRef CAS PubMed.
- A. F. Martin, Molecular Analysis of A2A Adenosine Receptor Regulation of NF-κb-dependent Inflammatory Responses, University of Glasgow, United Kingdom, 2004 Search PubMed.
- D. Allard, M. Turcotte and J. Stagg, Immunol. Cell Biol., 2017, 95, 333–339 CrossRef CAS PubMed.
- S. Phosri, A. Arieyawong, K. Bunrukchai, W. Parichatikanond, A. Nishimura, M. Nishida and S. Mangmool, Front. Pharmacol., 2017, 8, 428 CrossRef PubMed.
- S. Phosri, K. Bunrukchai, W. Parichatikanond, V. H. Sato and S. Mangmool, Purinergic Signalling, 2018, 14, 141–156 CrossRef CAS PubMed.
- B. Koscsó, B. Csóka, Z. Selmeczy, L. Himer, P. Pacher, L. Virág and G. Haskó, J. Immunol., 2012, 188, 445–453 CrossRef PubMed.
- S. Merighi, S. Bencivenni, F. Vincenzi, K. Varani, P. A. Borea and S. Gessi, Pharmacol. Res., 2017, 117, 9–19 CrossRef CAS PubMed.
- A. Chin, B. Svejda, B. I. Gustafsson, A. B. Granlund, A. K. Sandvik, A. Timberlake, B. Sumpio, R. Pfragner, I. M. Modlin and M. Kidd, Am. J. Physiol. Liver Physiol., 2012, 302, G397–G405 CAS.
- X. Yang, W. Xin, X. Yang, A. Kuno, T. C. Rich, M. V Cohen and J. M. Downey, Br. J. Pharmacol., 2011, 163, 995–1006 CrossRef CAS PubMed.
- Y. Sun and P. Huang, Front. Chem., 2016, 4, 37 Search PubMed.
- W. Wei, C. Du, J. Lv, G. Zhao, Z. Li, Z. Wu, G. Haskó and X. Xie, J. Immunol., 2013, 190, 138–146 CrossRef CAS PubMed.
- C. Cekic, D. Sag, Y. Li, D. Theodorescu, R. M. Strieter and J. Linden, J. Immunol., 2012, 188, 198–205 CrossRef CAS PubMed.
- P. Fishman, S. Bar-Yehuda, G. Ohana, F. Barer, A. Ochaion, A. Erlanger and L. Madi, Oncogene, 2004, 23, 2465–2471 CrossRef CAS PubMed.
- P. Fishman, S. Bar-Yehuda, B. T. Liang and K. A. Jacobson, Drug Discovery Today, 2012, 17, 359–366 CrossRef CAS PubMed.
- B. Suh, T. Kim, J. Lee, J. Seong and K. Kim, Br. J. Pharmacol., 2001, 134, 132–142 CrossRef CAS PubMed.
- S. Gessi, E. Cattabriga, A. Avitabile, R. Gafa’, G. Lanza, L. Cavazzini, N. Bianchi, R. Gambari, C. Feo and A. Liboni, Clin. Cancer Res., 2004, 10, 5895–5901 CrossRef CAS PubMed.
- S. Bar-Yehuda, S. M. Stemmer, L. Madi, D. Castel, A. Ochaion, S. Cohen, F. Barer, A. Zabutti, G. Perez-Liz and L. Del Valle, Int. J. Oncol., 2008, 33, 287–295 CAS.
- L. Madi, A. Ochaion, L. Rath-Wolfson, S. Bar-Yehuda, A. Erlanger, G. Ohana, A. Harish, O. Merimski, F. Barer and P. Fishman, Clin. Cancer Res., 2004, 10, 4472–4479 CrossRef CAS PubMed.
- S. Morello, A. Petrella, M. Festa, A. Popolo, M. Monaco, E. Vuttariello, G. Chiappetta, L. Parente and A. Pinto, Cancer Biol. Ther., 2008, 7, 278–284 CrossRef CAS PubMed.
