
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStarch as an efficient precursor for hard carbon anodes enables high-performance sodium-ion storage†
Yuyang Panab,
Zhihao Liab,
Rui Fangab,
Baoshu Chenab,
Ning Dangab and
Tianbao Zhao *ab
*ab
aSchool of Materials Science and Engineering, Xihua University, Chengdu, Sichuan 610039, China. E-mail: btz123@foxmail.com
bKey Laboratory of Materials and Surface Technology (Ministry of Education), School of Materials Science and Engineering, Xihua University, Chengdu, 610039, Sichuan, China. E-mail: 21077215@qq.com
First published on 6th May 2025
Abstract
Starch is an ideal choice for hard carbon anode precursors in sodium-ion batteries because of its diverse sources and high carbon content. Nevertheless, starch experiences melting and foaming in the initial stages of pyrolysis, which results in low yields and hinders the large-scale production and application of starch-derived hard carbon. Herein, we employed phenolic resin and maleic anhydride to esterify and cross-link starch to prepare hard carbon. The starch foaming was successfully inhibited through the synergistic effect among composite precursors. Benefiting from abundant micropores/mesopores and larger microcrystalline structure, the prepared hard carbon delivers a high specific capacity of up to 342.28 mA h g−1 and an impressive initial discharge capacity of 85.89%. The capacity retention is as high as 87.24% after 100 cycles of charge and discharge. Furthermore, the hard carbon demonstrates excellent rate performance with a capacity recovery rate of 95.11%. The stabilized starch-derived hard carbon anode possesses both high sodium storage performance and a lower cost, demonstrating potential for commercialization on a large scale.
1 Introduction
Lithium-ion batteries (LIBs) are well-known for their excellent energy density, which makes them a top choice for a variety of applications, including consumer electronics and electric vehicles.1,2 However, recent challenges have arisen for LIB technology. The limited availability of lithium resources has become a major concern, driving up production costs and raising questions about the long-term sustainability and scalability of LIBs. In contrast, sodium-ion batteries (SIBs) have gained considerable attention as a promising alternative. Sodium is abundant in the Earth's crust, with an abundance approximately 400 times that of lithium, making it an appealing option in terms of resource availability.3,4 The extraction and processing of sodium are also relatively straightforward and cost-effective. Additionally, SIBs can function efficiently in aqueous environments, allowing for the use of inexpensive electrolytes and simpler manufacturing processes.5–7 This not only significantly lowers the overall cost of battery production but also positions SIBs as a viable option for large-scale energy storage applications, potentially complementing or replacing LIBs.8–11Hard carbon has emerged as a leading candidate among various anode materials being explored for sodium-ion batteries (SIBs). The attractiveness of hard carbon is based on several key factors. These include its wide availability and cost-effectiveness, the environmental benefits arising from the non-toxic nature of its production process; the low working potential resulting from its unique sodium storage mechanism; the cycling stability due to the minimal volume change during the sodiation and desodiation processes; and the excellent electrochemical performance attributed to its unique microstructure composed of randomly stacked and curved graphene-like sheets.2,12–14 As a result, it is well-positioned for commercialization as an anode material for SIBs in the near future. During the pyrolysis of hard carbon precursors, graphite microcrystals form locally and aggregate in a disordered yet functionally important manner. This unique microstructure features a long-range disordered and short-range ordered arrangement, which gives hard carbon multiple reversible sodium storage sites.15–17 These sites include intercalation sites within the graphite layers, atomic clusters trapped within closed pores, capacitive adsorption sites on the surface that interacts with the electrolyte, and pseudocapacitive storage sites linked to internal defects.18–20
Starch, due to its simple elemental composition, high carbon content, and wide availability at a relatively low cost, has been considered a potential precursor for hard carbon.21 However, directly carbonizing starch presents several challenges. The crystalline regions of starch melt during this process, leading to the formation of a porous and foamy structure. This results in the cleavage of glycosidic linkages and the depolymerization of D-glucose units into small volatile molecules, leading to a reduced carbon yield and poor sodium storage performance of the resulting hard carbon.22–24 Despite such challenges, researchers have made many efforts to address them. For example, Yang et al.17 implemented a solvent heat stabilization pretreatment, which successfully addressed the aforementioned issues, producing a hard carbon with a stable sodium storage capacity of 110 mA h g−1 at a rate of 0.4 C. Additionally, chemical cross-linking has been explored as a method to improve the properties of starch-derived hard carbon.25 Maleic anhydride, a small organic compound, is frequently used to modify starch. When maleic anhydride esterifies starch, it consumes many hydroxyl groups within the starch molecular chains, disrupting the crystalline structure of the starch granules. This effectively reduces the problems associated with starch melting, foaming, and microstructure degradation during pyrolysis, leading to a significant improvement in carbon yield. Song et al.26 demonstrated the promising cycling performance of esterified starch-derived hard carbon as an anode material in lithium-ion batteries, following the initiation of the esterification reaction with maleic anhydride. However, despite these advancements, the sodium storage properties of maleic anhydride-esterified starch-derived hard carbon remain relatively underexplored, with a lack of comprehensive and representative studies in this area.
