
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione derivatives via the Diels–Alder reaction: molecular docking, antibacterial activity, ADMET analysis and photophysical properties†
Sonali Priyadarshini Paridaa,
Seetaram Mohapatra*a,
Suhasini Mohapatra a,
Tankadhar Beherab,
Sabita Nayak*a and
Chita Ranjan Sahooc
a,
Tankadhar Beherab,
Sabita Nayak*a and
Chita Ranjan Sahooc
aOrganic Synthesis Laboratory, Department of Chemistry, Ravenshaw University, Cuttack, 753003, Odisha, India. E-mail: seetram.mohapatra@gmail.com; sabitanayak18@gmail.com
bSchool of Chemistry, Sambalpur University, Jyoti Vihar, 768019, Sambalpur, Odisha, India
cICMR-Regional Medical Research Centre, Department of Health Research, Ministry of Health & Family Welfare, Govt. of India, Bhubaneswar, 751023, Odisha, India
First published on 6th May 2025
Abstract
A series of fused tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione derivatives was successfully synthesized via the Diels–Alder reaction. Molecular docking studies were conducted to understand the interaction modes between the synthesized hybrid compounds and the receptor bacterial strains of Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus). Notably, the in silico results demonstrated that compound 19l (−8.7 kcal mol−1 with E. coli and −8.4 kcal mol−1 with S. aureus) and 19p (−8.7 kcal mol−1 with E. coli and −9.1 kcal mol−1 with S. aureus) exhibited good binding values. Additionally, the in vitro antibacterial studies showed that compounds 19l and 19p demonstrated excellent antibacterial activities, with a zone of inhibition (ZI) of 17 mm and a minimum inhibitory concentration (MIC) of 12.5 μg mL−1 against both E. coli and S. aureus, which were comparable to the performance of the standard antibiotic ciprofloxacin. Further, the bioavailability was assessed through virtual ADMET parameters, which suggested that most of the compounds possessed favorable pharmacokinetic profiles. To further enrich the study, photophysical properties of all the synthesized molecules were also examined using UV-visible and fluorescent spectroscopies.
1. Introduction
Bacterial infections have become a major problem globally.1,2 The misuse and overuse of antibiotics have led to the emergence of antimicrobial resistance (AMR), resulting in rapid rise of antibiotic-resistant strains of bacteria known as.3,4 Antibiotic-resistant infections result in severe illnesses, prolonged hospital stays, higher medical costs, and treatment failures. Over the past two decades, microbial infections have significantly increased.5 Each year, there is a death range of approximately 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 worldwide due to antimicrobial resistance, and it is estimated that it may increase to 10 million cases per year by 2050.6–8 Despite the discovery and development of numerous new anti-microbial drugs, there is still an increase in bacterial infections, and their treatment is still a major challenge for public health.9,10 Therefore, the discovery of novel, cost-effective antimicrobial agents is necessary to mitigate the proliferation of these pathogens.
000 worldwide due to antimicrobial resistance, and it is estimated that it may increase to 10 million cases per year by 2050.6–8 Despite the discovery and development of numerous new anti-microbial drugs, there is still an increase in bacterial infections, and their treatment is still a major challenge for public health.9,10 Therefore, the discovery of novel, cost-effective antimicrobial agents is necessary to mitigate the proliferation of these pathogens.
Among the promising scaffolds for antibacterial drug development, the 2H-chromene core has garnered significant attention owing to its diverse biological activities.11–13 Studies have demonstrated that the 2H-chromene derivatives exhibit potent antibacterial properties, along with other pharmacological effects such as anticancer, anti-inflammatory, anti-tubercular, and anti-HIV activities.14–23 This versatile scaffold provides a robust platform for designing novel therapeutic agents with improved efficacy and selectivity against bacterial pathogens.
Similarly, maleimides are the nitrogen-containing heterocyclic motifs which have shown substantial promise in biomedical research. For instance, N-disubstituted maleimides represent a valuable structural motif in drug discovery, contributing to the development of novel therapeutic agents targeting bacterial infections.24–27 By combining the structural features of 2H-chromene and maleimide, there is an opportunity to enhance antibacterial efficacy through hybrid molecules that leverage the biological potency of both scaffolds (Fig. 1).24–27
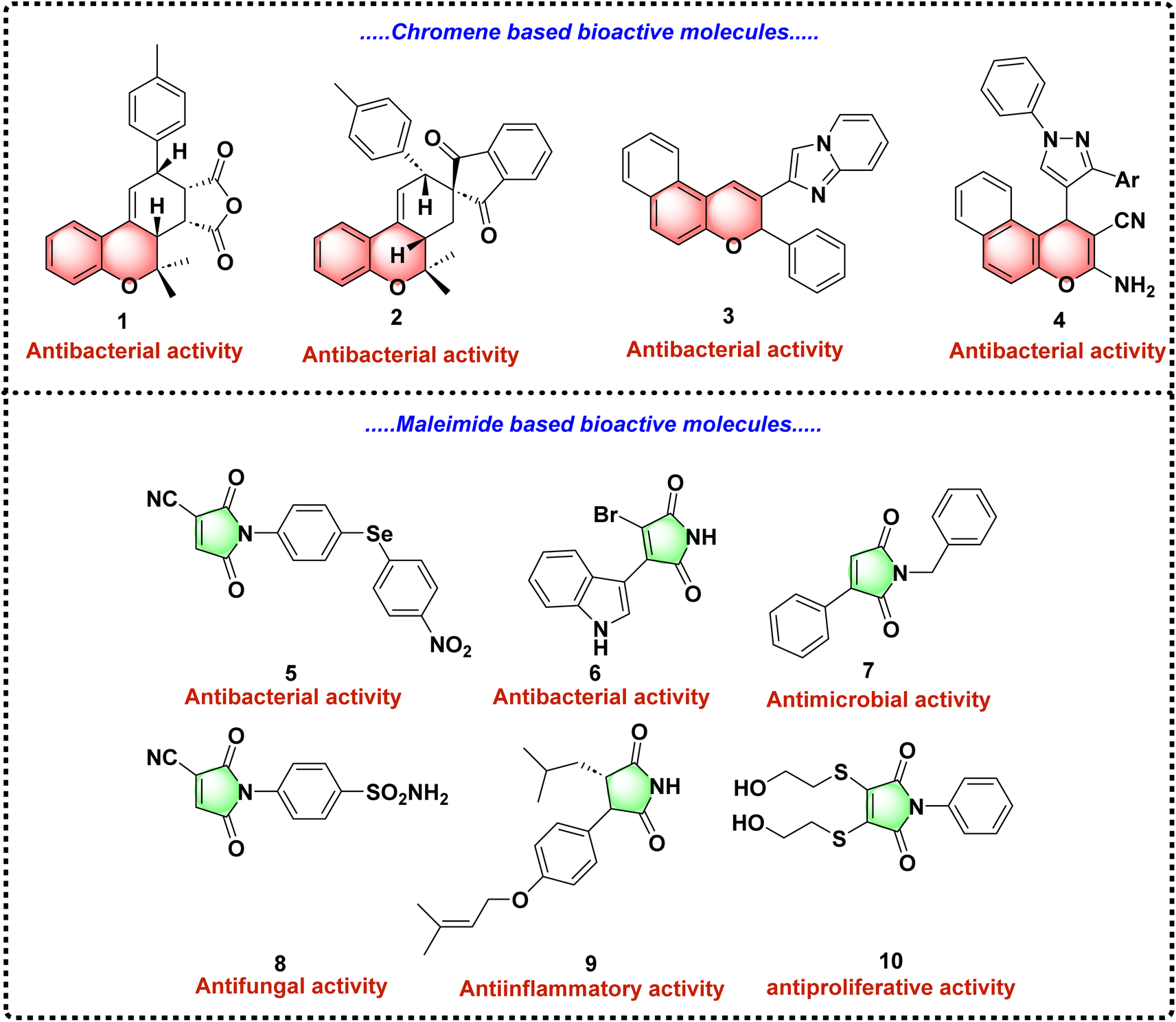 | ||
| Fig. 1 Structures of some selected bioactive molecules of 2H-chromenes and maleimide. | ||
Previous studies have explored the synthesis of chromene-based maleimide hybrids using various chemical transformations. In 2006, Park et al. first reported the synthesis of novel benzopyran-based scaffolds through Diels–Alder reactions.28 Subsequent work by the same research group in 2010 demonstrated the biological potential of these hybrids in treating prostate cancer, diabetes, and obesity.29,30 However, their synthetic methods relied on expensive and carcinogenic reagents, such as trifluoromethanesulfonic anhydride and tin catalysts, limiting their scalability and environmental compatibility.
To address these limitations and develop a more cost-effective and eco-friendly approach, we present an efficient synthesis of tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione derivatives. Our strategy involves phosphorus tribromide and palladium catalyst, which are more economical and environmentally benign alternatives. This method also explored diverse substituents on the chromene framework and the para position of the styrene ring, enhancing the scope of structural modifications.
As part of our ongoing efforts to develop tricyclic benzopyran derivatives as antibacterial agents, we recently synthesized a series of tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione derivatives and evaluated their potent antibacterial activities. Molecular docking studies provided insights into their binding affinities and possible mechanisms of action. Furthermore, in silico ADMET analyses were conducted to assess their pharmacokinetic properties, and in vitro antibacterial assays were performed against two pathogenic bacterial strains to evaluate their efficacy. Spectroscopic studies, including UV-visible and fluorescence analyses, were also carried out to investigate the impact of various substituents on the photophysical properties of the synthesized compounds. This comprehensive investigation aims to advance the development of potent, cost-effective antibacterial agents to combat the rising challenge of antibiotic resistance.