- S. Merighi, P. Mirandola, D. Milani, K. Varani, S. Gessi, K.-N. Klotz, E. Leung, P. G. Baraldi and P. A. Borea, J. Invest. Dermatol., 2002, 119, 923–933 CrossRef CAS PubMed.
- I. Azoulay-Alfaguter, R. Elya, L. Avrahami, A. Katz and H. Eldar-Finkelman, Oncogene, 2015, 34, 4613–4623 CrossRef CAS PubMed.
- S. Jajoo, D. Mukherjea, K. Watabe and V. Ramkumar, Neoplasia, 2009, 11, 1132 CrossRef CAS PubMed.
- M. R. Atkinson, A. Townsend-Nicholson, J. K. Nicholl, G. R. Sutherland and P. R. Schofield, Neurosci. Res., 1997, 29, 73–79 CrossRef CAS PubMed.
- L. Yan, J. C. Burbiel, A. Maaß and C. E. Müller, Expert Opin. Emerging Drugs, 2003, 8, 537–576 CrossRef CAS PubMed.
- Z.-G. Gao and K. A. Jacobson, Expert Opin. Emerging Drugs, 2007, 12, 479–492 CrossRef CAS PubMed.
- Z.-G. Gao and K. A. Jacobson, Expert Opin. Emerging Drugs, 2011, 16(4), 597–602 CrossRef CAS PubMed.
- R. F. Bruns, Biochem. Pharmacol., 1981, 30, 325–333 CrossRef CAS PubMed.
- B. K. Trivedi, A. J. Bridges and R. F. Bruns, in Adenosine and Adenosine Receptors, Springer, 1990, pp. 57–103 Search PubMed.
- S. Schenone, C. Brullo, F. Musumeci, O. Bruno and M. Botta, Curr. Top. Med. Chem., 2010, 10, 878–901 CrossRef CAS PubMed.
- K.-N. Klotz and N. Schmiedebergs, Arch. Pharmacol., 2000, 362, 382–391 CrossRef CAS PubMed.
- Z. Gao and K. A. Jacobson, Eur. J. Pharmacol., 2002, 443, 39–42 CrossRef CAS PubMed.
- C. E. Muller, Curr. Med. Chem., 2000, 7, 1269–1288 CrossRef CAS PubMed.
- Z.-G. Gao, J. B. Blaustein, A. S. Gross, N. Melman and K. A. Jacobson, Biochem. Pharmacol., 2003, 65, 1675–1684 CrossRef CAS PubMed.
- K. A. Jacobson, P. J. M. Van Galen and M. Williams, J. Med. Chem., 1992, 35, 407–422 CrossRef CAS PubMed.
- J. L. Carlin, S. Jain, E. Gizewski, T. C. Wan, D. K. Tosh, C. Xiao, J. A. Auchampach, K. A. Jacobson, O. Gavrilova and M. L. Reitman, Neuropharmacology, 2017, 114, 101–113 CrossRef CAS PubMed.
- P. Franchetti, L. Cappellacci, P. Vita, R. Petrelli, A. Lavecchia, S. Kachler, K.-N. Klotz, I. Marabese, L. Luongo and S. Maione, J. Med. Chem., 2009, 52, 2393–2406 CrossRef CAS PubMed.
- M. A. Jacobson, Expert Opin. Ther. Pat., 2002, 12, 489–501 CrossRef CAS.
- P. G. Nell and B. Albrecht-Küpper, Prog. Med. Chem., 2009, 47, 163–201 CAS.
- J. Blay, Encycl. Cancer, SpringerBerlin Heidelb., 2012, 49–52 Search PubMed.
- N. Etique, I. Grillier-Vuissoz, J. Lecomte and S. Flament, Oncol. Rep., 2009, 21, 977–981 CAS.
- M. Perez-Aso, A. Mediero, Y. C. Low, J. Levine and B. N. Cronstein, FASEB J., 2015, 30, 457 CrossRef PubMed.
- A. Ohta, E. Gorelik, S. J. Prasad, F. Ronchese, D. Lukashev, M. K. K. Wong, X. Huang, S. Caldwell, K. Liu and P. Smith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13132–13137 CrossRef CAS PubMed.