Phenolic resins have a chemically tunable structure, allowing for the synthesis of hard carbons with a variety of morphological and property characteristics. For example, Kamiyama et al.27 achieved a maximum specific capacity of 386 mA h g−1 at 10 mA g−1 by carefully adjusting the carbonization temperature of phenolic resin. Similarly, Wang et al.28 and Jin et al.29 produced hard carbon microspheres and materials with excellent cycling stability, respectively, using phenolic resin as a precursor. However, it is important to note that phenolic resins are relatively expensive precursors for hard carbon production. Therefore, there is an urgent need to develop strategies that combine the high sodium storage performance of certain components with the cost-effectiveness of biomass-derived starch to help reduce overall costs.
In this work, a combination of low-cost starch and high-performance phenolic resin has been used as precursors for HCs. Additionally, maleic anhydride was introduced to initiate an esterification reaction. The cross-linking between starch, maleic anhydride, and phenolic resin formed a larger and denser carbon structure. During the pyrolysis process, these precursors were transformed into graphite crystallites of larger size, leading to an increased sodium storage capacity. Due to the synergistic effect of the three components, the resulting HCs demonstrated excellent sodium storage properties. The discharge specific capacity reached up to 342.28 mA h g−1, with an initial coulombic efficiency as high as 85.89% at a rate of 0.05 C, which is superior to that of individual precursors made only from starch or phenolic resin. This discovery not only presents a novel method for synthesizing high-performance hard carbon materials but also paves the way for the large-scale utilization of low-cost SIBs.
2 Materials and methods
2.1 Materials synthesis
The precursors were prepared by ball milling food-grade tapioca starch with ash content ≤0.2% (Guangxi Gaoyuan Starch Co., Ltd); high-purity phenolic resin containing ≤0.1% free phenol (Shandong Shengquan New Materials Co., Ltd); maleic anhydride of analytical reagent (AR) grade with purity ≥99.5% (Chengdu Cologne Chemical Co., Ltd). The mass ratios were 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0:1, 0:5:1, 7:3:0, 7:3:1, 7:3:2, and 7:3:3, respectively. The rotational speed was 200 rpm and the duration time was 30 minutes. After thorough mixing the precursors, the samples were transferred to a stainless steel reactor and maintained at a constant temperature of 80 °C for a period of three hours. Afterward, the samples were placed into the stainless steel reactor and heated at a constant temperature of 220 °C for six hours. After cooling to room temperature, the samples were crushed. Finally, the samples were placed in a tube furnace and heated to 1300 °C under a nitrogen atmosphere at a heating rate of 3 °C min−1 for a duration of 3 hours. The resulting hard carbon materials were designated as ST-M-51, PR-M-51, ST-PR-73, ST-PR-M-731, ST-PR-M-732, and ST-PR-M-733, where ST represents starch, PR denotes phenolic resin, and M stands for maleic anhydride.
:0:1, 0:5:1, 7:3:0, 7:3:1, 7:3:2, and 7:3:3, respectively. The rotational speed was 200 rpm and the duration time was 30 minutes. After thorough mixing the precursors, the samples were transferred to a stainless steel reactor and maintained at a constant temperature of 80 °C for a period of three hours. Afterward, the samples were placed into the stainless steel reactor and heated at a constant temperature of 220 °C for six hours. After cooling to room temperature, the samples were crushed. Finally, the samples were placed in a tube furnace and heated to 1300 °C under a nitrogen atmosphere at a heating rate of 3 °C min−1 for a duration of 3 hours. The resulting hard carbon materials were designated as ST-M-51, PR-M-51, ST-PR-73, ST-PR-M-731, ST-PR-M-732, and ST-PR-M-733, where ST represents starch, PR denotes phenolic resin, and M stands for maleic anhydride.
