
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew nicotinamide–thiadiazol hybrids as VEGFR-2 inhibitors for breast cancer therapy: design, synthesis and in silico and in vitro evaluation†
Walid E. Elgammal a,
Hazem Elkady*b,
Reda G. Yousefbc,
Wagdy M. Eldehnade,
Dalal Z. Huseinf,
Fatma G. Aming,
Bshra A. Alsfoukh,
Eslam B. Elkaeedi,
Ibrahim H. Eissa*b and
Ahmed M. Metwaly*j
a,
Hazem Elkady*b,
Reda G. Yousefbc,
Wagdy M. Eldehnade,
Dalal Z. Huseinf,
Fatma G. Aming,
Bshra A. Alsfoukh,
Eslam B. Elkaeedi,
Ibrahim H. Eissa*b and
Ahmed M. Metwaly*j
aChemistry Department, Faculty of Science, Al-Azhar University, Nasr City, 11884, Cairo, Egypt
bPharmaceutical Medicinal Chemistry & Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo, 11884, Egypt. E-mail: Ibrahimeissa@azhar.edu.eg; Hazemelkady@azhar.edu.eg
cPharmaceutical Chemistry Department, Faculty of Pharmacy, Merit University, Sohag 82755, Egypt
dDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Kafrelsheikh University, P. O. Box 33516, Kafrelsheikh, Egypt
eDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Pharos University in Alexandria, Canal El Mahmoudia Street, Alexandria 21648, Egypt
fChemistry Department, Faculty of Science, New Valley University, El-Kharja, 72511, Egypt
gPhysics Department, Faculty of Science, Alexandria University, Alexandria, Egypt
hDepartment of Pharmaceutical Sciences, College of Pharmacy, Princess Nourah bint Abdulrahman University, P. O. Box 84428, Riyadh 11671, Saudi Arabia
iDepartment of Pharmaceutical Sciences, College of Pharmacy, AlMaarefa University, P.O. Box 71666, Riyadh 11597, Saudi Arabia
jPharmacognosy and Medicinal Plants Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo 11884, Egypt. E-mail: ametwaly@azhar.edu.eg
First published on 7th May 2025
Abstract
Vascular endothelial growth factor receptor-2 (VEGFR-2) is a key regulator of tumor angiogenesis and has become an important target in anticancer drug development. In this study, novel nicotinamide–thiadiazol hybrids were synthesized and evaluated for their anti-breast cancer potential through VEGFR-2 inhibition. The compounds were assessed in vitro for their cytotoxicity against MDA-MB-231 and MCF-7 cell lines. Among the nicotinamide–thiadiazol hybrids, 7a exhibited the most potent anticancer activity, with IC50 values of 4.64 ± 0.3 μM in MDA-MB-231 and 7.09 ± 0.5 μM in MCF-7, showing comparable efficacy to sorafenib. VEGFR-2 inhibition assays confirmed strong inhibitory potential with an IC50 of 0.095 ± 0.05 μM. In vitro cell cycle analysis indicated that 7a induced S-phase arrest, while apoptosis assays demonstrated a substantial increase in late apoptotic cells (44.01%). Other in vitro mechanistic studies further confirmed the activation of the intrinsic apoptotic pathway, as evidenced by caspase-3 activation (8.2-fold), Bax upregulation (6.9-fold), and Bcl-2 downregulation (3.68-fold). Computational studies, including molecular docking and 200 ns molecular dynamics (MD) simulations, confirmed the stable interaction of 7a with VEGFR-2, showing binding affinities comparable to sorafenib. Further validation through MM-GBSA, ProLIF, PCAT, and FEL analyses reinforced its strong binding capability. Additionally, ADMET predictions suggested favorable pharmacokinetic properties, including good absorption, high plasma protein binding, and non-CYP2D6 inhibition. Moreover, toxicity analysis classified 7a as non-mutagenic and non-carcinogenic, with a lower predicted toxicity than sorafenib. Finally, density functional theory (DFT) calculations highlighted the structural stability and reactivity of 7a, further supporting its potential as a VEGFR-2 inhibitor. These findings suggest that 7a is a promising VEGFR-2 inhibitor with significant anticancer potential, favorable pharmacokinetics, and an improved safety profile. Further preclinical studies and structural modifications are warranted to optimize its therapeutic potential.
1. Introduction
Cancer remains a leading cause of mortality worldwide,1,2 with breast cancer being the most frequently diagnosed malignancy among women. Despite significant advances in cancer treatment, the development of resistance to conventional chemotherapeutic agents and targeted therapies presents a major challenge in clinical oncology. Angiogenesis, the process of forming new blood vessels, plays a crucial role in tumor growth, progression, and metastasis.3 Among the various pro-angiogenic factors, VEGFR-2 is a key mediator of endothelial cell proliferation, migration, and survival, making it an attractive therapeutic target for antiangiogenic drug development.4,5Several VEGFR-2 inhibitors, including sorafenib, sunitinib, and pazopanib, have been approved for the treatment of various cancers.6 These inhibitors function by blocking the VEGF/VEGFR-2 signaling cascade, thereby reducing angiogenesis and limiting tumor growth.7 However, despite their clinical success, many of these agents are associated with significant drawbacks, including acquired resistance,8,9 off-target toxicities,10–12 and suboptimal pharmacokinetic properties.13 These limitations highlight the need for novel VEGFR-2 inhibitors with improved efficacy, selectivity, and safety profiles.
Nicotinamide, a derivative of vitamin B3, has gained considerable attention in drug discovery due to its diverse biological activities, including anticancer effects. Nicotinamide-based scaffolds have been explored as promising leads for kinase inhibitors, including those targeting VEGFR-2, due to their ability to form key interactions with the ATP-binding site of the receptor.14 Likewise, thiadiazole moieties are widely recognized for their ability to fight cancer by interactions with target proteins.15
Computational chemistry plays a crucial role in modern drug discovery, transforming how new medicines are designed and improved.16 Using advanced algorithms and molecular modeling, enables the prediction of how drugs interact with biological targets at the atomic level, saving both time and money compared to traditional experiments.17 Techniques like molecular docking, and MD simulations help evaluate how well drugs bind to their targets shedding light on the relationship between a compound's structure and its activity, guiding smarter drug design and refinement.18,19 ADMET predictions (covering absorption, distribution, metabolism, excretion, and toxicity) allow assessment of pharmacokinetics and safety early in development.20,21 By combining computational methods with lab experiments, drug discovery becomes more accurate and efficient, leading to better treatments for challenging targets like infections22,23 and cancer.24,25
Our team has successfully identified and developed a range of nicotinamide26–28 and thiadiazole29–32 derivatives that act as potent VEGFR-2 inhibitors and significant potential anti-cancer agents. Merging these two pharmacophores enables the development and synthesis of innovative hybrids capable of selectively inhibiting VEGFR-2 and demonstrating strong anticancer properties. The compounds were examined in vitro and in silico for their anticancer and VEGFR-2 inhibition.
1.1. Rationale
The ATP-binding site of VEGFR-2 consists of several distinct pockets, such as the hinge region, the linker region between the hinge and DFG domain, the DFG region, and an allosteric hydrophobic pocket (Fig. 1). VEGFR-2 inhibitors (I and II) typically display essential pharmacophoric characteristics: (i) a hetero-aromatic group that forms hydrogen bonds with Cys919 in the hinge region, (ii) a spacer group that aligns between the hinge region and the DFG domain to maintain proper orientation, (iii) a pharmacophore group with hydrogen bond donor and acceptor properties that interact with Glu885 and Asp1046 in the DFG domain, and (iv) a hydrophobic tail that binds to the allosteric hydrophobic pocket of the ATP-binding site.33–36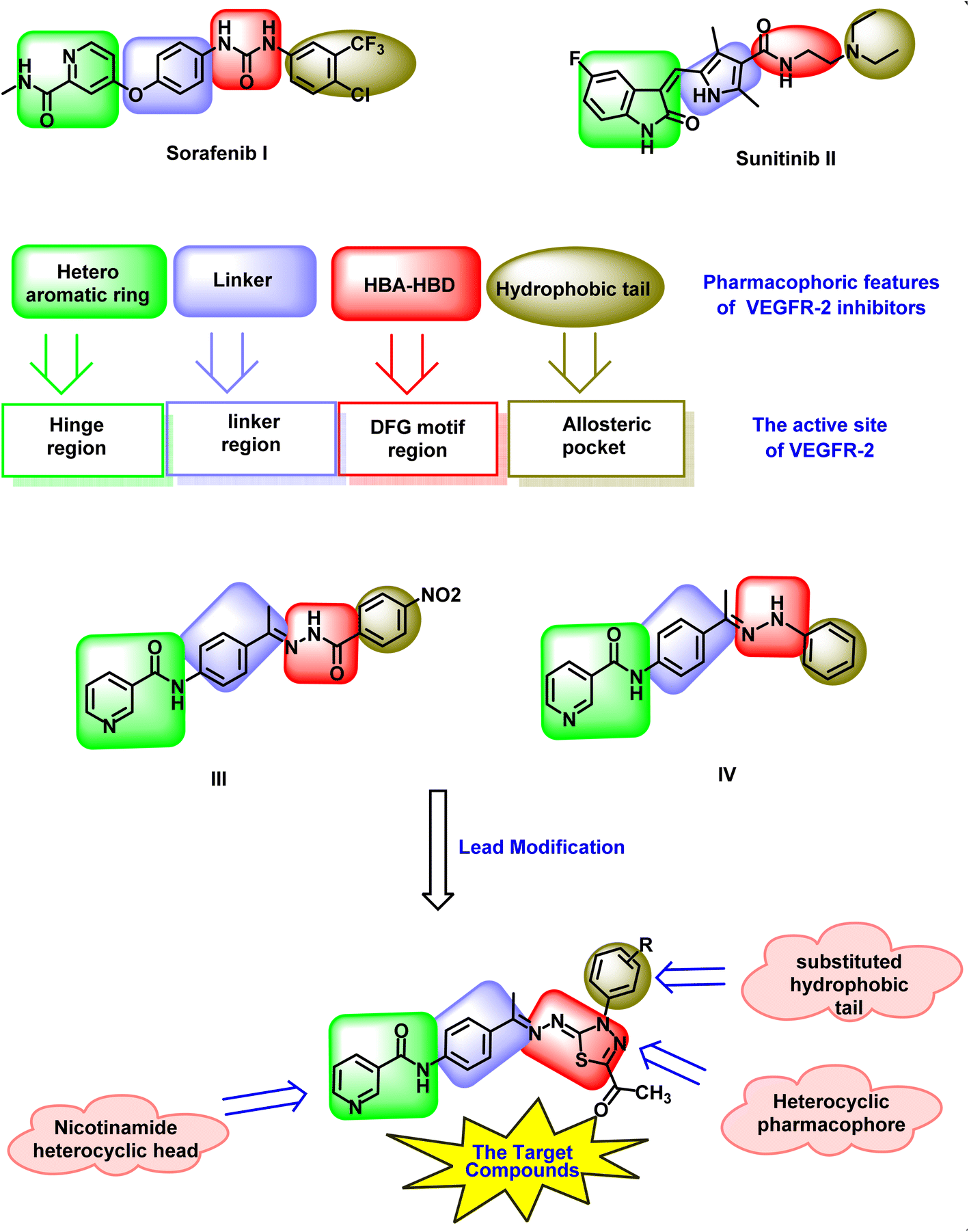 | ||
| Fig. 1 Rational design of the new nicotinamide–thiadiazol hybrids as VEGFR-2 inhibitors. Sorafenib and sunitinib as examples of FDA-approved VEGFR-2 inhibitors showing the essential pharmacophoric features. Compounds III and IV as reported highly effective VEGFR-2 inhibitors. The designed compounds have the main pharmacophoric features of VEGFR-2 inhibitors. | ||
Recently, our research team discovered several nicotinamide compounds (III (ref. 37) and IV (ref. 38)) (Fig. 1) as highly effective VEGFR-2 inhibitors. Compound III showed promising IC50 values of 2.1, 4.61, and 4.05 μM against MCF-7, HepG2, and HCT-116 cell lines, respectively. Its activities was stronger than those of sorafenib (IC50 = 6.72, 7.40, and 9.30 μM against MCF-7, HepG2, and HCT-116, respectively). The cytotoxic activity of compound IV against HCT-116 cells (IC50 = 3.08 μM) was almost double that of sorafenib (IC50 = 7.28 μM), while its activity against HepG-2 (IC50 = 4.09 μM) was about 1.2-fold more than sorafenib (IC50 = 5.28 μM). Compounds III and IV exhibited good VEGFR-2 inhibitory activities with IC50 values of 240 and 24.93 nM (Fig. 1).
