
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid-phase synthesis of aryl squaramides using Liebeskind–Srogl cross-coupling†
Jan Chasák and
Lucie Brulíková *
*
Department of Organic Chemistry, Faculty of Science, Palacký University, 17. listopadu 12, Olomouc, 77146, Czech Republic. E-mail: lucie.brulikova@upol.cz
First published on 2nd May 2025
Abstract
We present a method for the synthesis of aryl-substituted squaramides through the Liebeskind–Srogl cross-coupling reaction performed on solid support. This approach offers a unique application for the cross-coupling reaction, allowing for the rapid and efficient production of a diverse range of substituted analogs within a combinatorial framework. Through our technique, we successfully synthesized derivatives that were previously unattainable. Additionally, the optimized conditions have been effectively applied in a scale-up reaction. The derivatives show potential for the treatment of drug-resistant tuberculosis.
Introduction
Squaramides represent an important category of new antituberculosis agents that gained the attention of medicinal chemists in 2017 following the release of the pioneering study on this subject.1 These compounds inhibit mycobacterial ATP synthase and demonstrate superior efficacy against drug-resistant tuberculosis (Fig. 1).2–7 Moreover, they operate via a distinct mechanism compared to the clinically used bedaquiline, enhancing their potential as promising alternatives. | ||
| Fig. 1 Synthetic squaramides and their potential as antituberculosis agents. The structure of mycobacterial ATP synthase was created using mol* viewer and PDB: 7NJP. | ||
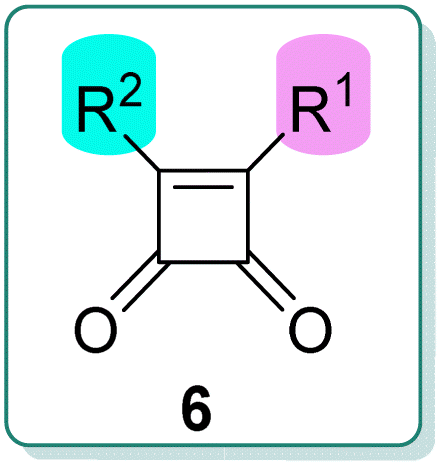
The carbon–carbon bond between the squaric cycle and the aryl substituent is crucial for antituberculosis activity, as shown in Fig. 1. However, synthesizing this bond poses significant challenges. Previous methods have faced various obstacles that hindered the preparation of all required derivatives.1–3 The Friedel–Crafts reaction, for instance, suffered from very low yields and was not applicable to various ranges of substrates.1,2 Alternatively, the Liebeskind–Srogl Pd0/CuI-mediated cross-coupling reaction offered several synthetic advantages, including mild, nonbasic conditions and high specificity for inorganic compounds. Nonetheless, these reactions have proven ineffective with certain boronic acids.3 The solution-phase synthesis of these compounds proved highly dependent on the electronic properties of the boronic acids used in the cross-coupling step. As a result, some derivatives were obtained in very low yields (below 1%), or not at all. For this reason, other methods were sought to allow the use of a wider range of substrates.
In our current research, we have utilized the Liebeskind–Srogl cross-coupling reaction on a solid support. This solid-phase synthetic approach offers many advantages over traditional solution synthesis, particularly in the field of medicinal chemistry. Solid-phase synthesis allows for the rapid and efficient generation of compounds without the necessity of isolating individual intermediates. This makes it a valuable method for developing new drug candidates and conducting extensive structure–activity relationship (SAR) studies. One of the key advantages of solid-phase synthesis is its compatibility with automated or high-throughput screenings. However, many resins used in this type of synthesis, including those studied here, can only work with a limited range of organic solvents. Polar protic solvents, such as alcohol and water, do not swell the resins adequately, which can lead to lower reactivity. Consequently, optimizing solid-phase synthetic sequences can be challenging, as only certain reaction parameters are adjustable. Additionally, the integration of solid-phase synthesis with cross-coupling reactions is relatively rare, making the Liebeskind–Srogl cross-coupling reaction particularly unique.
The Liebeskind–Srogl reaction involves the formation of a new carbon–carbon bond between a thioester and a boronic acid facilitated by a metal catalyst. This cross-coupling reaction follows a distinct mechanism. Initially, Cu(I) coordinates with the thioester, making it more electron-deficient and promoting the efficient oxidative addition of the palladium catalyst. This step is followed by transmetallation and reductive elimination, which are typical phases of cross-coupling reactions, ultimately yielding the final product. Performing the synthesis on a solid phase, where the molecule is attached to a resin, does not negatively affect any of these reaction phases, allowing the reaction to proceed smoothly. On the contrary, the inherent properties of this reaction allow for traceless cleavage from the resin when appropriate solid support is selected, which is particularly beneficial for substituents that may be unstable under the acidic or basic conditions typically used for resin cleavage.
From a medicinal chemistry perspective, it is highly desirable to develop a rapid and efficient method for synthesizing a diverse range of squaramides that can be used for biological testing. This study presents an effective and robust approach to achieving that goal.
Results and discussion
In the pilot studies, we selected two commercially available resins (Scheme 1) that featured suitable functional groups for conversion into the appropriate thioether: aminomethyl resin (AM resin) and Merrifield resin. The subsequent synthetic approach involves substituting the “right” part of the central scaffold with various nucleophiles, followed by a Liebeskind–Srogl cross-coupling reaction, which simultaneously facilitates traceless cleavage from the resin. This process is clearly illustrated in Scheme 1. | ||
| Scheme 1 Design of synthetic pathways using various resins. | ||
Initially, we concentrated on utilizing aminomethyl resin 1 as a solid support, which contains an amino functional group that can be readily substituted with an appropriate linker (Scheme 2). The first step in this reaction sequence involved the introduction of a suitable thio linker, specifically achieved through the acylation of 3-(tritylthio)propionic acid resin. We monitored the progress of this initial step using the bromophenol blue test, which yielded a negative result, confirming that all amino groups on the aminomethyl resin had reacted completely. It should be noted that all yields were calculated based on the loading of the starting aminomethyl resin.
 | ||
Scheme 2 Synthesis of squaramides 6 using aminomethyl resin. aReagents and conditions: (i) 3-(tritylthio)propionic acid, DIC, DCM/DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), rt, 18 h; (ii) TFA/TES (95:5), rt, 1 h; (iii) 3-(tert-butoxy)-4-methoxycyclobut-3-ene-1,2-dione, TEA, DCM, rt, 18 h; (iv) amines (or other nucleophiles), DCM, (TEA – for Nu–H·HCl), rt, 18 h; (v) R2–B(OH)2, CuTC, TFP, Pd2(dba)3, dioxane anhydrous, 50 °C, 18 h. :1), rt, 18 h; (ii) TFA/TES (95:5), rt, 1 h; (iii) 3-(tert-butoxy)-4-methoxycyclobut-3-ene-1,2-dione, TEA, DCM, rt, 18 h; (iv) amines (or other nucleophiles), DCM, (TEA – for Nu–H·HCl), rt, 18 h; (v) R2–B(OH)2, CuTC, TFP, Pd2(dba)3, dioxane anhydrous, 50 °C, 18 h. | ||
In the subsequent stage, the trityl-protecting group was removed, enabling a reaction of intermediate 3 with 3-(tert-butoxy)-4-methoxycyclobut-3-ene-1,2-dione (Scheme 2, synthesis of this precursor was carried out according to the publication by Shinada et al. and is described in ESI†).8 This was followed by reactions involving various nucleophiles, resulting in resin-bound precursors 5. The final step of the reaction sequence involved a cross-coupling reaction carried out in the presence of copper(I) thiophene-2-carboxylate (CuTC), tri(2-furyl)phosphine (TFP), and a Pd catalyst, which led to the cleavage of the product 6 from the resin. The individual steps are discussed in detail below.
To assess the relevance of the key cross-coupling step for the chosen resin, we initiated preliminary studies using the readily available intermediate 5a (see Table 1). At room temperature, the reaction showed only minimal conversion (Table 1, entry 1). Therefore, we raised the reaction temperature; however, even under these conditions, the conversion remained unsatisfactory (see Table 1, entry 2). The use of dioxane as a solvent in the reaction proved to be highly effective, resulting in a satisfactory yield of the desired product 6a, as indicated in Table 1, entry 3. Additionally, attempts to modify the catalyst (see entries 4 and 5) or prolong the reaction time (entry 6) did not yield significant improvements in the overall outcome.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Temperature | Solvent (anhydrous) | Time | Result |
| a The amount of all reagents is related to boronic acid. All components are in excess compared to AM resin (theoretical loading of AM resin (250 mg) is 0.28 mmol).b NI: not isolated, product detected only in trace amount.c NI: not isolated, complex mixture with many side products was formed.d Product isolated from ∼250 mg of resin. | |||||
| 1 | Pd2(dba)3 | RT | THF | 20 h | NIb |
| 2 | Pd2(dba)3 | 50 °C | THF | 20 h | NIb |
| 3 | Pd2(dba)3 | 50 °C | Dioxane | 20 h | 25.3 mg (33%)d |
| 4 | Pd(OAc)2 | 50 °C | Dioxane | 20 h | NIc |
| 5 | Pd(PPh3)4 | 50 °C | Dioxane | 20 h | 21.6 mg (29%)d |
| 6 | Pd2(dba)3 | 50 °C | Dioxane | 44 h | 23.1 mg (30%)d |
The developed methodology enabled us to effectively introduce a range of boronic acids (see Table 2). We achieved successful reactions with electron-rich, electron-poor, and heterocyclic boronic acids. Notably, vinylboronic acid (6m) was coupled successfully. However, the final coupling was less effective with five-membered heterocycles where the –B(OH)2 group was placed at the 2-position of the heterocycle. Furthermore, the reaction was unsuccessful with the sterically hindered 2,6-(dimethylphenyl)boronic acid (6n).
|
||||
|---|---|---|---|---|
| Compdh | R2 | R1 | Yielda (%) | Yieldb (%) |
|
|
|||
| a Yields for AM resin calculated from a theoretical loading of the resin (over five reaction steps).b Yields for Merrifield resin calculated from a theoretical loading of the resin (over three reaction steps).c NI: not isolated, product detected only in trace amount.d NI: not isolated, product was not detected after the coupling cleavage.e NI: not isolated, derivative 6aa was obtained instead.f ND: not determined.g Derivative 6p was previously prepared also via a solution-phase synthesis using Liebeskind–Srogl cross-coupling, achieving an overall yield of 40%.3h Derivatives with the citation noted in the left column have already been prepared in the past by solution-phase synthesis. | ||||
| 6a |  |
 |
33 | 40 |
| 6b |  |
 |
29 | NDf |
| 6c |  |
 |
17 | NDf |
| 6d10 |
 |
 |
32 | NDf |
| 6e |  |
 |
16 | 21 |
| 6f |  |
 |
26 | NDf |
| 6g |  |
 |
18 | NDf |
| 6h |  |
 |
23 | 27 |
| 6i |  |
 |
38 | NDf |
| 6j |  |
 |
NIc | NDf |
| 6k |  |
 |
28 | 35 |
| 6l |  |
 |
NIc | 14 |
| 6m |  |
 |
29 | 41 |
| 6n |  |
 |
NIc | 31 |
| 6o |  |
 |
20 | NDf |
| 6p3,g |
 |
 |
NIc | 50 |
| 6q |  |
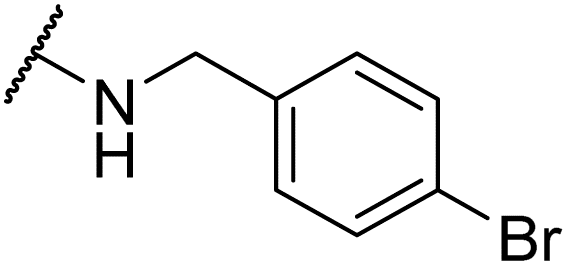 |
20 | 37 |
| 6r |  |
 |
34 | 53 |
| 6s11 |
 |
 |
34 | 49 |
| 6t11 |
 |
 |
NId | 50 |
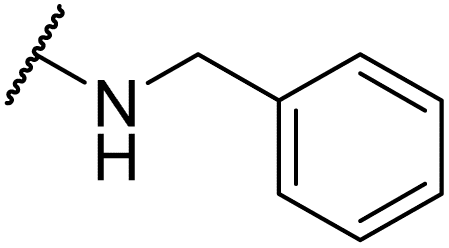
| 6u |  |
 |
NIc | 46 |
| 6v |  |
 |
NId | NId |
| 6w |  |
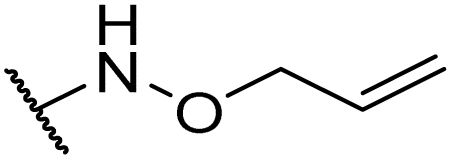 |
NId | NId |
| 6x |  |
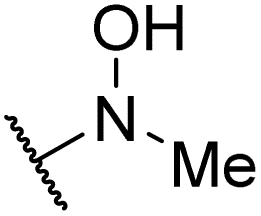 |
NId | NId |
| 6y |  |
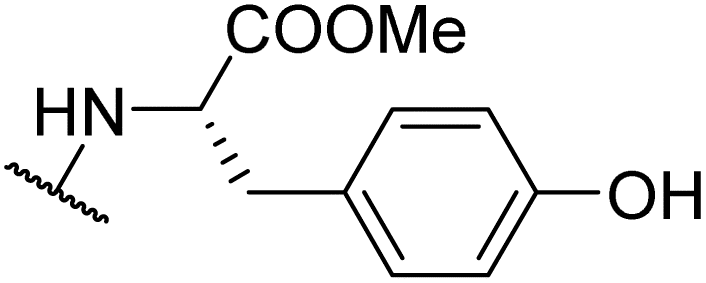 |
33 | 45 |
| 6z |  |
 |
NId | NIc |
| 6aa12 |
 |
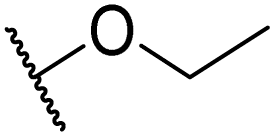 |
NDf | 58 |
| 6ab |  |
 |
NDf | 35 |
| 6ac |  |
 |
NDf | 34 |
| 6ad |  |
 |
NDf | 46 |
| 6ae | 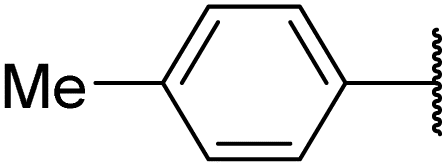 |
 |
NDf | 34 |
| 6af |  |
 |
NDf | 41 |
| 6ag13 |
 |
 |
NDf | 50 |
| 6ah |  |
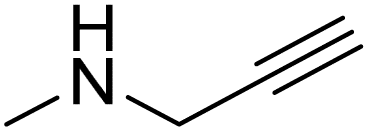 |
NDf | NId |
| 6ai |  |
 |
NDf | 42 |
| 6aj |  |
 |
NDf | 51 |
| 6ak |  |
 |
NDf | NId |
| 6al | 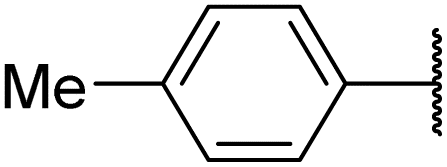 |
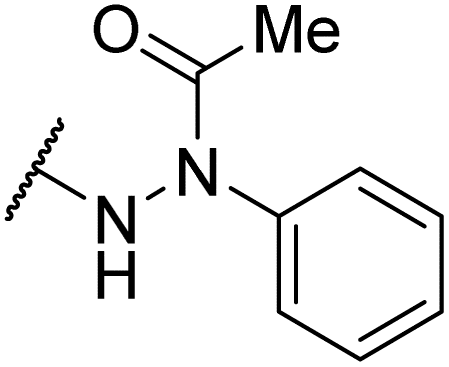 |
NDf | NIe |

Notable limitations of the developed methodology became evident when we attempted to introduce various nucleophiles onto the immobilized four-membered squarate skeleton. Only primary aliphatic amines yielded successful results. In contrast, heterocyclic amines 6p produced only trace amounts of product, while aromatic amines resulted in no detectable final products 6. Even elevating the temperature to 50 °C or changing the solvent to DMF during the substitution reaction did not enhance the outcome. Due to the absence of analysis following the substitution step, whether the challenge originated from the substitution itself or the subsequent coupling reaction remains uncertain. However, the findings suggest that the substitution step is more likely to be problematic, given that previous experiments have demonstrated the versatility and effectiveness of the coupling process.
Notably, the Libeskind–Srogl cross-coupling during the synthesis of derivative 6c surprisingly proceeded as anticipated, resulting in the formation of the final compound. This was achieved despite the presence of an additional sulfur atom in the structure, which could potentially participate in this type of reaction. While we noted higher impurities than reactions involving other substituents, the final compound 6c was still obtained in a slightly lower, yet acceptable, yield. The yields of the final compounds varied between 16% and 38%, primarily influenced by the electronic activation of the boronic acid in the final reaction step (Table 2).
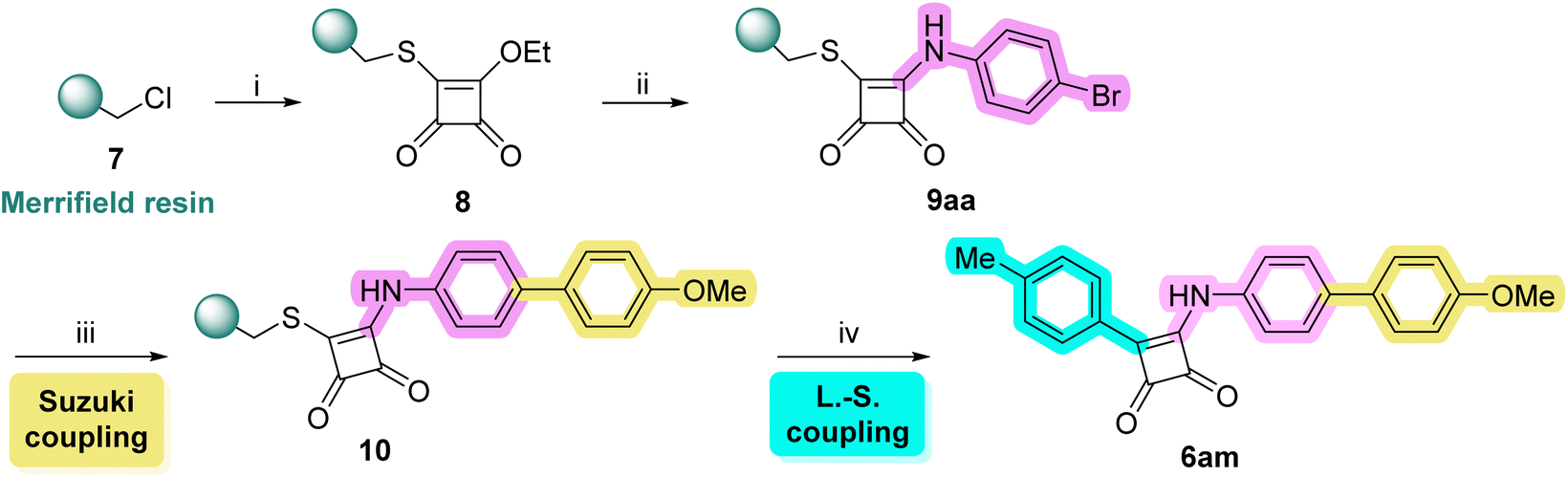
Due to certain limitations, we faced with aminomethyl resin, we were unable to prepare some derivatives 6 (see Table 2). Consequently, we have decided to explore Merrifield resin with a chloromethyl functional group. We aim to thoroughly evaluate the scope and limitations of Merrifield resin, which may deepen our understanding of its capabilities and potentially lead to the development of new synthetic methodologies that could address the challenges we faced previously.
Scheme 3 offers a succinct overview of the synthetic pathway leading to the proposed squaramides 6 performed on Merrifield resin. The initial step of the synthesis involves the straightforward reaction between Merrifield resin and 3-ethoxy-4-mercaptocyclobut-3-ene-1,2-dione (the synthesis of this precursor was carried out according to the publication by Srivastava et al. and is detailed in the ESI†).9 Then, reaction with various nucleophiles followed and the reaction sequence was concluded with a Liebeskind–Srogl cross-coupling reaction providing the final products 6. Similar to the previous case, the reaction sequence does not include a cleavable group that would aid in verifying successful reactions/sufficient conversions throughout the synthesis. As a result, the yields of the final products 6 are calculated based on the theoretical loading of the starting Merrifield resin, as outlined in the experimental section.
 | ||
| Scheme 3 Synthesis of squaramides 6 using Merrifield resin. aReagents and conditions: (i) 3-ethoxy-4-mercaptocyclobut-3-ene-1,2-dione, TEA, DMF, rt, 18 h; (ii) amines (or other nucleophiles), DCM, (TEA – for Nu–H·HCl), rt-50 °C, 18 h; (iii) R2-B(OH)2, CuTC, TFP, Pd2(dba)3, anhydrous dioxane, 50 °C, 18 h. | ||
Squaramides 6 with various aromatic substituents were successfully synthesized utilizing Merrifield resin through the Liebeskind–Srogl cross-coupling reaction (Table 2). Both electron-rich and electron-poor, as well as heteroaromatic boronic acids, were effectively incorporated. As previously observed, the reaction also proceeded successfully with a vinyl analog. Notably, the use of Merrifield resin facilitated the incorporation of 2-thienylboronic acid (6l), which had not been successfully achieved in the earlier synthesis using AM resin. Furthermore, unlike the AM resin approach, we were able to obtain derivative 6n, which contains the sterically hindered 2,6-dimethylphenyl substituent. However, the yields for these two derivatives, 6l and 6n, are relatively low, at 14% and 31% (Table 2).
In our investigations concerning nucleophilic reactions, Merrifield resin demonstrated superior effectiveness compared to the AM resin method. We successfully introduced a range of aliphatic primary and secondary amines onto the central squaric acid scaffold, including specific amino acids (6y and 6ad). The approach also yielded positive results with aromatic amines, even those that featured electron-withdrawing groups on the aromatic ring (6ab). Furthermore, we were able to incorporate various types of hydroxylamines, including N-substituted and N,O-disubstituted derivatives (6ae, 6af). It is noteworthy that the reaction conditions varied based on the nucleophiles employed. Aliphatic amines reacted efficiently at room temperature, as well as the two hydroxylamine derivatives, while aromatic amines necessitated an increase in temperature to 50 °C. Despite the method's apparent robustness and versatility, all our attempts to synthesize hydrazine derivatives 6z, 6ak, 6al were unsuccessful. Even with the addition of Zn(OTf)2 as an activator and elevated temperatures, we were unable to obtain any of the proposed hydrazine derivatives 6z, 6ak, 6al. This limitation is likely due to the necessity of a polar solvent to facilitate the introduction of hydrazine onto the squaric acid framework, as suggested by solution-phase reaction conditions. Unfortunately, this solvent requirement conflicts with solid-phase synthesis and the resins chosen for our synthetic efforts. In the case of hydrazine derivatives 6z, 6ak, 6al, difficulties arise during the substitution step rather than the coupling step, preventing us from obtaining the desired products.
It is important to note that we did not observe the formation of significant by-products or impurities during the synthesis of any derivatives after the cleavage step. The experiments conducted indicate that the final coupling step is not problematic. Instead, the issues arise during the reaction with nucleophiles. In the cases of unsuccessful synthesis of final derivatives, we observed the formation of a new C–C bond while the ethoxy group remained intact. This suggests that, although the final cleavage was successful, the preceding nucleophilic substitution did not occur. Furthermore, in the case of some hydrazine derivatives, we successfully isolated the “unsubstituted” derivative 6aa after completing the entire reaction sequence, which supports our hypothesis regarding the reactivity. As seen in the previous reaction sequence, when the substitution reaction step was successfully completed, we obtained the final products 6. The yield primarily depended on the electronic activation of the boronic acid, ranging from 14% to 51% (Table 2). Please note that we do not include derivative 6aa in this yield calculation, as the substitution reaction step was not performed in that case.
As confirmed by NMR analysis, three synthesized derivatives (6m, 6n, 6i) were isolated as mixtures of rotamers. The presence of bulky aromatic or vinyl substituents contributes to the formation of these rotameric mixtures, as their steric hindrance limits bond rotation. This occurrence has been documented in our previous studies involving structurally similar compounds.3 Our investigations indicated that the energy barrier between the two rotameric forms is significantly high, rendering thermal interconversion unfeasible under standard experimental conditions. Additionally, variations in the deuterated solvent did not impact the observed ratio of rotameric forms. This phenomenon should be considered when synthesizing biologically active compounds of this type, as restricted bond rotation can significantly affect biological activity.
As highlighted in the text above, the utilization of Merrifield resin enabled the successful synthesis of certain derivatives that could not be produced using AM resin. The findings suggest that the Merrifield resin approach facilitates a more efficient Liebeskind–Srogl cross-coupling reaction in the final step while also enabling the substitution of the central core with a broader spectrum of nucleophiles. Table 2 presents an apparent comparison between the products 6 obtained from AM and Merrifield resins, clearly demonstrating the advantages of the latter. For instance, when employing Merrifield resin, boronic acids that did not react in the AM resin synthesis (specifically compounds 6l and 6n) were effectively coupled. Moreover, the Merrifield resin method proved to be more effective in the substitution step, successfully introducing aromatic amines (6t and 6u) into the structure when the reaction was conducted at 50 °C. In contrast, these same derivatives were either undetectable or only present in trace amounts in the synthesis using AM resin.
Additionally, the Merrifield resin method consistently produced higher yields of the final products 6 than the AM resin approach. This difference may be attributed to the overall greater efficiency of the reaction sequence or the fact that the Merrifield resin synthesis involves one fewer reaction step.
To emphasize the distinct chemoselectivity of the Liebeskind–Srogl cross-coupling reaction, we carried out a solid-phase experiment involving the sequential modification of an intermediate using the Suzuki–Miyaura cross-coupling reaction (Scheme 4). Initially, 4-bromoaniline was introduced onto the central squaramide scaffold. This intermediate was then reacted with 4-methoxyphenylboronic acid under Suzuki coupling conditions, utilizing KOAc and a palladium catalyst in anhydrous DMF, following the procedures outlined by Piettre et al.14 After thoroughly washing the reaction slurry with DMF and DCM, the intermediate 10 was subsequently reacted with p-tolylboronic acid via the Liebeskind–Srogl cross-coupling reaction, which simultaneously cleaved the final product 6am from the resin. The successful isolation of the desired product 6am, achieved with a yield of 23% (25.3 mg from a starting resin of 250 mg), demonstrated that both couplings can proceed independently. This finding paves the way for broader modification possibilities of target molecules.
 | ||
| Scheme 4 Synthesis of squaramides via chemoselective approach. aReagents and conditions: (i) 3-ethoxy-4-mercaptocyclobut-3-ene-1,2-dione, TEA, DMF, rt, 18 h; (ii) 4-bromoaniline, DCM, 50 °C, 18 h; (iii) 4-(methoxyphenyl)boronic acid, KOAc, PdCl2(dppf), anhydrous DMF, 60 °C, 18 h; (iv) p-tolylboronic acid, CuTC, TFP, Pd2(dba)3, anhydrous dioxane, 50 °C, 18 h. | ||
Solid-phase synthesis and a combinatorial approach are commonly used to generate compound libraries due to their efficiency in producing structural diversity with minimal synthetic steps, thereby eliminating the need for intermediate isolation or purification. To enhance the potential applications of our developed methodology, we aimed to evaluate its scalability using Merrifield resin. To achieve this, we conducted a model reaction sequence using benzylamine as the nucleophile and p-tolyl boronic acid for the final Liebeskind–Srogl cross-coupling step, leading to the final compound 6a depicted in Scheme 5. In the coupling reaction, we reduced the ratios of the individual reagents compared to previous experiments. Assuming full utilization of the resin's theoretical loading capacity of 1.21 mmol g−1, the reaction employed 2.5 equivalents of p-tolyl boronic acid, 3 equivalents of CuTC, 5 mol% TFP, and 2.5 mol% Pd2(dba)3. A scale-up reaction was conducted using either 1 g or 5 g of Merrifield resin. As demonstrated in Scheme 5, the employed reaction methodology is applicable for all tested quantities.
 | ||
| Scheme 5 An overview of the scale-up process and the utilized instrumentation. | ||
Table 3 outlines the advantages and disadvantages of utilizing the Liebeskind–Srogl cross-coupling reaction for the synthesis of squaramides through a solid-phase synthetic approach, in comparison to traditional solution synthesis methods. Based on extensive data collected during our projects, solid-phase synthesis has demonstrated greater overall efficiency. This increased efficiency can be primarily attributed to higher product yields and improved compatibility with a wider range of boronic acids.
| Conventional synthesis (solution-phase) | Solid-phase synthesis | |
|---|---|---|
| Yields of target compounds |  |
 |
| Boronic acid scope |  |
 |
| Reaction time |  |
 |
| Overall synthesis time requirement |  |
 |
| Purification |  |
 |
| Economic demands |  |
 |
While the reaction time for the cross-coupling step is similar in both methods, solid-phase synthesis significantly shortens the overall time required to produce the final products 6. In this approach, intermediates are attached to the resin, allowing for simple washing between steps. This eliminates the lengthy isolation and purification processes typically needed in other methods. Notably, the lack of intermediate purification does not complicate the isolation of the final product 6. In fact, our experience shows that purifying the final compound 6 is often easier when it is synthesized on a solid support. This contributes to the higher yields of the final compounds 6 obtained.
One common limitation of solid-phase synthesis is its scalability. However, in this case, it seems feasible to scale up. As we have demonstrated, the synthetic approach remains effective for at least up to a 5 g resin scale. Nevertheless, one drawback of the solid-phase synthesis method is its higher cost. This approach typically requires reagents in excess and involves relatively expensive resins. Although we were able to reduce the excess of reagents in our scale-up experiments, the higher material costs continue to contribute to the overall expense of this method.
Ultimately, the choice of method depends on the specific goals of the synthesis. However, for the rapid and efficient generation of large compound series, such as in SAR studies, we consider solid-phase synthesis to be the more beneficial approach.
Conclusion
In summary, we have developed a traceless solid-phase synthetic approach that enables the preparation of a diverse array of new squaramides, which show significant promise for the treatment of drug-resistant tuberculosis. Our approach emphasizes simplicity, allowing for manual execution without the necessity of isolating and purifying individual intermediates, and it is applicable across a broad spectrum of starting materials. Moreover, the specific type of cleavage from the resin through the Liebeskind–Srogl coupling reaction allows the presence of acid-labile groups. Additionally, we have confirmed remarkable chemoselectivity and the feasibility of scaling up the synthesis. The presented synthesis brings a unique application of the Liebeskind–Srogl cross-coupling in solid-phase synthesis. We anticipate that our findings will serve as a foundation for further exploration of the biological implications of the discovered reaction and the development of innovative squaramides with exceptional antituberculosis activity.Experimental section
Materials and methods
Solvents and chemicals were purchased from Sigma-Aldrich (St. Louis, Missouri, USA) or Fluorochem (UK). Analytical thin-layer chromatography (TLC) was performed using aluminum plates precoated with silica gel 60 F254. Synthesis was carried out on Domino Blocks in disposable polypropylene reaction vessels (Torviq, Tucson, AZ). All reactions were carried out at room temperature (21 °C) unless stated otherwise.The LC-MS analyses were carried out on the UHPLC-MS system (Waters). This system consists of UHPLC chromatograph Acquity with a photodiode array detector and single quadrupole mass spectrometer and uses an XSelect C18 column (2.1 × 50 mm) at 30 °C and a flow rate of 600 μL min−1. The mobile phase was (A) 10 mM ammonium acetate in HPLC grade water and (B) HPLC grade acetonitrile. A gradient was formed from 10% A to 80% B in 2.5 minutes and kept for 1.5 minutes. The column was re-equilibrated with a 10% solution of B for 1 minute. The ESI† source operated at a discharge current of 5 μA, vaporizer temperature of 350 °C, and capillary temperature of 200 °C.
NMR 1H/13C spectra were recorded on a JEOL ECA400II (400 MHz) spectrometer at magnetic field strengths of 9.39 T (with operating frequencies 399.78 MHz for 1H and 100.53 MHz for 13C) at ambient temperature (∼21 °C). Chemical shifts (δ) are reported in parts per million (ppm), and coupling constants (J) are reported in Hertz (Hz). NMR spectra are recorded at rt (21 °C) and referenced to the residual signals of DMSO-d6.
HRMS analysis was performed on an LC chromatograph (Dionex UltiMate 3000, Thermo Fischer Scientific, MA, USA) with mass spectrometer Exactive Plus Orbitrap high-resolution (Thermo Fischer Scientific, MA, USA) operating in positive scan mode in the range of 1000–1500 m/z. Electrospray was used as a source of ionization. Samples were diluted to a final concentration of 0.1 mg mL−1 in a solution of water and acetonitrile (50:50, v/v). The samples were injected into the mass spectrometer following HPLC separation on a Phenomenex Gemini column (C18, 50 × 2 mm, 3 μm particle) using an isocratic mobile phase of 0.01 M MeCN/ammonium acetate (80/20) at a flow rate of 0.3 mL min−1.
Chemistry
Amidation of 3-(tritylthio)propanoic acid with AM resin resulting in 2. AM resin 1 (∼1 g) was washed three times with DCM, three times with DMF, and reacted with 10 mL of solution of DIPEA in DMF (1
:9; v:v) at rt for 20 min. The resin was washed five times with DMF, followed by an addition of solution of 3-(tritylthio)propanoic acid (3 mmol) and DIC (3 mmol) in 10 mL of DCM/DMF (1:1; v:v). The resin was reacted with the prepared mixture at rt for 18 h. Finally, the resin was washed three times with DMF and three times with DCM. A convenient conversion of the reaction was verified using the bromophenol blue test.
Deprotection of thiol resulting in 3. Resin 2 (∼1 g) was washed three times with DCM and reacted with 10 mL of TFA/TES solution (95
:5; v:v) at rt for 1 h. The resin was washed five times with DCM.
Immobilization of central four-membered skeleton resulting in 4. Resin 3 (∼1 g) was washed three times with DCM and reacted with a solution of 3-(tert-butoxy)-4-methoxycyclobut-3-ene-1,2-dione (3 mmol) and TEA (3 mmol) in DCM at rt for 18 h. The resin was washed five times with DCM.
Nucleophile introduction resulting in 5. Resin 4 (∼250 mg) was washed three times with DCM and reacted with a solution of appropriate nucleophile (1 mmol) in DCM at rt for 18 h. The resin was washed three times with DCM. If the nucleophile was added to the reaction as a hydrochloride, an equimolar amount of TEA (1 mmol) was added.
Liebeskind–Srogl cross-coupling cleavage resulting in 6 (procedure for both AM resin and Merrifield resin). Resin 5 (∼250 mg) was washed three times with DCM, three times with THF anhydrous, and once with anhydrous dioxane. Then, appropriate boronic acid (1.6 mmol), CuTC (2.4 mmol), TFP (0.08 mmol), and Pd2(dba)3 (0.04 mmol) were added. Finally, anhydrous dioxane (3 mL) was drawn into the syringe, and the reaction mixture was shaken for 18 h at 50 °C. The reaction mixture was poured into a saturated solution of NH4Cl (30 mL), and the syringe was washed three times with EtOAc (5 mL). After the separation of both layers, the EtOAc was collected, and the residual NH4Cl solution was extracted with another two portions of EtOAc (2 × 30 mL). Combined organic layers were washed with a saturated solution of NH4Cl (30 mL) and brine (30 mL) and dried over anhydrous MgSO4. The solvent was evaporated under reduced pressure. Product 6 was purified by LC using DCM/MeOH (grad). Product 6 was precipitated from DCM by the addition of hexane if repurification was needed.
Immobilization of squaric scaffold (nucleophilic substitution of Merrifield resin by thio derivative of squaric acid) resulting in 8. Merrifield resin 7 (∼1 g) was washed three times with DCM, three times with DMF and reacted with a solution of 3-ethoxy-4-mercaptocyclobut-3-ene-1,2-dione (3 mmol) and TEA (3 mmol) in DMF (10 mL) at rt for 18 h. The resin was washed three times with DMF and three times with DCM.
Nucleophile introduction resulting in 9. Resins 8 (∼250 mg) were washed three times with DCM and reacted with a solution of appropriate nucleophile (1 mmol) in DCM at rt for 18 h. The resins were washed three times with DCM. In the case of aromatic amines, the resins were shaken with reaction mixtures at 50 °C for 18 h. If the nucleophile was added to the reaction as a hydrochloride, an equimolar amount of TEA (1 mmol) was added.
Suzuki–Miyaura cross-coupling reaction resulting in 10aa. Resin 9aa (∼250 mg) was washed three times with DCM, three times with DMF anhydrous. Then, 4-methoxyphenyl boronic acid (1.8 mmol), KOAc (2.7 mmol), and PdCl2(dppf) (0.09 mmol) were added. Finally, DMF anhydrous (3 mL) was drawn into the syringe, and the reaction mixture was shaken for 18 h at 60 °C. The resin was washed five times with DMF and five times with DCM.
Scale-up of the reaction sequence. The scale-up procedures were consistent with the method used for smaller amounts of starting resin, differing only in the ratio of the reactants.
For a 1 g resin scale, immobilization was performed using a solution of 3-ethoxy-4-mercaptocyclobut-3-ene-1,2-dione (3 mmol) and TEA (3 mmol) in DMF (10 mL). The subsequent substitution step employed a solution of benzylamine (3 mmol) in DCM (10 mL). The Liebeskind–Srogl cross-coupling reaction was carried out with p-tolylboronic acid (3.02 mmol), CuTC (3.63 mmol), TFP (0.121 mmol), and Pd2(dba)3 (0.06 mmol) in anhydrous dioxane (10 mL).
For the 5 g resin scale, a solution of 3-ethoxy-4-mercaptocyclobut-3-ene-1,2-dione (15 mmol) and TEA (15 mmol) in DMF (50 mL) was used for immobilization. The substitution step utilized a solution of benzylamine (15 mmol) in DCM (50 mL). The Liebeskind–Srogl cross-coupling reaction was performed with p-tolylboronic acid (15.13 mmol), CuTC (18.15 mmol), TFP (0.3 mmol), and Pd2(dba)3 (0.15 mmol) in anhydrous dioxane (50 mL).
Analytical data for individual compounds
Product 6a was obtained as a white solid. Yield: 25.3 mg (33%) using AM resin, 33.2 mg (40%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 9.53 (br s, 1H), 7.93 (d, J = 7.9 Hz, 2H), 7.44–7.26 (m, 7H), 4.92 (s, 2H), 2.37 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.2, 188.7, 178.2, 162.1, 140.8, 138.2, 129.6, 128.6, 127.6, 127.5, 126.5, 126.2, 47.5, 21.2. HRMS: m/z: calcd for C18H16NO2+: 278.1176 [M + H]+; found: 278.1177.

Product 6b was obtained as a white solid. Yield: 19.2 mg (29%) using AM resin. 1H NMR (400 MHz, DMSO-d6) δ 9.47 (t, J = 6.3 Hz, 1H), 8.09–7.90 (m, 2H), 7.39 (d, J = 4.4 Hz, 4H), 7.34–7.25 (m, 1H), 7.16–7.03 (m, 2H), 4.91 (d, J = 6.1 Hz, 2H), 3.31 (s, 3H).13C NMR (101 MHz, DMSO-d6) δ 192.5, 188.6, 177.8, 162.2, 161.1, 138.3, 128.6, 128.2, 127.6, 127.5, 122.1, 114.5, 55.4, 47.4. HRMS: m/z: calcd for C18H16NO3+: 294.1125 [M + H]+; found: 294.1127.

Product 6c was obtained as a yellow solid. Yield: 15.1 mg (17%) using AM resin. 1H NMR (400 MHz, DMSO-d6) δ 9.56 (t, J = 6.2 Hz, 1H), 8.03–7.93 (m, 2H), 7.43–7.35 (m, 6H), 7.35–7.28 (m, 1H), 4.92 (d, J = 6.1 Hz, 2H), 2.54 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 192.8, 188.6, 178.0, 161.5, 142.2, 138.1, 128.6, 127.6, 127.6, 126.5, 125.5, 125.4, 47.5, 14.1. HRMS: m/z: calcd for C18H16NO2S+: 310.0896 [M + H]+; found: 310.0896.

Product 6d was obtained as a white solid. Yield: 23.8 mg (32%) using AM resin. 1H NMR (400 MHz, DMSO-d6) δ 9.61 (br s, 1H), 8.02 (d, J = 7.4 Hz, 2H), 7.64–7.28 (m, 8H), 4.93 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 193.5, 188.6, 178.5, 161.7, 138.1, 130.6, 129.2, 129.0, 128.6, 127.6, 127.6, 126.1, 47.5. HRMS: m/z: calcd for C17H14NO2+: 264.1019 [M + H]+; found: 264.1020.

Product 6e was obtained as a yellow solid. Yield: 13.7 mg (16%) using AM resin, 19.6 mg (21%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 9.99 (br s, 1H), 8.39–8.33 (m, 2H), 8.26–8.19 (m, 2H), 7.47–7.36 (m, 4H), 7.35–7.30 (m, 1H), 4.96 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 193.9, 188.3, 178.6, 157.7, 147.3, 137.6, 134.8, 128.6, 127.7, 126.8, 124.2, 47.8. HRMS: m/z: calcd for C17H13N2O4+: 309.0870 [M + H]+; found: 309.0868.

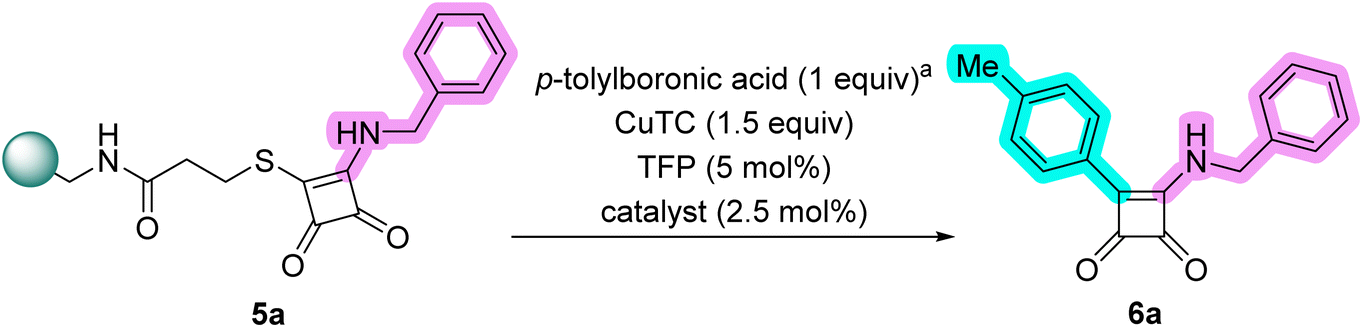
Product 6f was obtained as a white solid. Yield: 24.2 mg (26%) using AM resin. 1H NMR (400 MHz, DMSO-d6) δ 9.85 (s, 1H), 8.19 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 8.2 Hz, 2H), 7.44–7.36 (m, 4H), 7.35–7.28 (m, 1H), 4.95 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 194.4, 189.0, 179.2, 159.6, 138.3, 133.2, 130.1 (q, J = 31.9 Hz), 129.2, 128.2, 127.1, 126.4 (q, J = 3.6 Hz), 124.5 (q, J = 272.4 Hz), 48.2. HRMS: m/z: calcd for C18H13F3NO2+: 332.0893 [M + H]+; found: 332.0895.

Product 6g was obtained as a yellow solid. Yield: 14.4 mg (18%) using AM resin. 1H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H), 9.86 (br s, 1H), 8.19 (d, J = 8.3 Hz, 2H), 8.03 (d, J = 8.3 Hz, 2H), 7.44–7.37 (m, 4H), 7.36–7.29 (m, 1H), 4.95 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 193.8, 192.5, 188.5, 178.6, 159.3, 137.7, 136.4, 134.1, 129.9, 128.6, 127.7, 127.6, 126.4, 47.7. HRMS: m/z: calcd for C18H14NO3+: 292.0968 [M + H]+; found: 292.0972.

Product 6h was obtained as a white solid. Yield: 21.6 mg (23%) using AM resin, 27.7 mg (27%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 9.69 (br s, 1H), 7.98–7.93 (m, 2H), 7.79–7.72 (m, 2H), 7.42–7.35 (m, 4H), 7.35–7.29 (m, 1H), 4.92 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 193.4, 188.4, 178.3, 160.1, 137.9, 132.1, 128.6, 128.2, 127.9, 127.6, 127.6, 123.8, 47.6. HRMS: m/z: calcd for C17H13BrNO2+: 342.0124 [M + H]+; found: 342.0123.

Product 6i was obtained as a white solid. Yield: 21.3 mg (38%) using AM resin. 1H NMR (400 MHz, DMSO-d6),

 mixture of two rotamers. Major rotamer δ 9.34 (t, J = 6.3 Hz, 1H), 8.48 (t, J = 1.2 Hz, 1H), 7.91 (t, J = 1.7 Hz, 1H), 7.41–7.28 (m, 5H), 7.12 (dd, J = 2.0, 0.5 Hz, 1H), 4.90 (d, J = 6.2 Hz, 2H). Minor rotamer, characteristic signals: δ 9.73 (t, J = 6.0 Hz, 1H), 8.25 (s, 1H), 7.81 (t, J = 1.7 Hz, 1H), 4.72 (d, J = 6.3 Hz, 2H). 13C NMR (101 MHz, DMSO-d6),
mixture of two rotamers. Major rotamer δ 9.34 (t, J = 6.3 Hz, 1H), 8.48 (t, J = 1.2 Hz, 1H), 7.91 (t, J = 1.7 Hz, 1H), 7.41–7.28 (m, 5H), 7.12 (dd, J = 2.0, 0.5 Hz, 1H), 4.90 (d, J = 6.2 Hz, 2H). Minor rotamer, characteristic signals: δ 9.73 (t, J = 6.0 Hz, 1H), 8.25 (s, 1H), 7.81 (t, J = 1.7 Hz, 1H), 4.72 (d, J = 6.3 Hz, 2H). 13C NMR (101 MHz, DMSO-d6),  mixture of two rotamers. Major rotamer δ 192.3, 187.7, 177.7, 156.7, 145.0, 142.8, 138.0, 128.7, 127.6, 127.5, 114.7, 107.4, 47.3. Minor rotamer was not detected. HRMS: m/z: calcd for C15H12NO3+: 254.0812 [M + H]+; found: 254.0812.
mixture of two rotamers. Major rotamer δ 192.3, 187.7, 177.7, 156.7, 145.0, 142.8, 138.0, 128.7, 127.6, 127.5, 114.7, 107.4, 47.3. Minor rotamer was not detected. HRMS: m/z: calcd for C15H12NO3+: 254.0812 [M + H]+; found: 254.0812.Product 6k was obtained as a pale-yellow solid. Yield: 21.1 mg (28%) using AM resin, 28.4 mg (35%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 9.47 (t, J = 5.5 Hz, 1H), 8.37 (t, J = 2.4 Hz, 1H), 7.79 (d, J = 2.0 Hz, 2H), 7.39 (d, J = 4.4 Hz, 4H), 7.35–7.29 (m, 1H), 4.92 (d, J = 4.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 191.2, 186.6, 176.2, 156.8, 138.0, 131.5, 129.1, 128.6, 128.6, 128.3, 127.6, 47.5.HRMS: m/z: calcd for C15H12NO2S+: 270.0583 [M + H]+; found: 270.0585.

Product 6l was obtained as a yellow solid. Yield: 11.2 mg (14%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 9.56 (t, J = 6.2 Hz, 1H), 7.97 (d, J = 5.0 Hz, 1H), 7.88 (d, J = 3.8 Hz, 1H), 7.44–7.27 (m, 6H), 4.92 (d, J = 6.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 191.8, 187.1, 176.7, 157.4, 138.6, 132.0, 129.6, 129.2, 129.1, 128.9, 128.2, 48.1. HRMS: m/z: calcd for C15H12NO2S+: 270.0583 [M + H]+; found: 270.0580.

Product 6m was obtained as a yellow solid. Yield: 23.8 mg (29%) using AM resin, 35.6 mg (41%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6),

 mixture of two rotamers. Major rotamer δ 9.5 (t, J = 6.0 Hz, 1H), 7.6 (d, J = 16.3 Hz, 1H), 7.6–7.5 (m, 3H), 7.4–7.3 (m, 7H), 7.3 (d, J = 16.2 Hz, 1H), 4.9 (d, J = 5.3 Hz, 2H). Minor rotamer, characteristic signals: δ 9.8 (d, J = 6.3 Hz, 1H), 7.8 (d, J = 15.8 Hz, 1H), 7.1 (d, J = 15.8 Hz, 1H), 4.8 (d, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6),
mixture of two rotamers. Major rotamer δ 9.5 (t, J = 6.0 Hz, 1H), 7.6 (d, J = 16.3 Hz, 1H), 7.6–7.5 (m, 3H), 7.4–7.3 (m, 7H), 7.3 (d, J = 16.2 Hz, 1H), 4.9 (d, J = 5.3 Hz, 2H). Minor rotamer, characteristic signals: δ 9.8 (d, J = 6.3 Hz, 1H), 7.8 (d, J = 15.8 Hz, 1H), 7.1 (d, J = 15.8 Hz, 1H), 4.8 (d, J = 5.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6),  mixture of two rotamers. Signals are not specified for major and minor rotamer. δ 192.9, 192.2, 189.3, 188.6, 178.8, 162.3, 161.2, 137.8, 137.4, 136.1, 136.0, 135.9, 129.4, 129.2, 129.0, 128.9, 128.7, 128.7, 127.7, 127.7, 127.5, 127.4, 127.2, 116.7, 115.4, 47.4, 47.2. HRMS: m/z: calcd for C19H16NO2+: 290.1176 [M + H]+; found: 290.1177.
mixture of two rotamers. Signals are not specified for major and minor rotamer. δ 192.9, 192.2, 189.3, 188.6, 178.8, 162.3, 161.2, 137.8, 137.4, 136.1, 136.0, 135.9, 129.4, 129.2, 129.0, 128.9, 128.7, 128.7, 127.7, 127.7, 127.5, 127.4, 127.2, 116.7, 115.4, 47.4, 47.2. HRMS: m/z: calcd for C19H16NO2+: 290.1176 [M + H]+; found: 290.1177.Product 6n was obtained as a brown solid. Yield: 27.3 mg (31%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6),

 mixture of two rotamers. Major rotamer δ 9.37 (t, J = 6.3 Hz, 1H), 7.66–7.03 (m, 7H), 6.83 (dd, J = 6.1, 2.5 Hz, 1H), 4.82 (d, J = 6.3 Hz, 2H), 2.20 (s, 6H). Minor rotamer, characteristic signals: δ 9.92 (t, J = 6.4 Hz, 1H), 4.13 (d, J = 6.3 Hz, 2H), 2.02 (s, 6H). 13C NMR (101 MHz, DMSO-d6),
mixture of two rotamers. Major rotamer δ 9.37 (t, J = 6.3 Hz, 1H), 7.66–7.03 (m, 7H), 6.83 (dd, J = 6.1, 2.5 Hz, 1H), 4.82 (d, J = 6.3 Hz, 2H), 2.20 (s, 6H). Minor rotamer, characteristic signals: δ 9.92 (t, J = 6.4 Hz, 1H), 4.13 (d, J = 6.3 Hz, 2H), 2.02 (s, 6H). 13C NMR (101 MHz, DMSO-d6), 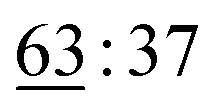 mixture of two rotamers. Signals are not specified for major and minor rotamer. δ 194.3, 193.4, 189.0, 188.3, 181.8, 180.8, 167.6, 166.3, 138.1, 137.2, 135.8, 135.3, 129.2, 129.1, 129.0, 128.6, 128.4, 128.0, 127.5, 127.5, 127.3, 127.1, 127.0, 47.4, 47.1, 19.9, 19.7. HRMS: m/z: calcd for C19H18NO2+: 292.1332 [M + H]+; found: 292.1332.
mixture of two rotamers. Signals are not specified for major and minor rotamer. δ 194.3, 193.4, 189.0, 188.3, 181.8, 180.8, 167.6, 166.3, 138.1, 137.2, 135.8, 135.3, 129.2, 129.1, 129.0, 128.6, 128.4, 128.0, 127.5, 127.5, 127.3, 127.1, 127.0, 47.4, 47.1, 19.9, 19.7. HRMS: m/z: calcd for C19H18NO2+: 292.1332 [M + H]+; found: 292.1332.Product 6o was obtained as a pale-yellow solid. Yield: 19.6 mg (20%) using AM resin. 1H NMR (400 MHz, DMSO-d6) δ 9.63 (br s, 1H), 8.51 (d, J = 1.9 Hz, 1H), 8.08 (dd, J = 8.6, 1.7 Hz, 1H), 7.91 (t, J = 8.9 Hz, 2H), 7.45–7.36 (m, 5H), 7.35–7.29 (m, 1H), 7.23 (dd, J = 8.9, 2.5 Hz, 1H), 4.98 (d, J = 4.1 Hz, 2H), 4.18 (q, J = 7.0 Hz, 2H), 1.41 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.1, 188.8, 178.3, 162.3, 158.0, 138.2, 135.2, 130.3, 128.7, 128.0, 127.6, 127.4, 125.9, 124.5, 123.7, 119.8, 106.9, 63.4, 47.6, 14.6. HRMS: m/z: calcd for C23H20NO3+: 358.1438 [M + H]+; found: 358.1437.

Product 6p was obtained as a brown solid. Yield: 42.1 mg (50%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 9.55 (t, J = 6.0 Hz, 1H), 8.55 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H), 7.99–7.89 (m, 2H), 7.81 (td, J = 7.7, 1.8 Hz, 1H), 7.45 (dt, J = 7.9, 1.1 Hz, 1H), 7.37 (d, J = 7.9 Hz, 2H), 7.32 (ddd, J = 7.6, 4.8, 1.1 Hz, 1H), 5.01 (d, J = 6.0 Hz, 2H), 2.38 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.0, 188.9, 179.0, 162.0, 157.1, 149.2, 140.8, 137.0, 129.6, 126.6, 126.2, 122.7, 121.6, 48.9, 21.2. HRMS: m/z: calcd for C17H15N2O2+: 279.1128 [M + H]+; found: 279.1125.
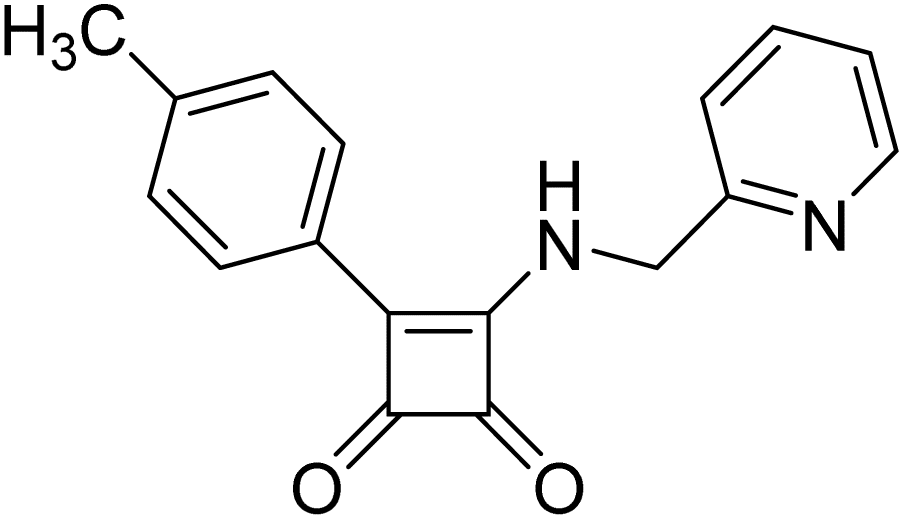
Product 6q was obtained as a white solid. Yield: 20.1 mg (20%) using AM resin, 40.1 mg (37%) using Merrifield resin1 H NMR (400 MHz, DMSO-d6) δ 9.51 (t, J = 6.2 Hz, 1H), 8.00–7.85 (m, 2H), 7.61–7.54 (m, 2H), 7.41–7.32 (m, 4H), 4.88 (d, J = 6.0 Hz, 2H), 2.37 (s, 3H) 13C NMR (101 MHz, DMSO-d6) δ 193.1, 188.7, 178.3, 162.2, 140.9, 137.6, 131.5, 129.9, 129.6, 126.5, 126.2, 120.7, 46.8, 21.2.

Product 6r was obtained as a white solid. Yield: 22.1 mg (34%) using AM resin, 36.8 mg (53%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 9.04 (t, J = 6.2 Hz, 1H), 7.94–7.86 (m, 2H), 7.36 (d, J = 8.1 Hz, 2H), 3.66 (q, J = 6.7 Hz, 2H), 2.37 (s, 3H), 1.64 (h, J = 7.3 Hz, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.1, 188.6, 178.4, 161.6, 140.6, 129.5, 126.7, 126.0, 45.9, 23.7, 21.2, 10.7. HRMS: m/z: calcd for C14H16NO2+: 230.1176 [M + H]+; found: 230.1180.

Product 6s was obtained as a pale-yellow solid. Yield: 25.7 mg (34%) using AM resin, 39.6 mg (49%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 8.83 (br s, 1H), 7.94 (d, J = 8.2 Hz, 2H), 7.35 (d, J = 7.9 Hz, 2H), 4.14 (t, J = 10.7 Hz, 1H), 2.37 (s, 3H), 1.92 (dd, J = 12.1, 3.9 Hz, 2H), 1.78 (dt, J = 13.2, 3.4 Hz, 2H), 1.63 (dt, J = 12.7, 3.5 Hz, 1H), 1.46 (qd, J = 12.3, 3.4 Hz, 2H), 1.37–1.20 (m, 2H), 1.18–1.04 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 192.9, 188.5, 177.4, 161.5, 140.5, 129.4, 126.6, 126.2, 54.1, 33.3, 24.7, 21.2. HRMS: m/z: calcd for C17H20NO2+: 270.1489 [M + H]+; found: 270.1490.
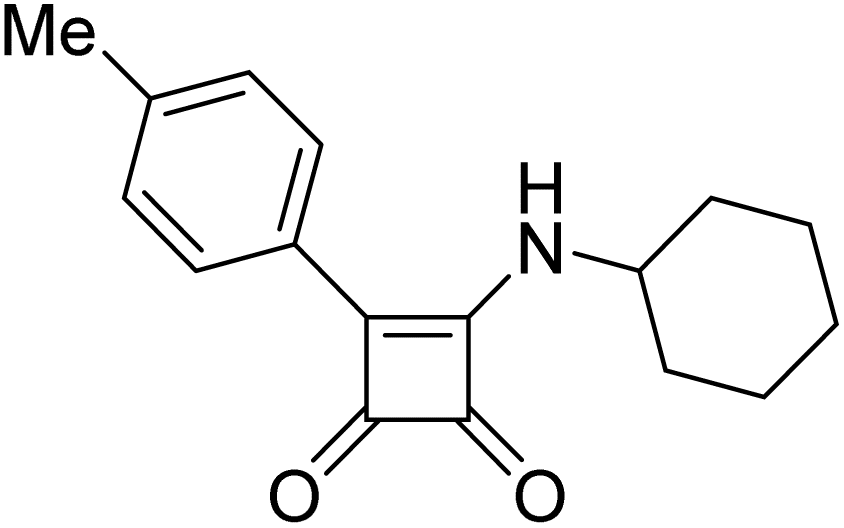
Product 6t was obtained as a yellow solid. Yield: 39.8 mg (50%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (br s, 1H), 9.14–6.65 (m, 9H), 2.39 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 190.8, 190.1, 177.7, 163.8, 141.1, 137.2, 129.4, 128.6, 127.1, 126.0, 125.6, 122.7, 21.3. HRMS: m/z: calcd for C17H14NO2+: 264.1019 [M + H]+; found: 264.1019.

Product 6u was obtained as a yellow solid. Yield: 41.1 mg (46%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 11.44–10.17 (m, 1H), 8.27–6.06 (m, 8H), 3.78 (s, 3H), 2.39 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 190.6, 189.8, 177.5, 163.0, 157.3, 141.0, 130.2, 129.5, 126.6, 126.2, 124.3, 113.8, 55.3, 21.3. HRMS: m/z: calcd for C18H16NO3+: 294.1125 [M + H]+; found: 294.1126.

Product 6x was obtained as an orange solid. Yield: 33.6 mg (33%) using AM resin, 49.2 mg (45%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 9.29 (d, J = 9.0 Hz, 1H), 9.20 (br s, 1H), 7.90 (d, J = 8.2 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 7.21–6.96 (m, 2H), 6.75–6.55 (m, 2H), 5.16 (ddd, J = 10.6, 9.2, 4.5 Hz, 1H), 3.73 (s, 3H), 3.23 (dd, J = 14.0, 4.6 Hz, 1H), 3.00 (dd, J = 13.9, 10.7 Hz, 1H), 2.38 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 192.2, 188.6, 178.4, 170.7, 162.3, 156.0, 141.2, 130.2, 129.6, 126.6, 126.4, 126.1, 115.1, 58.3, 52.5, 36.5, 21.3. HRMS: m/z: calcd for C21H20NO5+: 366.1336 [M + H]+; found: 366.1336.

Product 6z was obtained as a yellow solid. Yield: 38.2 mg (58%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 7.83 (d, J = 8.3 Hz, 2H), 7.41 (d, J = 7.9 Hz, 2H), 4.90 (q, J = 7.1 Hz, 2H), 2.39 (s, 3H), 1.48 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 194.4, 192.8, 191.9, 170.7, 143.0, 130.0, 126.8, 124.9, 71.3, 21.4, 15.4. HRMS: m/z: calcd for C13H13O3+: 217.0859 [M + H]+; found: 217.0857.
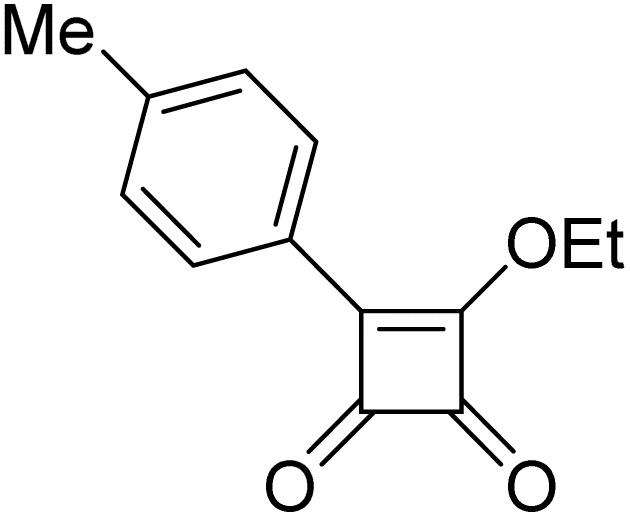
Product 6aa was obtained as a yellow solid. Yield: 36.6 mg (35%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 10.83 (br s, 1H), 7.95 (br s, 1H), 7.70–7.15 (m, 7H), 2.39 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 190.5, 190.1, 177.6, 164.1, 141.3, 136.7, 131.5, 129.5, 127.1, 125.9, 124.6, 117.9, 21.3. HRMS: m/z: calcd for C17H13BrNO2+: 342.0124 [M + H]+; found: 342.0124.

Product 6ab was obtained as a brown solid. Yield: 35.5 mg (34%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 11.35–10.39 (m, 1H), 8.19–6.55 (m, 8H), 3.75 (t, J = 4.7 Hz, 4H), 3.12 (t, J = 4.7 Hz, 4H), 2.40 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 190.6, 189.7, 177.3, 162.9, 149.0, 141.0, 129.5, 129.0, 126.6, 126.3, 123.7, 114.8, 66.0, 48.4, 21.3. HRMS: m/z: calcd for C21H21N2O3+: 349.1547 [M + H]+; found: 349.1545.

Product 6ac was obtained as a white solid. Yield: 39.9 mg (46%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 9.00 (br s, 1H), 7.88 (d, J = 8.2 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 4.05 (q, J = 7.1 Hz, 2H), 3.96 (t, J = 7.0 Hz, 2H), 2.72 (t, J = 6.7 Hz, 2H), 2.37 (s, 3H), 1.15 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 192.8, 188.7, 178.6, 170.6, 161.5, 140.7, 129.6, 126.5, 126.0, 60.1, 38.9, 34.6, 21.2, 14.0. HRMS: m/z: calcd for C16H18NO4+: 288.1230 [M + H]+; found: 288.1233.

Product 6ad was obtained as a white solid. Yield: 29.8 mg (34%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 11.70 (s, 1H), 8.01 (d, J = 8.3 Hz, 2H), 7.45–7.33 (m, 5H), 7.29 (d, J = 8.1 Hz, 2H), 5.10 (s, 2H), 2.34 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 188.6, 185.8, 172.7, 160.3, 140.2, 134.7, 129.2, 128.7, 128.5, 128.3, 128.2, 126.2, 57.1, 21.1. HRMS: m/z: calcd C18H16NO3+: 294.1125 [M + H]+; found: 294.1124.
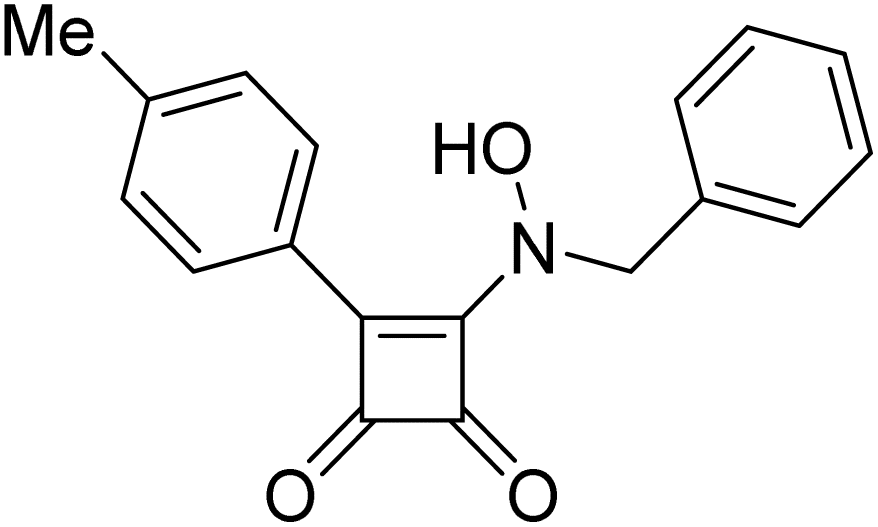
Product 6ae was obtained as a pale-yellow solid. Yield: 28.8 mg (41%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 7.89 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 3.72 (s, 3H), 3.65 (s, 3H), 2.36 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 188.1, 186.1, 172.9, 160.3, 140.4, 129.5, 127.8, 125.8, 61.6, 37.2, 21.1. HRMS: m/z: calcd C13H14NO3+: 232.0968 [M + H]+; found: 232.0968.

Product 6af was obtained as a white solid. Yield: 38.8 mg (50%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 7.48 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 7.9 Hz, 2H), 3.98 (br s, 2H), 3.75 (br s, 4H), 3.55 (br s, 2H), 2.36 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 192.8, 187.9, 177.4, 162.6, 139.7, 129.3, 127.5, 125.8, 65.8, 65.4, 49.0, 47.0, 21.0. HRMS: m/z: calcd C15H16NO3+: 258.1125 [M + H]+; found: 258.1123.

Product 6ah was obtained as a white solid. Yield: 45.5 mg (42%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 7.49 (d, J = 8.1 Hz, 2H), 7.34 (d, J = 7.9 Hz, 2H), 3.95 (br s, 2H), 3.52 (br s, 6H), 2.36 (s, 3H), 1.42 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 192.7, 187.9, 177.7, 162.8, 153.6, 139.7, 129.4, 127.5, 125.8, 79.4, 48.5, 46.5, 42.7, 27.9, 21.0. HRMS: m/z: calcd C20H25N2O4+: 357.1809 [M + H]+; found: 357.1810.
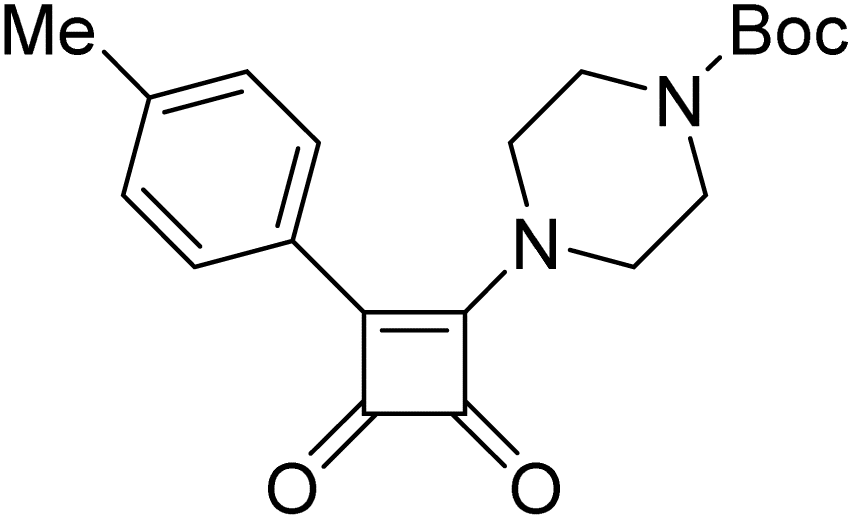
Product 6ai was obtained as a white solid. Yield: 40.9 mg (51%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 9.53 (br s, 1H), 7.92 (d, J = 8.2 Hz, 2H), 7.65 (dd, J = 1.5, 0.7 Hz, 1H), 7.34 (d, J = 8.0 Hz, 2H), 6.46–6.38 (m, 2H), 4.91 (s, 2H), 2.36 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 192.8, 188.8, 178.4, 162.3, 150.9, 143.0, 140.9, 129.6, 126.4, 126.2, 110.7, 108.2, 40.5, 21.2. HRMS: m/z: calcd C16H14NO3+: 268.0968 [M + H]+; found: 268.0968.
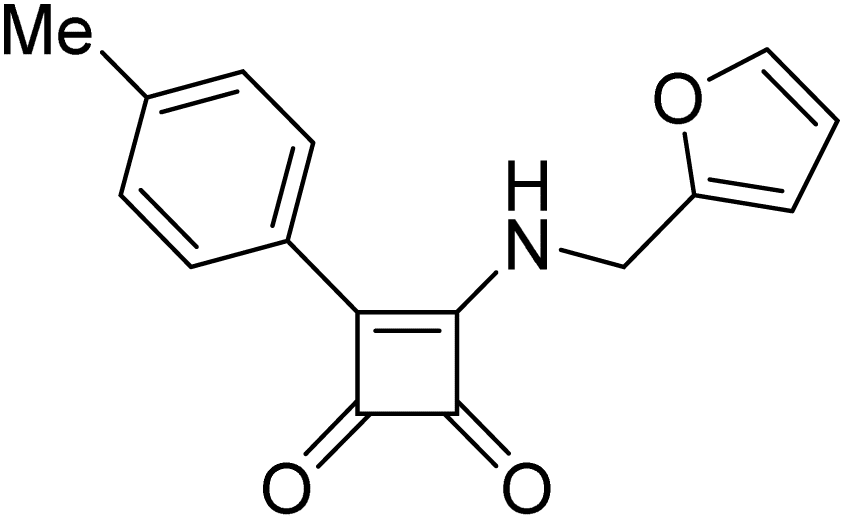
Product 6am was obtained as a yellow solid. Yield: 25.3 mg (23%) using Merrifield resin. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (br s, 1H), 8.28–7.23 (m, 10H), 7.11–6.94 (m, 2H), 3.80 (s, 3H), 2.40 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 190.6, 190.1, 177.6, 163.8, 158.9, 141.2, 136.9, 136.0, 131.6, 129.4, 127.6, 127.1, 126.2, 126.1, 123.0, 114.4, 55.2, 21.3. HRMS: m/z: calcd C24H20NO3+: 370.1438 [M + H]+; found: 370.1439.

Data availability
The data supporting the findings of this study are available in the published article as well as in its ESI.†Author contributions
J. C. designed and performed the synthetic experiments, analyzed the experimental data, and wrote the experimental section. L. B. initiated the project, designed the synthetic experiments, analyzed the results, and was responsible for funding acquisition. L. B. and J. C. wrote the paper. Both authors have approved the final version of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by Palacky University Olomouc (project IGA_PrF_2024_028 and IGA_PrF_2025_014).References
- S. J. Tantry, S. D. Markad, V. Shinde, J. Bhat, G. Balakrishnan, A. K. Gupta, A. Ambady, A. Raichurkar, C. Kedari, S. Sharma, N. V. Mudugal, A. Narayan, C. N. Naveen Kumar, R. Nanduri, S. Bharath, J. Reddy, V. Panduga, K. R. Prabhakar, K. Kandaswamy, R. Saralaya, P. Kaur, N. Dinesh, S. Guptha, K. Rich, D. Murray, H. Plant, M. Preston, H. Ashton, D. Plant, J. Walsh, P. Alcock, K. Naylor, M. Collier, J. Whiteaker, R. E. McLaughlin, M. Mallya, M. Panda, S. Rudrapatna, V. Ramachandran, R. Shandil, V. K. Sambandamurthy, K. Mdluli, C. B. Cooper, H. Rubin, T. Yano, P. Iyer, S. Narayanan, S. Kavanagh, K. Mukherjee, V. Balasubramanian, V. P. Hosagrahara, S. Solapure, S. Ravishankar and P. S. Hameed, Discovery of imidazo[1,2-a]pyridine ethers and squaramides as selective and potent inhibitors of mycobacterial adenosine triphosphate (ATP) synthesis, J. Med. Chem., 2017, 60(4), 1379–1399 CrossRef CAS PubMed.
- J. Chasák, L. Oorts, M. Dak, V. Šlachtová, V. Bazgier, K. Berka, L. De Vooght, N. Smiejkowska, K. Van Calster, L. Van Moll, D. Cappoen, P. Cos and L. Brulíková, Expanding the squaramide library as mycobacterial ATP synthase inhibitors: innovative synthetic pathway and biological evaluation, Bioorg. Med. Chem., 2023, 95, 117504 CrossRef PubMed.
- J. Chasák, L. Van Moll, A. Matheeussen, L. De Vooght, P. Cos and L. Brulíková, The liebeskind–srogl cross-coupling reaction as a crucial step in the synthesis of new squaramide-based antituberculosis agents, ACS Omega, 2024, 9(32), 34808–34828 CrossRef PubMed.
- J. Chasák, V. Šlachtová, M. Urban and L. Brulíková, Squaric acid analogues in medicinal chemistry, Eur. J. Med. Chem., 2021, 209, 112872 CrossRef PubMed.
- J. Chen and D. C. Ekiert, A tale of two inhibitors: diarylquinolines and squaramides, EMBO J., 2023, 42(15), e114912 CrossRef PubMed.
- G. M. Courbon, P. R. Palme, L. Mann, A. Richter, P. Imming and J. L. Rubinstein, Mechanism of mycobacterial ATP synthase inhibition by squaramides and second generation diarylquinolines, EMBO J., 2023, 42(15), e113687 CrossRef CAS PubMed.
- S. A. E. P. Ahmed and A. K. Saxena, Molecular docking-based interaction studies on imidazo[1,2-a] pyridine ethers and squaramides as anti-tubercular agents, SAR QSAR Environ. Res., 2023, 34(6), 435–457 CrossRef CAS PubMed.
- T. Shinada, A. Yamasaki, Y. Kiniwa, K. Shimamoto and Y. Ohfune, Thiol addition to T-butyl methyl squarate. efficient synthesis of novel sulfur-linked squaryl group-containing glutamate analogs, Tetrahedron Lett., 2009, 50(30), 4354–4357 CrossRef CAS.
- D. Srivastava, A. Kushwaha, G. Kociok-Köhn, S. W. Gosavi, R. Chauhan, A. Kumar and M. Muddassir, The supramolecular frameworks and electrocatalytic properties of two new structurally diverse tertiary phosphane-appended nickel(II) and copper(I) Thiosquarates, CrystEngComm, 2023, 25(48), 6822–6836 RSC.
- W. Schmidt and H. A. Ried, Die präparative chemie der cyclobutendione; I. Synthese von cyclobutendion und dessen alkyl-, alkenyl-und aryl-derivaten, Synthesis, 1978, 1978(01), 1–22 CrossRef.
- S. A. Ivanovsky, V. D. Mikhail, V. K. Dmitry and A. V. Ivachtchenko, Synthesis of the substituted 3-cyclobutene-1,2-diones, Synth. Commun., 2007, 37(15), 2527–2542 CrossRef CAS.
- D. J. Asby, M. G. Radigois, D. C. Wilson, F. Cuda, C. L. L. Chai, A. Chen, A. S. Bienemann, M. E. Light, D. C. Harrowven and A. Tavassoli, Triggering apoptosis in cancer cells with an analogue of Cribrostatin 6 that elevates intracellular ROS, Org. Biomol. Chem., 2016, 14(39), 9322–9330 RSC.
- D. Schmidt Günter and A. H. S. Heike, Oxokohlenstoffe und verwandte verbindungen; 15. mitteilung. 1Meerwein-arylierung von semiquadratsäure und semiquadratsäureamiden. Eine allgemeine darstellungsmethode von 3-aryl-4-hydroxy-und 3-amino-4-aryl-3-cyclobuten-1,2-dionen, Synthesis, 1990, 1990(07), 579–582 CrossRef.
- S. R. Piettre and S. Baltzer, A new approach to the solid-phase suzuki coupling reaction, Tetrahedron Lett., 1997, 38(7), 1197–1200 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02225h |
| This journal is © The Royal Society of Chemistry 2025 |