2.2 Material characterization
The microscopic morphology and structure were observed by scanning electron microscope (SEM, JSM-IT500LV, JEOL). The crystal structure was analyzed using an X-ray diffractometer (DX-2700BH) with a Cu target Kα-ray source (wave length λ = 0.15406 nm) and scanning rate of 8° min−1. Raman spectra were recorded by using a Thermo Raman spectrometer (HE Evolution, HORIBA) with a laser wavelength of 532 nm and a spectral range of 200–2500 cm−1. The functional groups present in the samples were identified and analyzed using a Fourier transform infrared spectrometer (FT-IR, Nicolet iS20, Thermo Fisher Scientific Ltd). A thermogravimetric analyzer (TG209F3) was used to study the pyrolysis behavior of the samples under a nitrogen atmosphere within the temperature range of 25–900 °C at a heating rate of 10 °C min−1. The specific surface area and pore size distribution of the samples were determined by nitrogen adsorption measurements using a specific surface area and pore size analyzer (V-Sorb 4800TP, Ultrametrics).2.3 Electrochemical tests
For the electrochemical tests, an electrode slurry was prepared by dispersing 80 wt% of the active substance, 10 wt% of the conductive agent (Super-P), and 10 wt% of the binder (NaCMC:SBR = 1:1) in deionized water. The mixture was stirred thoroughly to ensure homogeneity and then cast onto a current collector to form pole pieces. The fabricated discs were vacuum dried at 80 °C for 12 hours in a vacuum oven. The CR2032 button cells were assembled in an argon-filled glove box. A glass fiber diaphragm (GF/D, Whatman) was used as the separator, and the electrolyte consisted of 1 M NaPF6 dissolved in a ternary solvent mixture of ethylene carbonate (EC), dimethyl carbonate (DMC), and ethyl methyl carbonate (EMC) in a volume ratio of 1:2:2. Galvanostatic charge and discharge (GCD) testing was carried out on a battery test system (Newell, China) at 25 °C with potential range of 0.005–2.5 V versus Na+/Na. Cyclic voltammetry (CV) tests were performed on a potentiostat (CHI660E, Chenhua, Shanghai, China) with a sweep rate of 0.1 mV s−1.
3 Results and discussion
The preparation process of HCs with composite precursors is depicted in Fig. 1. The apparent states of each stabilized sample are shown in Fig. S1.† It is clear that in comparison to the HC from pure starch, HCs from composite precursors are less foaming. Fig. 2 presents the SEM images of different HCs. As depicted in Fig. 2a, ST-M-51 retains the original morphology of natural starch (Fig. S2†). The HC particles are approximately 10 μm in size and display a tendency to agglomerate. In contrast, PR-M-51 in Fig. 2b possesses an irregular lumpy morphology, with a particle size ranging from 10 to 20 μm. The ST-RC-73 sample, obtained through the physical mixing and carbonization of starch and phenolic resin, displays a morphology composed of a mixture of spherical particles with a diameter of around 10 μm and irregular lumpy particles of approximately 20 μm (Fig. 2c). Notably, ST-PR-M-731 (Fig. 2d), ST-PR-M-732 (Fig. 2e), and ST-PR-M-733 (Fig. 2f) reveal a more pronounced transformation in the morphology. With increasing maleic anhydride content, the spherical hard carbon particles derived from starch and the irregular lumpy hard carbon particles derived from phenolic resin gradually coalesced. This led to the disappearance of the distinct spherical particle characteristics and the emergence of pores with a diameter of 10 μm or less on the surface of the HCs. These newly formed pore structures may provide more pathways for the transportation of Na+, thereby achieving considerable sodium storage performance. In addition, we found that except for sample ST-M-51, in all other samples, there are nanoscale small particles attached to hard carbon. The difference common to ST-M-51 and the other samples is the addition of phenolic resin, and irregular phenolic resin is likely to form local stress concentration points during ball milling. Therefore, we speculate that the small particles originate from the rupture of phenolic resin. To further verify that the small particles are not due to the introduction of impurities, we conducted SEM-EDS tests (Fig. S3†) on each sample. The tests found that there is no difference in element composition between the small particles and their adjacent hard carbon blocks. | ||
| Fig. 1 Scheme of preparation of composite precursors derived HCs. | ||
 | ||
| Fig. 2 SEM images of HCs. (a) ST-M-51, (b) PR-M-51, (c) ST-PR-73, (d) ST-PR-M-731, (e) ST-PR-M-732, (f) ST-PR-M-733. | ||
Taking starch and phenolic resin as units, maleic anhydride is introduced to enable the three components to undergo nucleophilic substitution and form ester groups. In order to verify these esterification reactions, FT-IR analysis was performed on HCs. As shown in Fig. 3a, the peak observed around 3300 cm−1 is ascribed to the O–H stretching vibration associated with hydrogen bonding. Notably, upon the addition of maleic anhydride, a decrease in the intensity of this peak is discernible. This demonstrates that the precursor has engaged in a chemical reaction, leading to the consumption of the hydroxyl groups. Subsequent to the introduction of maleic anhydride, the absorption peaks at 1720 cm−1 in the spectra of ST-PR-M-731, ST-PR-M-732, ST-PR-M-733, PR-M-51, and ST-M-51 are attributed to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond within the ester linkage. In contrast, the ST-PR-73 did not exhibit a characteristic peak. This disparity unequivocally suggests that the incorporation of maleic anhydride instigated an inter-precursor esterification reaction among the constituents. As shown in Fig. 1 the introduced ester groups act as a bridge to cross-link the different precursors. Additionally in Fig. 3a, it can be observed that the intensity of the peaks near 520 cm−1 significantly decreases in the sample with maleic anhydride incorporation. These peaks typically correspond to local lattice distortions caused by sp3 hybridization in disordered carbon structures of high-temperature carbonized samples. This phenomenon suggests that the three-dimensional cross-linked network formed by maleic anhydride likely guides a more ordered sp2 carbon arrangement during carbonization, thereby reducing sp3 defects.