2. Result and discussion
2.1. Chemistry
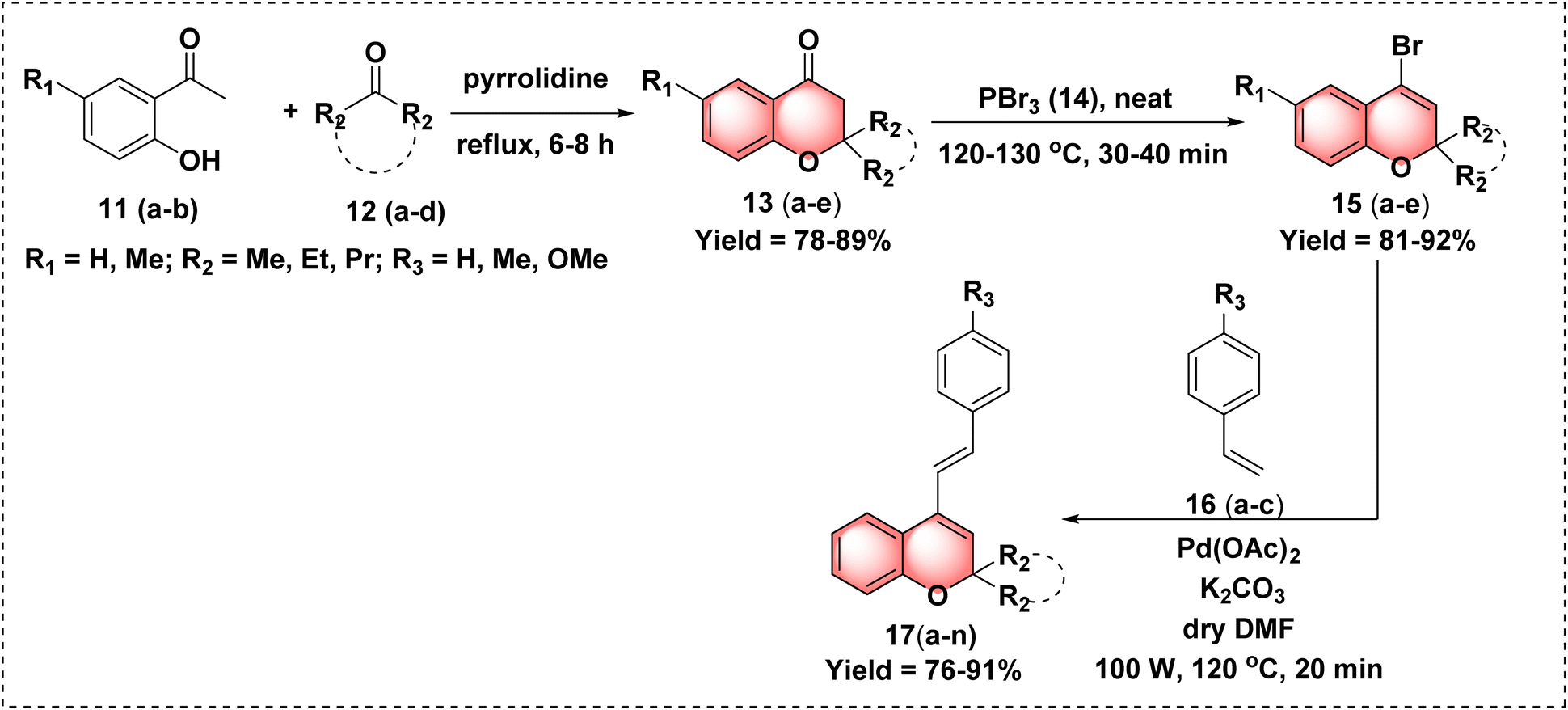 | ||
| Scheme 1 Synthesis of 2,2-disubstituted chromones 13(a–e), 4-bromo-2,2-dialkylsubstituted-2H-chromenes 15(a–e) and 2,2-disubstituted-4-styryl-2H-chromene 17(a–n). | ||
After successfully synthesizing the parent pharmacophore 17a, we synthesized the target hybrid molecule 19a using commercially available N-methyl maleimide 18a. To synthesize the fused carbacycle 19a through a Diels–Alder reaction, we screened the reaction conditions by altering the solvent, time and temperature.
According to the previously reported literature,14,15 the reaction was first carried out in different solvents, including DCM, THF, 1,4-dioxane, DMF and benzene at their respective boiling points for about 12–24 hours, which resulted in poor to moderate yield (entries 1–6). However, the reaction did not proceed in H2O (entry 7). We observed an increase in the product yield when the reaction temperature was raised from 80 °C to 100 °C using toluene as the solvent while maintaining the same reaction time. However, the starting materials were not fully consumed at this stage. Upon further increasing the temperature to 120 °C, we achieved excellent yield within 4 hours. This improvement may be due to the energy barrier of the reactants being overcome at higher temperatures, thereby facilitating the reaction (entries 8–10). The results are illustrated in Table 1. After the successful completion of the reaction, product 19a was characterized using 1H NMR, 13C NMR and HRMS analyses.
|
|||||
|---|---|---|---|---|---|
| Entrya | Maleimide equiv. | Solvent | Temp. (°C) | Time (h) | Yield (%) |
| a Reaction conditions: 17a (1 equiv.), 18 (3 equiv.), toluene (2–3 mL); isolated yield; n.r. = no reaction. | |||||
| 1 | 1 | DCM | 35 | 24 | 12 |
| 2 | 2 | DCM | 35 | 24 | 22 |
| 3 | 2 | THF | 60 | 24 | 28 |
| 4 | 2 | 1,4-Dioxane | 100 | 24 | 32 |
| 5 | 2 | DMF | 150 | 24 | 37 |
| 6 | 2 | Benzene | 80 | 12 | 51 |
| 7 | 2 | H2O | 100 | 24 | NR |
| 8 | 2 | Toluene | 80 | 12 | 59 |
| 9 | 2 | Toluene | 100 | 12 | 76 |
| 10 | 3 | Toluene | 120 | 04 | 91 |

 | ||
| Fig. 2 1H and 13C NMR spectral data analysis of compound 19a. | ||
After optimizing the reaction conditions and spectroscopic characterization, we next explored the scope and generality of the reaction on various substrates, as shown in Scheme 2.
 | ||
| Scheme 2 Synthesis of a library of tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione derivatives 19(a–q). | ||
2,2-Disubstituted-4-styryl-2H-chromene 17(a–n) initially reacted with N-methyl maleimide 18a, resulting in tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione derivatives in good to excellent yield. From Scheme 2, it is observed that the yield of the cyclized product decreases with an increase in the alkyl chain length at the R2 position of the chromene moiety. Compounds having 2,2-dimethyl substituents at the R2 position of 19(a–c) displayed excellent yield (91–94%). Further, by modifying the methyl group to ethyl group 19(d–f), there is a slight decrease in the yield of the cyclized product (88–91%). Similarly, having 2,2-dipropyl substituents in 19(g–i), the yield decreases significantly (83–89%). However, compounds 19(j–n) show remarkable yields (84–90%) when a methyl group is introduced at the R1 position of the chromene framework.
Furthermore, to increase the substrate scope and check the yield of the cyclized product, 2,2-disubstituted-4-styryl-2H-chromene 17(o–q) was further reacted with N-phenyl maleimide 18b to give the desired derivatives in good yield (73–79%). It has been concluded that N-methyl maleimide derivatives give higher yield as compared to N-phenyl maleimide, as shown in Scheme 2. The structures of cycloadducts 19(a–q) were confirmed by 1H, 13C, and HRMS spectroscopic techniques.
