
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, characterization and mechanistic studies of an activated carbon-supported nickel (Ni/C) catalyst for the effective amidation of aldehydes†
Mohammad Kamrojjaman a,
Ashutosh Nathb,
Rifat Jahanc,
Mehdi Ahmed Bhuiyanc,
Shakhawat Ullah Siyamc and
Md Ayub Ali*ad
a,
Ashutosh Nathb,
Rifat Jahanc,
Mehdi Ahmed Bhuiyanc,
Shakhawat Ullah Siyamc and
Md Ayub Ali*ad
aDepartment of Chemistry, Bangladesh University of Engineering and Technology (BUET), Dhaka, Bangladesh. E-mail: shuvro070@chem.buet.ac.bd
bDepartment of Chemistry, University of Massachusetts Boston, MA 02125, USA
cDepartment of Chemistry, Govt. Hazi Muhammad Mohsin College, Chattogram, Bangladesh
dDepartment of Chemistry, University of Iowa, Iowa City, IA 52242-1294, USA
First published on 30th May 2025
Abstract
A novel reusable activated carbon-supported nickel (Ni/C) catalyst was prepared, characterized and used as a heterogeneous catalyst for the synthesis of amide compounds through the amidation of aldehydes with amines. Mechanistic studies were also performed. This heterogeneous catalyst was reusable and could be employed three times without any notable degradation in its catalytic efficacy. Mechanistic studies showed that the catalytic approach efficiently activated both the carbonyl group and the N–H bond. In addition, Gaussian 16 software was employed to perform density functional theory (DFT) calculations to determine the interactions between the hydrogen and nickel in the transition state and the energy of the transition complexes in the reactions.
Introduction
Amides are well known and have attracted significant attention for their tremendous application in pharmaceuticals, agrochemicals, and in other chemical industries (Fig. 1).1,2 About 25% of the available drugs contain amide bonds.3 Usually, amides are synthesized from various amines through the reactions of carboxylic acid derivatives,4 alcohols,5 and aldehydes6 and the hydration of nitriles,7 which often require stoichiometric amounts of coupling reagents and produce toxic chemical wastes.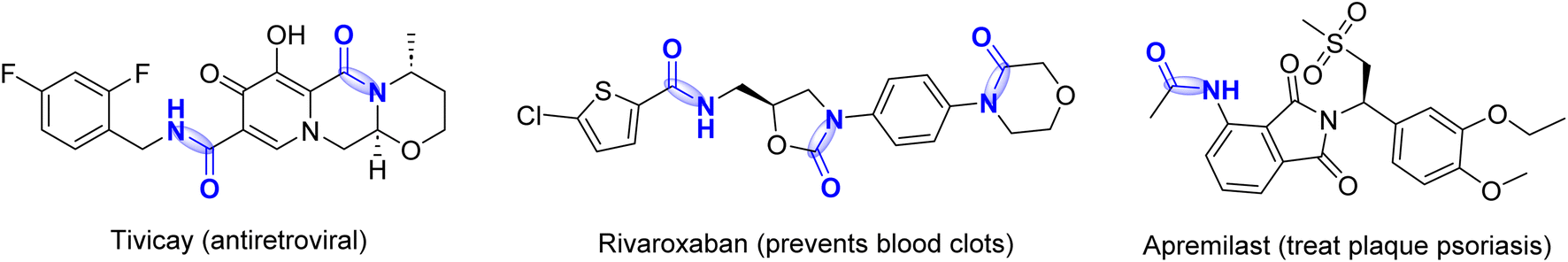 | ||