O bond within the ester linkage. In contrast, the ST-PR-73 did not exhibit a characteristic peak. This disparity unequivocally suggests that the incorporation of maleic anhydride instigated an inter-precursor esterification reaction among the constituents. As shown in Fig. 1 the introduced ester groups act as a bridge to cross-link the different precursors. Additionally in Fig. 3a, it can be observed that the intensity of the peaks near 520 cm−1 significantly decreases in the sample with maleic anhydride incorporation. These peaks typically correspond to local lattice distortions caused by sp3 hybridization in disordered carbon structures of high-temperature carbonized samples. This phenomenon suggests that the three-dimensional cross-linked network formed by maleic anhydride likely guides a more ordered sp2 carbon arrangement during carbonization, thereby reducing sp3 defects.
 | ||
| Fig. 3 Physicochemical characterization of HCs. (a) FTIR spectrum of HCs. (b) TG-DTG curves of ST-PR-M-733. (c) X-ray diffraction patterns and (d) Raman spectrum of HCs. | ||
To investigate the influence of esterification-based cross-linking reactions on the pyrolytic behavior of starch, the thermogravimetric analysis was applied. The TG and DTG curves of the HCs are depicted in Fig. 3b and S4.† The direct carbonization of starch alone yielded a relatively meager carbon yield of ca. 5.4%. However, the combined addition of maleic anhydride and phenolic resin led to a significant transformation. A more intricate and densely intertwined network of molecular chains was formed, and the crystalline regions of the starch were partially disrupted. This structural modification culminated in a maximum carbon yield of 41.87%, signifying a substantial improvement in the carbon yield.
Fig. 3c illustrates the X-ray diffraction (XRD) patterns of all the HC samples. All samples exhibit two typical amorphous broad peaks near 23° and 43°, which correspond to the crystallographic planes (002) and (100), respectively. These broad peaks are indicative of the amorphous nature of the materials. When the mass ratio of the carbon source to maleic anhydride is less than or equal to 5:1, the position of the diffraction peak corresponding to the (002) crystal plane remains relatively stable with changes in the maleic anhydride content. However, when the mass ratio reaches 10:3, a shift in the diffraction peak towards a higher angle is observed. This shift can be attributed to the further increase in the maleic anhydride content during the pretreatment process, which leads to a more extensive esterification reaction. As a result, a denser carbon chain structure is formed. Upon pyrolysis, the carbon layers are compressed against each other, leading to a reduction in the interlayer spacing and, consequently, a shift in the diffraction peak position to a higher angle.30 To further understand the structure of the HCs, several key parameters, including the layer spacing, the apparent microcrystalline width on the a-axis, the apparent microcrystalline thickness on the c-axis, and the number of graphene sheet stack, were determined using the following equations:31
|
2dsinθ = nλ
| (1) |
|
L = κλ/βcosθ
| (2) |
| N = Lc/d002 + 1 | (3) |
| d002 (nm) | Lc (nm) | La (nm) | N | ID/IG | Surface area (m2 g−1) | Total pore volume (cm3 g−1) | |
|---|---|---|---|---|---|---|---|
| ST-PRC-73 | 0.390 | 1.22 | 1.53 | 1.31 | 1.03 | 1.28 | 0.003 |
| ST-PR-M-731 | 0.389 | 1.32 | 1.70 | 1.34 | 1.05 | 1.4 | 0.004 |
| ST-PR-M-732 | 0.387 | 1.88 | 1.75 | 1.48 | 1.11 | 1.5 | 0.007 |
| ST-PR-M-733 | 0.375 | 1.52 | 2.04 | 1.40 | 1.10 | 2.86 | 0.013 |
| PR-M-51 | 0.390 | 1.70 | 1.64 | 1.44 | 1.06 | 1.2 | 0.007 |
| ST-M-51 | 0.386 | 1.69 | 1.56 | 1.44 | 1.08 | 1.2 | 0.005 |
The Raman spectra of the HCs, shown in Fig. 3d, display two characteristic peaks at 1350 cm−1 and 1580 cm−1, the former representing the A1g vibration mode of the sp2 carbon ring caused by defects, and the latter representing the E2g vibration mode of the sp2 carbon atom.29 The degree of disorder in the carbon materials is commonly quantified by the ratio of the intensities of the D-band (associated with defects) to the G-band (representing graphitic carbon), denoted as ID/IG. As shown in Table 1, all the HCs exhibit a relatively low degree of graphitization. The ST-PR-M-732 and ST-PR-M-733 samples show the highest degree of disorder, which implies a greater concentration of defects in these samples. Previous studies demonstrated that the slope region at the high potential in the discharging curve is related to the Na+ adsorption at the edge and defect sites of graphite microcrystals.34–36 Therefore, it can be hypothesized that the ST-PR-M-732 and ST-PR-M-733 samples may possess a greater slope capacity for sodium storage.