 | ||
| Fig. 3 Single crystal X-ray representation of compound 19i. | ||
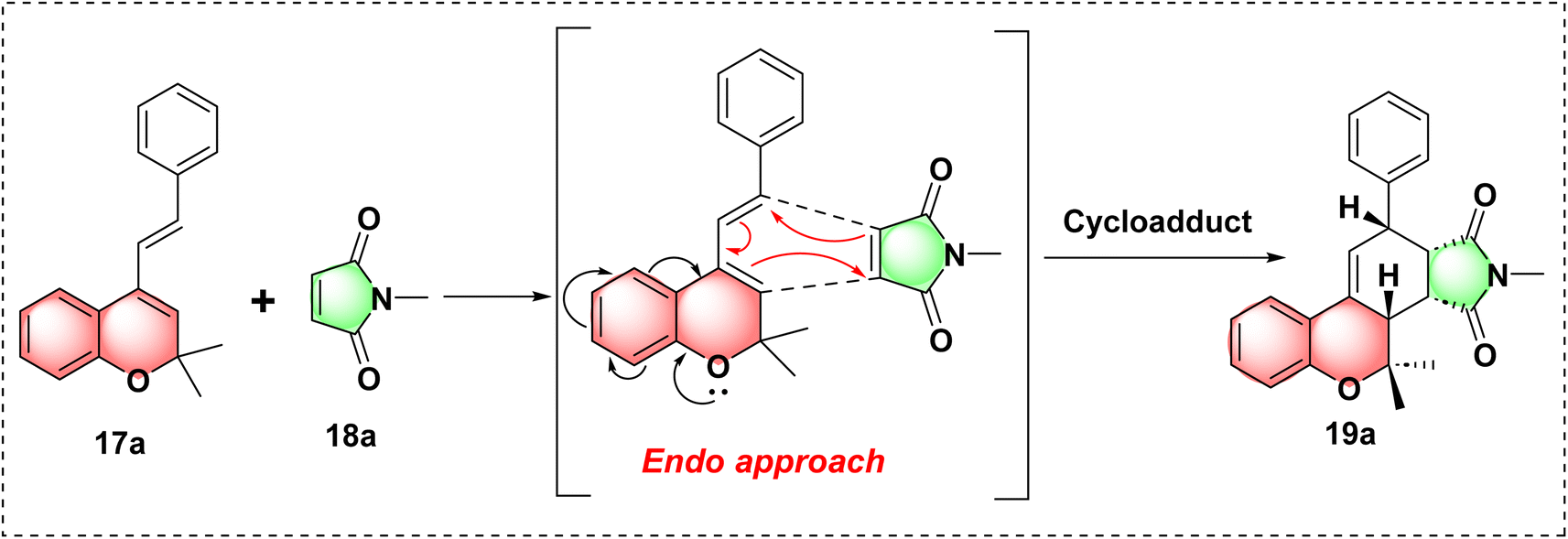 | ||
| Scheme 3 Proposed mechanism of 19a. | ||
 | ||
| Scheme 4 Synthesis of 11-(4-methoxyphenyl)-2-methyl-4,4-dipropylchromeno[3,4-e]isoindole-1,3(2H,4H)-dione 21. | ||
3. Biological studies
3.1. Molecular docking study
Molecular docking studies of the synthesized hybrid tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione 19(a–q) derivatives were performed to identify the binding interaction with the DNA gyrase inhibitor. The Gram-negative bacterial strain E. coli and Gram-positive bacterial strain S. aureus are two human pathogenic bacterial strains of the DNA gyrase inhibitor used for molecular modelling. The crystal structure of E. coli (PDB ID: 3G7E) and S. aureus (PDBID: 3G7B) were retrieved, and the heteroatoms and water molecules were removed during the docking procedure. Autodock tool 4.2.0 was used to determine the binding affinity scores, and the Discovery Studio v 2017 program was employed in visualizing the intermolecular binding interaction of the docked complex. In this study, seventeen newly designed hybrid molecules were successfully docked, and the binding affinity values ranged from −5.6 kcal mol−1 to −8.7 kcal mol−1 in the case of E. coli and −6.9 kcal mol−1 to −9.1 kcal mol−1 in the case of S. aureus. The current investigation revealed that compound 19l possessing diethyl at the R2 position of the chromene ring, methyl at the R1 position, hydrogen at the R3 position and methyl at the R4 position showed very good yield −8.7 kcal mol−1 with E. coli DNA gyrase and −8.4 kcal mol−1 with S. aureus DNA gyrase. Furthermore, compound 19p possesses dimethyl at the R2 position of the chromene ring, methyl at the R3 position and phenyl group at the R4 position, demonstrating robust binding interactions with a binding energy value of −8.7 kcal mol−1 for E. coli (PDB ID: 3G7E) and −9.1 kcal mol−1 for S. aureus (PDBID: 3G7B). The synthesized compounds interacted with bacterial DNA gyrase through various interactions such as van der Waals, conventional hydrogen bond, carbon–hydrogen bond, alkyl, π–alkyl, π–anion, π–cation, and π–sigma. The docking scores and their binding interactions are displayed in Table 2.| Sl no. | E. coli (PDB ID: 3G7E) | S. aureus (PDBID: 3G7B) | ||
|---|---|---|---|---|
| Score (kcal mol−1) | Binding interactions | Score (kcal mol−1) | Binding interactions | |
| 19a | −7.1 | Arg62, Ala39, Pro65, Lys89 | −8.1 | Ile79, Ile63, Ala38 |
| 19b | −6.2 | Asn32, Phe90, Ile80, Ala76, Pro65, Ile64 | −7.8 | Asn31, Ile63, Ala38 |
| 19c | −7.9 | Pro65, Asp35, Phe90, His102, Ala39, Lys89, Ile64, Ala76, Ile80 | −8.2 | Arg61, Ser32, Ala38, Asn31, Ile79, Pro64, Ile63 |
| 19d | −7.9 | Arg62, Pro65, Lys89, Ala39, Ile64 | −8.3 | Asn31, Pro64 |
| 19e | −7.4 | Arg62, Leu38, Lys89, Val97 | −8.3 | Asn31, Glu35, Arg61, Ile63, Ala38 |
| 19f | −6.5 | Phe90, Arg62, Leu38, Pro65, Ala39, Lys89 | −7.0 | Asn31, Glu35, Pro64, Ile79, Ile63, Ala75 |
| 19g | −6.7 | Arg62, Leu38, His41, Ala39, Lys89 | −7.9 | Glu35, Arg61, Ile79, Ile63, Pro64 |
| 19h | −5.6 | Phe90, Glu36, Arg62, Ile80, Pro65, Ile64, Ala39, Lys89 | −7.9 | Arg61, Ile63, Ile79, Pro64, Ala38 |
| 19i | −8.2 | Asp35, Pro65, Ile64, Ile80, Leu38, Ala39, Lys89 | −6.9 | Asn31, Pro64, Ile63 |
| 19j | −6.9 | Leu38, Glu36, Lys89, Ala39, Asp35, Val97 | −9.1 | Glu35, Arg61, Asn31, Ile63, Ile79, Ile129, Pro64 |
| 19k | −6.9 | Ala39, Arg62, Glu36, Pro65, Ala39 | −8.2 | Arg61, Ile63, Ile79, Ala38 |
| 19l | −8.7 | Thr151, Ile64, Pro65, Ala33, Glu36, Arg62, Phe90, Lys89, Ala39, Ile80 | −8.4 | Asn31, Arg61, Ile79, Pro64, Ile63, Ala38 |
| 19m | −7.1 | Arg62, Glu36, Asp35, Lys89, His102, Ala39 | −8.4 | Asn31, Arg61, Glu35, Gly62, Pro64, Ile63 |
| 19n | −8.3 | Arg62, Ala86, Asp59, Lys89, Val106, Val29, Ala33, Ile64, Pro65, Ile80 | −8.4 | Asn31, Ile79, Ile63 |
| 19o | −6.4 | Arg62, Lys89, Asn32, Pro65, Ile64, Phe90 | −8.6 | Arg61, Ile63, Asn31, Ile79, Pro64 |
| 19p | −8.7 | Arg62, Val97, Lys89, Ala39, Ile64, Pro65 | −9.1 | Asn31, Glu35, Arg61, Ile63, Ile79, Pro64 |
| 19q | −7.5 | Arg62, Glu36, Asp35, Ala39, Lys89, Val97 | −8.7 | Glu35, Arg61, Ile79, Ile63, Pro64 |
From Table 2, it is noted that compound 19l has very good binding interactions with the active pocket site of E. coli and S. aureus DNA gyrase. The docked complex with DNA gyrase E. coli displays a series of van der Waals interactions with specific amino acid residues Asp92, Arg122, Asp91, Ala76, Asp59, Gly63, Gly61, Gly88, Ala86, Gly87, His102, Asp35, Leu38, carbon hydrogen bonds with Pro65, Thr151, Ile64, Ala33, π–cation and π–anion interactions with Arg62, Glu36, alkyl interaction with Ala39, Phe90, Lys89, Pro65, Ile64, and π–alkyl interaction with Ile80, Ala39, Lys89 for compound 19l. Similarly, compound 19l held into the active cavities with the DNA gyrase enzymes of S. aureus (PDBID: 3G7B) via van der Waals interactions with Ala75, Asp34, Glu35, Ser32, Gly62, Ser83, conventional hydrogen bond with Asn31, π–cation interaction with Arg61, π–sigma interaction with ILE79, alkyl interaction with Ile63, Pro64 and π-alkyl interaction with Ala38, Ile63 and Pro64. Compound 19p also displayed a remarkable result for both the docked complexes: van der Waals interactions with Gly99, Ser98, Gly100, His102, Asp35, Gly88, Ile80, Ala76, Phe90, Glu36, Asp91, conventional hydrogen bond with Arg62, alkyl interaction with Val97, Lys89, π–alkyl interaction with Ala39, Pro65, Ile64 in the case of E. coli DNA gyrase. Alternatively, S. aureus shows van der Waals interactions with Arg98, Gly62, Ser32, Asp58, Thr127, Ile129, Ile28, Asp34, conventional hydrogen bond with Asn31, π–cation and π–anion interactions with Arg61, Glu35, π–sigma with Ile63, alkyl and π–alkyl interaction with Ile79 and Pro64 for compound 19p. From the docking score, it is observed that the compounds 19l and 19p display promising results in terms of antibacterial activities. The 2D and 3D diagrams of the most potent compounds, 19l and 19p, against E. coli DNA gyrase and S. aureus DNA gyrase, are represented in Fig. 4 and 5, respectively.