- C. E. Muller and S. Ferré, Front. CNS Drug Discovery, 2010, 1, 304–341 Search PubMed.
- U. Shah and R. Hodgson, Curr. Opin. Drug Discovery Dev., 2010, 13, 466–480 CAS.
- S. Marwein, B. Mishra, U. C. De and P. C. Acharya, Curr. Pharm. Des., 2019, 25, 2842–2858 CrossRef CAS PubMed.
- F. Gatta, M. R. Del Giudice, A. Borioni, P. A. Borea, S. Dionisotti and E. Ongini, Eur. J. Med. Chem., 1993, 28, 569–576 CrossRef CAS.
- L. S. Jeong, D. Z. Jin, H. O. Kim, D. H. Shin, H. R. Moon, P. Gunaga, M. W. Chun, Y.-C. Kim, N. Melman and Z.-G. Gao, J. Med. Chem., 2003, 46, 3775–3777 CrossRef CAS PubMed.
- R. Iannone, L. Miele, P. Maiolino, A. Pinto and S. Morello, Am. J. Cancer Res., 2014, 4, 172 Search PubMed.
- P. A. Beavis, U. Divisekera, C. Paget, M. T. Chow, L. B. John, C. Devaud, K. Dwyer, J. Stagg, M. J. Smyth and P. K. Darcy, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(36), 14711–14716 CrossRef CAS PubMed.
- D. Jin, J. Fan, L. Wang, L. F. Thompson, A. Liu, B. J. Daniel, T. Shin, T. J. Curiel and B. Zhang, Cancer Res., 2010, 70, 2245–2255 CrossRef CAS PubMed.
- P. A. Beavis, N. Milenkovski, M. A. Henderson, L. B. John, B. Allard, S. Loi, M. H. Kershaw, J. Stagg and P. K. Darcy, Cancer Immunol. Res., 2015, 3, 506–517 CrossRef CAS PubMed.
- P. G. Baraldi, S. Manfredini, D. Simoni, L. Zappaterra, C. Zocchi, S. Dionisotti and E. Ongini, Bioorg. Med. Chem. Lett., 1994, 4, 2539–2544 CrossRef CAS.
- S. Willingham, P. Ho, R. Leone, C. Choy, J. Powell, I. McCaffery and R. Miller, Ann. Oncol., 2016, 27, vi366 CrossRef.
- I. McCaffery, G. Laport, A. Hotson, S. Willingham, A. Patnaik, M. Beeram and R. Miller, Ann. Oncol., 2016, 27, vi124 CrossRef.
- K. N. Venugopala, Pharmaceuticals, 2022, 15, 1475 CrossRef CAS PubMed.
- A. Churov and G. Zhulai, Hum. Immunol., 2021, 82, 270–278 CrossRef CAS PubMed.
- M. Mediavilla-Varela, J. Castro, A. Chiappori, D. Noyes, D. C. Hernandez, B. Allard, J. Stagg and S. J. Antonia, Neoplasia, 2017, 19, 530–536 CrossRef CAS PubMed.
- S. Merighi, E. Battistello, L. Giacomelli, K. Varani, F. Vincenzi, P. A. Borea and S. Gessi, Expert Opin. Ther. Targets, 2019, 23, 669–678 CrossRef CAS PubMed.
- K. E. de Goede, A. J. M. Driessen and J. Van den Bossche, Biology, 2020, 9, 380 CrossRef CAS PubMed.
- P. K. Deb, P. Maity, B. Sarkar, K. N. Venugopala, R. K. Tekade and S. Batra, ACS Pharmacol. Transl. Sci. Search PubMed.
- S. Subudhi, G. S. Falchook, M. A. Salkeni, A. El-Khoueiry, J. Grewal, W. Tester, R. Pachynski, S. Upadhaya, A. R. S. Ibanez and S. Kumar, BMJ Specialist Journals, 2023 Search PubMed.
- A. Chiappori, C. Williams, B. Creelan, T. Tanvetyanon, J. Gray, E. Haura, D. T. Chen, R. Thapa, A. Beg and T. Boyle, J. Thorac. Oncol., 2018, 13, S538 CrossRef.