It is well-known that the pore structure is of paramount importance in dictating the behaviors of Na+ diffusion and storage.32,35,37–39 Na+ can be stored by filling the pores, thereby contributing to a high platform capacity. The specific surface areas and pore size distributions of HC samples were characterized through nitrogen isothermal adsorption tests. The N2 adsorption/desorption isotherms and pore size distribution of HCs are presented in Fig. 4. The corresponding specific surface areas of the HCs were measured to be 1.28, 1.43, 1.56, 2.86, 1.21, and 1.20 m2 g−1 (Table 1). The pores were predominantly micropores and mesopores with pore sizes less than 10 nm, exhibiting a relatively narrow distribution. This abundant “microporous plus mesoporous” system is favorable for the efficient transport and storage of Na+, thus facilitating the attainment of enhanced sodium storage performance.40–42 Among the samples, ST-PR-M-733, ST-PR-M-732, and ST-M-51 display relatively high cumulative pore volumes. Given a similar pore size distribution, a higher cumulative pore volume implies a greater number of pores. This suggests that these samples may possess a more pronounced advantage in the platform region, potentially leading to superior sodium storage capacities.
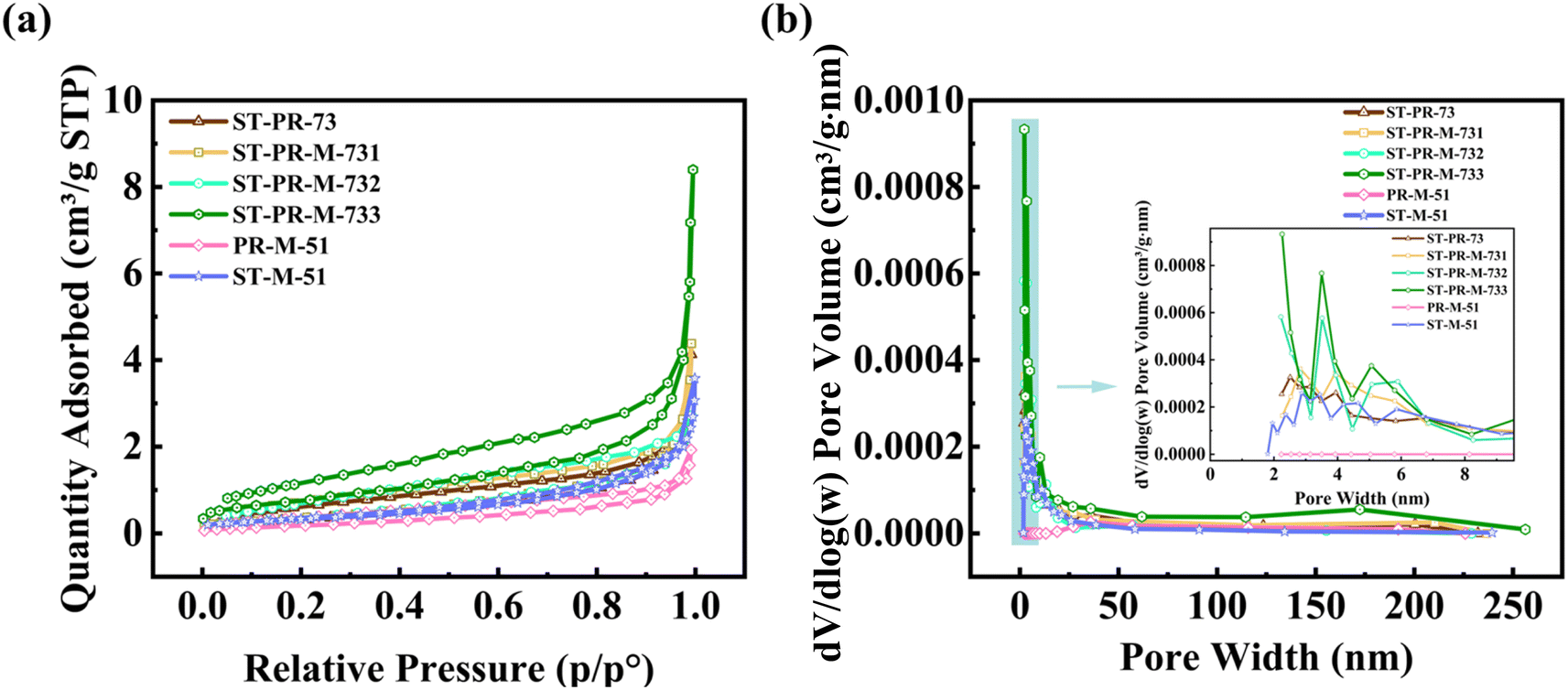 | ||
| Fig. 4 (a) N2 adsorption/desorption isotherms and (b) pore size distribution of HCs. | ||
Fig. 5a shows the CV curves of the hard carbon ST-PR-M-733 from the first to the fiffth cycle at a sweep rate of 0.1 mV s−1. The pair of redox peaks observed below 0.1 V and at 0.3 V correspond to the embedding/pore-filling and extraction of Na+, respectively. However, after the first cycle, the complete deposition of the SEI film typically causes irreversible deactivation of some sodium storage sites on the material surface, thereby reducing the peak current of the oxidation peak at 0.3 V. It should be noted that peak currents in CV curves do not directly reflect material capacity but rather the reaction kinetics. The peak current at 0.3 V gradually stabilizes by the third cycle. The stabilization of the SEI film reduces interfacial resistance and enhances sodium-ion diffusion rates. Since CV scanning rates are faster than GCD testing, complete SEI formation is not achieved during the first CV cycle. Additionally, the reaction of the negative electrode of the sodium ion battery is not composed of a pair of reversible REDOX reactions.43 Starting from the second cycle, a pair of broad redox peaks appears in the 0.1–0.6 V and 0.3–0.5 V ranges, corresponding to Na+ adsorption/desorption in the slope region. The gradual increase in peak intensity from the first to the third cycle aligns with the progressive stabilization of the SEI film.