 | ||
| Fig. 4 2D interactions of the most potent compounds (A) compound 19l and (B) Compound 19p with Gram-negative bacterial DNA gyrase (PDBID: 3 G7E). (C) Compound 19l and (D) compound 19p with Gram-positive bacterial DNA gyrase (PDBID: 3G7B). | ||
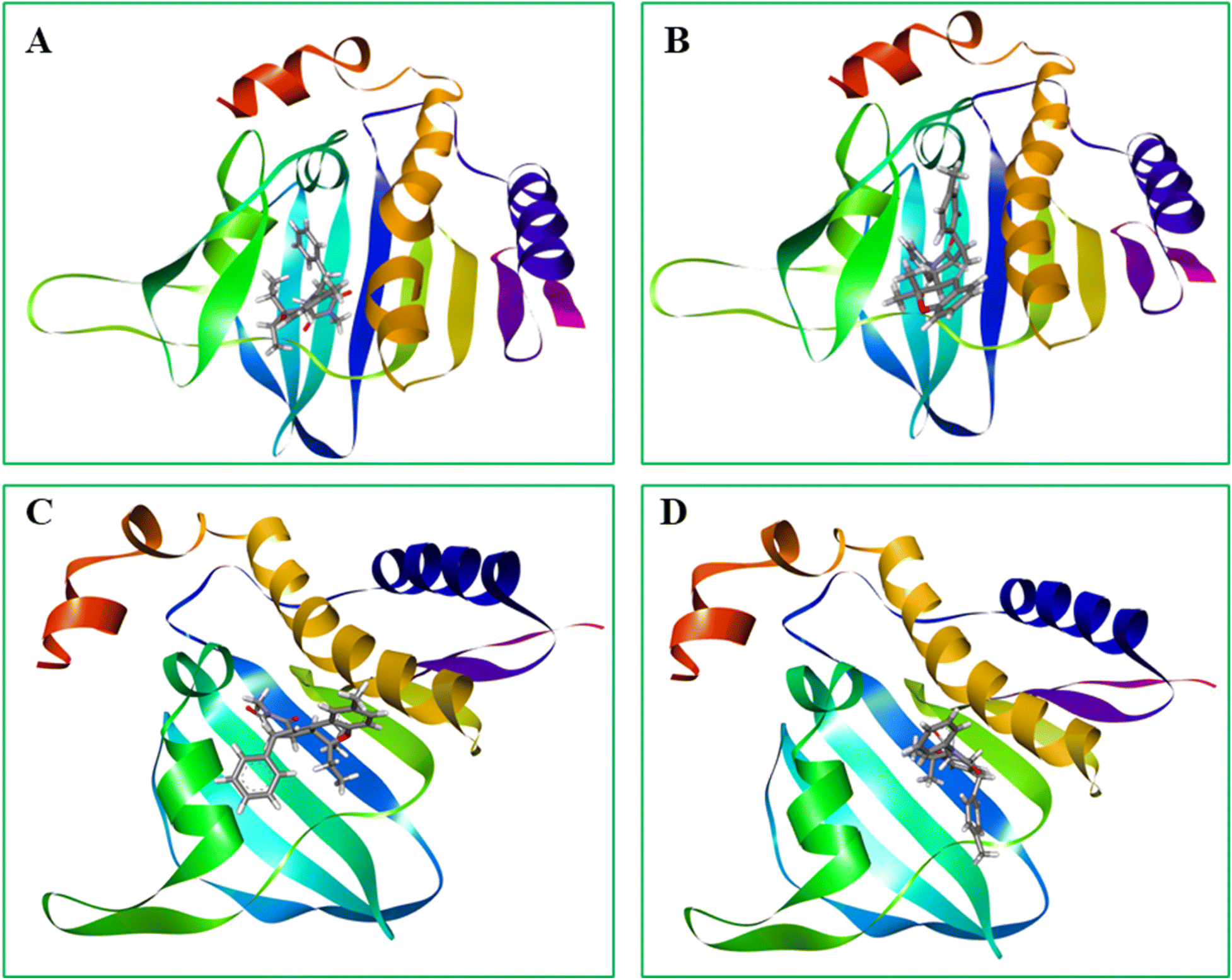 | ||
| Fig. 5 Most potent molecular docking studies of maleimide derivatives. The images depict the protein and ball-stick models. (A) Compound 19l and (B) compound 19p with Gram-negative bacterial DNA gyrase (PDBID: 3G7E). (C) Compound 19l and (D) Compound 19p with Gram-positive bacterial DNA gyrase (PDBID: 3G7B). | ||
3.2. Antibacterial evaluation
A series of tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione derivatives 19(a–q) were investigated for in vitro antibacterial activities against two human bacterial strains. All the synthesized derivatives were evaluated against Gram-negative E. coli and Gram-positive S. aureus via the agar well diffusion method using the standard drug ciprofloxacin. The ZI (zone of inhibition) and MIC (minimum inhibitory concentration) values of those molecules against these bacteria are also determined and the molecules are found to be active against both the bacterial strains, as represented in Table 3.| Sl. no. | E. coli | S. aureus | ||
|---|---|---|---|---|
| ZI (mm) | MIC (μg mL−1) | ZI (mm) | MIC (μg mL−1) | |
| 19a | 15 | 25 | 16 | 25 |
| 19b | 13 | 50 | 15 | 25 |
| 19c | 16 | 25 | 16 | 25 |
| 19d | 16 | 25 | 16 | 25 |
| 19e | 15 | 25 | 16 | 25 |
| 19f | 14 | 50 | 14 | 50 |
| 19g | 14 | 50 | 15 | 25 |
| 19h | 12 | 50 | 15 | 25 |
| 19i | 16 | 50 | 14 | 50 |
| 19j | 14 | 50 | 17 | 12.5 |
| 19k | 14 | 50 | 16 | 25 |
| 19l | 17 | 12.5 | 17 | 12.5 |
| 19m | 15 | 25 | 16 | 25 |
| 19n | 16 | 25 | 16 | 25 |
| 19o | 14 | 50 | 17 | 12.5 |
| 19p | 17 | 12.5 | 17 | 12.5 |
| 19q | 15 | 25 | 16 | 25 |
| StdCiprofloxacin | — | 6.25 | — | 6.25 |
Among all the tested compounds, compound 4,4-diethyl-2,8-dimethyl-11-phenyl-3b,4,11,11a-tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione 19l and 4,4-dimethyl-2-phenyl-11-(p-tolyl)-3b,4,11,11a-tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione 19p were found to be the most potent compounds with a Z1 of 17 mm against both E. coli and S. aureus bacterial strains. Furthermore, we performed a cell viability assay to determine the minimum inhibitory concentration (MIC) of the compounds. The MIC results revealed that both E. coli and S. aureus displayed potent activity with an MIC value of 12.5 μg mL−1 for both compounds 19l and 19p. However, from the antibacterial study, we found that compound 2-methyl-4,4-dipropyl-11-(p-tolyl)-3b,4,11,11a-tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione 19h possessed notably poor antimicrobial activity with a ZI value of 12 mm and 15 mm for E. coli and S. aureus, respectively. The respective ZI values and MIC values of all the tested compounds are illustrated graphically in Fig. 6 and 7, respectively.
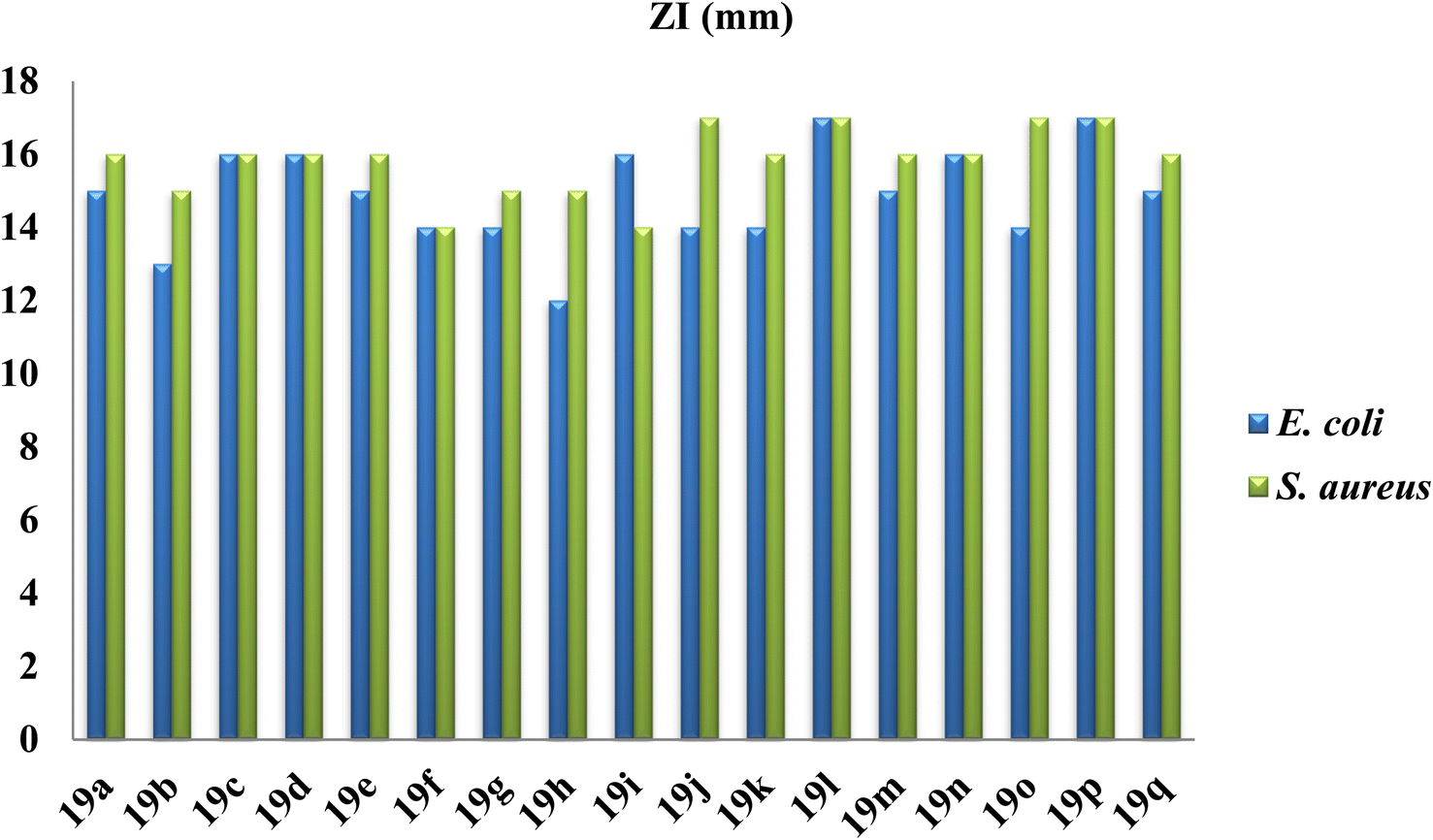 | ||
| Fig. 6 Graphical representation of the in vitro antimicrobial (ZI) assay of the synthesized tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione derivatives. | ||
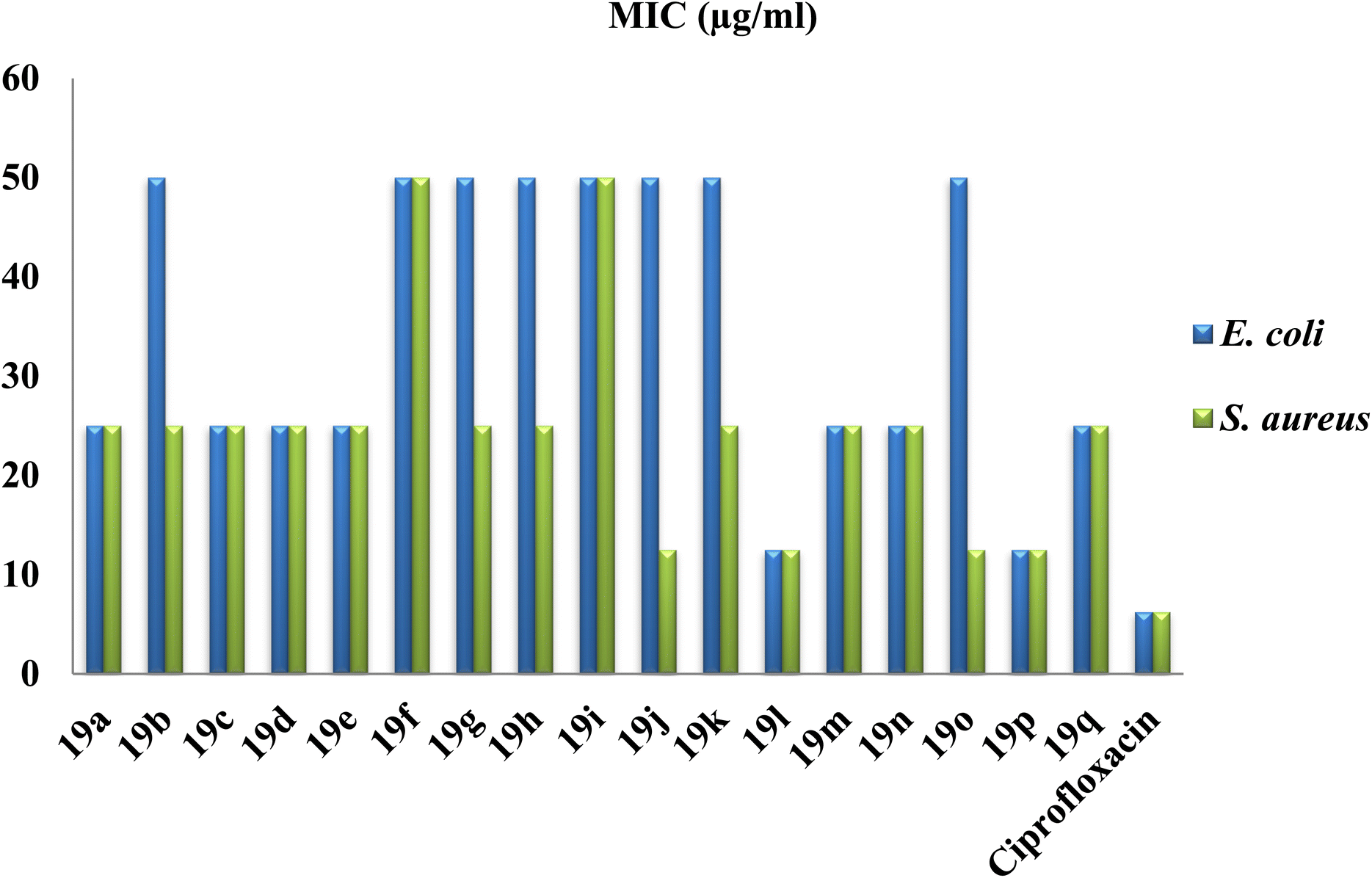 | ||
| Fig. 7 Graphical representation of the in vitro antimicrobial (MIC) assay of the synthesized tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione derivatives compared with the standard drug ciprofloxacin. | ||
3.3. Structure activity relationship (SAR) studies