- L. C. Harshman, M. Chu, S. George, B. G. M. Hughes, B. C. Carthon, L. Fong, J. R. Merchan, L. Kwei, A. N. Hotson, M. Mobasher and R. A. Miller, J. Clin. Oncol., 2020, 129 Search PubMed.
- J. Powderly, A. Spira, R. Gutierrez, D. DiRenzo, A. Udyavar, J. J. Karakunnel, A. Rieger, J. Colabella, D. W. Lai and P. de Souza, Ann. Oncol., 2019, 30, v493 CrossRef.
- J. Zhang, W. Yan, W. Duan, K. Wüthrich and J. Cheng, Pharmaceuticals, 2020, 13, 237 CrossRef CAS PubMed.
- E. A. Lim, J. C. Bendell, G. S. Falchook, T. M. Bauer, C. G. Drake, J. H. Choe, D. J. George, J. L. Karlix, S. Ulahannan and K. F. Sachsenmeier, Clin. Cancer Res., 2022, 28, 4871–4884 CrossRef CAS PubMed.
- O. M. Abo-Salem, A. M. Hayallah, A. Bilkei-Gorzo, B. Filipek, A. Zimmer and C. E. Müller, J. Pharmacol. Exp. Ther., 2004, 308, 358–366 CrossRef CAS PubMed.
- G. Schneider, T. Glaser, C. Lameu, A. Abdelbaset-Ismail, Z. P. Sellers, M. Moniuszko, H. Ulrich and M. Z. Ratajczak, Mol. Cancer, 2015, 14, 1–15 CrossRef PubMed.
- R. Dungo and E. D. Deeks, Drugs, 2013, 73, 875–882 CrossRef CAS PubMed.
- P. K. Deb, B. Chandrasekaran, R. Mailavaram, R. K. Tekade and A. M. Y. Jaber, Drug Discovery Today, 2019, 24, 1854–1864 CrossRef CAS PubMed.
- D. Zeng, T. Maa, U. Wang, I. Feoktistov, I. Biaggioni and L. Belardinelli, Drug Dev. Res., 2003, 58, 405–411 CrossRef CAS.
- A. Jemal, R. Siegel, J. Xu and E. Ward, Ca-Cancer J. Clin., 2010, 60, 277–300 CrossRef PubMed.
- D. Mittal, D. Sinha, D. Barkauskas, A. Young, M. Kalimutho, K. Stannard, F. Caramia, B. Haibe-Kains, J. Stagg and K. K. Khanna, Cancer Res., 2016, 76, 4372–4382 CrossRef CAS PubMed.
- A. W.-H. Cheung, J. Brinkman, F. Firooznia, A. Flohr, J. Grimsby, M. Lou Gubler, K. Guertin, R. Hamid, N. Marcopulos and R. D. Norcross, Bioorg. Med. Chem. Lett., 2010, 20, 4140–4146 CrossRef CAS PubMed.
- W. Kaji, S. Tanaka, M. Tsukimoto and S. Kojima, J. Toxicol. Sci., 2014, 39, 191–198 CrossRef CAS PubMed.
- L. Seitz, L. Jin, M. Leleti, D. Ashok, J. Jeffrey, A. Rieger, R. G. Tiessen, G. Arold, J. B. L. Tan and J. P. Powers, Invest. New Drugs, 2019, 37, 711–721 CrossRef CAS PubMed.
- J. V Evans, S. Suman, M. U. L. Goruganthu, E. E. Tchekneva, S. Guan, R. R. Arasada, A. Antonucci, L. Piao, I. Ilgisonis and A. A. Bobko, JNCI, J. Natl. Cancer Inst., 2023, 115, 1404–1419 CrossRef PubMed.
- K. A. Jacobson, A. M. Klutz, D. K. Tosh, A. A. Ivanov, D. Preti and P. G. Baraldi, Medicinal Chemistry of the A3 Adenosine Receptor: Agonists, Antagonists, and Receptor Engineering, Adenosine Receptors in Health and Disease, 2009, pp. 123–159 Search PubMed.