| Fig. 1 Some FDA-approved drugs bearing amide bonds (blue). | ||
These conventional methods typically suffer from a low atom efficiency and produce a large number of byproducts. It is generally accepted that the catalytic synthesis of amides from readily available starting materials is a priority area for the pharmaceutical industry.8 Homogeneous catalysts, such as Ni-complexes9 and Ru-complexes,10 have been developed for amidation reactions. However, employing these homogeneous catalysts leads to difficulties in catalyst/product separation and catalyst reuse.11 On the other hand, heterogeneous catalysts, such as hydrotalcite-supported gold nanoparticles (Au/HTs)12 and alumina-supported silver cluster (Ag/Al2O3),13 were used to synthesize amides via the dehydrogenation of alcohols with amines. Nickel, supported on microwave-activated Ni/carbon (Ni/CSC),14 was used as a catalyst for the selective hydrogenation of nitrobenzene to cyclohexylamine. Most recently, Zhang et al. reported the amidation of aldehydes with hydroxyl amines using TBAF·3H2O as a promotor. In their study, they used an equivalent (1.5 eq.) amount of TBAF·3H2O in the presence of KOH (Scheme 1).15
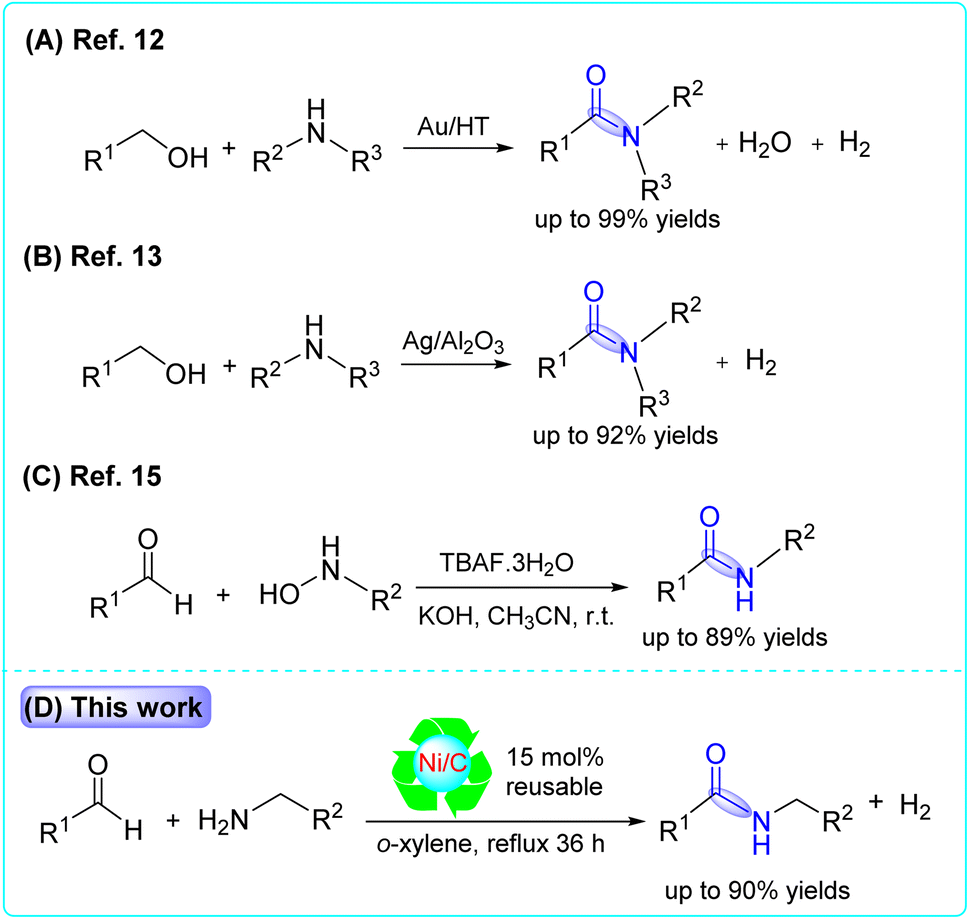 | ||
| Scheme 1 Some synthetic methods for the preparation of amide bonds (blue). | ||
However, these reported methods showed limited substrate scope and were applied only for one reaction. So, it is highly desired to develop a new heterogeneous catalytic method for the synthesis of amides by the coupling of aldehydes and amines followed by a dehydrogenation reaction. In this work, a reusable new heterogeneous activated carbon-supported nickel (Ni/C) catalyst was prepared by pouring the support (activated carbon) over nickel chloride solution for the synthesis of amides from aldehydes and amines.