 | ||
| Fig. 5 Electrochemical performance of HCs. (a) CV curves of ST-PR-M-732 in the first three cycles with the scan rate of 0.1 mV s−1 (b)The first discharge/charge profiles of HCs at 0.05C (15 mA g−1). (c) Slope and plateau capacities of HCs at 0.05C (15 mA g−1). (d) Discharge capacity at various rates (1C = 300 mA g−1). (e) Cycling performance of HCs at 0.2C. | ||
The electrochemical performance of HCs was further evaluated through a galvanostatic charge–discharge (GCD) test. As shown in Fig. 5b, a “step” at approximately 0.6 V is evident in the first discharge curve of the HCs, which is attributed to the formation of the SEI. This observation is consistent with the CV curves depicted in Fig. 5a. Regarding the discharge specific capacity and initial Coulomb efficiency at a rate of 0.05C, the materials ST-PR-73, ST-PR-M-731, ST-PR-M-732, ST-PR-M-733, PR-M-51, and ST-M-51 exhibited the following values: 302.11 mA h g−1/86.98%, 310.97 mA h g−1/87.10%, 342.28 mA h g−1/85.89%, 345.16 mA h g−1/88.68%, 329.99 mA h g−1/84.46%, and 333.05 mA h g−1/90.40%, respectively. Notably, ST-PR-M-732 and ST-PR-M-733 exhibited the highest discharge-specific capacities. This can be explained by the increased content of maleic anhydride, which intensifies the esterification reaction. As a result, the precursor forms larger carbon chain structures with a highly cross-linked carbon network. During the carbonization process, the formation of larger graphite microcrystals occurs, leading to the creation of more sodium storage sites. This enhanced structural development contributes to a higher sodium storage capacity, which aligns well with the X-ray diffraction (XRD) results.
In Fig. 5c, a comparison of the slope capacity and plateau capacity of hollow carbons (HCs) during the initial discharge at a rate of 0.05 C is presented. Both ST-PR-M-732 and ST-PR-M-733 show excellent slope capacities. Typically, the Na+ ions in the slope region are stored through adsorption on the surfaces, edges, and defect sites of the material.38 From Raman spectroscopy tests, it can be observed that ST-PR-M-732 and ST-PR-M-733 exhibit high ID/IG values. This indicates that these two HCs can provide more active sites for Na+ storage. As expected, these two samples demonstrate relatively high slope capacities of 218.08 mA h g−1 and 215.66 mA h g−1, respectively. Additionally, ST-PR-M-733 also shows a significant plateau capacity of 129.5 mA h g−1 at 0.05 C. This can be attributed to the abundance of pores in the HC structure (as indicated by BET results). Na+ ions can occupy these pores, resulting in the observed plateau capacity at low voltage.
The rate capacities of the HCs are presented in Fig. 5d. Both ST-PR-M-732 and ST-PR-M-733 show satisfactory rate performance. Specifically, after reverting to a current of 0.1C following a high current of 1C, their capacity recovery rates are 95.11% and 96.94%, respectively. Fig. 5e illustrates the cycling performance of the HCs when cycled at 0.2C for 100 cycles. ST-PR-M-733 maintains a specific discharge capacity of 177.42 mA h g−1, with a capacity retention of 87.24% after 100 cycles. The other components also demonstrate good cycling properties. In comparison to the hard carbon derived solely from phenolic resin, the combination of starch with phenolic resin, along with the addition of maleic anhydride for esterification, significantly enhances the sodium storage properties of the resulting hard carbon. During the pyrolysis process of the starch/phenolic resin composite-derived hard carbon, the molecular structure of the composite precursor rearranges, triggering an aromatization reaction. This reaction leads to the aggregation of macromolecules into aromatic compounds, which then undergo polycondensation. This entire process contributes to the mechanical stability of the carbon network. The denser carbon network of the composite system facilitates the formation of larger graphite layers, providing more active sites for sodium ion storage. This effect is further enhanced by the addition of maleic anhydride to the starch/phenolic resin composite system, which promotes the esterification reaction. As a result, the hard carbons derived from the composite system exhibit superior sodium storage properties compared to those obtained from either starch or phenolic resin alone.
4 Conclusion
In summary, an efficient and scalable method has been developed to prepare starch-based hard carbon (HC) with high sodium storage properties. The starch was esterified and cross-linked by maleic anhydride and phenolic resin. The foaming of starch was successfully inhibited then producing a larger and denser carbon framework with abundant micropores/mesopores. In particular, the material, known as ST-PR-M-733 demonstrates excellent performance, achieving a first discharge-specific capacity of up to 345.16 mA h g−1 with an excellent initial charge efficiency of 88.68%. When the current is increased to 1C and then returned to 0.1C, ST-PR-M-733 shows a high capacity recovery rate of 95.11%. This work provides a new starch pyrolysis stabilization strategy. The prepared HCs have advantages such as high sodium storage performance and low cost, showing the potential for large-scale commercialization.Conflicts of interest
There are no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The work was supported by the National Natural Science Foundation of China (Grant No. 52373046), Liangshan Science and Technology Program (Grant No. 23ZDYF0008), Natural Science Foundation of Sichuan Province (Grant No. 2025ZNSFSC1348), and Xihua University Science and Technology Innovation Competition Project for Postgraduate Students (Grant No. RC2400002136, RC2400002139, RC2400002132 and RC2400002152).Notes and references
- F. Xie, Z. Xu, Z. Guo and M.-M. Titirici, Prog. Energy, 2020, 2, 042002 CrossRef.
- M. Liu, Y. Wang, F. Wu, Y. Bai, Y. Li, Y. Gong, X. Feng, Y. Li, X. Wang and C. Wu, Adv. Funct. Mater., 2022, 32, 2203117 CrossRef CAS.
- W. Zhang, J. Peng, W. Hua, Y. Liu, J. Wang, Y. Liang, W. Lai, Y. Jiang, Y. Huang, W. Zhang, H. Yang, Y. Yang, L. Li, Z. Liu, L. Wang and S.-L. Chou, Adv. Energy Mater., 2021, 11, 2100757 CrossRef CAS.
- A. M. Escamilla-Pérez, A. Beda, L. Simonin, M.-L. Grapotte, J. M. Le-Meins and C. Matei Ghimbeu, ACS Appl. Energy Mater., 2023, 6, 7419–7432 CrossRef.
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- Y. Zheng, Y. Yao, J. Ou, M. Li, D. Luo, H. Dou, Z. Li, K. Amine, A. Yu and Z. Chen, Chem. Soc. Rev., 2020, 49, 8790–8839 RSC.
- R. Rohan, T.-C. Kuo, C.-Y. Chiou, Y.-L. Chang, C.-C. Li and J.-T. Lee, J. Power Sources, 2018, 396, 459–466 CrossRef CAS.
- W. Cao, E. Zhang, J. Wang, Z. Liu, J. Ge, X. Yu, H. Yang and B. Lu, Electrochim. Acta, 2019, 293, 364–370 CrossRef CAS.
- J. Wang, X. Guo, P. Apostol, X. Liu, K. Robeyns, L. Gence, C. Morari, J.-F. Gohy and A. Vlad, Energy Environ. Sci., 2022, 15, 3923–3932 RSC.
- Q. Liu, Y. Hu, X. Yu, Y. Qin, T. Meng and X. Hu, Nano Res. Energy, 2022, 1, 9120037 CrossRef.
- T. Lu, H. Du, N. Kong, H. Li, S. Xu, Z. Li and X. Du, J. Mater. Sci.: Mater. Electron., 2023, 34, 797 CrossRef CAS.
- L.-F. Zhao, Z. Hu, W.-H. Lai, Y. Tao, J. Peng, Z.-C. Miao, Y.-X. Wang, S.-L. Chou, H.-K. Liu and S.-X. Dou, Adv. Energy Mater., 2021, 11, 2002704 CrossRef CAS.
- Q. Li, J. Zhang, L. Zhong, F. Geng, Y. Tao, C. Geng, S. Li, B. Hu and Q.-H. Yang, Adv. Energy Mater., 2022, 12, 2201734 CrossRef CAS.
- K. Niitani, S. Ushiroda, H. Kuwata, H. N. Ohata, Y. Shimo, M. Hozumi, T. Matsunaga and S. Nakanishi, ACS Energy Lett., 2022, 7, 145–149 CrossRef CAS.
- O. Fromm, A. Heckmann, U. C. Rodehorst, J. Frerichs, D. Becker, M. Winter and T. Placke, Carbon, 2018, 128, 147–163 CrossRef CAS.
- M. Liu, J. Zhang, S. Guo, B. Wang, Y. Shen, X. Ai, H. Yang and J. Qian, ACS Appl. Mater. Interfaces, 2020, 12, 17620–17627 CrossRef CAS PubMed.
- B. Yang, J. Wang, Y. Zhu, K. Ji, C. Wang, D. Ruan and Y. Xia, J. Power Sources, 2021, 492, 229656 CrossRef CAS.
- Y. Chen, X. Shi, B. Lu and J. Zhou, Adv. Energy Mater., 2022, 12, 2202851 CrossRef CAS.
- Y. Cao, M. K. Majeed, Y. Li, G. Ma, Z. Feng, X. Ma and W. Ma, J. Alloys Compd., 2019, 775, 1286–1292 CrossRef CAS.
- S. Lee, G. Kwon, K. Ku, K. Yoon, S.-K. Jung, H.-D. Lim and K. Kang, Adv. Mater., 2018, 30, 1704682 CrossRef PubMed.
- J. Huang, E. Li, B. Dai, T. Lu, J. Teng, X. Tang, K. Zhang and J. Li, Carbon, 2024, 229, 119527 CrossRef CAS.
- M. Li, Z. Bi, L. Xie, G. Sun and C. M. Chen, ACS Sustain. Chem. Eng., 2019, 7, 14796–14804 CrossRef CAS.
- X. Liu, H. Liu, R. Yuan, L. Shang, T. Liu, W. Wang, Y. Miao, X. Chen and H. Song, Solid State Ionics, 2024, 406, 116475 CrossRef CAS.
- S. Zhao, C.-y. Wang, M.-m. Chen, Z.-q. Shi and N. Liu, New Carbon Mater., 2010, 25, 438–443 CrossRef CAS.
- Y. Sun, T. Shen, Z. He and S. Wang, J. Colloid Interface Sci., 2025, 678, 1142–1150 Search PubMed.
- M.-X. Song, L.-J. Xie, J.-Y. Cheng, Z.-L. Yi, G. Song, X.-Y. Jia, J.-P. Chen, Q.-G. Guo and C.-M. Chen, J. Energy Chem., 2022, 66, 448–458 CrossRef CAS.
- A. Kamiyama, K. Kubota, T. Nakano, S. Fujimura, S. Shiraishi, H. Tsukada and S. Komaba, ACS Appl. Energy Mater., 2020, 3, 135–140 Search PubMed.
- H.-l. Wang, Z.-q. Shi, J. Jin, C.-b. Chong and C.-y. Wang, J. Electroanal. Chem., 2015, 755, 87–91 CrossRef CAS.
- Y. Jin, S. Sun, M. Ou, Y. Liu, C. Fan, X. Sun, J. Peng, Y. Li, Y. Qiu, P. Wei, Z. Deng, Y. Xu, J. Han and Y. Huang, ACS Appl. Energy Mater., 2018, 1, 2295–2305 CrossRef CAS.
- A. Adamson, R. Vli, M. Paalo, J. Aruvli and A. Jnes, RSC Adv., 2020, 10, 20145–20154 RSC.
- J. Wang, L. Yan, Q. Ren, L. Fan, F. Zhang and Z. Shi, Electrochim. Acta, 2018, 291, 188–196 CrossRef CAS.
- Y. Cao, L. Xiao, M. L. Sushko, W. Wang, B. Schwenzer, J. Xiao, Z. Nie, L. V. Saraf, Z. Yang and J. Liu, Nano Lett., 2012, 12, 3783–3787 CrossRef CAS PubMed.
- L. Zhou, Y. Cui, P. Niu, L. Ge, R. Zheng, S. Liang and W. Xing, Carbon, 2025, 231, 119733 Search PubMed.
- Y. Li, Y.-S. Hu, M.-M. Titirici, L. Chen and X. Huang, Adv. Energy Mater., 2016, 6, 1600659 CrossRef.
- X. Chen, J. Tian, P. Li, Y. Fang, Y. Fang, X. Liang, J. Feng, J. Dong, X. Ai, H. Yang and Y. Cao, Adv. Energy Mater., 2022, 12, 2200886 Search PubMed.
- C. Bommier, T. W. Surta, M. Dolgos and X. Ji, Nano Lett., 2015, 15, 5888–5892 CrossRef CAS PubMed.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271 Search PubMed.
- X. Chen, C. Liu, Y. Fang, X. Ai, F. Zhong, H. Yang and Y. Cao, Carbon Energy, 2022, 4, 1133–1150 CrossRef CAS.
- Y. Li, Y. Lu, Q. Meng, A. C. S. Jensen, Q. Zhang, Q. Zhang, Y. Tong, Y. Qi, L. Gu, M.-M. Titirici and Y.-S. Hu, Adv. Energy Mater., 2019, 9, 1902852 Search PubMed.
- Q. Meng, Y. Lu, F. Ding, Q. Zhang, L. Chen and Y.-S. Hu, ACS Energy Lett., 2019, 4, 2608–2612 CrossRef CAS.
- T. Meenatchi, V. Priyanka, R. Subadevi, W.-R. Liu, C.-H. Huang and M. Sivakumar, Carbon Lett., 2021, 31, 1033–1039 CrossRef.
- M. Vincent, S. S. Kumar and D. Kowalski, Electrochim. Acta, 2023, 469, 143161 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00896d |
| This journal is © The Royal Society of Chemistry 2025 |