- C. Gallo-Rodriguez, X. Ji, N. Melman, B. D. Siegman, L. H. Sanders, J. Orlina, B. Fischer, Q. Pu and M. E. Olah, J. Med. Chem., 1994, 37, 636–646 CrossRef CAS PubMed.
- H. O. Kim, X. Ji, S. M. Siddiqi, M. E. Olah, G. L. Stiles and K. A. Jacobson, J. Med. Chem., 1994, 37, 3614–3621 CrossRef CAS PubMed.
- P. Fishman, K. A. Jacobson, A. Ochaion, S. Cohen and S. Bar-Yehuda, Immunol., Endocr. Metab. Agents Med. Chem., 2007, 7, 298–303 CrossRef CAS PubMed.
- B. V Joshi and K. A. Jacobson, Curr. Top. Med. Chem., 2005, 5, 1275–1295 CrossRef PubMed.
- S. Bar-Yehuda, L. Madi, D. Silberman, S. Gery, M. Shkapenuk and P. Fishman, Neoplasia, 2005, 7, 85–90 CrossRef CAS PubMed.
- S. Merighi, A. Benini, P. Mirandola, S. Gessi, K. Varani, E. Leung, S. Maclennan and P. A. Borea, J. Biol. Chem., 2005, 280, 19516–19526 CrossRef CAS PubMed.
- J. Lu, A. Pierron and K. Ravid, Cancer Res., 2003, 63, 6413–6423 CAS.
- G. Ohana, S. Bar-Yehuda, A. Arich, L. Madi, Z. Dreznick, L. Rath-Wolfson, D. Silberman, G. Slosman and P. Fishman, Br. J. Cancer, 2003, 89, 1552–1558 CrossRef CAS PubMed.
- G. D. Kim, J. Oh, L. S. Jeong and S. K. Lee, Biochem. Biophys. Res. Commun., 2013, 437, 79–86 CrossRef CAS PubMed.
- S. Cohen, S. M. Stemmer, G. Zozulya, A. Ochaion, R. Patoka, F. Barer, S. Bar-Yehuda, L. Rath-Wolfson, K. A. Jacobson and P. Fishman, J. Cell. Physiol., 2011, 226, 2438–2447 CrossRef CAS PubMed.
- E.-J. Lee, H.-Y. Min, H.-J. Chung, E.-J. Park, D.-H. Shin, L. S. Jeong and S. K. Lee, Biochem. Pharmacol., 2005, 70, 918–924 CrossRef CAS PubMed.
- H. Chung, J.-Y. Jung, S.-D. Cho, K.-A. Hong, H.-J. Kim, D.-H. Shin, H. Kim, H. O. Kim, D. H. Shin and H. W. Lee, Mol. Cancer Ther., 2006, 5, 685–692 CrossRef CAS PubMed.
- K. Nakamura, N. Yoshikawa, Y. U. Yamaguchi, S. Kagota, K. Shinozuka and M. Kunitomo, Anticancer Res., 2006, 26, 43–47 CAS.
- K. Nakamura, K. Shinozuka and N. Yoshikawa, J. Pharmacol. Sci., 2015, 127, 53–56 CrossRef CAS PubMed.
- C. C. Blad, J. K. von Frijtag Drabbe Künzel, H. de Vries, T. Mulder-Krieger, S. Bar-Yehuda, P. Fishman and A. P. IJzerman, Purinergic Signalling, 2011, 7, 453–462 CrossRef CAS PubMed.
- E. Polycarpou, L. B. Meira, S. Carrington, E. Tyrrell, H. Modjtahedi and M. A. Carew, Mol. Nutr. Food Res., 2013, 57, 1708–1717 CrossRef CAS PubMed.
- V. Aires, E. Limagne, A. K. Cotte, N. Latruffe, F. Ghiringhelli and D. Delmas, Mol. Nutr. Food Res., 2013, 57, 1170–1181 CrossRef CAS PubMed.