Results and discussion
For the catalyst preparation, the supported precursor was prepared by pouring the support (activated carbon) over nickel chloride solution with an appropriate concentration. After 15 min of rotation, the mixture was heated and dried at 383 K for 1 h. The reduction of the supported precursors was performed in a two-necked reaction flask fitted with a reflux condenser in a sand bath for heating. The reaction mixture was heated at 353 K under continuous stirring in an excess hydrazine solution to maintain the pH at a desired value of 9 to 11. After reduction the solid obtained was filtrated off, washed with distilled water and dried to obtain the expected Ni/AC catalyst. Then, it was characterized using several characterization techniques such as scanning electron microscopy (SEM), energy dispersive X-ray (XRD) spectroscopy and X-ray diffraction. The surface morphology of the Ni and supported Ni was investigated by SEM, as shown in Fig. 2a–d. Fig. 2a shows the pure nickel nanoparticles were spherical with an average size of 53 nm. Ni/C had a similar average size of 53 nm, as shown in Fig. 2b, demonstrating that the carbon support did not appreciably change the particle size at this ratio. Fig. 2c shows that a separate Ni/C sample had bigger particles averaging 121 nm, suggesting aggregation or increased Ni loading. Fig. 2d shows Ni/Al2O3 with a distinct, distributed morphology with an average particle size of 67 nm, indicating its good distribution over the alumina surface. Overall, the catalyst composition and support material can affect the morphology and particle size, and thereby can affect the catalytic performance. | ||
| Fig. 2 (a–d) SEM images of the Ni and supported Ni catalysts. | ||
Further elemental analysis of the nickel and supported nickel catalysts was performed by energy dispersive X-ray (EDX) spectroscopy. Fig. 3a shows the results for 100% nickel. The EDX patterns for the nickel–carbon systems with an increasing Ni concentration (10.4% and 13.36%, respectively) are shown in Fig. 3b and c. Fig. 3d shows the EDX pattern for a nickel–alumina composite with Al (23.87%) and O (67.02%). Nickel incorporates in to the carbon or alumina matrices for diverse material applications.
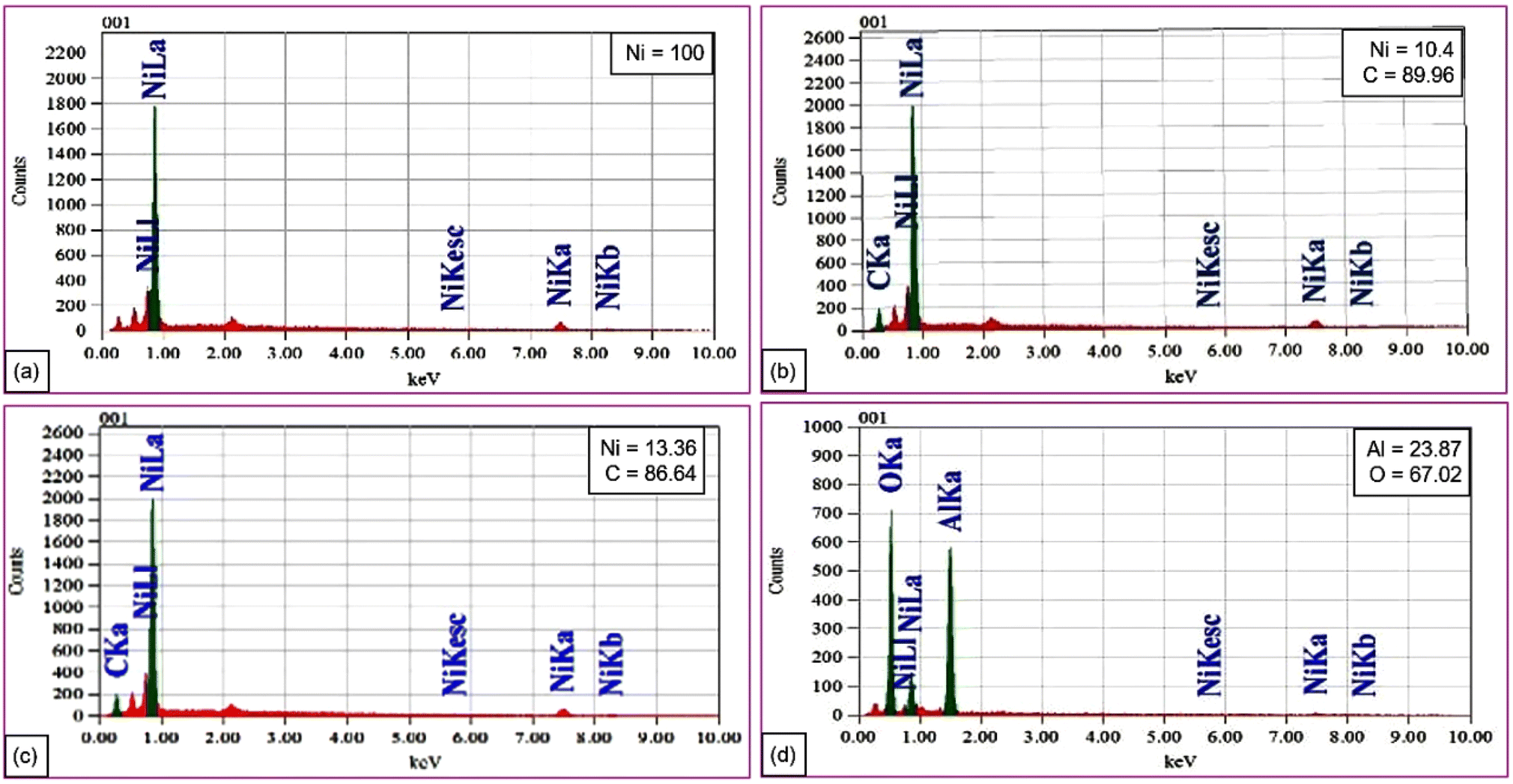 | ||
| Fig. 3 (a–d) EDX spectral analysis of the Ni and supported Ni catalysts. | ||
The XRD pattern of the 10% Ni/C catalyst is shown in Fig. 4. Characteristic peaks for activated carbon 2θ = 23.5° and for Ni metal (2θ = 44.3°, 51.6°, and 75.7°) were observed for the 10% Ni/C catalyst, in which the peak at 23.5° was attributed to the [003] diffraction peak of AC and three peaks at 44.3°, 51.6°, and 75.7° were attributed to the [111], [200], [220] diffraction peaks of Ni0. The results show that the structure of the AC had not been destroyed while the majority of Ni2+ was reduced to Ni0 during the preparation. The average crystalline size of the 10% Ni/C catalyst particles was 27 nm.
 | ||
Fig. 4 XRD pattern of the 10% Ni/C (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12:12 reduction ratio) catalyst. :12:12 reduction ratio) catalyst. | ||
The nickel and supported Ni catalysts had great magnetic properties due to their highly magnetic Ni particles.16 We could observe the magnetic properties of the catalysts and proved that the Ni2+ was converted to Ni0. This magnetic property enables the easy separation of the catalyst from the reaction mixture.
In this study, two reactants, namely benzaldehyde 1 and benzylamine 2, were added to a round-bottom flask in a 1:2 ratio to the Ni/C catalyst in o-xylene solvent (Scheme 2). This reaction mixture was heated at 140 °C for 36 h with continuous stirring using a magnetic bar. The reaction progress and completion of the reaction was monitored by thin-layer chromatography (TLC) with appropriate solvents. Once the reaction was completed, the catalyst was separated from the mixture by centrifugation, washed with acetone, and dried at 90 °C for 3 h. The recovered catalyst could be reused in several cycles without a significant reduction in product yield. Through the use of a rotatory evaporator, the solvent was removed from the mixture. Product 3 was purified by column chromatography separation using an ethyl acetate:n-hexane (70:30) solvent. The synthesized pure amide products were identified by FT-IR, 1H NMR, and 13C NMR.
 | ||
| Scheme 2 Synthesis of amide 3. | ||
Synthesis of N-benzyl benzamide 3a
Molecular weight: 212 g mol−1; molecular formula: C14NOH13; solubility: soluble in chloroform; melting point: 104–107 °C; FT-IR (ν KBr): 3457.52, 3063.06, 2850.88, 1644.37, 1601.93, 1550.82, 1454.38 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.074–8.076 (m, 2H, C-3), 7.390–7.458 (m, 3H, C-2), 7.227–7.287 (m, 5H, C-1), 5.279 (s, 1H, –NH, N-5), 4.038 (S, 2H, C-4); 13C-NMR (100 MHz, CDCl3): δ 170.62 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 1C, C-9), 134.31 (1C, C-8), 131.46 (1C, C-7), 128.81 (2C, C-1, C-6), 128.75 (4C, C-4, C-5), 127.04 (4C, C-2, C-3), 50.70 (1C, C-9); GC-MS (CDCl3): the GC-MS spectrum exhibited a retention time of 7.091 min; mass spectrum (in m/z): molecular ion peak (m/z): 211, base peak (m/z): 105 and others (m/z) at 91, 77.
O, 1C, C-9), 134.31 (1C, C-8), 131.46 (1C, C-7), 128.81 (2C, C-1, C-6), 128.75 (4C, C-4, C-5), 127.04 (4C, C-2, C-3), 50.70 (1C, C-9); GC-MS (CDCl3): the GC-MS spectrum exhibited a retention time of 7.091 min; mass spectrum (in m/z): molecular ion peak (m/z): 211, base peak (m/z): 105 and others (m/z) at 91, 77.
Synthesis of N-phenyl benzamide 3b
Molecular weight: 197 g mol−1; molecular formula: C13NOH11; solubility: soluble in chloroform; melting point: 162–164 °C; FT-IR (ν KBr): 3450, 3063, 1650, 1529.29, 1550.82, 1628.94, 1183.70 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.598 (s, 1H, –NH, N-7), 8.119–8.126 (m, 2H, C-4), 7.875–7.885 (m, 2H, C-3), 7.440–7.611 (m, 3H, C-5, C-6), 7.250–7.421 (m, 2H, C-2), 6.709–6.720 (3, 1H, C-1); 13C-NMR (100 MHz, CDCl3): δ 167.193 (1C, CO, C-5), 138.909 (1C, C-4), 136.012 (1C, C-6), 131.801 (1C, C-9), 127.383–129.438 (4C, C-2, C-8) 126.10 (2C, C-7), 124.80 (1C, C-1), 121.30 (2C, C-3).
Both the solvent and catalyst composition had a considerable impact on the yield, according to the optimization study for the amidation reaction that yielded compound 3a (Table 1). The best solvent–catalyst combination was o-xylene with Ni/C (1:12:12), which produced the highest yield (90%, entry 2) out of all the circumstances evaluated. No product was produced when there was no solvent or catalyst present (entries 1 and 6). Moderate to low yields were obtained from other solvents, such as toluene, triethylamine, and benzene (entries 3–5). The typical Ni/C (1:12:12) formulation in o-xylene was the most effective condition for this transformation, as seen by the lower yields obtained from the different catalyst, compositions, such as utilizing Ni alone, Ni/GO, or modified Ni/C ratios (entries 7–11).
|
|||
|---|---|---|---|
| Entry | Solvent | Catalyst (reduction ratio) | % yield |
| 1 | No solvent | Ni/C (1:12:12) |
0 |
| 2 | o-Xylene | Ni/C (1:12:12) |
90 |
| 3 | Benzene | Ni/C (1:12:12) |
20 |
| 4 | Triethylamine | Ni/C (1:12:12) |
14 |
| 5 | Toluene | Ni/C (1:12:12) |
29 |
| 6 | o-Xylene | No catalyst | 0 |
| 7 | o-Xylene | Ni (1:12:12) |
13 |
| 8 | o-Xylene | Ni/GO (1:12:12) |
34 |
| 9 | o-Xylene | Ni/C (1:4:4) |
28 |
| 10 | o-Xylene | Ni/C (1.35:4.5:3.6) |
14 |

The reusability of the Ni/C catalyst was checked for the reaction of benzaldehyde with benzylamine under the optimized conditions. After the reaction, the catalyst was separated by centrifugation, washed with acetone, and then dried in an oven at 110 °C for 3 h. The recovered Ni/C catalyst was reused for three cycles without any significant loss of catalytic activity. However, after the three cycles, there was a noticeable decrease in the product yield, as shown in Table S5.†
To understand and determine the mechanism of the amidation of an aldehyde group with an amine group, a probable mechanism is proposed. Then the proposed scheme was tested using DFT calculations to crossmatch the ideas with the energy diagram of some common reaction mechanisms, from which it could be observed if the rise and fall of the energy cliff of those transitory states satisfied the expected curves or not. All the relevant information is provided in the following sections with the necessary graphs.
In the proposed mechanism, benzaldehyde reacts with benzylamine in the presence of a nickel-on-carbon (Ni/C) catalyst under reflux in o-xylene, leading to the formation of N-benzyl benzamide as the final product along with the release of hydrogen gas.
The Ni/C catalyst likely plays a crucial role in facilitating the reaction by acting as a hydrogenation agent.17 The mechanism can be divided into several steps, as shown in Scheme 3. Initially, benzaldehyde and benzylamine undergo condensation to form an imine intermediate. This imine is then reduced by the Ni/C catalyst, which provides the necessary hydrogen to convert the imine into the amine.
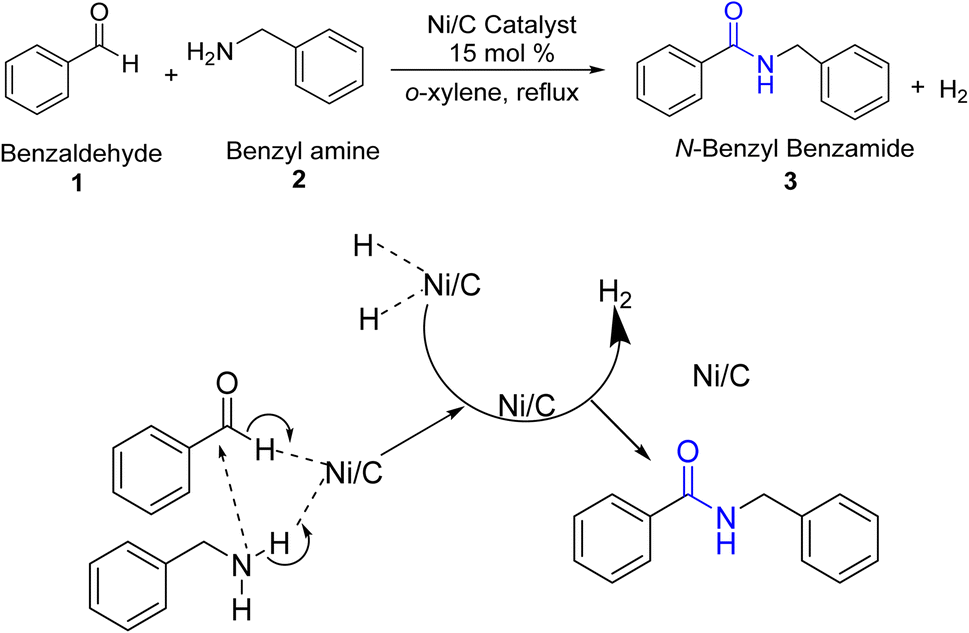 | ||
| Scheme 3 Proposed mechanism for the synthesis of amide 3a. | ||
The energy diagram in Fig. 5 illustrates the energy profile of this catalytic pathway, identifying the key transition states (TS1, TS2, TS3, and TS4) and intermediates. The starting materials (SM1 and SM2) combine to reach a higher energy transition state (TS1), which is associated with the condensation step. This is followed by subsequent steps that lead to further increases in energy at TS2 and TS3, likely corresponding to hydrogen transfer and amide bond formation, respectively. Finally, at TS4, the system transitions to the product formation step, where the energy decreases significantly as the stable N-benzyl benzamide is formed.
 | ||
| Fig. 5 Relative optimized energy plot of amide 3a. | ||
Each transition state represents an energetic barrier that must be overcome for the reaction to proceed, and the role of the Ni/C catalyst in stabilizing these intermediates and facilitating hydrogen transfer is crucial. The energy diagram suggests that the most challenging step energetically may be associated with TS2, indicating that hydrogenation and formation of the amide bond are likely the rate-determining steps of the reaction. The presence of multiple energy barriers signifies a stepwise mechanism, with each transition state marking a critical transformation in the reaction pathway.18
Overall, the Ni/C catalyst enabled the direct transformation of benzaldehyde and benzylamine into the amide product by providing hydrogen and facilitating the required bond rearrangements, resulting in a straightforward synthetic route to obtain benzamides from simple starting materials.
Conclusion
This study presents an effective and sustainable approach for amide synthesis via the coupling19 of aldehydes and amines, facilitated by a reusable Ni/C catalyst. The Ni/C catalyst efficiently promoted the process, demonstrating elevated catalytic activity20 and stability across numerous cycles without any substantial performance degradation. The results highlight the benefits of heterogeneous catalysis,21 including the facile separation and reusability of the catalyst, which are advantageous for industrial applications. Furthermore, computational analysis elucidated the reaction mechanism, emphasizing the function of the Ni/C catalyst in stabilizing the transition states and diminishing the activation energy. This method offers a viable avenue for the synthesis of green amides, with significant advantages for pharmaceutical and chemical production.Data availability
The datasets generated and/or analyzed in this current study are available from the corresponding author upon reasonable request.Author contributions
MK and AN: writing – original draft, review & editing, visualization, software, formal analysis, data curation, software, resources. MAA: writing – review & editing, resources, methodology, investigation, supervision. RJ, MAB and SUS: software, methodology, review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We are grateful to the Bangladesh University of Engineering and Technology (BUET), Dhaka, Dhaka-1000, Bangladesh, for all kinds of experimental and financial supports.References
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS PubMed.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
- R. M. Lanigan, P. Starkov and T. D. Sheppard, J. Org. Chem., 2013, 78, 4512–4523 CrossRef CAS PubMed.
- R. M. Lanigan and T. D. Sheppard, Eur. J. Org Chem., 2013, 2013, 7453–7465 CrossRef CAS.
- L. U. Nordstrøm, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672–17673 CrossRef PubMed.
- J. Liang, J. Lv and Z.-c. Shang, Tetrahedron, 2011, 67, 8532–8535 CrossRef CAS.
- J. H. Hall and M. Gisler, J. Org. Chem., 1976, 41, 3769–3770 CrossRef CAS.
- D. J. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. Leazer, L. Johnnie, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman and A. Wells, Green Chem., 2007, 9, 411–420 RSC.
- E. L. Baker, M. M. Yamano, Y. Zhou, S. M. Anthony and N. K. Garg, Nat. Commun., 2016, 7, 11554 CrossRef PubMed.
- S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS.
- K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196–4197 CrossRef CAS PubMed.
- J. Zhu, Y. Zhang, F. Shi and Y. Deng, Tetrahedron Lett., 2012, 53, 3178–3180 CrossRef CAS.
- K. i. Shimizu, K. Ohshima and A. Satsuma, Chem.–Eur. J., 2009, 15, 9977–9980 CrossRef CAS PubMed.
- X. Lu, J. He, R. Jing, P. Tao, R. Nie, D. Zhou and Q. Xia, Sci. Rep., 2017, 7, 2676 CrossRef PubMed.
- H. Zhang, N. Xu, B. Su, J. Zhang, C. Zhang, Z. Zhang, B. Guo, S. Xu, S. Wang and R. Tang, J. Org. Chem., 2024, 7579–7590 CrossRef CAS PubMed.
- Z. Ji, X. Shen, G. Zhu, H. Zhou and A. Yuan, J. Mater. Chem., 2012, 22, 3471–3477 RSC.
- M. Fan, H. Tian, Y. Shao, L. Zhang, S. Zhang, G. Hu and X. Hu, J. Environ. Chem. Eng., 2023, 11, 110013 CrossRef CAS.
- A. C. West, M. W. Schmidt, M. S. Gordon and K. Ruedenberg, J. Phys. Chem. A, 2015, 119, 10360–10367 CrossRef CAS PubMed.
- V. Arun, K. Mahanty and S. De Sarkar, ChemCatChem, 2019, 11, 2243–2259 CrossRef CAS.
- C. L. Allen, Catalytic Approaches to the Synthesis of Amide Bonds, Doctoral dissertation, University of Bath, 2012.
- C. Bai, X. Yao and Y. Li, ACS Catal., 2015, 5, 884–891 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02647d |
| This journal is © The Royal Society of Chemistry 2025 |