From the in silico molecular docking study, we found that compounds, 4,4-diethyl-2,8-dimethyl-11-phenyl-3b,4,11,11a-tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione 19l and 4,4-dimethyl-2-phenyl-11-(p-tolyl)-3b,4,11,11a-tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione 19p, have good binding scores of −8.7 kcal mol−1 and −8.7 kcal mol−1 for E. coli and −8.4 kcal mol−1 and −9.1 kcal mol−1 for S. aureus DNA gyrase, respectively. It also demonstrated effective antibacterial activity with a MIC value of 12.5 μg mL−1 against both E. coli and S. aureus bacterial strains for both molecules 19l and 19p. Furthermore, it is observed that compound 2-methyl-4,4-dipropyl-11-(p-tolyl)-3b,4,11,11a-tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione 19h tivity with a MIC value of 50 μg mL−1 for E. coli and 25 μg mL−1 for S. aureus bacterial strains. Also, compound 19h has a poor binding affinity score (−5.6 kcal mol−1 for E. coli and −7.9 kcal mol−1 for S. aureus), as compared to all the synthesized molecules.The structure–activity relationship studies reveal that the compound is more favorable towards antibacterial activity, particularly when the chromene scaffold is associated with a diethyl group at the R2 position with a methyl group at the R1 position, hydrogen at the R3 position with N-methyl maleimide (19l) as well as with dimethyl at the R2 position, methyl at the R3 position with N-phenyl maleimide (19p). However, very poor results were observed in the case of compound 19h, which is associated with the dipropyl group at the R2 position and methyl at the R3 position with N-methyl maleimide. The pictorial representation of the SAR study is illustrated in Fig. 8.
 | ||
| Fig. 8 Pictorial representation of the SAR of the tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3 aH)-dione derivatives 19(a–q). | ||
3.4. Estimation of physicochemical, pharmacokinetic and ADMET properties
The in silico pharmacokinetic study using ADMET was carried out for drug development cascades. The pharmacokinetic and physicochemical properties of seventeen synthesized molecules 19(a–q) were evaluated using the SwissADME tool, comparing them to the standard drug ciprofloxacin.40,41 Subsequently, the Lipinski's Rule of Five (RO5) was used for the physicochemical properties.42 It states that molecular weight (MW) should be ≤500 g mol−1, octanol–water partition coefficient (cLogP) should be ≤5, number of hydrogen bond acceptors (HBA) should be ≤10, number of hydrogen bond donors (HBD) should be ≤5 and topological polar surface area (tPSA) should be ≤140 Å for a potential compound to be an orally active drug molecule.43–45
Additionally, the topological polar surface area (TPSA), which is inversely related to the percentage absorption (%ABS = 109–0.345 × TPSA), was proposed as a potential alternative to counting hydrogen bonding groups for estimating the absorption percentages. The analysis of the results indicates that the synthesized derivatives exhibit favorable physicochemical properties, with hydrogen bond acceptors (HBA) ranging from 3 to 4, TPSA values between 36.13 and 44.06 Å, and an acceptable number of hydrogen bond donors.
Again, the toxicity of the potent molecules 19l and 19p was determined using ProTox II online software. ProTox II tool determines the toxicity levels and the 50% lethal dose (LD50).20,46–48 From Table 4, compound 19l showed a Class 5 toxicity level with an estimated LD50 of 2200 mg kg−1, whereas compound 19p showed a Class 5 toxicity level with an estimated LD50 of 3900 mg kg−1 (Fig. 9). Consequently, the blood–brain-barrier (BBB), human-intestinal-absorption (HIA), and Caco-2-permeability were evaluated. From the result, it was concluded that most of the compounds are less toxic and suitable as oral bioactive drug molecules.
| Sl. no | Compound code | RO5 | % ABSf | Toxicity | Pharmacokinetics | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MWa | HBAb | HBDc | mlogPd | tPSAe | Class | LD50g (mg kg−1) | BBBh | HIAi | Caco-2j | |||
| a Molecular weight in g mol−1.b Number of H-bond acceptors.c Number of H-bond donors.d Partition coefficient (lipophilicity).e Topological polar surface area in Å.f % of Absorbance.g 50% lethal dose.h Blood brain barrier permeant.i Human-intestinal-absorption.j Caco-2-permeability. | ||||||||||||
| 1 | 19a | 373 | 3 | 0 | 3.69 | 36.13 | 96.53 | 5 | 2200 | 0.93 | 1 | 0.63 |
| 2 | 19b | 387 | 3 | 0 | 4.13 | 36.13 | 96.53 | 5 | 2200 | 0.93 | 1 | 0.63 |
| 3 | 19c | 403 | 4 | 0 | 3.65 | 43.67 | 93.93 | 5 | 2200 | 0.87 | 1 | 0.60 |
| 4 | 19d | 401 | 3 | 0 | 4.35 | 36.52 | 96.40 | 5 | 2200 | 0.97 | 1 | 0.63 |
| 5 | 19e | 415 | 3 | 0 | 4.79 | 36.52 | 96.40 | 5 | 2200 | 0.97 | 1 | 0.63 |
| 6 | 19f | 431 | 4 | 0 | 4.31 | 44.06 | 93.79 | 5 | 2200 | 0.93 | 1 | 0.60 |
| 7 | 19g | 429 | 3 | 0 | 5.37 | 36.52 | 96.40 | 5 | 2200 | 0.98 | 1 | 0.63 |
| 8 | 19h | 443 | 3 | 0 | 5.81 | 36.52 | 96.40 | 5 | 2200 | 0.98 | 1 | 0.63 |
| 9 | 19i | 459 | 4 | 0 | 5.32 | 44.06 | 93.79 | 5 | 2200 | 0.97 | 1 | 0.60 |
| 10 | 19j | 387 | 3 | 0 | 4.13 | 36.13 | 96.53 | 5 | 2200 | 0.93 | 1 | 0.63 |
| 11 | 19k | 417 | 4 | 0 | 4.09 | 43.67 | 93.93 | 5 | 2200 | 0.87 | 1 | 0.60 |
| 12 | 19l | 415 | 3 | 0 | 4.79 | 36.52 | 96.40 | 5 | 2200 | 0.97 | 1 | 0.63 |
| 13 | 19m | 429 | 3 | 0 | 5.24 | 36.52 | 96.40 | 5 | 2200 | 0.97 | 1 | 0.63 |
| 14 | 19n | 445 | 4 | 0 | 4.75 | 44.06 | 93.79 | 5 | 2200 | 0.93 | 1 | 0.60 |
| 15 | 19o | 435 | 3 | 0 | 4.98 | 35.48 | 96.75 | 5 | 3900 | 0.95 | 1 | 0.59 |
| 16 | 19p | 449 | 3 | 0 | 5.42 | 35.48 | 96.75 | 5 | 3900 | 0.94 | 1 | 0.58 |
| 17 | 19q | 463 | 3 | 0 | 5.64 | 35.87 | 96.62 | 5 | 3900 | 0.98 | 1 | 0.61 |
| 18 | Ciprofloxacin | 331 | 5 | 2 | 1.28 | 74.57 | 83.27 | 4 | 1000 | 0.72 | 0.97 | 0.64 |
 | ||
| Fig. 9 Bioavailability radar plot of the standard antibiotic ciprofloxacin compared with synthesized compounds 19l and 19p. | ||
A radar chart was plotted to describe the six physicochemical parameters: lipophilicity (LIPO), size (SIZE), polarity (POLAR), solubility (INSOLU), saturation (INSATU), and flexibility (FLEX). As illustrated in Fig. 9, the physicochemical properties of compounds 19l and 19p generally fall within the acceptable range (pink region) (Fig. 10).2,49
 | ||
| Fig. 10 Theoretical toxicity results of ciprofloxacin and synthesized compounds 19l and 19p calculated using ProTox 3.0. | ||
After the ADMET analysis, from the literature survey, we found that both 2H-chromene and maleimide show fluorescent behavior.50–52 Hence, we studied the UV and fluorescence activity of all the synthesized tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione derivatives 19(a–q).
4. Photophysical properties
4.1. UV-visible studies
The UV-visible spectra of seventeen synthesized tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dionederivatives 19(a–q) were recorded in a Shimadzu single monochromator UV-2600 spectrophotometer at 15 μm concentration. Initially, the cyclic derivative 19a was dissolved in DMSO at 15 μM concentration, and the spectra showed two peaks at 262 nm (n to π*) and 313 nm (π to π*) transitions. Notably, compounds 19b and 19c showed peaks at 262 nm and 313 nm, 263 nm and 312 nm, respectively. The graph also revealed an isosbestic point at 291 nm, as illustrated in Fig. 11. Additionally, the spectra for compounds 19d, 19e, and 19f showed absorption maxima at 262 nm for the n to π* transition and 312 nm for the π to π* transition, with the same isosbestic point at 291 nm. Similarly, compounds 19g, 19h and 19i displayed λmax at 262 nm, 262 nm and 263 nm for the n to π* transition and 313 nm, 312 nm and 313 nm for the π to π* transition, respectively. It was observed that increasing the chain length at the R2 position in the chromene moiety resulted in a shift from hypochromic to hyperchromic in the intensity of the UV-visible spectra. | ||
| Fig. 11 UV-visible spectra of compounds 19(a–q) in DMSO medium. | ||
On the other hand, a blue shift to red shift (from 312 to 321 nm) was observed in compound 19j when a methyl group was introduced at the R1 position of the chromene framework, accompanied by a gradual decrease in optical density (OD). Similarly, a slight change in absorption maxima was noted in compound 19k compared to 19j. In a similar fashion, the absorption maxima and optical density of compounds 19l, 19m, and 19n were compared, showing peaks at 265 nm, 265 nm, and 266 nm for the n to π* transition, and 320 nm, 321 nm, and 320 nm for the π to π* transition, respectively. An isosbestic point appeared at 297 nm for compounds 19l, 19m, and 19n. Furthermore, the UV-visible spectra of three different synthesized molecules (19o, 19p, and 19q), where a phenyl group was introduced at the R4 position, showed a slight decrease in intensity compared to compounds 19a, 19b and 19c. The overall UV-visible spectra are shown in Fig. 11.
4.2. Fluorescence studies
To examine the fluorescence properties of the series of tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione derivatives 19(a–q), fluorescence experiments were conducted using fluorescence spectrometry Hitachi-7000 keeping the fluorescence slit width at 2.5 nm for each. A specific concentration (15 μm) was maintained for all synthesized compounds during the assay. At the beginning of our experiment, compound 19a showed a wavelength of 352 nm with an optical density of 6151. Compound 19b, which has a methyl group introduced at the R3 position of the 4-styrylaromatic ring, showed a recorded wavelength of 352 nm with a slight increase in OD to 8421. Similarly, compounds 19c, 19d, 19e, 19f, 19g, 19h, and 19i showed the same λmax (352 nm) value with a gradual increase in OD values ranging from 8053 to 9784. It is noted that the increase in the chain length at the R2 position in the chromene analogue led to a corresponding increase in the optical density value (6151–9784).However, a blue shift to red shift (from 352 to 363 nm) was observed for compound 19j, which exhibited notably strong fluorescence intensity amongst all the synthesized derivatives. Also, the compounds 19k, 19l and 19n have the same wave number at 361 nm with OD values of 6615, 6516 and 5127, respectively. Consequently, compounds 19o, 19p, and 19q displayed similar λmax at 353 nm, comparable to compounds 19a, 19b, and 19c. Finally, it was observed that all the synthesized derivatives demonstrated good fluorescent activity, as illustrated in Fig. 12.
 | ||
| Fig. 12 Fluorescence spectra of compounds 19(a–q) in DMSO medium. | ||
5. Conclusion
In this study, a class of fused tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione derivatives were synthesized with good to excellent yields without the need for column chromatography. The reaction was carried out under mild conditions, yielding compounds with good binding affinities, potent antibacterial activity, and favorable photophysical properties, making this method highly efficient and practical. The in vitro results revealed that compounds 19l and 19p showed the most potent antibacterial activity against the tested pathogenic strains. Also, these results have been explained by the binding energy parameters determined from molecular docking studies, which showed that the selected compounds can bind with bacterial DNA gyrase more efficiently, similar to the antibiotic ciprofloxacin. Interestingly, the ADMET parameter prediction indicated that the majority of the synthesized conjugates have an acceptable pharmacokinetic profile with a nontoxic characteristic. Moreover, the photophysical properties are suitable for in vivo high-resolution microscope imaging. We propose that in the future, this strategy can be applied to medicinal chemistry for the synthesis of natural products with pharmacological activity and drug-containing structure of tetrahydrochromeno isoindole dione derivatives.6. Experimental section
The commercially available reactants and reagents required for this reaction were purchased from the standard supplier (Sigma-Aldrich, TCI) and were used without further purification. All the reactions were carried out in anhydrous conditions. The progress of each reaction was monitored by thin-layer chromatography (TLC) on silica gel 60 (F254) coated with aluminum plates. Visualization of spots on the TLC plate was accomplished with UV light (254 nm). 1H NMR spectra were recorded on a 400 MHz (100 MHz for 13C NMR) JEOL NMR spectrometer using CDCl3 as solvent. High-resolution mass spectra (HRMS) were recorded using a Bruker micro TOF-QII mass spectrometer at IISER-Berhampur. The chemical shift was calibrated to tetramethyl silane (TMS) as an internal reference. Chemical shifts (δ) are given in ppm and coupling constants (J) in Hertz (Hz). The following abbreviations are used to indicate the signal multiplicity: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Melting points were determined using a SMP-10 digital apparatus and were uncorrected. The UV-visible and fluorescent spectra were taken in a Shimadzu single monochromator UV-2600 spectrophotometer and HITACHI F-7000 fluorescence spectrometer machine at Sambalpur University.7. Experimental procedure
7.1. General experimental procedure for the preparation of substituted chroman-4-one 13(a–e)
In a 100 mL clean and dry round bottom (RB) flask, substituted o-hydroxyacetophenone (1.0 equiv.) 11(a–b), substituted ketone (3 equiv.) 12(a–c) and pyrrolidine (0.5 equiv.) were taken. The mixture was stirred under a reflux medium for about 6–8 h. The reaction progress was monitored by TLC and was found to be completed after 6–8 h. After cooling to room temperature, the reaction mixture was quenched using water and extracted with ethyl acetate. The organic layer was washed with brine solution, dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude product was purified by column chromatography using ethyl acetate/hexane to afford the pure compound 13(a–e). All compounds were characterized by NMR.7.2. General experimental procedure for the synthesis of 4-bromo-2H-chromene derivatives 15(a–e)
Substituted chroman-4-one, (1.0 equiv.) 13(a–e) and phosphorus tribromide (PBr3) 14 (3 equiv.) were taken in a clean and dry round bottom flask and stirred under reflux condition for 30–40 min. Set up by a trap using cotton and anhydrous calcium chloride. After completion, the reaction mixture was quenched using ice water and extracted with ethyl acetate. The organic layer was washed with brine, separated, dried over anhydrous Na2SO4 and concentrated. The crude product was purified by column chromatography using ethyl acetate/hexane to furnish the pure compound 15(a–e). All compounds were characterized by NMR data.7.3. General experimental procedure for the synthesis of 4-styryl-2H-chromene derivatives 17(a–n)
Substituted 4-bromo-2H-chromene, (1.0 equiv.) 15(a–e), substituted styrene (2.0 equiv.) 16(a–c), palladium(II) complex (0.1 equiv.), K2CO3 (3.0 equiv.) and 1.5 mL of dry DMF were taken in the same vial and subjected to microwave irradiation at 120 °C, 100 W for about 20 min. The reaction was monitored by TLC and was found to be completed after 20 min. After completion, the reaction mixture was quenched using water and extracted with ethyl acetate. The organic layer was washed with brine, separated, dried over anhydrous Na2SO4 and concentrated. The crude product was purified by flask column chromatography using ethyl acetate/hexane to afford the pure compound 17(a–n). It gives two isomer which is not separated.7.4. General experimental procedure for the synthesis of tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3aH)-dione derivatives 19(a–q)
Substituted 4-styryl-2H-chromene, (1.0 equiv.) 17(a–n), maleimide (3.0 equiv.) 18(a–b), and 3 mL of dry toluene were taken in a sealed tube and stirred under reflux condition at 120 °C for 4 h. The reaction was monitored by TLC and was found to be completed after 4 h. Then, the reaction was cooled at room temperature, washed with hexane and filtered to afford a pure white solid compound 19(a–q). The compound was characterized by NMR and HRMS spectral data.7.5. General experimental procedure for the synthesis of 11-(4-methoxyphenyl)-2-methyl-4,4-dipropylchromeno[3,4-e]isoindole-1,3(2H,4H)-dione (21)
11-(4-Methoxyphenyl)-2-methyl-4,4-dipropyl-3b,4,11,11a-tetrahydrochromeno[3,4-e]isoindole-1,3(2H,3 aH)-dione(1.0 equiv.) 19i, DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) (1.5 equiv.) and 2 mL of dry toluene were taken in a clean and dry sealed tube and allowed to stir at 100 °C for 30 min. The progress of the reaction was monitored by TLC and after completion of the reaction, the pure compound 21 was obtained using column chromatography using ethyl acetate/hexane. After synthesis, the compound was characterized using1H NMR, 13C NMR and HRMS spectroscopic techniques.8. Method of crystal growth
Compound 19i was first dissolved in a minimal amount of ethyl acetate, followed by the addition of drops of hexane. This solution was then placed in a sample vial and stored in a dark area for 4 to 5 weeks to enable gradual evaporation. After 5 weeks, white needle-like crystals of compound 19i were formed.9. Molecular docking studies of compound 19(a–q) against bacterial DNA gyrase of E. coli and S. aureus
All the synthesized derivatives 19(a–q) were carefully drawn with proper stereochemistry and the 2D structures were converted to optimized 3D structures. Molecular docking studies were conducted using Autodock Tools version 4.2.0. The host proteins, including the bacterial DNA gyrase inhibitors for Gram-negative bacterial strain E. coli (PDB ID: 3 G7E) and Gram-positive bacterial strain S. aureus (PDB ID: 3 G7B), were downloaded and retrieved from the RCSB Protein Data Bank (https://www.rcsb.org/). Before docking the complex, water molecules and other hetero-atoms were removed from these host protein structures using PyMOL (https://www.pymol.org/).53,54 Also, polar hydrogen bonds were added, and a grid box was set to 60 × 60 × 60 for the xyz axes (covering ligand binding pocket). 0.375 Å is the grid space for 100 genetic algorithm runs.2,55 The two programs, ‘autodock’ and ‘autogrid’, were used to determine the binding affinity scores for the host–guest interaction. The 2D structures of ligand–receptor interactions were visualized using PyMOL and BIOVIA Discovery Studio v 2017.10. Antibacterial evaluation of compound 19(a–q) against bacterial DNA gyrase of E. coli and S. aureus
The in vitro antibacterial activity of all the synthesized molecules was tested using the agar-well diffusion method against E. coli (MTCC614) and S. aureus (MTCC7443) bacterial strains. Ciprofloxacin served as the standard antibiotic. Initially, the compounds were dissolved in DMSO and tested on Mueller–Hinton agar plates to assess the zones of inhibition (ZI). For the minimum inhibitory concentration (MIC) assay, the compounds were prepared at various concentrations (6.25, 12.5, 25, 50 and 100 μg mL−1 and 200 μg mL−1) and tested in a 96-well plate format to evaluate their bacterial inhibitory properties.11. Physicochemical, pharmacokinetic and ADME studies
The ADME and pharmacokinetic properties of all the synthesized compounds were studied on the free online server Swiss ADME (https://www.swissadme.ch/). Initially, chemical structures were drawn on Marvin to generate SMILE and inserted directly on the webpage to initiate the prediction process.56–58 The toxicity evaluation of the synthesized compounds 19(a–q) was determined using ProTox II free software.12. Photophysical study
The photophysical (UV-visible and fluorescence) studies were performed for all the synthesized compounds using the Shimadzu single monochromator UV-2600 spectrophotometer and HITACHI F-7000 fluorescence spectrometer. Initially, the compounds were dissolved in DMSO at a uniform concentration (15 μm), and the spectra were recorded.59Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Sonali Priyadarshini Parida: conceptualization, investigation, methodology, formal analysis, data correction, writing-review & editing; Seetaram Mohapatra: supervision, resources, funding acquisition, editing; Suhasini Mohapatra: writing original draft, conceptualization, writing-review & editing; Tankadhar Behera: UV-visible & fluorescent study; Sabita Nayak: conceptualization, validation, supervision, funding acquisition, editing; software, resources; Chita Ranjan Sahoo: software, antibacterial studies.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The author, Seetaram Mohapatra, expresses gratitude to the Department of Science and Technology (DST), Odisha (ST-SCST-MISC-0011-2023, Bhubaneswar, 25-05-2023). He also acknowledges the Centre of Excellence in Environment and Public Health, Department of Higher Education, Government of Odisha (Grant no 26913/HED/HE-PTC-WB-02-17 OHEPEE). Author Sabita Nayak extends thanks to Mukhyamantri Research Innovation (MRI) for Extramural Research Grant-2024(24 EM/CH/35). The author, Sonali Priyadarshini Parida, also acknowledges DST, Odisha program (2692/ST, Bhubaneswar, 16-7-2020) for providing the financial support as a project assistant. Author Suhasini Mohapatra acknowledges the financial support from SERB-SURE, New Delhi (Ref. SUR/2022/000257). Additionally, the author is thankful to DST-FIST, New Delhi (SR/FST/CSI-243/2012) for the NMR facility.References
- A. F. Al-burgus, O. T. Ali and O. Y. Al-abbasy, Chim. Techno Acta, 2024, 11, 202411308 CrossRef CAS.
- C. R. Sahoo, S. K. Paidesetty, S. Sarathbabu, B. Dehury, N. S. Kumar and R. N. Padhy, J. Biomol. Struct. Dyn., 2022, 40, 11653–11663 CrossRef CAS.
- A. C. Singer, H. Shaw, V. Rhodes and A. Hart, Front. Microbiol., 2016, 7, 1728 Search PubMed.
- B. M. Marshall and S. B. Levy, Clin. Microbiol. Rev., 2011, 24, 718–733 CrossRef CAS PubMed.
- M. E. Faydy, L. Lakhrissi, N. Dahaieh, K. Ounine, B. Tuzun, N. Chahboun, A. Boshaala, A. AlObaid, I. Warad, B. Lakhrissi and A. Zarrouk, ACS Omega, 2024, 9, 25395–25409 CrossRef PubMed.
- M. F. A. Mohamed, A. M. Soliman, O. Alshazly, A. Nafady and R. A. Soomro, Med. Chem. Res., 2025, 34, 172–182 CrossRef CAS.
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. Robles Aguilar and A. Gray, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
- M. D. Vaghasiya, J. V. Mendapara, I. Ahmad, H. Patel, D. P. Rajani and P. Kumari, J. Iran. Chem. Soc., 2024, 21, 1531–1545 CrossRef CAS.
- A. R. Zala, D. P. Rajani and P. Kumari, J. Biochem. Mol. Toxicol., 2023, 37, e23231 CrossRef CAS PubMed.
- M. Magana, M. Pushpanathan, A. L. Santos, L. Leanse, M. Fernandez, A. Ioannidis, M. A. Giulianotti, Y. Apidianakis, S. Bradfute, A. L. Ferguson, A. Cherkasov, M. N. Seleem, C. Pinilla, C. de la Fuente-Nunez, T. Lazaridis, T. Dai, R. A. Houghten, R. E. W. Hancock and G. P. Tegos, Lancet Infect. Dis., 2020, 20, e216–e230 CrossRef CAS PubMed.
- N. Baral, D. R. Mishra, N. P. Mishra, S. Mohapatra, B. P. Raiguru, P. Panda, S. Nayak, M. Nayak and P. S. Kumar, J. Heterocycl. Chem., 2019, 57, 575–589 CrossRef.
- M. A. Ahemad, A. Patra, L. Muduli, S. Nayak, S. Mohapatra, J. Panda and C. R. Sahoo, Carbohydr. Res., 2024, 543, 109222 CrossRef CAS PubMed.
- P. Panda, S. Nayak, S. Bhakta, S. Mohapatra and T. R. Murthy, J. Chem. Sci., 2018, 130, 127 CrossRef.
- S. P. Parida, S. Mohapatra, S. Nayak, S. Mohapatra, J. Panda and C. R. Sahoo, ChemistrySelect, 2024, 9, e202402223 CrossRef.
- J. Panda, B. Brahma, S. Nayak, S. Mohapatra, B. P. Raiguru, S. Rout, S. P. Parida and K. Naithani, J. Heterocycl. Chem., 2025, 62, 99–121 CrossRef CAS.
- T. Das, S. Mohapatra, A. K. Pradhan and S. Nayak, ChemistrySelect, 2023, 8, e202300477 CrossRef CAS.
- M. S. K. Youssef, A. A. O. Abeedand and T. I. El-Emary, Heterocycl. Commun., 2017, 23, 55–64 CrossRef CAS.
- N. Baral, S. Mohapatra, B. P. Raiguru, N. P. Mishra, P. Panda, S. Nayak, S. K. Pandey, P. S. Kumar and C. R. Sahoo, J. Heterocycl. Chem., 2018, 56, 552–565 CrossRef.
- K. Alseud, T. Ostlund, M. Durymanov, J. Reineke and F. Halaweish, Bioorg. Med. Chem., 2024, 103, 117678 CrossRef CAS PubMed.
- B. S. Panda, B. Samanta, S. S. S. S. Sudha Ambadipudi, S. Nayak, L. VNayak, S. Ramakrishna, S. Mohapatra, P. M. Behera and L. Samanta, ChemistrySelect, 2024, 9, e202400115 CrossRef.
- S. Ghomashi, R. Ghomashi and M. S. Damavandi, Sci. Rep., 2024, 14, 12878 CrossRef CAS PubMed.
- N. P. Mishra, S. Mohapatra, C. R. Sahoo, B. P. Raiguru, S. Nayak, S. Jena and R. N. Padhy, J. Mol. Struct., 2021, 1246, 131183 CrossRef CAS.
- D. R. Mishra, B. S. Panda, S. Nayak, N. K. Rauta, S. Mohapatra, C. R. Sahoo and R. N. Padhy, Tetrahedron, 2022, 124, 133015 CrossRef CAS.
- L. Xiao, D. T. Shen, W. X. Zou, J. L. Song, J. Wei, X. Liu, S. S. Zhang and L. Zhanga, Adv. Synth. Catal., 2024, 366, 3646–3652 CrossRef CAS.
- J. E. Barker, J. D. Tibbetts, C. T. J. Ferguson, Y. Xie and R. K. O'Reilly, Angew. Chem., Int. Ed., 2024, 63, e202410550 CAS.
- M. S. Sherikar and K. R. h Prabhu, Org. Lett., 2019, 21, 4525–4530 CrossRef CAS.
- (a) S. G. Stewart, L. A. Ho, M. E. Polomska, A. Percival and G. C. T. Yeoh, ChemMedChem, 2009, 4, 1657–1667 CrossRef CAS; (b) Q. Guan, D. Zuo, N. Jiang, H. Qi, Y. Zhai, Z. Bai, D. Feng, L. Yang, M. Jiang, K. Bao, C. Li, Y. Wu and W. Zhang, Org. Lett., 2019, 21, 4525–4530 CrossRef PubMed.
- S. K. Ko, H. J. Jang, E. Kim and S. B. Park, Chem. Commun., 2006, 2962–2964 RSC.
- S. Oh, H. J. Nam, J. Park, S. H. Beak and S. B. Park, ChemMedChem, 2010, 5, 529–533 CrossRef CAS PubMed.
- S. Oh, S. J. Kim, J. H. Hwang, H. Y. Lee, M. J. Ryu, J. Park, S. J. Kim, Y. S. Jo, Y. K. Kim, C. H. Lee, K. R. Kweon, M. Shong and S. B. Park, J. Med. Chem., 2010, 53, 7405–7413 CrossRef CAS PubMed.
- D. Ashok, R. S. Kumar, D. M. Gandhi and A. Jayashree, Russ. J. Gen. Chem., 2016, 86, 1396–1404 CrossRef CAS.
- D. Ashok, R. S. Kumar, D. M. Gandhi, M. Sarasija, A. Jayashree and S. Adam, Heterocycl. Commun., 2016, 22, 363–368 CrossRef CAS.
- C. D. Gabbutt, D. J. Hartley, J. D. Hepworth, B. M. Heron, M. Kanjia and M. M. Rahman, Tetrahedron, 1994, 50, 2507–2522 CrossRef CAS.
- R. Singh and G. Panda, RSC Adv., 2013, 3, 19533–19544 RSC.
- R. Singh, M. K. Parai, S. Mondal and G. Panda, Synth. Commun., 2013, 43, 253–259 CrossRef CAS.
- Ri. Singh and G. Panda, Org. Biomol. Chem., 2011, 9(13), 4782–4790 RSC.
- P. Singh, S. K. Dinda, Shagufta and G. Panda, RSC Adv., 2013, 3, 12100–12103 RSC.
- CCDC2321501 (19i) contain the supplementary crystallographic data for this paper. The data can be obtained free of charge from the Cambridge Crystallographic Data Center via https://www.ccdc.cam.ac.uk/structures/?.
- H. M. T. Albuquerque, C. M. M. Santos, C. F. R. A. C. Lima, L. M. N. B. F. Santos, J. A. S. Cavaleiro and A. M. S. Silva, Eur. J. Org Chem., 2017, 2017, 87–101 CrossRef CAS.
- U. Norinder and C. A. Bergstrom, ChemMedChem, 1, 920–937 CrossRef CAS PubMed.
- T. Aycan, F. Özturk and T. Doruk, J. Mol. Struct., 2024, 1306, 137865 CrossRef CAS.
- E. Lipinski, Semitic Languages: Outline of a Comparative GrammarBIUX’X@ Peeters, Publisher, Leuven, Belgium, 2001 Search PubMed.
- C. R. Sahoo, S. K. Paidesetty and R. N. Padhy, Indian J. Pharm. Educ. Res., 2020, 54, 669–675 Search PubMed.
- S. Mamidala, R. K. Aravilli, G. Ramesh, S. Khajavali, R. Chedupaka, V. Manga and R. R. Vedula, J. Mol. Struct., 2021, 1236, 130356 CrossRef CAS.
- B. M. Mlodawska, M. Jelen, E. Martula and R. Korlacki, Int. J. Mol. Sci., 2023, 24, 6970 CrossRef PubMed.
- P. Banerjee, A. O. Eckert, A. K. Schrey and R. Preissner, Nucleic Acids Res., 2018, 46, W257–W263 CrossRef CAS PubMed.
- C. O. Nnadi, L. M. U. Ozioko, G. C. Eneje, C. M. Onah and W. O. Obonga, Trop. J. Pharm. Res., 2020, 19, 2369–2375 CAS.
- S. Sestito, A. Bacci, S. Chiarugi, M. Runfola, F. Gado, E. Margheritis, S. Gul, M. E. Riveiro, R. Vazquez, S. Huguet and C. Manera, Eur. J. Med. Chem., 2021, 226, 113895 CrossRef CAS PubMed.
- M. S. Musa, M. S. Miah, Y. A. Munni, M. A. M. Patwary, M. Kazi and M. M. Matin, RSC Adv., 2024, 14, 30469–30481 RSC.
- R. Roy, S. Rakshit, T. Bhowmik, S. Khan, A. Ghatak and S. Bhar, J. Org. Chem., 2014, 79, 6603–6614 CrossRef CAS PubMed.
- (a) C. Ranjith, K. K Vijayan, V. K. Praveen and N. S. S. Kumar, Spectrochim. Acta, 2010, 75, 1610–1616 CrossRef PubMed; (b) X. Yi, J. Zhao, W. Wu, D. Huang, S. Ji and J. Sun, Dalton Trans., 2012, 41, 8931–8940 RSC; (c) J. Sun, J. Zhao, H. Guo and W. Wu, Chem. Commun., 2012, 48, 4169–4171 RSC; (d) X. Yi, P. Yang, D. Huang and J. Zhao, Dyes Pigm., 2013, 96, 104–115 CrossRef CAS; (e) K. H. Lee, M. H. Park, B. M. Seo, J. H. Seo, Y. K. Kim and S. S. Yoon, Mol. Cryst. Liq. Cryst., 2011, 550, 260–269 CrossRef CAS; (f) A. Kobayashi, K. Takehira, T. Yoshihara, S. Uchiyama and S. Tobita, Photochem. Photobiol. Sci., 2012, 11, 1368–1376 CrossRef CAS PubMed; (g) M. Wei, P. Yin, Y. Shen, L. Zhang, J. Deng, S. Xue, H. Li, B. Guo, Y. Zhang and S. Yao, Chem. Commun., 2013, 49, 4640–4642 RSC.
- T. Lu, B. Wang, W. Chang, L. Liu and J. Li, J. Org. Chem., 2023, 88, 818–827 CrossRef CAS PubMed.
- S. P. Parida, A. Bhuyan, P. P. Dash, D. Mohanta, A. Pati, S. Swain, M. Palsaniya, M. Das and B. K. Kundu, J. Clin. Trials, 2021, 4, 257–266 Search PubMed.
- M. Palsaniya, B. Patel, N. Panigrahi, D. Mohanta, S. P. Parida, D. K. Patel, M. Das and B. K. Kundu, J. Biomed. Res. Environ. Sci., 2021, 2, 383–391 CrossRef.
- C. R. Sahoo, S. K. Paidesetty, B. Dehury and R. N. Padhy, J. Biomol. Struct. Dyn., 2024, 42, 2539–2549 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2012, 64, 3–26 CrossRef.
- H. M. Faidallah and S. A. F. Rostom, Arch. Pharm., 2017, 350, 1700025 CrossRef PubMed.
- T. Behera, S. Sethi, J. Rout, B. P. Bag and N. Behera, Phys. Chem. Chem. Phys., 2024, 26, 26431 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2321501. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra02212f |
| This journal is © The Royal Society of Chemistry 2025 |