In this study, compounds III and IV served as lead structures for further modifications aimed at developing more potent VEGFR-2 inhibitors. The nicotinamide scaffold (heterocyclic head) was retained. The 1-phenylethylidene linker remained unchanged. The hydrazine (pharmacophore) moiety was replaced by a 2-hydrazono-2,3-dihydro-1,3,4-thiadiazole moiety. However, the hydrophobic tail (4-nitrophenyl and phenyl groups) of compounds III and IV, respectively were replaced with different substituted phenyl rings to increase the hydrophobic interaction at the allosteric pocket (Fig. 1).
2. Results and discussions
2.1. Chemistry
This study builds on prior findings and concentrates on the synthesis of newly designed nicotinamide compounds 7a–g. The synthesis of these compounds was conducted according to the pathways depicted in Scheme 1. Compound 3 was synthesized by the addition of nicotinoyl chloride 2 to 4-aminoacetophenone 1 under stirring in an ice bath. The produced compound 3 was subsequently refluxed with methyl hydrazinecarbodithioate 4 in absolute ethanol, yielding methyl (E)-2-(1-(4-(nicotinamido)phenyl)ethylidene)hydrazine-1-carbodithioate 5 in a good yield. Finally, compound 5 subsequently reacted with the hydrazono derivatives 6a–g in ethanol under reflux conditions, utilizing triethylamine as a catalyst, resulting in the formation of the target nicotinamide derivatives 7a–g, respectively. The IR, NMR, and CHN data provide detailed insights into the structure of the compounds. The FT-IR spectra show characteristic absorption bands that reflect key functional groups: the NH stretch around 3300 cm−1, indicative of the amide or hydrazone group, along with aromatic C–H stretches in the range of 3000–3050 cm−1. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations are observed around 1650–1700 cm−1, confirming the presence of a carbonyl group, and C–H aliphatic stretches are seen in the 2800–3000 cm−1 region. The 1H NMR spectra exhibit well-resolved signals, with the amide NH proton appearing as a singlet around δ 10.60–10.66 ppm. Aromatic protons are found in the δ 7–9 ppm range, with splitting patterns such as doublets (d), double doublets (dd), and multiplets (m), which suggest a highly substituted aromatic system. Additionally, the acetyl group (–COCH3) protons appear as singlets around δ 2.20–2.60 ppm. The 13C NMR spectra further support the structure with distinct chemical shifts corresponding to the carbonyl (δ 164–199 ppm), aromatic carbons (δ 120–160 ppm), and methyl groups (δ 14–40 ppm). The CHN analysis confirms the elemental composition, showing close agreement with the calculated values for carbon, hydrogen, and nitrogen content, which supports the molecular formulas and structural assignments.
O stretching vibrations are observed around 1650–1700 cm−1, confirming the presence of a carbonyl group, and C–H aliphatic stretches are seen in the 2800–3000 cm−1 region. The 1H NMR spectra exhibit well-resolved signals, with the amide NH proton appearing as a singlet around δ 10.60–10.66 ppm. Aromatic protons are found in the δ 7–9 ppm range, with splitting patterns such as doublets (d), double doublets (dd), and multiplets (m), which suggest a highly substituted aromatic system. Additionally, the acetyl group (–COCH3) protons appear as singlets around δ 2.20–2.60 ppm. The 13C NMR spectra further support the structure with distinct chemical shifts corresponding to the carbonyl (δ 164–199 ppm), aromatic carbons (δ 120–160 ppm), and methyl groups (δ 14–40 ppm). The CHN analysis confirms the elemental composition, showing close agreement with the calculated values for carbon, hydrogen, and nitrogen content, which supports the molecular formulas and structural assignments.
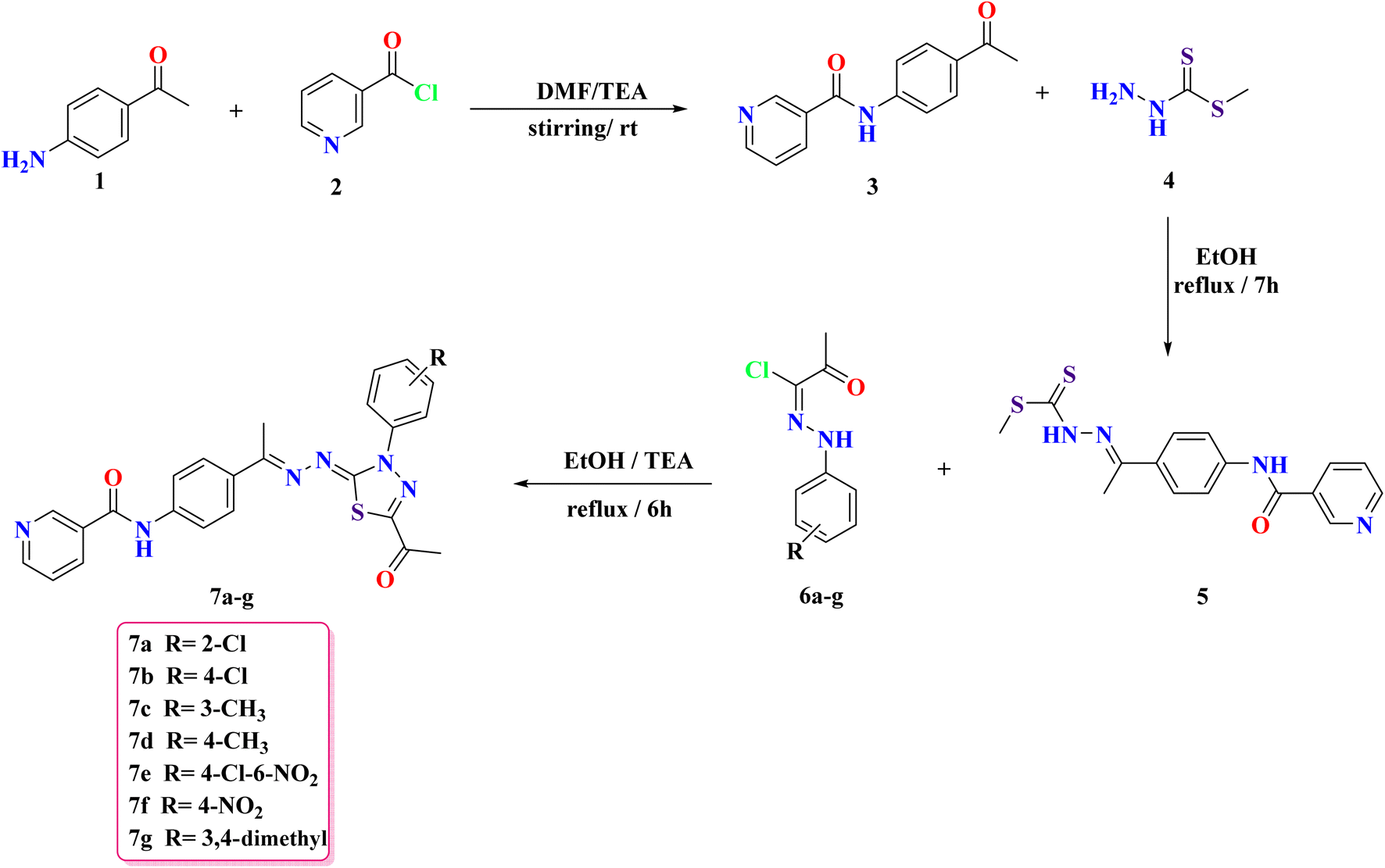 | ||
| Scheme 1 The synthetic methodology exploited for the synthesis of nicotinamide derivatives 7a–g. | ||
A reasonable mechanism for forming 7a–g from derivative 5 is illustrated in Scheme 2. Wherein the hydrazonyl chlorides 6a–g react with a di-thioester derivative 5 (thiol form, –SH) to yield the hydrazonic thioanhydride (i), it undergoes intermolecular cyclization to form intermediate (iii), or through 1,3-dipolar cycloaddition of nitrileimine (ii) [prepared from the hydrazonyl chlorides 6a–g in situ under basic conditions (triethylamine)], to the activated double bond (thiocarbonyl group (CS)) of di-thioester derivative 5, which then cyclizes, followed by losing methyl thiol (CH3SH), from cycloadduct (iii) to give 7a–g. The constitution of compounds 7a–g was verified through analytical and spectroscopic data and synthesis approaches.
 | ||
| Scheme 2 Anticipated mechanism for the synthesis of the target nicotinamide derivatives 7a–g. | ||
2.2. In vitro evaluations
Among the tested compounds, 7a (IC50 = 4.64 ± 0.3 μM in MDA-MB-231 and 7.09 ± 0.5 μM in MCF-7) exhibited the most potent cytotoxicity, surpassing the activity of SOR (IC50 = 7.61 ± 0.4 μM in MDA-MB-231 and 7.29 ± 0.3 μM in MCF-7). More importantly, 7a demonstrated a very weak cytotoxicity against WI-38 and WISH normal cell lines (IC50 = 73.58 ± 3.8 μM and 57.70 ± 3.3 μM, respectively), indicating a high degree of safety.
Additionally, compounds 7c (IC50 = 13.83 ± 1.2 μM in MDA-MB-231, 20.52 ± 1.4 μM in MCF-7) and 7e (IC50 = 7.90 ± 0.6 μM in MDA-MB-231, 10.37 ± 0.8 μM in MCF-7) also demonstrated notable anticancer activity, with 7e exhibiting a cytotoxic profile similar to sorafenib. The lower IC50 values of these compounds suggest effective inhibition of VEGFR-2-mediated tumor growth and angiogenesis.
Conversely, compounds 7f and 7g displayed higher IC50 values (>47 μM in both cancer cell lines), indicating weaker cytotoxic effects. Similarly, 7d and 7b exhibited moderate activity, with IC50 values ranging between 22 and 36 μM, suggesting that structural modifications could enhance their potency.
The selectivity index (SI) is a critical parameter in evaluating the therapeutic potential of anticancer agents, as it reflects the ability of a compound to target cancer cells while minimizing toxicity to normal cells. As shown in Table 1, 7a exhibited significantly higher SI values compared to sorafenib, with SI = 15.86 for MDA-MB-231 and SI = 8.14 for MCF-7, whereas sorafenib demonstrated much lower selectivity (SI = 1.40 and 1.84, respectively). These results suggest that 7a possesses a strong preference for cancer cells over normal cells, indicating a potentially safer profile with reduced off-target cytotoxicity.
| Comp. | In vitro cytotoxicity IC50a (μM) | |||
|---|---|---|---|---|
| MDA-231 | MCF-7 | WI-38 | WISH | |
| a The values are expressed as the mean ± SEM from three independent experiments. | ||||
| 7a | 4.64 ± 0.3 | 7.09 ± 0.5 | 73.58 ± 3.8 | 57.70 ± 3.3 |
| 7b | 22.68 ± 1.5 | 36.78 ± 2.2 | — | — |
| 7c | 13.83 ± 1.2 | 20.52 ± 1.4 | — | — |
| 7d | 31.77 ± 2.1 | 28.81 ± 1.9 | — | — |
| 7e | 7.90 ± 0.6 | 10.37 ± 0.8 | — | — |
| 7f | 47.74 ± 2.7 | 58.25 ± 3.3 | — | — |
| 7g | 60.53 ± 3.4 | 49.51 ± 2.8 | — | — |
| SOR | 7.61 ± 0.4 | 7.29 ± 0.3 | 10.64 ± 0.8 | 13.44 ± 1.1 |
 | ||
| Fig. 2 Structure–activity relationship (SAR) of the synthesized compounds based on the cytotoxic evaluation against MDA-MB-231 and MCF-7 cell lines. | ||
2.2.2.1. Effect of hydrophobic versus hydrophilic substituents. Hydrophobic substituents generally improve cytotoxic activity, while hydrophilic groups tend to reduce it. For example, compound 7b (IC50: 22.68 μM for MDA-MB-231, 36.78 μM for MCF-7), which contains a hydrophobic 4-Cl group, exhibited significantly better activity than compound 7f (IC50: 47.74 μM for MDA-MB-231, 58.25 μM for MCF-7), which has a hydrophilic 4-NO2 group. This suggests that increased hydrophobicity enhances the interaction with the biological target.
Interestingly, compound 7e (IC50: 7.90 μM for MDA-MB-231, 10.37 μM for MCF-7), which contains both a hydrophobic (4-Cl) and a hydrophilic (6-NO2) group, demonstrated greater cytotoxic activity than compound 7b (IC50: 22.68 μM for MDA-MB-231, 36.78 μM for MCF-7), which has only the hydrophobic 4-Cl group. This indicates that a balanced hydrophobic–hydrophilic environment can enhance activity, possibly by improving bioavailability or target binding.
2.2.2.2. Effect of mono- versus di-substitution. Mono-substituted compounds showed superior activity compared to di-substituted compounds. Compound 7c (IC50: 13.83 μM for MDA-MB-231, 20.52 μM for MCF-7) and compound 7d (IC50: 31.77 μM for MDA-MB-231, 28.81 μM for MCF-7), which feature single methyl groups at the 3- and 4-positions, respectively, exhibited significantly stronger activity than compound 7g (IC50: 60.53 μM for MDA-MB-231, 49.51 μM for MCF-7), which contains two methyl groups at the 3- and 4-positions. This suggests that di-substitution at adjacent positions (3,4-di-CH3) may introduce steric hindrance, reducing the binding efficiency and lowering the overall cytotoxic effect.
2.2.2.3. Effect of substituent position (meta versus para). The position of the substituent also influences activity. Meta-substitution, as seen in compound 7c (IC50: 13.83 μM for MDA-MB-231, 20.52 μM for MCF-7), resulted in better cytotoxicity than para-substitution, as in compound 7d (IC50: 31.77 μM for MDA-MB-231, 28.81 μM for MCF-7). This suggests that meta-positioning may enhance receptor binding, possibly due to steric or electronic interactions.
2.2.2.4. Effect of ortho versus para substitution. Ortho-substitution proved to be significantly more beneficial than para-substitution. Compound 7a (IC50: 4.64 μM for MDA-MB-231, 7.09 μM for MCF-7), which contains a 2-Cl (ortho) substituent, exhibited much stronger activity than compound 7b (IC50: 22.68 μM for MDA-MB-231, 36.78 μM for MCF-7), which has a 4-Cl (para) substituent. This indicates that placing chloro atom at the ortho position enhances interactions with the biological target, leading to increased potency.
The strong VEGFR-2 inhibition observed for 7a likely contributes to its ability to suppress angiogenesis and tumor progression. This result aligns with its potent cytotoxic effects against cancer cells, reinforcing our design.
| Sample | G0/G1 | S | G2/M |
|---|---|---|---|
| Control MDA-MB-231 | 12% | 45% | 43% |
| Comp. 7a/MDA-MB-231 | 33% | 59% | 8% |
 | ||
| Fig. 3 The effect of comp. 7a on different phases of MDA-MB-231 cell cycle. | ||
| Sample | Viable cells | Early apoptosis | Late apoptosis | Necrosis |
|---|---|---|---|---|
| Control | 99.30% | 0.48% | 0.03% | 0.19% |
| Comp. 7a | 49.99% | 1.06% | 44.01% | 4.94% |
 | ||
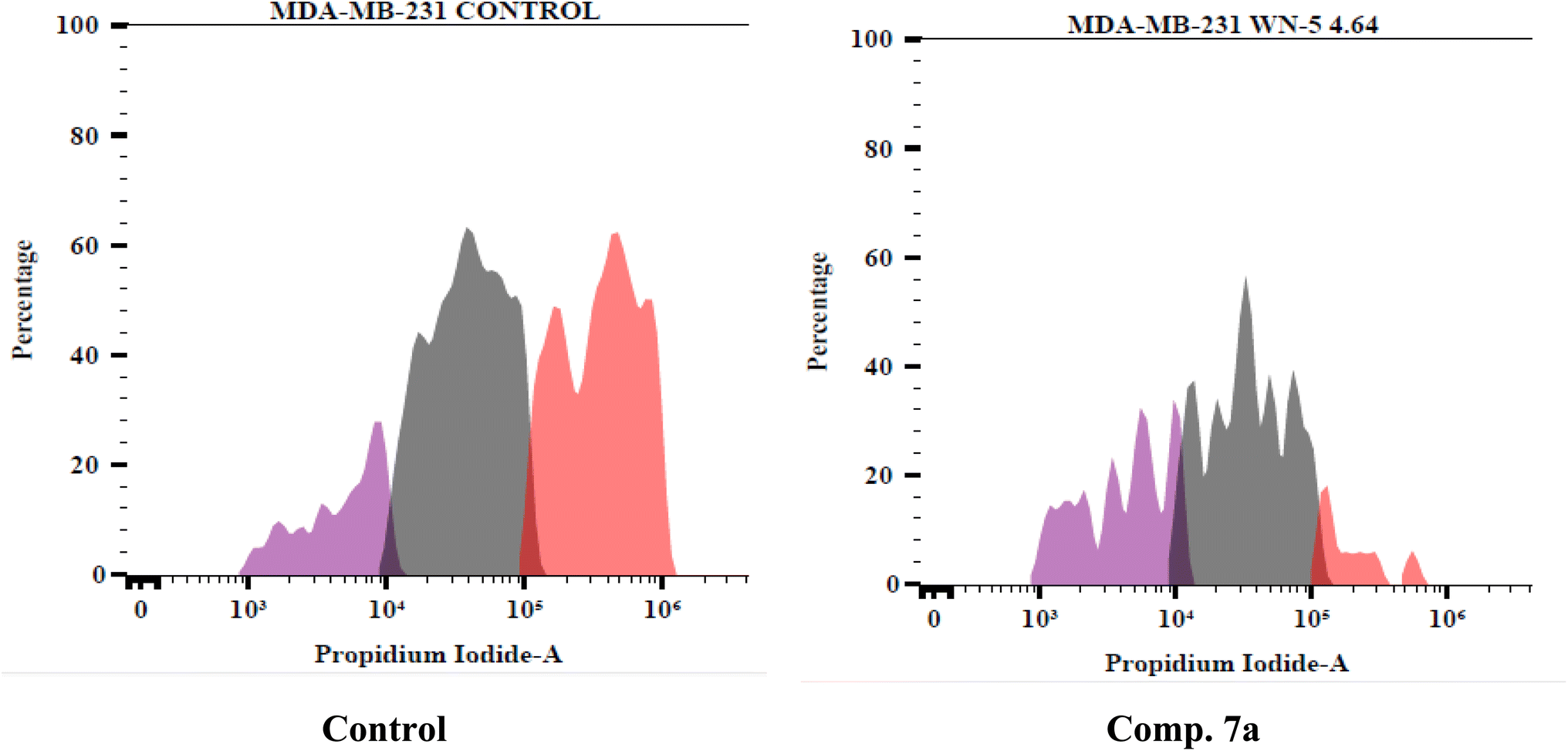
| Fig. 4 Apoptosis and necrosis rates of MDA-MB-231 cells treated with compound 7a compared to control. | ||
In the untreated control group, 99.30% of the cells remained viable, with minimal signs of apoptosis (0.48% early apoptosis, 0.03% late apoptosis) or necrosis (0.19%). However, treatment with 7a drastically reduced cell viability to 49.99%, while markedly increasing late apoptosis (44.01%) and necrosis (4.94%). The minimal increase in early apoptosis (1.06%) suggests that 7a induces rapid apoptotic progression, leading to a higher proportion of cells in the late apoptotic stage.
The substantial rise in late apoptosis indicates that 7a effectively triggers cell death pathways, possibly through VEGFR-2 inhibition and downstream disruption of pro-survival signaling. The slight increase in necrotic cells suggests that a subset of cells undergoes non-apoptotic death, which may be due to high-dose cytotoxic effects or secondary necrosis following prolonged apoptosis.
As shown in Table 4, caspase-3, a key effector protease in the execution phase of apoptosis, was significantly upregulated in 7a-treated cells (441.93; FLD = 8.2) compared to the untreated control (53.61; FLD = 1). This substantial increase suggests that 7a effectively activates caspase-dependent apoptotic pathways, leading to programmed cell death. Conversely, the anti-apoptotic protein Bcl-2 was markedly downregulated following 7a treatment (1.53; FLD = 3.68) compared to control cells (5.64; FLD = 1). This decrease in Bcl-2 expression indicates suppression of survival signaling, thereby promoting apoptosis. In contrast, the pro-apoptotic protein Bax was significantly upregulated (394.72; FLD = 6.9) compared to control cells (56.9; FLD = 1), further supporting the induction of apoptosis through the mitochondrial (intrinsic) pathway. The observed Bax upregulation, Bcl-2 downregulation, and caspase-3 activation collectively suggest that 7a triggers apoptosis through intrinsic mitochondrial signaling.
| Code | Caspase-3 conc. | FLD | Bcl-2 conc. | FLD | Bax conc. | FLD |
|---|---|---|---|---|---|---|
| Comp. 7a | 441.93 | 8.2 | 1.53 | 3.68 | 394.72 | 6.9 |
| Cont. MDA-MB-231 | 53.61 | 1 | 5.64 | 1 | 56.9 | 1 |
2.3. In silico evaluations
Validation of the docking protocol was first carried out by re-docking the co-crystallized sorafenib (PDB ID 4ASD) into its specific binding site. The docked conformation was compared to its native conformation, resulting in a notably low RMSD value (RMSD = 1.15 Å) as shown in Fig. 5. This result confirmed the reliability of the docking approach and validated the correctness of the binding evaluation.
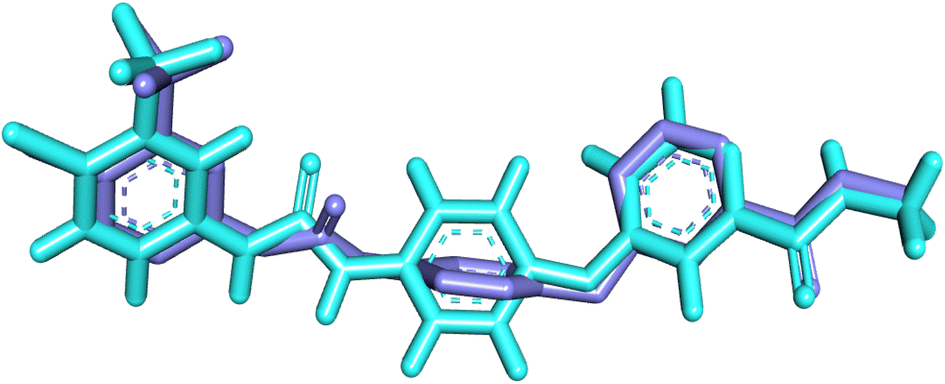 | ||
| Fig. 5 Validation within VEGFR-2 active pocket through the superimposition of the docked ligand (purple) upon the original ligand (turquoise). | ||
The docking of a reference inhibitor (sorafenib) demonstrated binding interactions with Cys917, Asp1044, and Glu883, which are key residues in the active pocket, as illustrated in Fig. S2.† The main four areas of the VEGFR-2 pocket were likewise shown to be occupied by compound 7a. Compound 7a's nicotinamide moiety formed several hydrophobic interactions, including an important one with Cys917, inside the hinge area. Additionally, two pi–pi interactions were accomplished by the central phenyl (spacer) moiety with Val916 and Val848. An additional two crucial hydrogen bonds were formed in the inner DFG-motif region by compound 7a using its ethylidenehydrazine moiety with Asp1046 and Glu885. Furthermore, the hydrophobic region's amino acids reacted with the active candidate's 2-acetyl-4-phenyl-4,5-dihydro-1,3,4-thiadiazol group (Fig. 6).
 | ||
| Fig. 6 MS, 3D, and 2D images of the most promising nicotinamide derivative 7a interacted with the VEGFR-2 active site. | ||
 | ||
| Fig. 7 (A) RMSD values from the trajectory for the VEGFR-2, 7a RMSD values, RoG for the VEFR-2 protein in the VEGFR-2_7a complex, SASA for the VEGFR-2 protein in the VEGFR-2_7a complex, change in the number of hydrogen bonds and distance from the center of mass of 7a compound and VEGFR-2 protein. (B) RMSF for the VEFR-2 protein in VEGFR-2_ 7a complex. | ||
 | ||
| Fig. 8 Energetic components and their values for the VEGFR-2_7a complex. | ||
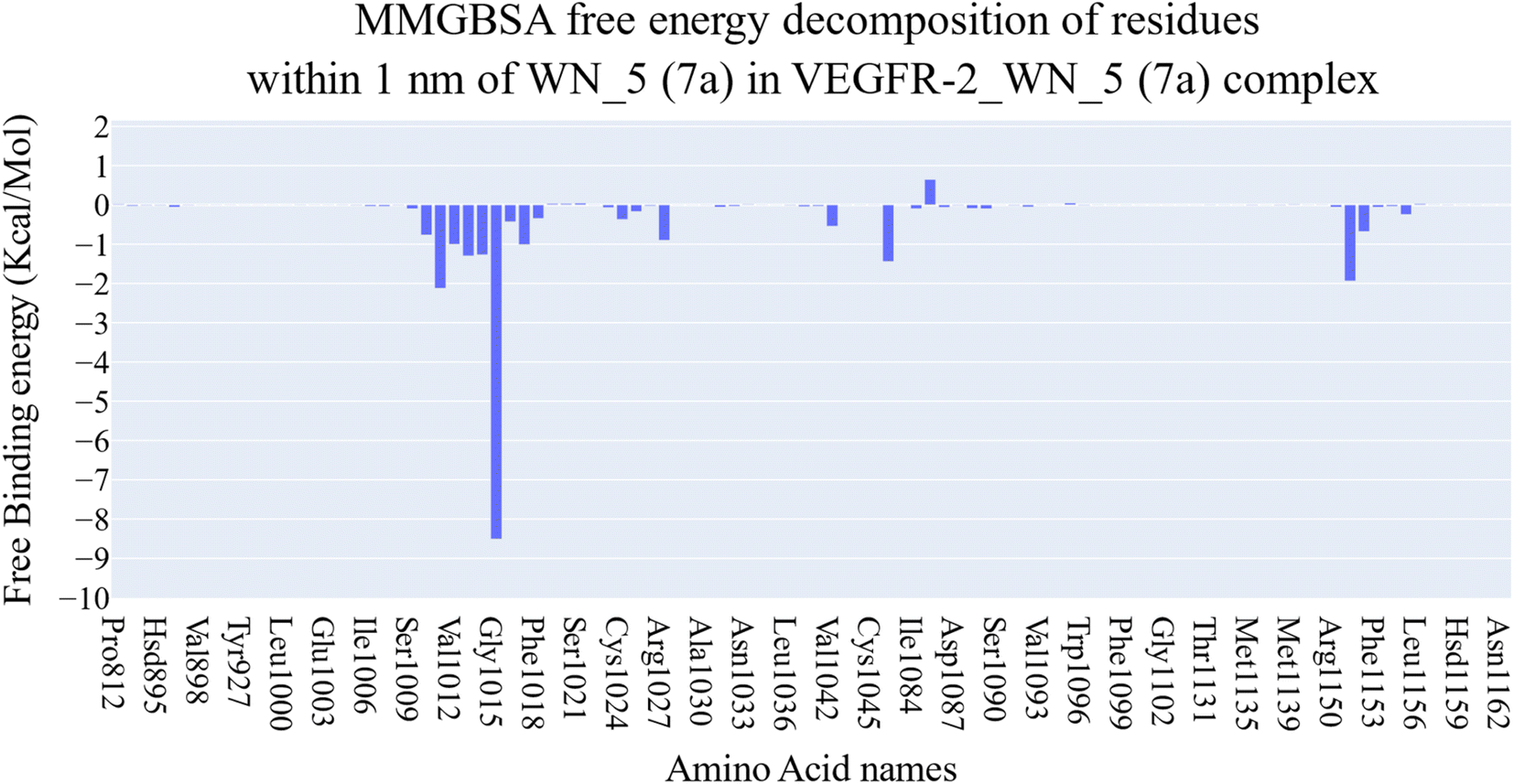 | ||
| Fig. 9 Binding free energy decomposition of the VEGFR-2_7a complex. | ||
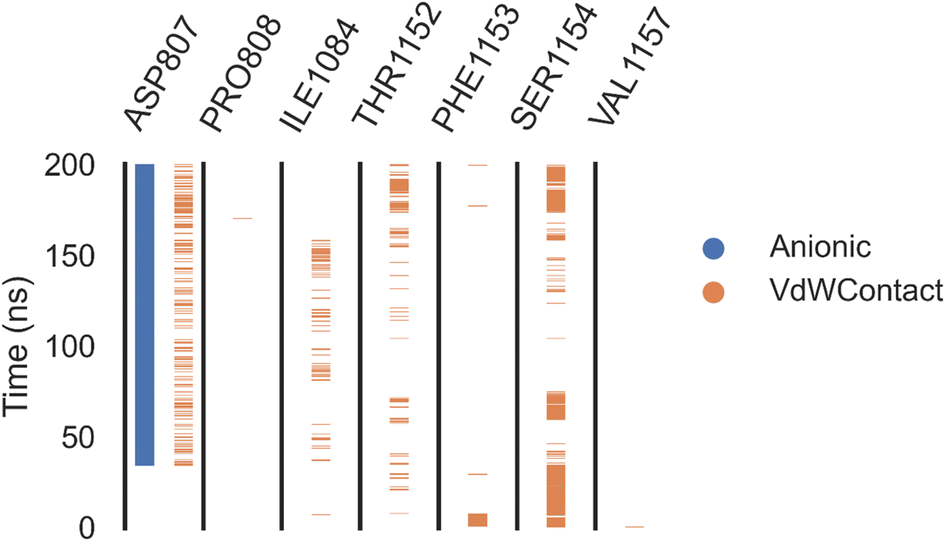 | ||
| Fig. 10 Amino acids, interactions types of VEGFER-2 and 7a. | ||
 | ||
| Fig. 11 Eigenvalues' changes showing an increase in eigenvectors (blue) and retained cumulative variance in the eigenvectors (red). | ||
 | ||
| Fig. 12 The initial ten eigenvectors' distribution. | ||
 | ||
| Fig. 13 Cosine content values of the initial ten eigenvectors for the trajectory. | ||
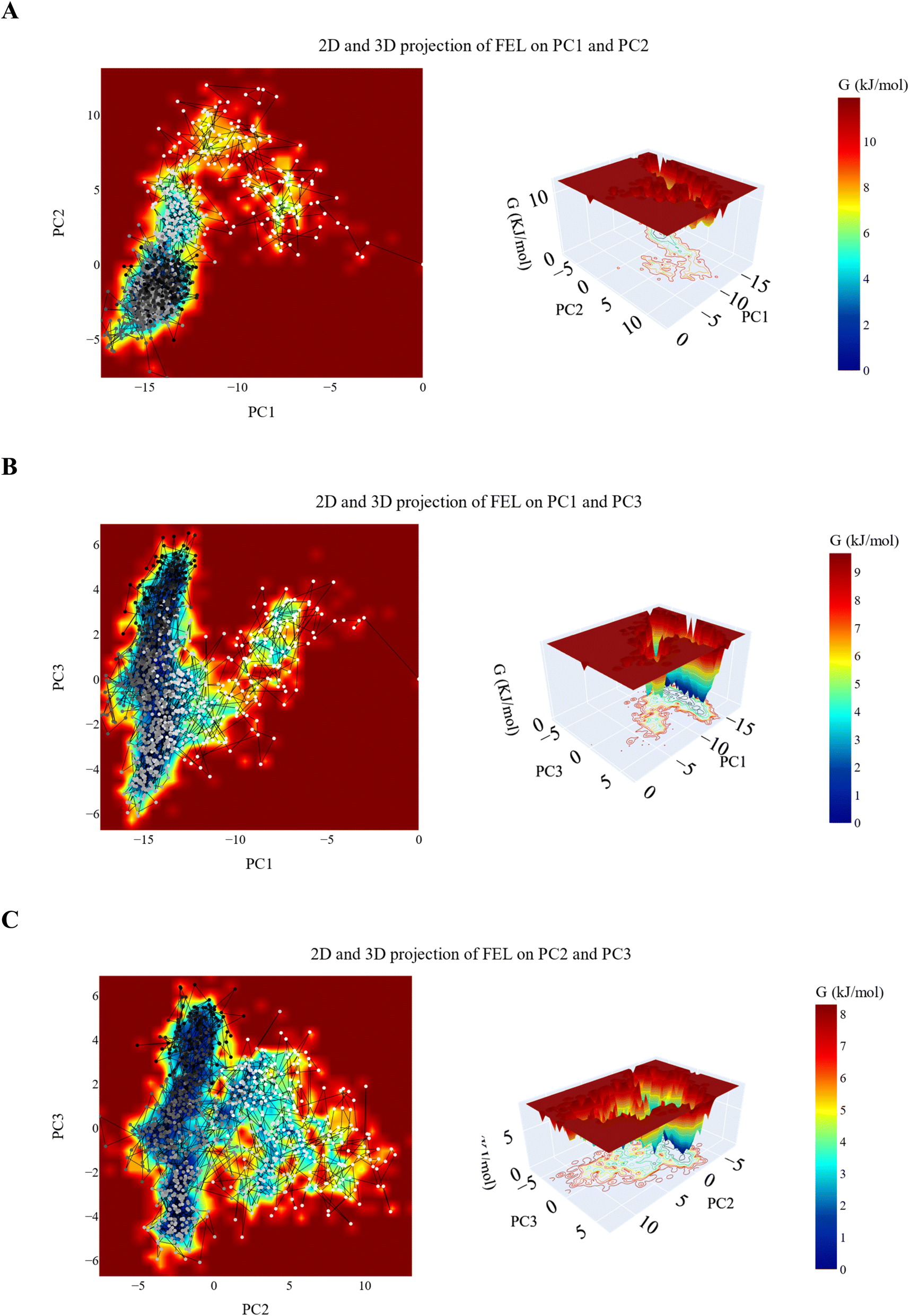 | ||
| Fig. 14 2D and 3D projection of the VEGFR-2_7a trajectory's FEL on (A) first two, (B) first and third and (C) second and third eigenvectors. | ||
| IP | EA | μ (eV) | χ (eV) | η (eV) | σ (eV) | ω (eV) | Dm (debye) | TE (eV) | ΔNmax | ΔE (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| 5.846 | 2.531 | −4.189 | 4.189 | 1.658 | 0.603 | 14.542 | 8.796 | −61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 589.613 589.613 |
2.527 | −14.542 |
 | ||
| Fig. 15 The optimized structure (A), the Mulliken charge distribution color scale (B), the HOMO/LUMO distribution functions and energy gap (C), the TDOS analysis (D), and the molecular electrostatic potential map (E) at B3LYP/6-31G(d,p) level for 7a. | ||
As demonstrated in Fig. 15A, the two terminal benzene rings are almost perpendicular to the rest of compounds which affect the lobes of HOMO and LUMO density function as shown in Fig. 15C. The two terminal benzene rings are slanted to the remaining compounds, influencing the HOMO and LUMO density function lobes. Table 5 calculates the energy gap, Egap, between the border orbitals, HOMO and LUMO, as well as all related HOMO/LUMO characteristics. To understand the reactivity and possible anticancer activity, the energy gap is an essential quantity.39–42 7a's band gap is relatively low (Egap = 3.315 eV) which points to a reactive medication. The 7a, and the protein target interact as a result of the electron transport between HOMO and LUMO being facilitated by a reduced Egap value. Fig. 15D shows the energy gap and total electron density surrounding HOMO/LUMO orbitals. Electronic densities increase under HOMO orbitals, indicating the drug's capacity to give electrons. Compound 7a is soft and reactive, according to the reactivity values in Table 5.
Drug–target interactions and selectivity are shown by 7a's electrostatic surface potential, shown in Fig. 15E. The red-colored, electron-rich sites that are targeted at the oxygen atoms would be the target of the electrophilic attack, as shown in Fig. 15E. The sites appear in blue, have positive potential values. These positive sites are found over hydrogen atoms and are electron deficient. Hydrogen bonding with the protein target may involve both positive and negative potential sites. Hydrophobic interactions can occur in the green zones over conjugated systems.
As Table 6 shows, all tested compounds, including 7a, exhibited very low BBB permeability, indicating minimal central nervous system penetration. This is a favorable property for anticancer drugs, as it reduces the risk of off-target CNS toxicity. However, their low solubility levels suggest potential limitations in bioavailability, which may require formulation strategies such as nanoparticle encapsulation or prodrug modifications to enhance solubility and systemic circulation.
| Comp. | Structure | BBB level | Solubility level | Absorption level | CYP2D6 prediction | PPB prediction |
|---|---|---|---|---|---|---|
| 7a |  |
Very low | Low | Good | Non inhibitor | More than 90% |
| 7b |  |
|||||
| 7c |  |
|||||
| 7d | 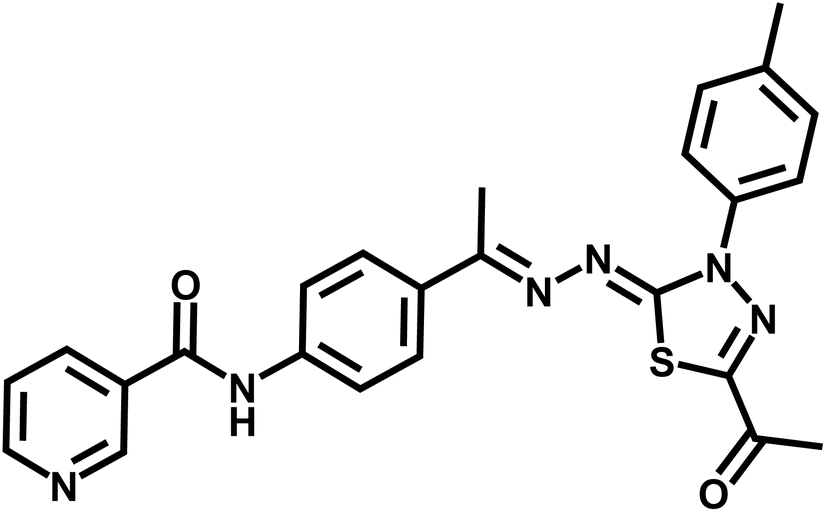 |
|||||
| 7e |  |
Poor | ||||
| 7f |  |
Less than 90% | ||||
| 7g | 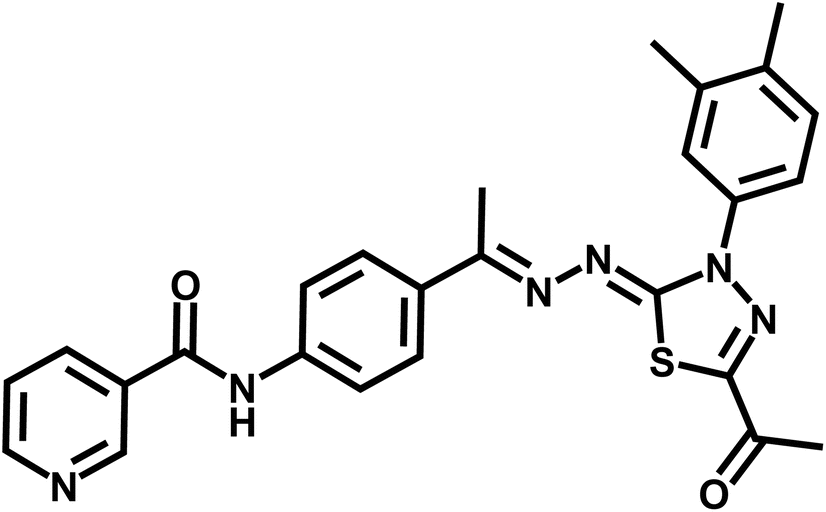 |
Moderate | More than 90% | |||
| Sorafenib |  |
Very low | Good |
In terms of intestinal absorption, most compounds, including 7a, showed good absorption, except 7e and 7f, which demonstrated poor absorption. This suggests that 7a has a favorable absorption profile, similar to sorafenib, making it a viable candidate for oral administration.
Regarding metabolic stability, all compounds were classified as non-inhibitors of CYP2D6, indicating a lower risk of drug–drug interactions through this metabolic pathway. Additionally, 7a displayed high plasma protein binding (PPB > 90%), similar to sorafenib, which may contribute to a longer half-life in systemic circulation but could also impact free drug availability for therapeutic action.
Overall, 7a demonstrates promising pharmacokinetic properties, particularly in terms of absorption and metabolic stability, while its low solubility presents a potential challenge for bioavailability. Future studies should focus on improving its formulation for better solubility and evaluating its in vivo pharmacokinetics to confirm these predictions.
As Table 7 shows, all tested compounds, including 7a, were classified as non-mutagenic except for 7f, which was identified as a potential mutagen. Additionally, 7a was predicted to be a non-carcinogen, while sorafenib was classified as a single carcinogen, highlighting the improved safety profile of 7a compared to the standard VEGFR-2 inhibitor.
| Comp. | Ames prediction | Mouse-female FDA | DTP | Rat oral LD50a | Rat chronic LOAELa | Skin irritancy | Ocular irritancy |
|---|---|---|---|---|---|---|---|
| a Unit: g kg−1 body weight. | |||||||
| 7a | Non-mutagen | Non-carcinogen | Non-toxic | 0.531148 | 0.0618943 | None | Mild |
| 7b | 0.247088 | 0.0276375 | |||||
| 7c | 0.418376 | 0.049371 | |||||
| 7d | 0.329557 | 0.0383128 | |||||
| 7e | 0.353069 | 0.0303718 | |||||
| 7f | Mutagen | 0.271895 | 0.034525 | ||||
| 7g | Non-mutagen | 0.331188 | 0.0368119 | ||||
| Sorafenib | Single-carcinogen | Toxic | 0.822583 | 0.00482816 | |||
Regarding acute toxicity, 7a exhibited an oral LD50 value of 0.531 mg kg−1, suggesting a relatively low toxicity level compared to sorafenib (0.822 mg kg−1). Similarly, the rat chronic lowest observed adverse effect level (LOAEL) for 7a (0.0618 mg kg−1) was higher than that of sorafenib (0.0048 mg kg−1), indicating lower chronic toxicity risk. Skin and ocular irritation assessments predicted that 7a poses no skin irritation risk and only mild ocular irritation, which is consistent with the safety profiles of other derivatives.
These findings suggest that 7a possesses a favorable toxicity and safety profile, making it a promising VEGFR-2 inhibitor with a lower carcinogenic and mutagenic risk than sorafenib. Future studies should include in vivo toxicity evaluations and long-term safety assessments to confirm these computational predictions and ensure its suitability for clinical development.
2.3.9.1. Pharmacokinetic profiling study. A computational in silico analysis was carried out to assess the physicochemical characteristics and pharmacokinetic profiles of the synthesized compounds, using sorafenib as a reference. Drug molecules are more likely to exhibit good oral absorption if they meet at least three out of the four criteria outlined in Lipinski's rule of five: (1) no more than 5 hydrogen bond donors (OH, NH, SH), (2) no more than 10 hydrogen bond acceptors (N, O, S), (3) a molecular weight under 500 Da, and (4) a log
P value below 5. Compounds that violate more than one of these rules are typically associated with poor oral bioavailability. Additionally, limited molecular flexibility—quantified by the number of rotatable bonds—and a low polar surface area have also been identified as key indicators of favorable oral bioavailability.45 Specifically, molecules with 10 or fewer rotatable bonds and a polar surface area of 140 Å2 or less are considered likely to be well absorbed. According to the data presented in Table 8, all the synthesized compounds, along with sorafenib, complied with both Lipinski's and Veber's guidelines.45,46
| Compound | Lipinski's rule of five | Veber's rule | |||||
|---|---|---|---|---|---|---|---|
| logP |
Mol. wt | HBD | HBA | Violation of Lipinski's rule | Number of rotatable bonds | TPSA | |
| 7a | 4.258 | 490.965 | 1 | 8 | 0 | 6 | 124.68 |
| 7b | 4.258 | 490.965 | 1 | 8 | 0 | 6 | 124.68 |
| 7c | 4.08 | 470.546 | 1 | 8 | 0 | 6 | 124.68 |
| 7d | 4.08 | 470.546 | 1 | 8 | 0 | 6 | 124.68 |
| 7g | 4.566 | 484.573 | 1 | 8 | 0 | 6 | 124.68 |
| Sorafenib | 4.175 | 464.825 | 3 | 7 | 0 | 6 | 92.35 |
3. Conclusion
This study demonstrates the potential of 7a as a promising VEGFR-2 inhibitor with strong anticancer activity against breast cancer cells. The compound exhibited potent cytotoxic effects, inducing S-phase arrest and apoptosis, with significant activation of caspase-3 and upregulation of Bax, confirming its role in intrinsic apoptotic pathway activation. Additionally, VEGFR-2 inhibition assays revealed its high kinase inhibitory potential, comparable to the clinically approved inhibitor sorafenib. Computational studies, including molecular docking, MD simulations (200 ns), MM-GBSA, ProLIF, PCAT, and FEL analyses, further validated the strong and stable binding of 7a to VEGFR-2. Moreover, ADMET predictions indicated good absorption, high plasma protein binding, and non-CYP2D6 inhibition, suggesting a favorable pharmacokinetic profile. Toxicity assessments classified 7a as non-mutagenic and non-carcinogenic, with a lower predicted toxicity compared to sorafenib, highlighting its potential as a safer alternative. Furthermore, Density Functional Theory (DFT) calculations confirmed the structural stability and reactivity of 7a, reinforcing its suitability as a lead candidate for further development. Given its potent anticancer activity, VEGFR-2 selectivity, and favorable safety profile, 7a represents a strong candidate for further in vivo evaluation. Future studies should focus on preclinical validation, pharmacokinetic profiling, and structural modifications to enhance its efficacy and therapeutic potential, ultimately advancing it as a novel targeted anticancer agent.4. Experimental
4.1. Chemistry
Standard chemical procedures were used to synthesize and characterize the target molecules. The ESI† include more details about the experimental procedures. Compounds 3, 5, and 6a–g were obtained using the published procedures.30,47–504.1.1.1. N-(4-((E)-1-(((Z)-5-Acetyl-3-(2-chlorophenyl)-1,3,4-thiadiazol-2(3H)-ylidene)hydrazono)ethyl)phenyl)nicotinamide 7a. Yellowish white powder (yield, 78%); mp = 205–207 °C; FT-IR (νmax, cm−1): 3322 (NH), 3069, 3037 (C–H aromatic), 2998, 2939, 2877 (C–H aliphatic), 1677, 1657 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 10.61 (s, 1H), 9.11 (dd, J = 2.4, 0.9 Hz, 1H), 8.76 (dd, J = 4.8, 1.7 Hz, 1H), 8.31–8.28 (m, 1H), 7.86 (d, J = 3.5 Hz, 4H), 7.81–7.75 (m, 2H), 7.62–7.56 (m, 3H), 2.52 (s, 3H), 2.20 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 199.70, 189.72, 164.26, 159.89, 152.30, 150.72, 148.78, 140.44, 135.66, 135.23, 132.82, 131.82, 131.17, 130.61, 130.22, 128.60, 127.27, 127.03, 123.65, 120.04, 120.01, 25.06, 14.82; mass (m/z): 490 (M+, 49.82%), 90 (100%, base peak); anal. calcd for C24H19ClN6O2S (490.97): C, 58.71; H, 3.90; N, 17.12. Found: C, 58.94; H, 4.06; N, 17.31%.
4.1.1.2. N-(4-((E)-1-(((Z)-5-Acetyl-3-(4-chlorophenyl)-1,3,4-thiadiazol-2(3H)-ylidene)hydrazono)ethyl)phenyl)nicotinamide 7b. Yellowish white powder (yield, 75%); mp = 212–214 °C; HPLC purity 96%; FT-IR (νmax, cm−1): 3398 (NH), 3061 (C–H aromatic), 2952, 2925, 2853 (C–H aliphatic), 1711, 1665 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 10.61 (s, 1H), 9.13 (s, 1H), 8.77 (d, J = 4.8 Hz, 1H), 8.31 (dt, J = 8.0, 2.1 Hz, 1H), 8.18–8.09 (m, 2H), 7.94–7.84 (m, 4H), 7.69–7.62 (m, 2H), 7.58 (dd, J = 8.0, 4.8 Hz, 1H), 2.59 (s, 3H), 2.43 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 189.77, 164.21, 163.93, 160.26, 152.22, 151.01, 148.74, 140.55, 137.67, 135.56, 132.53, 131.02, 129.12, 127.11, 123.53, 123.36, 119.88, 25.05, 15.33; mass (m/z): 520 (M+, 44.02%), 387 (100%, base peak); anal. calcd for C24H19ClN6O2S (490.97): C, 58.71; H, 3.90; N, 17.12. Found: C, 58.55; H, 3.75; N, 17.43%.
4.1.1.3. N-(4-((E)-1-(((Z)-5-Acetyl-3-(m-tolyl)-1,3,4-thiadiazol-2(3H)-ylidene)hydrazono)ethyl) phenyl)nicotinamide 7c. White powder (yield, 80%); mp = 197–199 °C; HPLC purity 100%; FT-IR (νmax, cm−1): 3338 (NH), 2981, 2977, 2918 (C–H aliphatic), 1744, 1657 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 10.61 (s, 1H), 9.13 (d, J = 2.3 Hz, 1H), 8.77 (dd, J = 4.9, 1.7 Hz, 1H), 8.31 (dt, J = 8.2, 2.1 Hz, 1H), 7.93–7.85 (m, 6H), 7.58 (dd, J = 8.0, 4.8 Hz, 1H), 7.45 (t, J = 7.8 Hz, 1H), 7.22 (d, J = 7.6 Hz, 1H), 2.58 (s, 3H), 2.41 (d, J = 1.7 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 189.89, 164.32, 159.95, 152.30, 148.79, 140.50, 138.85, 138.75, 135.66, 132.75, 130.52, 129.01, 127.89, 127.12, 123.64, 122.50, 119.99, 119.23, 40.20, 25.11, 21.23, 15.26; mass (m/z): 470 (M+, 23.76%), 88 (100%, base peak); anal. calcd for C25H22N6O2S (470.55): C, 63.81; H, 4.71; N, 17.86. Found: C, 63.65; H, 4.85; N, 18.03%.
4.1.1.4. N-(4-((E)-1-(((Z)-5-Acetyl-3-(p-tolyl)-1,3,4-thiadiazol-2(3H)-ylidene)hydrazono)ethyl)phenyl)nicotinamide 7d. White powder (yield, 80%); mp = 195–197 °C; HPLC purity 100%; FT-IR (νmax, cm−1): 3333 (NH), 2971, 2869 (C–H aliphatic), 1736, 1680 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 10.61 (s, 1H), 9.13 (d, J = 2.3 Hz, 1H), 8.77 (dd, J = 4.8, 1.7 Hz, 1H), 8.31 (dt, J = 8.1, 2.0 Hz, 1H), 7.95–7.87 (m, 6H), 7.58 (dd, J = 8.0, 4.8 Hz, 1H), 7.38 (d, J = 8.3 Hz, 2H), 2.57 (s, 3H), 2.40 (s, 3H), 2.38 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 189.71, 164.25, 164.19, 159.71, 152.21, 150.39, 148.74, 140.43, 136.76, 136.40, 135.54, 132.66, 130.45, 129.52, 127.02, 123.51, 122.04, 119.88, 25.01, 20.62, 15.18; mass (m/z): 470 (M+, 10.12%), 91 (100%, base peak); anal. calcd for C25H22N6O2S (470.55): C, 63.81; H, 4.71; N, 17.86. Found: C, 63.59; H, 4.85; N, 18.09%.
4.1.1.5. N-(4-((E)-1-(((Z)-5-Acetyl-3-(4-chloro-2-nitrophenyl)-1,3,4-thiadiazol-2(3H)-ylidene)hydrazono)ethyl)phenyl)nicotinamide 7e. Yellowish powder (yield, 70%); mp = 185–187 °C; FT-IR (νmax, cm−1): 3248 (NH), 3003 (C–H aromatic), 2931, 2838 (C–H aliphatic), 1691, 1643 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 10.60 (s, 1H), 9.12 (s, 1H), 8.76 (d, J = 4.3 Hz, 1H), 8.34 (d, J = 2.3 Hz, 1H), 8.30 (dt, J = 8.0, 2.0 Hz, 1H), 8.06 (dd, J = 8.7, 2.4 Hz, 1H), 7.99 (d, J = 8.6 Hz, 1H), 7.86 (s, 4H), 7.56 (dd, J = 7.9, 4.8 Hz, 1H), 2.55 (s, 3H), 2.20 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 189.52, 164.22, 163.08, 160.72, 152.22, 152.01, 148.77, 144.21, 140.62, 135.56, 134.54, 134.02, 132.41, 130.45, 129.87, 128.99, 127.11, 125.43, 123.52, 119.85, 25.04, 14.68; mass (m/z): 535 (M+, 4.46%), 367 (100%, base peak); anal. calcd for C24H18ClN7O4S (535.96): C, 53.78; H, 3.39; N, 18.29. Found: C, 53.91; H, 3.43; N, 18.50%.
4.1.1.6. N-(4-((E)-1-(((Z)-5-Acetyl-3-(4-nitrophenyl)-1,3,4-thiadiazol-2(3H)-ylidene)hydrazono)ethyl)phenyl)nicotinamide 7f. Orange powder (yield, 77%); mp = 190–192 °C; HPLC purity 100%; FT-IR (νmax, cm−1): 3256 (NH), 3046 (C–H aromatic), 2989, 2945, 2921 (C–H aliphatic), 1684, 1667 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 1H), 9.15 (s, 1H), 8.83–8.77 (m, 1H), 8.48 (s, 4H), 8.33 (s, 1H), 7.97–7.90 (m, 4H), 7.61 (s, 1H), 2.66 (s, 3H), 2.60 (s, 3H); mass (m/z): 501 (M+, 31.99%), 267 (100%, base peak); anal. calcd for C24H19N7O4S (501.52): C, 57.48; H, 3.82; N, 19.55. Found: C, 57.39; H, 4.06; N, 19.73%.
4.1.1.7. N-(4-((E)-1-(((Z)-5-Acetyl-3-(3,4-dimethylphenyl)-1,3,4-thiadiazol-2(3H)-ylidene)hydrazono)ethyl)phenyl)nicotinamide 7g. White powder (yield, 82%); mp = 195–197 °C; HPLC purity 100%; FT-IR (νmax, cm−1): 3420 (NH), 3057 (C–H aromatic), 2992, 2966, 2941 (C–H aliphatic), 1640 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 10.60 (s, 1H), 9.12 (d, J = 2.3 Hz, 1H), 8.77 (dd, J = 4.9, 1.6 Hz, 1H), 8.31 (dt, J = 8.1, 2.0 Hz, 1H), 7.86 (s, 4H), 7.81 (d, J = 2.4 Hz, 1H), 7.73 (dd, J = 8.2, 2.4 Hz, 1H), 7.57 (dd, J = 8.0, 4.8 Hz, 1H), 7.29 (d, J = 8.3 Hz, 1H), 2.54 (s, 3H), 2.37 (s, 3H), 2.29 (s, 3H), 2.26 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 189.68, 164.24, 164.18, 159.59, 152.22, 150.22, 148.75, 140.42, 137.06, 136.63, 135.55, 135.43, 132.67, 130.45, 129.85, 126.99, 123.52, 122.85, 119.85, 119.35, 24.97, 19.68, 19.00, 15.10; mass (m/z): 484 (M+, 1.89%), 414 (100%, base peak); anal. calcd for C26H24N6O2S (484.58): C, 64.44; H, 4.99; N, 17.34. Found: C, 64.27; H, 5.12; N, 17.48%.
4.2. In vitro studies
4.3. Computational studies
Data availability
Data are available with corresponding authors upon request.Conflicts of interest
The authors confirm that they have no conflicts of interest to disclose.Acknowledgements
This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2025R142), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors thank Research Center at AlMaarefa University for funding this work.References
- L. Wilkinson and T. Gathani, Understanding breast cancer as a global health concern, Brit. J. Radiol., 2022, 95(1130), 20211033 Search PubMed.
- S. M. Schwartz, Epidemiology of Cancer, Clin. Chem., 2024, 70(1), 140–149 Search PubMed.
- A. A. Shah, M. A. Kamal and S. Akhtar, Tumor Angiogenesis and VEGFR-2: Mechanism, Pathways and Current Biological Therapeutic Interventions, Curr. Drug Metab., 2021, 22(1), 50–59 Search PubMed.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, A. B. Mehany, B. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, In silico, in vitro VEGFR-2 inhibition, and anticancer activity of a 3-(hydrazonomethyl) naphthalene-2-ol derivative, J. Biomol. Struct. Dyn., 2023, 41(16), 7986–8001 Search PubMed.
- M. S. Taghour, H. Elkady, W. M. Eldehna, N. El-Deeb, A. M. Kenawy, A. E. Abd El-Wahab, E. B. Elkaeed, B. A. Alsfouk, A. M. Metwaly and I. H. Eissa, Discovery of new quinoline and isatine derivatives as potential VEGFR-2 inhibitors: design, synthesis, antiproliferative, docking and MD simulation studies, J. Biomol. Struct. Dyn., 2023, 41(21), 11535–11550 Search PubMed.
- I. H. Eissa, R. G. Yousef, H. Elkady, E. B. Elkaeed, A. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, M. A. Elhendawy, M. Godfrey and A. M. Metwaly, Design, semi-synthesis, anti-cancer assessment, docking, MD simulation, and DFT studies of novel theobromine-based derivatives as VEGFR-2 inhibitors and apoptosis inducers, Comput. Biol. Chem., 2023, 107, 107953 Search PubMed.
- X. J. Liu, H. C. Zhao, S. J. Hou, H. J. Zhang, L. Cheng, S. Yuan, L. R. Zhang, J. Song, S. Y. Zhang and S. W. Chen, Recent development of multi-target VEGFR-2 inhibitors for the cancer therapy, Bioorg. Chem., 2023, 133, 106425 Search PubMed.
- T. Arao, K. Matsumoto, K. Furuta, K. Kudo, H. Kaneda, T. Nagai, K. Sakai, Y. Fujita, D. Tamura, K. Aomatsu, F. Koizumi and K. Nishio, Acquired drug resistance to vascular endothelial growth factor receptor 2 tyrosine kinase inhibitor in human vascular endothelial cells, Anticancer Res., 2011, 31(9), 2787–2796 Search PubMed.
- Q. Li, K. Chen, T. Zhang, D. Jiang, L. Chen, J. Jiang, C. Zhang and S. Li, Understanding sorafenib-induced ferroptosis and resistance mechanisms: implications for cancer therapy, Eur. J. Pharmacol., 2023, 955, 175913 Search PubMed.
- L. Cosmai, M. Gallieni, W. Liguigli and C. Porta, Renal toxicity of anticancer agents targeting vascular endothelial growth factor (VEGF) and its receptors (VEGFRs), J. Nephrol., 2017, 30(2), 171–180 Search PubMed.
- S. Lai, A. Molfino, P. Seminara, F. Longo, G. Innico, B. Coppola, D. Mastroluca, A. Galani, M. Dimko, P. Aceto and C. Lai, Vascular Endothelial Growth Factor Inhibitor Therapy and Cardiovascular and Renal Damage in Renal Cell Carcinoma, Curr. Vasc. Pharmacol., 2018, 16(2), 190–196 Search PubMed.
- S. Lai, M. I. Amabile, S. Mazzaferro, A. P. Mitterhofer, A. Mazzarella, A. Galani, G. Imbimbo, R. Cianci, M. Pasquali and A. Molfino, Effects of sunitinib on endothelial dysfunction, metabolic changes, and cardiovascular risk indices in renal cell carcinoma, Cancer Med., 2020, 9(11), 3752–3757 Search PubMed.
- A. A. Abdelgalil, H. M. Alkahtani and F. I. Al-Jenoobi, Sorafenib, Profiles of Drug Substances, Excipients, and Related Methodology, 2019, vol. 44, pp. 239–266 Search PubMed.
- A. M. Metwaly, M. A. Abu-Saied, I. M. Gobaara, A. M. Lotfy, B. A. Alsfouk, E. B. Elkaeed and I. H. Eissa, Nicotinamide Loaded Chitosan Nanocomplex Shows Improved Anticancer Potential: Molecular Docking, Synthesis, Characterization and In vitro Evaluations, Curr. Org. Chem., 2024, 28(1), 46–55 Search PubMed.
- M. Szeliga, Thiadiazole derivatives as anticancer agents, Pharmacol. Rep., 2020, 72(5), 1079–1100 Search PubMed.
- S. Grimme and P. R. Schreiner, Computational Chemistry: The Fate of Current Methods and Future Challenges, Angew. Chem., Int. Ed. Engl., 2018, 57(16), 4170–4176 Search PubMed.
- A. Varnek and I. I. Baskin, Chemoinformatics as a Theoretical Chemistry Discipline, Mol. Inform., 2011, 30(1), 20–32 Search PubMed.
- S. A. Hollingsworth and R. O. Dror, Molecular Dynamics Simulation for All, Neuron, 2018, 99(6), 1129–1143 Search PubMed.
- A. M. Metwaly, E. M. El-Fakharany, A. A. Alsfouk, I. M. Ibrahim, E. B. Elkaeed and I. H. Eissa, Integrated study of Quercetin as a potent SARS-CoV-2 RdRp inhibitor: Binding interactions, MD simulations, and In vitro assays, PLoS One, 2024, 19(12), e0312866 Search PubMed.
- L. L. G. Ferreira and A. D. Andricopulo, ADMET modeling approaches in drug discovery, Drug Discov. Today, 2019, 24(5), 1157–1165 Search PubMed.
- A. M. Metwaly, M. S. Alesawy, B. A. Alsfouk, I. M. Ibrahim, E. B. Elkaeed and I. H. Eissa, Computer-Assisted Drug Discovery of Potential African Anti-SARS-CoV-2 Natural Products Targeting the Helicase Protein, Nat. Prod. Commun., 2024, 19(4), 1–23 Search PubMed.
- A. L. Jenner, R. A. Aogo, C. L. Davis, A. M. Smith and M. Craig, Leveraging Computational Modeling to Understand Infectious Diseases, Curr. Pathobiol. Rep., 2020, 8(4), 149–161 Search PubMed.
- M. M. Ahmed, M. E.-F. Esmail, A. A. Aisha, M. I. Ibrahim, B. E. Eslam and H. E. Ibrahim, Integrated in Silico and in Vitro Studies of Rutin's Potential against SARS-CoV-2 through the Inhibition of the RNA-dependent RNA Polymerase, Curr. Med. Chem., 2025, 32, 1–27 Search PubMed.
- U. Gupta and D. Paliwal, Discovery, current trends in computational chemistry for breast cancer, Lett. Drug Des. Discov., 2023, 20(1), 2–15 Search PubMed.
- A. M. Metwaly, H. Abd-El-Azim, M. Zewail, A. A. Alsfouk, E. B. Elkaeed and I. H. Eissa, Chitosomal Encapsulation Enhances the Anticancer Efficacy of a Theobromine Analogue: An Integrated In Silico and In Vitro Study, J. Comput. Biophys. Chem., 2025, 557-573, 1–17 Search PubMed.
- I. H. Eissa, R. G. Yousef, M. Sami, E. B. Elkaeed, B. A. Alsfouk, I. M. Ibrahim, D. Z. Husein, H. Elkady and A. M. Metwaly, Exploring the anticancer properties of a new nicotinamide analogue: investigations into in silico analysis, antiproliferative effects, selectivity, VEGFR-2 inhibition, apoptosis induction, and migration suppression, Pathol., Res. Pract., 2023, 252, 154924 Search PubMed.
- I. H. Eissa, M. A. E. Bkrah, R. G. Yousef, H. Elkady, E. B. Elkaeed, B. A. Alsfouk, I. M. Ibrahim, A. M. Metwaly and D. Z. Husein, Design and In Silico and In Vitro Evaluations of a Novel Nicotinamide Derivative as a VEGFR-2 Inhibitor, J. Chem., 2024, 2024(1), 2176512 Search PubMed.
- R. G. Yousef, H. Elkady, E. B. Elkaeed, I. M. M. Gobaara, H. A. Al-Ghulikah, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, (E)-N-(3-(1-(2-(4-(2,2,2-Trifluoroacetamido)benzoyl)hydrazono)ethyl)phenyl)nicotinamide: A Novel Pyridine Derivative for Inhibiting Vascular Endothelial Growth Factor Receptor-2: Synthesis, Computational, and Anticancer Studies, Molecules, 2022, 27(22), 7719–7743 Search PubMed.
- H. Elkady, W. E. Elgammal, H. A. Mahdy, S. Zara, S. Carradori, D. Z. Husein, A. A. Alsfouk, I. M. Ibrahim, E. B. Elkaeed, A. M. Metwaly and I. H. Eissa, Anti-proliferative 2,3-dihydro-1,3,4-thiadiazoles targeting VEGFR-2: design, synthesis, in vitro, and in silico studies, Comput. Biol. Chem., 2024, 113, 108221 Search PubMed.
- I. H. Eissa, H. Elkady, W. E. Elgammal, H. A. Mahdy, E. B. Elkaeed, A. A. Alsfouk, I. M. Ibrahim, D. Z. Husein and A. M. Metwaly, Integrated in silico and in vitro discovery of a new anticancer thiadiazole analog targeting VEGFR-2, J. Mol. Struct., 2024, 1312, 138641 Search PubMed.
- I. H. Eissa, W. E. Elgammal, H. A. Mahdy, S. Zara, S. Carradori, D. Z. Husein, M. N. Alharthi, I. M. Ibrahim, E. B. Elkaeed, H. Elkady and A. M. Metwaly, Design, synthesis, and evaluation of novel thiadiazole derivatives as potent VEGFR-2 inhibitors: a comprehensive in vitro and in silico study, RSC Adv., 2024, 14(48), 35505–35519 Search PubMed.
- W. E. Elgammal, H. Elkady, H. A. Mahdy, D. Z. Husein, A. A. Alsfouk, B. A. Alsfouk, I. M. Ibrahim, E. B. Elkaeed, A. M. Metwaly and I. H. Eissa, Rationale design and synthesis of new apoptotic thiadiazole derivatives targeting VEGFR-2: computational and in vitro studies, RSC Adv., 2023, 13(51), 35853–35876 Search PubMed.
- M. M. Alanazi, H. Elkady, N. A. Alsaif, A. J. Obaidullah, W. A. Alanazi, A. M. Al-Hossaini, M. A. Alharbi, I. H. Eissa and M. A. Dahab, Discovery of new quinoxaline-based derivatives as anticancer agents and potent VEGFR-2 inhibitors: design, synthesis, and in silico study, J. Mol. Struct., 2021, 132220 Search PubMed.
- M. M. Alanazi, H. Elkady, N. A. Alsaif, A. J. Obaidullah, H. M. Alkahtani, M. M. Alanazi, M. A. Alharbi, I. H. Eissa and M. A. Dahab, New quinoxaline-based VEGFR-2 inhibitors: design, synthesis, and antiproliferative evaluation with in silico docking, ADMET, toxicity, and DFT studies, RSC Adv., 2021, 11(48), 30315–30328 Search PubMed.
- R. G. Yousef, H. M. Sakr, I. H. Eissa, A. B. Mehany, A. M. Metwaly, M. A. Elhendawy, M. M. Radwan, M. A. ElSohly, H. S. Abulkhair and K. El-Adl, New quinoxaline-2 (1 H)-ones as potential VEGFR-2 inhibitors: design, synthesis, molecular docking, ADMET profile and anti-proliferative evaluations, New J. Chem., 2021, 45(36), 16949–16964 Search PubMed.
- I. H. Eissa, R. G. Yousef, H. Elkady, E. B. Elkaeed, A. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, M. A. Elhendawy, M. Godfrey and A. M. Metwaly, Identification of new theobromine-based derivatives as potent VEGFR-2 inhibitors: design, semi-synthesis, biological evaluation, and in silico studies, RSC Adv., 2023, 13(33), 23285–23307 Search PubMed.
- R. G. Yousef, I. H. Eissa, H. Elkady, A. B. M Mehany, M. A. Abo-Saif, M. M. Radwan, M. A. ElSohly, I. M. Ibrahim, A. Elwan and M. A. El-Zahabi, Design and synthesis of new nicotinamides as immunomodulatory VEGFR-2 inhibitors and apoptosis inducers, Future Med. Chem., 2024, 1–16 Search PubMed.
- R. G. Yousef, H. Elkady, E. B. Elkaeed, I. M. Gobaara, H. A. Al-Ghulikah, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, (E)-N-(3-(1-(2-(4-(2, 2, 2-Trifluoroacetamido) benzoyl) hydrazono) ethyl) phenyl) nicotinamide: A Novel Pyridine Derivative for Inhibiting Vascular Endothelial Growth Factor Receptor-2: Synthesis, Computational, and Anticancer Studies, Molecules, 2022, 27(22), 7719 Search PubMed.
- B. W. Matore, P. P. Roy and J. Singh, Discovery of novel VEGFR2-TK inhibitors by phthalimide pharmacophore based virtual screening, molecular docking, MD simulation and DFT, J. Biomol. Struct. Dyn., 2023, 41(22), 13056–13077 Search PubMed.
- P. J. Chaudhari, A. R. Nemade and A. A. Shirkhedkar, Recent updates on potential of VEGFR-2 small-molecule inhibitors as anticancer agents, RSC Adv., 2024, 14(45), 33384–33417 Search PubMed.
- I. Fleming, Molecular Orbitals and Organic Chemical Reactions, John Wiley & Sons, 2011 Search PubMed.
- T. Yoshida and S. Hirono, A 3D-QSAR analysis of CDK2 inhibitors using FMO calculations and PLS regression, Chem. Pharm. Bull., 2019, 67(6), 546–555 Search PubMed.
- S. A. El-Metwally, H. Elkady, M. Hagras, D. Z. Husein, I. M. Ibrahim, M. S. Taghour, H. A. El-Mahdy, A. Ismail, B. A. Alsfouk and E. B. Elkaeed, Design, synthesis, anti-proliferative evaluation, docking, and MD simulation studies of new thieno [2, 3-d] pyrimidines targeting VEGFR-2, RSC Adv., 2023, 13(33), 23365–23385 Search PubMed.
- H. Elkady, O. A. El-Dardir, A. Elwan, M. S. Taghour, H. A. Mahdy, M. A. Dahab, E. B. Elkaeed, B. A. Alsfouk, I. M. Ibrahim and D. Z. Husein, Synthesis, biological evaluation and computer-aided discovery of new thiazolidine-2, 4-dione derivatives as potential antitumor VEGFR-2 inhibitors, RSC Adv., 2023, 13(40), 27801–27827 Search PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45(12), 2615–2623 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev., 2012, 64, 4–17 Search PubMed.
- W. E. Elgammal, H. Elkady, H. A. Mahdy, D. Z. Husein, A. A. Alsfouk, B. A. Alsfouk, I. M. Ibrahim, E. B. Elkaeed, A. M. Metwaly and I. H. Eissa, Rationale design and synthesis of new apoptotic thiadiazole derivatives targeting VEGFR-2: computational and in vitro studies, RSC Adv., 2023, 13(51), 35853–35876 Search PubMed.
- H. Elkady, W. E. Elgammal, H. A. Mahdy, S. Zara, S. Carradori, D. Z. Husein, A. A. Alsfouk, I. M. Ibrahim, E. B. Elkaeed and A. M. Metwaly, Anti-proliferative 2, 3-dihydro-1, 3, 4-thiadiazoles targeting VEGFR-2: design, synthesis, in vitro, and in silico studies, Comput. Biol. Chem., 2024, 113, 108221 Search PubMed.
- H. A. Mahdy, H. Elkady, W. E. Elgammal, E. B. Elkaeed, A. A. Alsfouk, I. M. Ibrahim, D. Z. Husein, M. A. Elkady, A. M. Metwaly and I. H. Eissa, Design, synthesis, in vitro, and in silico studies of new thiadiazol derivatives as promising VEGFR-2 inhibitors and apoptosis inducers, J. Mol. Struct., 2024, 139019 Search PubMed.
- I. H. Eissa, W. E. Elgammal, H. A. Mahdy, S. Zara, S. Carradori, D. Z. Husein, M. N. Alharthi, I. M. Ibrahim, E. B. Elkaeed and H. Elkady, Design, synthesis, and evaluation of novel thiadiazole derivatives as potent VEGFR-2 inhibitors: a comprehensive in vitro and in silico study, RSC Adv., 2024, 14(48), 35505–35519 Search PubMed.
- A. R. Kotb, D. A. Bakhotmah, A. E. Abdallah, H. Elkady, M. S. Taghour, I. H. Eissa and M. A. El-Zahabi, Design, synthesis, and biological evaluation of novel bioactive thalidomide analogs as anticancer immunomodulatory agents, RSC Adv., 2022, 12(52), 33525–33539 Search PubMed.
- A. R. Kotb, A. E. Abdallah, H. Elkady, I. H. Eissa, M. S. Taghour, D. A. Bakhotmah, T. M. Abdelghany and M. A. El-Zahabi, Design, synthesis, anticancer evaluation, and in silico ADMET analysis of novel thalidomide analogs as promising immunomodulatory agents, RSC Adv., 2023, 13(16), 10488–10502 Search PubMed.
- H. A. Mahdy, H. Elkady, M. S. Taghour, A. Elwan, M. A. Dahab, M. A. Elkady, E. G. Elsakka, E. B. Elkaeed, B. A. Alsfouk and I. M. Ibrahim, New theobromine derivatives inhibiting VEGFR-2: design, synthesis, antiproliferative, docking and molecular dynamics simulations, Future Med. Chem., 2023, 15(14), 1233–1250 Search PubMed.
- H. Qin and E. N. Benveniste, ELISA methodology to quantify astrocyte production of cytokines/chemokines in vitro, Astrocytes: Methods and Protocols, 2012, pp. 235–249 Search PubMed.
- Z. Darzynkiewicz, E. Bedner and P. Smolewski, Flow cytometry in analysis of cell cycle and apoptosis, Seminars in Hematology, Elsevier, 2001, pp. 179–193 Search PubMed.
- D. S. Biovia, Discovery studio modeling environment, Release, San Diego, CA, 2017 Search PubMed.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell Jr, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels and S. Boresch, CHARMM: the biomolecular simulation program, J. Comput. Chem., 2009, 30(10), 1545–1614 Search PubMed.
- S. Jo, X. Cheng, S. M. Islam, L. Huang, H. Rui, A. Zhu, H. S. Lee, Y. Qi, W. Han and K. Vanommeslaeghe, CHARMM-GUI PDB manipulator for advanced modeling and simulations of proteins containing nonstandard residues, Adv. Protein Chem. Struct. Biol., 2014, 96, 235–265 Search PubMed.
- T. Tuccinardi, What is the current value of MM/PBSA and MM/GBSA methods in drug discovery?, Expet Opin. Drug Discov., 2021, 16(11), 1233–1237 Search PubMed.
- M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente and E. Moreno, gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS, J. Chem. Theor. Comput., 2021, 17(10), 6281–6291 Search PubMed.
- A. M. Metwaly, E. B. Elkaeed, B. A. Alsfouk, A. M. Saleh, A. E. Mostafa and I. H. Eissa, The Computational Preventive Potential of the Rare Flavonoid, Patuletin, Isolated from Tagetes patula, against SARS-CoV-2, Plants, 2022, 11(14), 1886–1905 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01223f |
| This journal is © The Royal Society of Chemistry 2025 |