- B. M. Ayoub, Y. M. Attia, M. S. Ahmed and J. Enzyme, Inhib. Med. Chem., 2018, 33, 858–866 CrossRef CAS PubMed.
- S. Gessi, S. Merighi, K. Varani, E. Leung, S. Mac Lennan and P. A. Borea, Pharmacol. Ther., 2008, 117, 123–140 CrossRef CAS PubMed.
- Z. Gao, B.-S. Li, Y.-J. Day and J. Linden, Mol. Pharmacol., 2001, 59, 76–82 CAS.
- L. M. Kreckler, T. C. Wan, Z.-D. Ge and J. A. Auchampach, J. Pharmacol. Exp. Ther., 2006, 317, 172–180 CrossRef CAS PubMed.
- Z.-G. Gao, R. R. Suresh and K. A. Jacobson, Purinergic Signalling, 2021, 17, 737–746 CrossRef CAS PubMed.
- H. Kim, J. W. Kang, S. Lee, W. J. Choi, L. S. Jeong, Y. Yang, J. T. Hong and Y. Do Young, Anticancer Res., 2010, 30, 2823–2830 CAS.
- A. Spinaci, M. Buccioni, D. Dal Ben, F. Maggi, G. Marucci, B. Francucci, G. Santoni, C. Lambertucci and R. Volpini, Pharmaceuticals, 2022, 15, 164 CrossRef CAS PubMed.
- S. Federico, M. Persico, L. Trevisan, C. Biasinutto, G. Bolcato, V. Salmaso, T. Da Ros, T. Gianferrara, F. Prencipe and S. Kachler, ChemMedChem, 2023, 18, e202300299 CrossRef CAS PubMed.
- K. A. Jacobson, K.-S. Park, J.-L. Jiang, Y.-C. Kim, M. E. Olah, G. L. Stiles and X.-D. Ji, Neuropharmacology, 1997, 36, 1157–1165 CrossRef CAS PubMed.
- G. Kim, X. Hou, W. S. Byun, G. Kim, D. B. Jarhad, G. Lee, Y. E. Hyun, J. Yu, C. S. Lee and S. Qu, J. Med. Chem., 2023, 66, 12249–12265 CrossRef CAS PubMed.
- C. Sun, B. Wang and S. Hao, Front. Immunol., 2022, 13, 837230 CrossRef CAS PubMed.
- S. B. Willingham, P. Y. Ho, A. Hotson, C. Hill, E. C. Piccione, J. Hsieh, L. Liu, J. J. Buggy, I. McCaffery and R. A. Miller, Cancer Immunol. Res., 2018, 6, 1136–1149 CrossRef CAS PubMed.
- R. A. Hodgson, R. Bertorelli, G. B. Varty, J. E. Lachowicz, A. Forlani, S. Fredduzzi, M. E. Cohen-Williams, G. A. Higgins, F. Impagnatiello and E. Nicolussi, J. Pharmacol. Exp. Ther., 2009, 330, 294–303 CrossRef CAS PubMed.
- A. Borodovsky, Y. Wang, M. Ye, J. C. Shaw, K. F. Sachsenmeier, N. Deng, K. J. DelSignore, A. J. Fretland, J. D. Clarke and R. J. Goodwin, Cancer Res., 2017, 77, 5580 CrossRef.
- L. Buisseret, S. Rottey, J. S. De Bono, A. Migeotte, B. Delafontaine, T. Manickavasagar, C. Martinoli, N. Wald, M. Rossetti, E. A. Gangolli and E. Wiegert, J. Clin. Oncol., 2021, 2562 Search PubMed.
- D. R. E. Pastore, K. Mookhtiar, B. Schwartz, S. Kumar, R. Nagaraj and A. V Meru, Cancer Res., 2022, 82, 3454 CrossRef.
- C. Xia, S. Yin, K. K. W. To and L. Fu, Mol. Cancer, 2023, 22, 44 CrossRef CAS PubMed.
- K. Sachsenmeier, E. Sult, C. Hay and E. Poon, Therapeutic combinations comprising anti-cd73 antibodies and uses thereof, US Pat., US14/937565, MedImmune Ltd, 2016 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |