
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into the synthesis and structural properties of pro-chiral 2-acetyl-N-aryl-2-(prop-2-yn-1-yl)pent-4-ynamides/-2-allyl-4-enamide derivatives through kinetics and energy frameworks†
Disha P. Vala a,
Bhavesh N. Sochab,
Victoria G. Collinsc,
Mehul P. Parmara,
Chirag D. Patela,
Savan S. Bhalodiyaa,
Subham G. Patela,
Sourav Banerjee*c and
Hitendra M. Patel*a
a,
Bhavesh N. Sochab,
Victoria G. Collinsc,
Mehul P. Parmara,
Chirag D. Patela,
Savan S. Bhalodiyaa,
Subham G. Patela,
Sourav Banerjee*c and
Hitendra M. Patel*a
aDepartment of Chemistry, Sardar Patel University, Vallabh Vidyanagar-388 120, Gujarat, India. E-mail: hm_patel@spuvvn.edu
bDepartment of Materials sciences, Sardar Patel University, Vallabh Vidyanagar-388 120, Gujarat, India
cDivision of Cancer Research, School of Medicine, University of Dundee, Dundee DD1 9SY, UK. E-mail: s.y.banerjee@dundee.ac.uk
First published on 12th May 2025
Abstract
In this study, we successfully synthesized a series of pro-chiral 2-acetyl-N-aryl-2-(prop-2-yn-1-yl)pent-4-ynamide/-2-allyl-4-enamide derivatives 5(a–n) starting from 4-enamide-N-(prop-2-yn-1-yl)pent-4-ynamide/-N-allyl-4-enamide 4 through a bimolecular nucleophilic substitution (SN2) reaction. We report, for the first time, a novel C–C bond formation between allyl and acetoacetanilide derivatives, supported by detailed mechanistic analysis involving alkyne interactions. The reaction mechanism, characterized by a nucleophilic attack at the substrate's carbon center leading to displacement of the leaving group, was validated through intrinsic reaction coordinate (IRC) analysis, providing deeper insights into its pathway and dynamics.
1 Introduction
Pro-chiral dienes/diynes are extensively implemented in various areas such as drug synthesis, agrochemicals, and pharmacy as well as in asymmetric organic synthesis as they also act as potential ligands in many metal-complex formations.1 A library of pro-chiral diynes/dienes with high structural diversity is useful for reaction development.2 The acetylene group has numerous elaboration options, such as nucleophilic additions, cyclizations, and metal-catalyzed coupling reactions ensuing optically active pro-chiral synthons are extremely important.3,4 Innovative approaches, such as diastereoselective desymmetrization, have enabled the transformation of symmetric dienes into chiral building blocks with high stereochemical control.5,6 The development of chiral pincer complexes has transformed enantioselective processes such as hydrogenation and alkynylation, while the stereoselective production of conjugated dienes has opened up new avenues for their application in polymers and materials science.7 Furthermore, photoredox catalysis and biocatalysis are gaining popularity because of their environmentally friendly and precise methods for producing chiral compounds.8–10 Computational chemistry has made a significant contribution by refining synthetic techniques, predicting reaction outcomes, and studying molecular orbitals, thereby driving reaction development.11 These breakthroughs pave the way for the development of a structurally diverse library of pro-chiral dienes and diynes, which will spur innovation in medicine, agrochemical, and material research.12 Although acetylene groups can be chemically modified to create optically active pro-chiral synthons, enantioselective catalytic synthesis is challenging owing to a lack of efficient ways for forming stereocenters with an acetylene group.13–15 The SN2 reaction is important and useful in organic transformation.16 Computational chemistry has emerged as an extremely valuable tool in organic chemistry17 for learning and identifying reaction intermediates and transition states, evaluating bond distances and atomic charges,18 and analyzing molecular orbitals. Furthermore, energy calculations aid in understanding the thermodynamic and kinetic factors of a reaction coordinate.19In continuation of our previous work,20–25 herein, we synthesised pro-chiral diynes/dienes 5(a–n) using acetoacetanilide derivatives 1(a–g) and propargyl/allyl bromide 2(a–b). We synthesised 14 analogues of pro-chiral allyl and alkyne containing acetoacetanilide 5(a–n) through the –NH functionalization of acetoacetanilides. Density functional theory (DFT) studies were conducted to understand the reaction mechanism, highlighting that the –NH moiety is less active in the reaction pathway than the –CH moiety.
2 Results and discussion
2.1 Chemistry
These pro-chiral diynes/dienes were synthesised using acetoacetanilide derivatives 1(a–g) and propargyl/allyl bromide 2(a–b). In this transformation (Scheme 1), the base first deprotonates the active methylene –H of acetoacetanilide 1(a–g), leading to the in situ formation of a racemic mixture of 2-acetyl-N-phenylpent-4-enamide/-ynamide 3, which immediately undergoes for the reaction led to the pro-chiral diynes/dienes 5(a–n) instead of deprotonation from the amide proton (–NH–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 4, as well as the mechanistic approach of both pathways (A & B). To confirm its formation and structure, 3 was also isolated and characterized using NMR and Mass spectroscopy. This SN2 transformation was carried out for better understanding reaction pathways, stability of intermediates and transition states. DFT studies were performed to elucidate the reaction mechanism, revealing why the –NH moiety does not participate as actively in the reaction pathway compared to the –CH moiety.
O) 4, as well as the mechanistic approach of both pathways (A & B). To confirm its formation and structure, 3 was also isolated and characterized using NMR and Mass spectroscopy. This SN2 transformation was carried out for better understanding reaction pathways, stability of intermediates and transition states. DFT studies were performed to elucidate the reaction mechanism, revealing why the –NH moiety does not participate as actively in the reaction pathway compared to the –CH moiety.
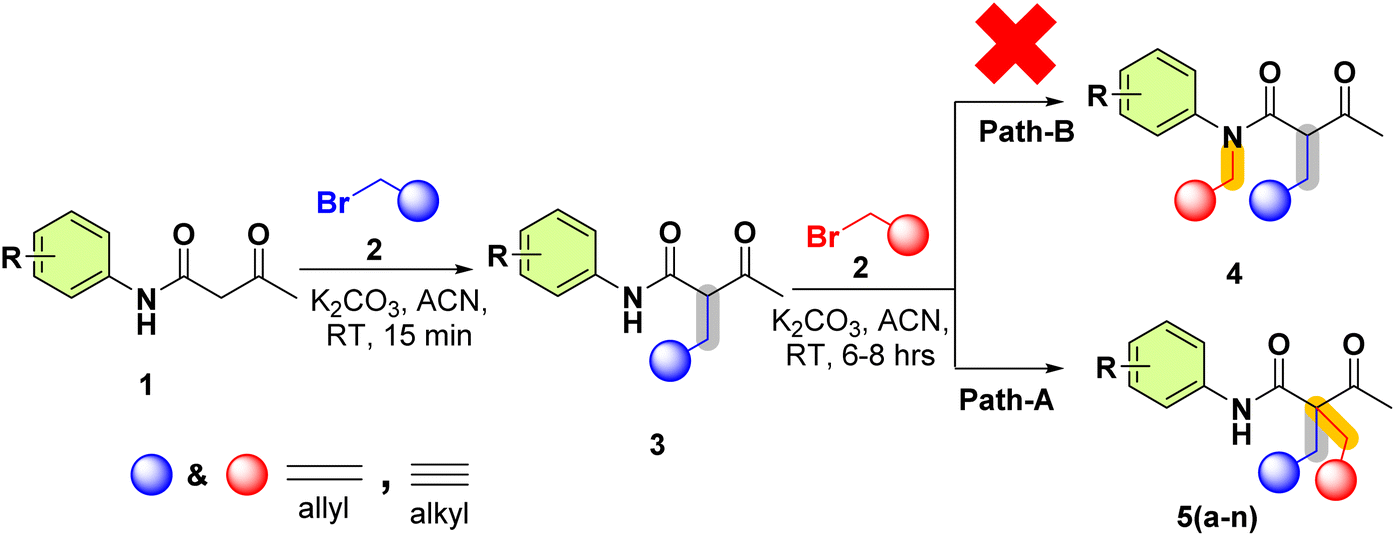 | ||
| Scheme 1 Synthesis of -acetyl-N-aryl-2-(prop-2-yn-1-yl)pent-4-ynamide/-2-allyl-4-enamide 5(a–n) over 4-enamide-N-(prop-2-yn-1-yl)pent-4-ynamide/-N-allyl-4-enamide. | ||
For optimization, acetoacetanilide 1a and propargyl bromide 2a were initially treated in the presence of K2CO3 (Table 1), which gave multiple spot-on TLC analyses (Entries 1 & 2). CS2CO3 also led to a mixture (Entry 3). Subsequently, 2 equiv. of 2a were taken. After 30 min, a mixture of products was generated and acetoacetanilide was still unreacted (Entry 4). After 10 hours, the reaction gave a single spot (Entry 5). 3 equiv. of 2a led to the single spot after 10 hours (Entry 6) (Table 1).
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Amount of the base | Amount of 2a | Time (min) t | % Conversion of 1a |
| a Reaction condition: 1a (1 mmol), 2a, base, 5–7 ml ACN, RT.b Observed from TLC analysis.c Isolated yield. | |||||
| 1 | K2CO3 | 2 equiv. | 1 equiv. | 30 min | Mixture |
| 2 | K2CO3 | 2 equiv. | 1 equiv. | 18 h | Mixture |
| 3 | Cs2CO3 | 1.5 equiv. | 1 equiv. | 18 h | Mixture |
| 4 | K2CO3 | 2 equiv. | 2 equiv. | 30 min | Mixture |
| 5 | K2CO3 | 2 equiv. | 2 equiv. | 10 h | 100% |
| 6 | K2CO3 | 3 equiv. | 3 equiv. | 10 h | 100% |
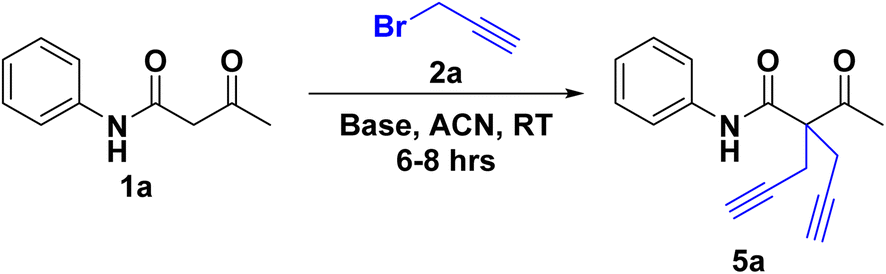
The reaction of 2(a–b) with different acetoacetanilide 1(a–g) using commercially available K2CO3 achieved 100% conversion yield for 1a within 8–10 h using acetonitrile. The desired crude product precipitated out with water after stirring for 1–1.5 h. This crude product was filtered and washed first with (2 × 20 mL) water, followed by (2 × 10 mL) n-hexane. All acetoacetanilide derivatives gave excellent yields (Scheme 2). First, the facial synthesis of the reaction between acetoacetanilide 1a and propargyl bromide 2a in the presence of K2CO3 for the generation of compound 3a, which further reacted with another molecule of propargyl bromide, led to the formation of prochiral dialkyne 5a.
 | ||
| Scheme 2 Substrate scope of 2-acetyl-N-aryl-2-(prop-2-yn-1-yl)pent-4-ynamide/-2-allyl-4-enamide 5(a–n). | ||
Here, K2CO3 deprotonates the nucleophile, increasing the nucleophilicity as it converts into the more reactive conjugate-base, which are the keto–enol tautomers INT-Ia & INT-Ib (Scheme 3). This Nu-attacks the electrophilic carbon of 2, where the carbon is partially bonded to both the nucleophile and the leaving group (-Br)-formed transition state-I. -Br then departs, resulting in the formation of 3. Subsequently, these 3 further undergo SN2 reaction, resulting in 5 and 4. Here, we explore the reaction mechanism of the formation of 5 and 4 via DFT using Gaussian 09.26
 | ||
| Scheme 3 Mechanism for possible pathways for active and inactive sites. | ||
2.2 Crystallographic investigation
For the structure–function relationship, single crystals of the final product 5f were obtained using a slow evaporation method (Fig. 1a). The crystal crystallized in the monoclinic system with space group P21/c, comprising four asymmetric molecules per unit cell (Table S1†). The two allyl moieties were arranged in such a way that one moiety was displaced by an angle of 110° and then rotated around an angle of 109°, with torsional angles of approximately −25.69° and 7.27°. The intra and intermolecular interactions, involving N–H⋯O, O–H⋯O, and C–H⋯O hydrogen bonds, played a critical role in stabilizing the molecule (Fig. 1b). The oxygen atoms facilitated binding between four molecules in the unit cell via intermolecular interactions, including C9–H9A⋯O9, N1–H1A⋯O3, and C18–H18⋯O4. In addition, the two C–H⋯O interactions between two 5f molecules form the graph set motif (Fig. 1d). The crystal structure was stabilized via carbon atoms (C9 and C17) from the allyl moieties and another carbon from the O–CH3 group on the ring, which participate in a C–H⋯π interaction (Fig. 1c), where the π system is sandwiched between two other molecules positioned above and below the π ring (Fig. S27†). | ||
| Fig. 1 (a) ORTAP view of the 5f molecule with 50% probability, (CCDC 2337812). (b) C–H⋯O intermolecular interaction, (c) C–H⋯π interaction and the (d) graph set motif. | ||
2.3 Computational analysis
All geometry optimizations and frequency calculations were performed using the B3LYP functional with the 6–311++G(d,p) basis set. The ‘++’ indicates that diffuse functions were included on all atoms, which is particularly important for accurately modeling the anionic species and long–range interactions. Solvent effects were incorporated using the PCM solvation model with acetonitrile as the solvent [SCRF = (solvent = acetonitrile)]. The optimization of the molecular structures using DFT showed excellent convergence for most of the systems, with all key parameters well within the defined thresholds. The mean maximum force across the systems was approximately 0.00003, significantly below the threshold of 0.000450. The RMS force also averaged at around 0.00052, comfortably meeting the threshold of 0.000300. For displacements, the mean maximum displacement was calculated at 0.00102, which was well within the allowable limit of 0.001800. The mean RMS displacement was 0.00067, which also satisfied the threshold of 0.001200. These results indicate successful structural optimization for the majority of the systems, with the exception of a few intermediates (INT-IIa, INT-IIb, INT-IIIb) and a transition state (TS-III), where slight deviations in force and displacement values were noted. Despite these exceptions, the overall convergence performance was robust and in line with the expectations for accurate molecular structure optimization. Solvent effects were incorporated using the Polarizable Continuum Model (PCM) with acetonitrile as the solvent via the scrf = (solvent = acetonitrile) approach (Table S3†).
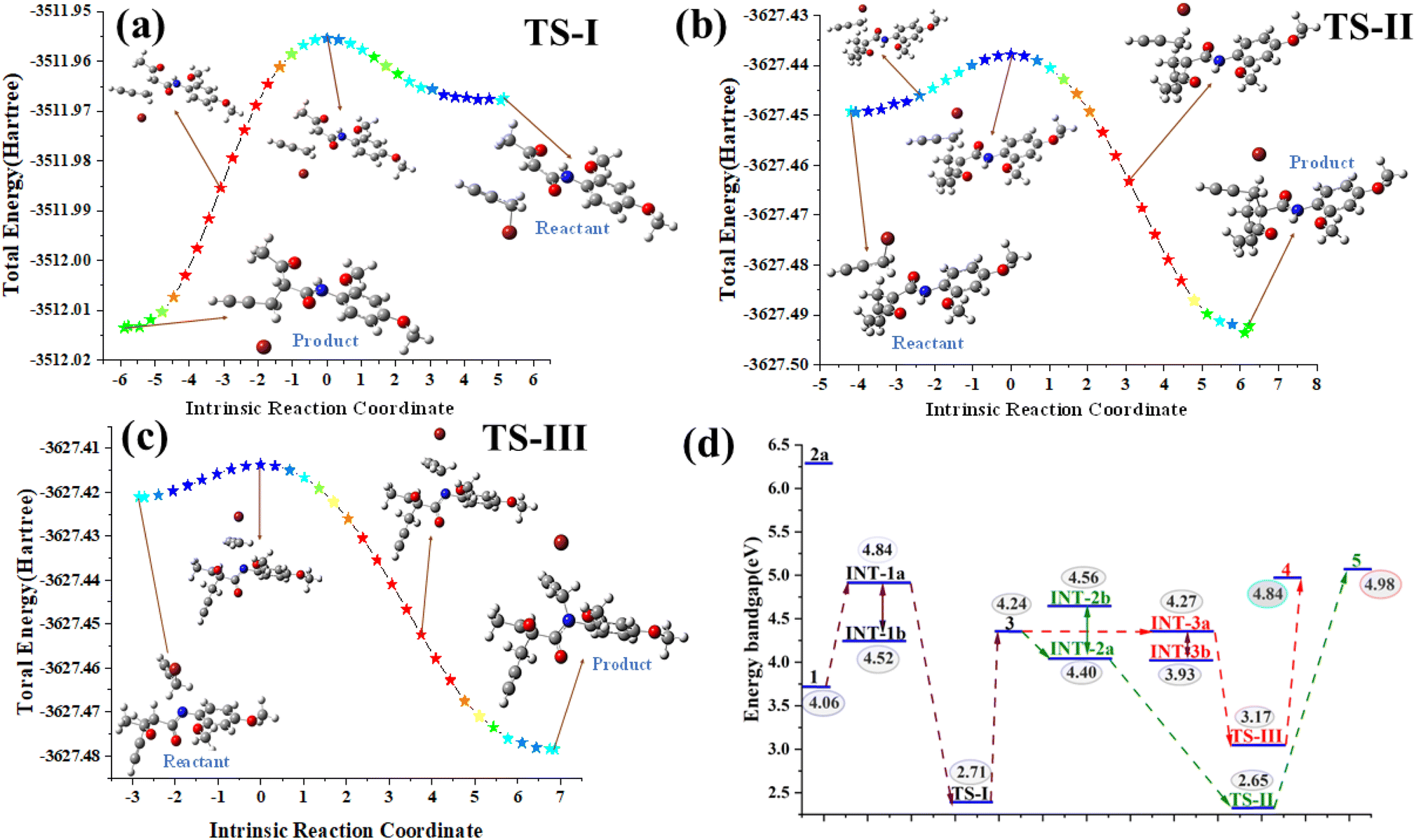 | ||
| Fig. 2 (a–c) Intrinsic reaction coordinate (IRC) pathways illustrating the progression from the reactant to product through various transition states (TS-I-TS-III); (d) band-gap analysis. | ||
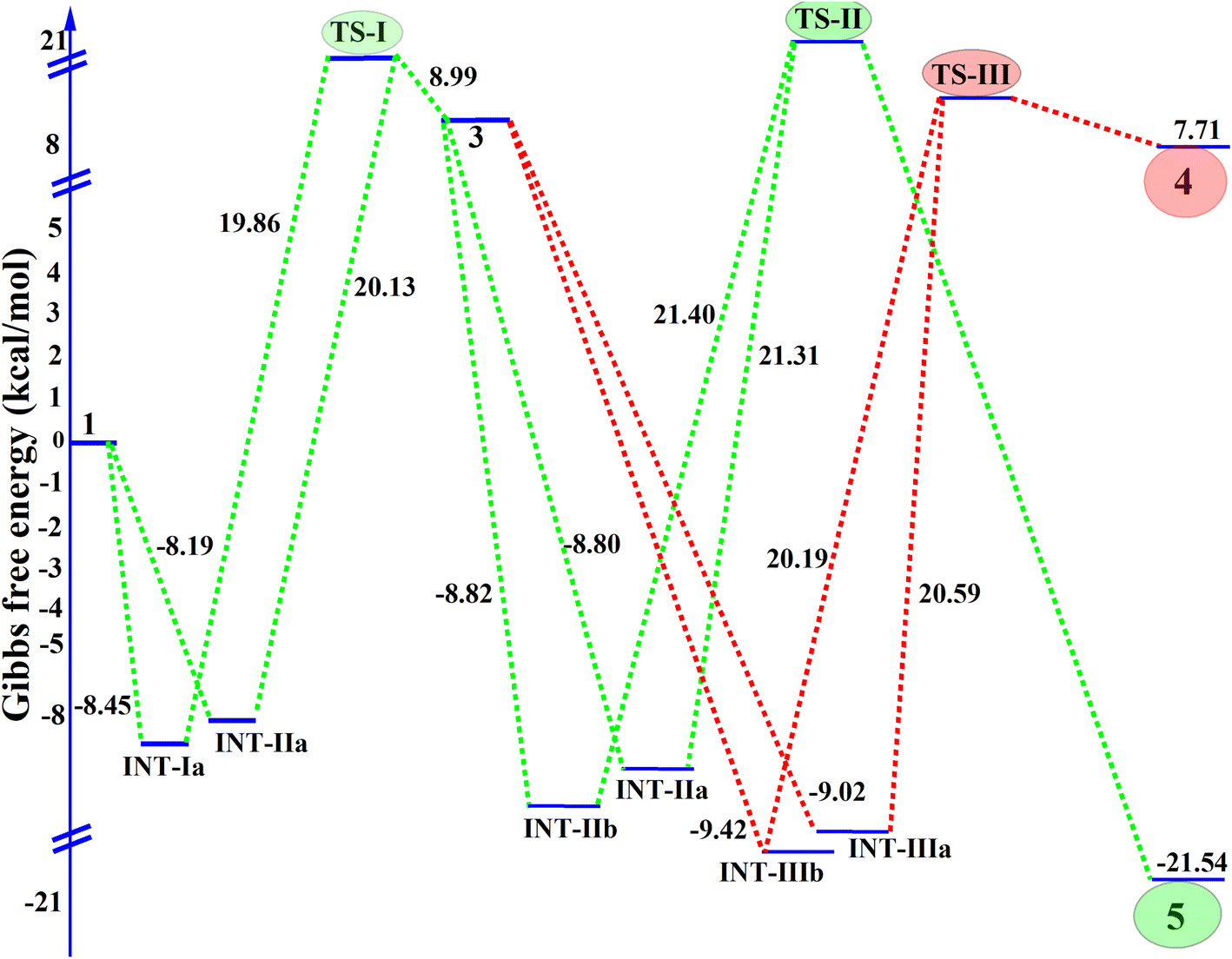 | ||
| Fig. 3 Gibbs free energy profile (kcal mol−1) of the active and inactive reaction molecules, along with the activation energy for the conversion of reactants into the final product. | ||
Overall, the computational energy profile strongly supports the reaction pathway 1 → INT-1a → TS-I → 3 → INT-Iia → TS-II → 5 as the dominant and most stable route. This mechanistic insight is further corroborated by experimental observations, including single-crystal X-ray data and electronic structure analyses, thereby providing a comprehensive understanding of the reaction's progression and the stability of the key species involved.37,38
3 Intra and intermolecular interaction studies
3.1 Hirshfeld surface analysis
The Hirshfeld surface analysis was carried using the Crystal Explorer 17.5 software with the same method and basis set that was used in DFT calculation. The Hirshfeld surface analysis of molecule 5f provides a comprehensive understanding of the intermolecular interactions governing its crystal packing (Fig. 4). The dnorm surface reveals significant red spots near the oxygen atoms and some carbon atoms, indicating regions of close intermolecular contacts, such as hydrogen bonding or van der Waals interactions. Additionally, a small red area is observed near the nitrogen atom, signifying its involvement in weaker interactions. The rest of the molecule displays white and blue regions, corresponding to neutral and less significant contacts, respectively. On the di surface, the nitrogen and carbon atoms exhibit prominent red spots, further corroborating their roles as interaction sites. Conversely, the de surface highlights red areas primarily around the oxygen atoms, emphasizing their accessibility and participation in intermolecular interaction. The shape index surface analysis identifies red and blue triangular patterns on the carbon atoms that are not part of the benzene ring, indicative of these regions' predisposition to participate in C–H⋯π interactions. This observation suggests that these carbons play a critical role in stabilizing the molecular assembly through π-cloud interactions. The curvedness surface analysis shows flat areas corresponding to the benzene ring in molecule 5f. These flat regions suggest a high propensity for π⋯π stacking interactions, which are essential for the molecule's crystal stability and packing. The Hirshfeld surface analysis thus elucidates the intricate balance of intermolecular forces in molecule 5f, providing valuable insights into its structural behavior. The analysis revealed a globularity value of 0.286 and an asphericity of 0.324, indicating that the voids possess a slightly irregular and non-spherical geometry. These results highlight the anisotropic nature of the pores, which may contribute to selective guest inclusion and functional versatility.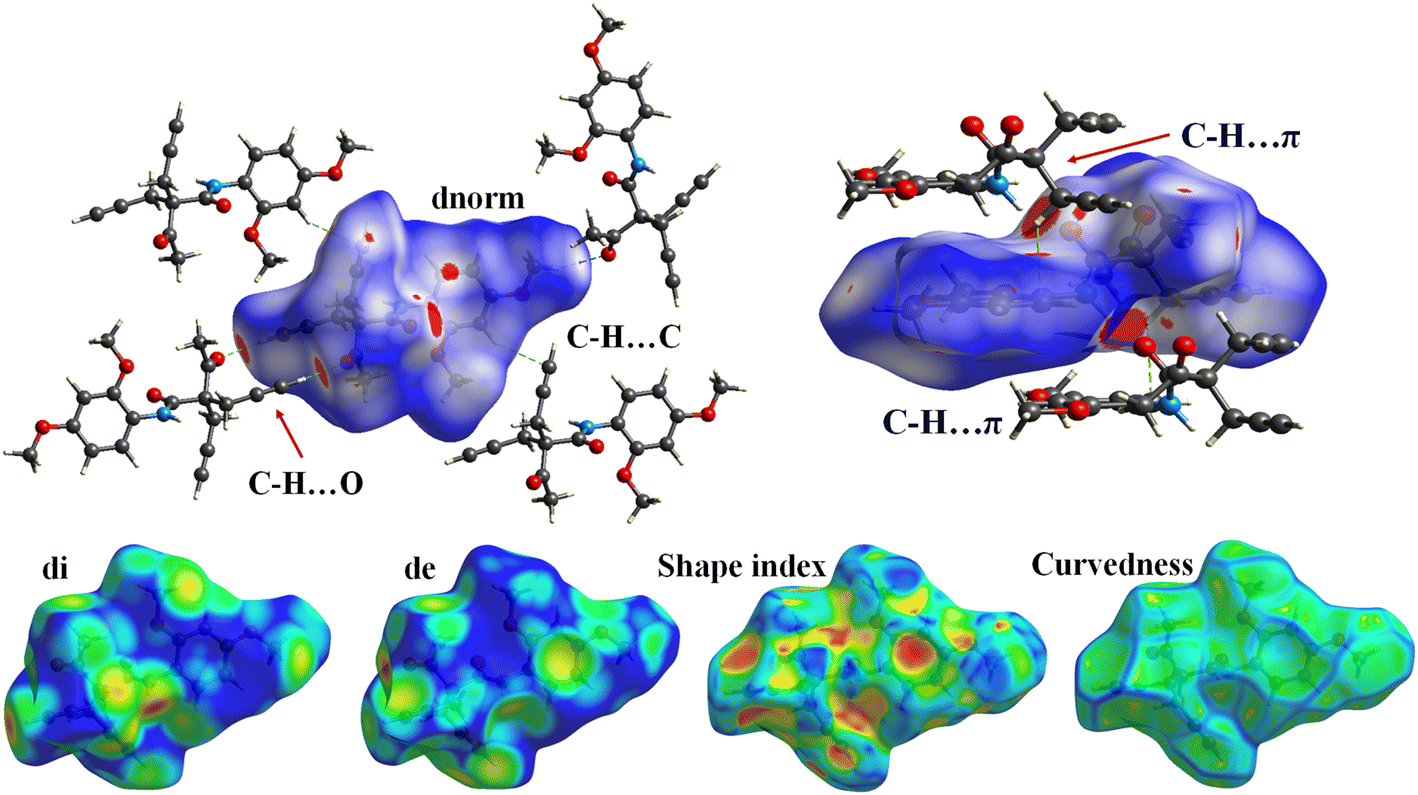 | ||
| Fig. 4 Hirshfeld surface analysis of the 5f molecule: dnorm, di, de, shape index and curvedness. | ||
In the 2D fingerprint plot analysis, the interaction contributions of various atomic pairs are represented in the form of distinctive wings. The plot reveals that hydrogen–hydrogen (H⋯H) interactions dominate the molecular interactions, contributing 48.3% to the total interactions. Hydrogen-carbon (H⋯C) interactions are also significant, accounting for 28.1%. The hydrogen-oxygen (H⋯O) interactions show a contribution of 22.1%, which is prominently displayed as a distinct wing in the plot. These H⋯O interactions are particularly important as they suggest potential hydrogen bonding or other intermolecular interactions that could influence the overall stability and properties of the system. Carbon–carbon (C⋯C) and hydrogen-nitrogen (H⋯N) interactions have minimal contributions, with 0.3% and 0.1%, respectively (Fig. 5).
 | ||
| Fig. 5 2D Fingerprint plot with the qualitative interaction % contribution in molecular packing. | ||
3.2 Energy framework
The intermolecular interaction energies were calculated using the B3LYP/6-311G(d,p) energy model available in Crystal Explorer 17.5, where a cluster of molecules is generated by applying crystallographic symmetry operations with respect to a selected central molecule within the radius of 3.8 Å by default. The total intermolecular energy Etot is the sum of electrostatic Eele, polarization Epol, dispersion Edis and exchange-repulsion Erep energies, respectively. The molecular packing, Coulomb energy, dispersion energy and total energy are shown in Fig. 6a–d, respectively. The thickness of the tubes correlates with the magnitude of the interaction energy. The tube thickness is large toward the −x, −y, −z symmetry molecular axes. Two symmetry-related molecules participate in interactions with the original molecule via C–H⋯O hydrogen bonds one involving the propyne moiety and the other from the acetyl group (Fig. 6d). | ||
| Fig. 6 Three energy frameworks of 5a molecular with molecular packing with visualization of interaction strength (a) molecular packing, (b) Coulomb energy, (c) dispersion energy and (d) total energy. | ||
Energy framework analysis reveals key intermolecular interactions contributing to the stability of the crystal structure. Table 2 highlights the interactions, symmetry operations, and corresponding energy components, including electrostatic (Eele), polarization (Epol), dispersion (Edis), repulsion (Erep), and total interaction energy (Etot). For the interaction at R = 9.57 Å, symmetry operation x, −y + 1/2, z + 1/2 describes a C–H⋯C interaction involving an aryl ring. This interaction exhibits a moderate dispersion energy (Edis = −4.13 kcal mol−1) and repulsion (Erep = 2.0 kcal mol−1), resulting in a total energy of Etot = −3.75 kcal mol−1 (Fig. 7b). The second interaction, also at symmetry operation x, −y + 1/2, z + 1/2 with R = 4.53 Å, involves a combination of C–H⋯O, N–H⋯O (amide), and C–H⋯π (prop-2-yne moiety) interactions. This interaction is characterized by strong electrostatic (Eele = −2.46 kcal mol−1) and dispersion (Edis = −17.11 kcal mol−1) contributions, resulting in the most significant total interaction energy (Etot) of −18.71 kcal mol−1. The strong electrostatic nature of this interaction, supported by substantial dispersion contributions, underscores its importance in stabilizing the crystal (Fig. 7c).
| N | Symop | R | E_ele | E_pol | E_dis | E_rep | E_tot | Interactions |
|---|---|---|---|---|---|---|---|---|
| 2 | x, −y + 1/2, z + 1/2 | 9.57 | −1.07 | −0.33 | −4.13 | 2.0 | −3.75 | C–H⋯C (aryl ring) |
| 2 | x, −y + 1/2, z + 1/2 | 4.53 | −10.32 | −3.60 | −17.1 | 15.82 | −18.71 | C–H⋯O, N–H⋯O (an amide) and C–H⋯π (prop-2-yne moiety) |
| 1 | −x, −y, −z | 12.39 | −6.81 | −1.33 | −3.03 | 6.78 | −6.64 | C–H⋯O (prop-2-yne moiety) C–H⋯O (acetyl moiety) |
 | ||
| Fig. 7 Coulombic energy analysis of various intermolecular interactions and their respective interaction strengths. (a) C–H⋯O interaction, where the C–H group (donor) from a symmetry-related molecule interacts with an oxygen atom from the original molecule (Edis = –17.1 kcal mol−1), along with an N–H⋯O (amide) interaction. (b and c) C–H⋯π interactions between the aryl ring of a symmetry-related molecule and a C–H group from the original molecule (Edis = –4.13 kcal mol−1). (d) Intermolecular C–H⋯O interaction between a C–H group from the acetyl moiety and the oxygen atom of a carbonyl group. | ||
Lastly, the interaction at R = 12.39 Å with symmetry operation −x, −y, −z features C–H⋯O interactions involving both the prop-2-yne and acetyl moieties. This interaction demonstrates a balance between electrostatic (Eele = −6.8 kcal mol−1), dispersion (Edis = −3.03 kcal mol−1), and repulsion energies (Erep = 6.78 kcal mol−1), leading to a total energy of Etot = −6.64 kcal mol−1. Among the analyzed interactions, the C–H⋯O, N–H⋯O (amide), and C–H⋯π (prop-2-yne moiety) interaction at R = 4.53 Å is the strongest, as evidenced by its highly negative total energy (Etot = −18.71 kcal mol−1). This highlights the dominant role of hydrogen bonding and π interactions in driving the stability of the crystal lattice (Fig. 7b).
4 Conclusion
We synthesized a series of pro-chiral 2-acetyl-N-subtituted-phenyl-2-(prop-2-yn-1-yl)pent-4-ynamide/-2-allyl-4-enamide analogues 5(a–n) using a K2CO3 promoted SN2 reaction in ACN at RT. The reaction of diverse acetoacetanilides 1(a–g) with propargyl/allyl bromide 2(a–b) efficiently achieved an excellent yield of up to 96%, a significant contribution to the field of asymmetric synthesis with fascinating implications for future applications. This work not only gives insight into the reaction pathways, but also lays the groundwork for the construction of structurally diverse libraries of pro-chiral compounds, using approaches such as SN2 reactions under optimal circumstances and DFT calculations. DFT calculations provided further insight into the reaction path of formation of 5(a–n), which undergoes SN2 to give a pro-chiral diyne/diene product instead of deprotonation from the amide proton (–NH–CO). The reaction mechanism, structural stability, and energy profile of INT & TS were thoroughly examined using the DFT platform, single crystal analysis, and energy band gap evaluation compilations with synthetic compounds. Slow evaporation yielded well-defined single crystals of product 5f, crystallized in the monoclinic system, stabilized by intermolecular interactions including hydrogen bonds and C–H⋯π interactions. DFT optimizations confirmed structural stability across intermediates, with notable favorability observed in the INT-1a pathway due to the Gibbs free energy differences. The Gibbs free energy profile illustrates the stability of the intermediate and unstable molecules during the transition to the final product, along with the activation energy for the conversion from reactants to the final product. Transition states TS-I and TS-II exhibited identical low activation energies, confirming an energetically favorable pathway toward 5 as the most stable product. The electrostatic potential analysis highlighted critical reactive sites, while band gap differences reinforced 5's thermodynamic stability over 4, corroborating crystallographic and theoretical findings. Consequently, desymmetrization of these pro-chiral diynes/dienes to 1,2,3-triazole will be carried out.
Data availability
The data supporting this article, including DFT analysis carried out using Gaussian 09,26 are included as part of the ESI.† Crystallographic data for compound 5f have been deposited at the CCDC under 2337812.Conflicts of interest
The authors declare no competing interests.Acknowledgements
All the authors are thankful to the Department of Chemistry, Sardar Patel University, Vallabh Vidyanagar, for providing the necessary research facilities and computational study software Gaussian 09. DPV and MPP are thankful to the Knowledge Consortium of Gujarat for the SHODH fellowship (student reference No. 2021016429 and 2021016434), CDP is thankful to the National fellowship for the scheduled Tribe (Award No. 202223-NFST-GUJ-00003). SGP is grateful to the UGC-New Delhi for UGC-JRF (NTA ref. No. 201610157514; dated: 01.04.2021). SSB is thankful to the CSIR-NET fellowship (reference No. Nov/06/2020(i)EU-V). SB was funded by the United Kingdom Research and Innovation Future Leader Fellowship MR/W008114/1.References
- T. Imamoto, Synthesis and applications of high-performance P-chiral phosphine ligands, Proc. Jpn. Acad. Ser. B Phys. Biol. Sci., 2021, 97(9), 520–542, DOI:10.2183/pjab.97.026.
- J. Meng, Y.-L. Zhao, C.-Q. Ren, Y. Li, Z. Li and Q. Liu, Highly Efficient Access to Bi- and Tricyclic Ketals through Gold-Catalyzed Tandem Reactions of 4-Acyl-1,6-diynes, Chem.–Eur. J., 2009, 15(8), 1830–1834, DOI:10.1002/chem.200802304.
- R. Chauvin and C. Lepetit, Theoretical Studies on Acetylenic Scaffolds, Acetylene Chem., 2004, 1–50 Search PubMed.
- B. M. Trost and A. H. Weiss, Catalysis, The enantioselective addition of alkyne nucleophiles to carbonyl groups, Adv. Synth. Catal., 2009, 351, 7–8, DOI:10.1002/adsc.200800776.
- K. Nakahara and H. Fujioka, Diastereoselective Desymmetrization of Symmetric Dienes and its Synthetic Application, Symmetry, 2010, 437–454, DOI:10.3390/sym2020437.
- M. De Paolis, I. Chataigner and J. Maddaluno, Recent Advances in Stereoselective Synthesis of 1,3-Dienes. in Stereoselective Alkene Synthesis, Wang, J., ed. Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 87–146, DOI:10.1007/128_2012_320.
- S. Musa, Y. Peretz and G. Dinnar, Advances in Chiral Pincer Complexes: Insights and Applications in Catalytic Asymmetric Reactions, Int. J. Mol. Sci., 2024, 25(19), 10344, DOI:10.3390/ijms251910344.
- L. Dai, Q. Zhu, J. Zeng, Y. Liu, G. Zhong, X. Han and X. Zeng, Asymmetric synthesis of chiral imidazolidines by merging copper and visible light-induced photoredox catalysis, Org. Chem. Front., 2022, 9(11), 2994–2999, 10.1039/D2QO00303A.
- Y. Yin, X. Zhao, B. Qiao and Z. Jiang, Cooperative photoredox and chiral hydrogen-bonding catalysis, Org. Chem. Front., 2020, 7(10), 1283–1296, 10.1039/D0QO00276C.
- S. Majee, K. Anjali, S. Yadav and D. Ray, Synthesis and Functionalization of Natural Products with Light-Driven Reactions, in Emerging Trends in Photoredox Synthetic Transformation, Sen, M. and Ray, D., ed. Springer Nature Singapore, Singapore, 2025, pp. 217–247. DOI:10.1007/978-981-97-8205-5_8.
- P. Nakliang, S. Yoon and S. Choi, Emerging computational approaches for the study of regio- and stereoselectivity in organic synthesis, Org. Chem. Front., 2021, 8(18), 5165–5181, 10.1039/D1QO00531F.
- W. Chen, Z. Qu, S. Qu and A. Duan, Chiral phosphoric acid catalyzed intermolecular [4 + 2] cycloaddition for the synthesis of chiral azomethine imines: mechanism and stereochemical model, Org. Chem. Front., 2025, 12(8), 2694–2703, 10.1039/D5QO00203F.
- Y. Miyake, S. Uemura and Y. Nishibayashi, Catalytic Propargylic Substitution Reactions, ChemCatChem, 2009, 1(3), 342–356, DOI:10.1002/cctc.200900214.
- R.-Y. Zhu, L. Chen, X.-S. Hu, F. Zhou and J. Zhou, Enantioselective synthesis of P-chiral tertiary phosphine oxides with an ethynyl group via Cu(i)-catalyzed azide–alkyne cycloaddition, Chem. Sci., 2020, 11(1), 97–106, 10.1039/C9SC04938J.
- D. P. Vala, R. M. Vala and H. M. Patel, Versatile Synthetic Platform for 1,2,3-Triazole Chemistry, ACS Omega, 2022, 7(42), 36945–36987, DOI:10.1021/acsomega.2c04883.
- M. Capurso, R. Gette, G. Radivoy and V. Dorn, The SN2 Reaction: A Theoretical-Computational Analysis of a Simple and Very Interesting Mechanism, Proceedings, 2019, 41(1), 81, DOI:10.3390/ecsoc-23-06514.
- S. M. Bachrach, Computational organic chemistry, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2012, 108, 334–352, 10.1039/C2OC90002E.
- G.-J. Cheng, X. Zhang, L. W. Chung, L. Xu and Y.-D. Wu, Computational Organic Chemistry: Bridging Theory and Experiment in Establishing the Mechanisms of Chemical Reactions, J. Am. Chem. Soc., 2015, 137(5), 1706–1725, DOI:10.1021/ja5112749.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry Part A: Structure and Mechanisms. in Advanced Organic Chemistry, Springer, 5 ed., New York, NY, 2007 Search PubMed.
- D. P. Vala, A. Dunne Miller, A. Atmasidha, M. P. Parmar, C. D. Patel, D. B. Upadhyay, S. S. Bhalodiya, A. González-Bakker, A. N. Khan, J. Nogales, J. M. Padrón, S. Banerjee and H. M. Patel, Click-chemistry mediated synthesis of OTBN-1,2,3-Triazole derivatives exhibiting STK33 inhibition with diverse anti-cancer activities, Bioorg. Chem., 2024, 149, 107485, DOI:10.1016/j.bioorg.2024.107485.
- S. G. Patel, I. Sharma, M. P. Parmar, J. Nogales, C. D. Patel, S. S. Bhalodiya, D. P. Vala, N. V. Shah, S. Banerjee and H. M. Patel, Alkoxy-functionalised dihydropyrimido[4,5-b]quinolinones enabling anti-proliferative and anti-invasive agents, Chem. Commun., 2024, 60(55), 7093–7096, 10.1039/D4CC01219D.
- M. P. Parmar, D. P. Vala, S. S. Bhalodiya, D. B. Upadhyay, C. D. Patel, S. G. Patel, S. R. Gandholi, A. H. Shaik, A. D. Miller, J. Nogales, S. Banerjee, J. M. Padrón, N. Amri, N. K. Kandukuri and H. M. Patel, A green bio-organic catalyst (taurine) promoted one-pot synthesis of (R/S)-2-thioxo-3,4-dihydropyrimidine(TDHPM)-5-carboxanilides: chiral investigations using circular dichroism and validation by computational approaches, RSC Adv., 2024, 14(13), 9300–9313, 10.1039/D4RA01391C.
- S. G. Patel, A. González-Bakker, R. M. Vala, P. J. Patel, A. Puerta, A. Malik, R. K. Sharma, J. M. Padrón and H. M. Patel, Microwave-assisted multicomponent synthesis of antiproliferative 2,4-dimethoxy-tetrahydropyrimido[4,5-b]quinolin-6(7H)-ones, RSC Adv., 2022, 12(47), 30404–30415, 10.1039/D2RA04669E.
- S. G. Patel, P. J. Patel, D. B. Upadhyay, A. Puerta, A. Malik, N. K. Kandukuri, R. K. Sharma, J. M. Padrón and H. M. Patel, Insights into microwave assisted synthesis of spiro-chromeno[2,3-d]pyrimidines using PEG-OSO3H catalyst: DFT study and their antiproliferative activity, J. Mol. Struct., 2023, 1292, 136174, DOI:10.1016/j.molstruc.2023.136174.
- P. J. Patel, D. M. Patel, R. M. Vala, S. G. Patel, D. B. Upadhyay, Y. Pannerselvam and H. M. Patel, Catalyst-Free, Room-Temperature Accessible Regioselective Synthesis of Spiroquinolines and Their Antioxidant Study, ACS Omega, 2023, 8(1), 444–456, DOI:10.1021/acsomega.2c05020.
- M. Caricato, M. J. Frisch, J. Hiscocks and M. J. Frisch, Gaussian 09: IOps Reference, Gaussian Wallingford, CT, USA, 2009 Search PubMed.
- B. N. Socha, S. B. Pandya, U. H. Patel and R. H. Patel, Structural investigation, quantum chemical calculation, energy framework analysis and MIC studies of silver and cobalt complexes of 4-amino-N-(4, 6-dimethyl-2 pyrimidinyl) benzenesulfonamide in presence of secondary ligand, Inorg. Chem. Commun., 2023, 154, 110936, DOI:10.1016/j.inoche.2023.110936.
- B. N. Socha, S. B. Pandya, U. H. Patel, R. H. Patel, B. S. Bhatt, P. Thakor and A. B. Thakkar, Silver-Based 3D-Supramolecular Metal–Organic Framework: Crystallographic Evaluation, Antibiotic, Anticancer, and Visible-Light-Driven Photocatalytic Activity, J. Inorg. Organomet. Polym., 2024, 34(5), 2042–2063, DOI:10.1007/s10904-023-02946-9.
- B. N. Socha, S. B. Pandya, U. H. Patel, R. H. Patel, B. S. Bhatt, S. Bhakhar, N. Vekariya, J. Valand, P. Thakor and A. B. Thakkar, 1-D MOF [Ag2(C10H10N3O3S)2(C4H8N)2]n: photocatalytic treatment, crystallographic evaluation, ADMET parameters, CT-DNA and anticancer activity, J. Biomol. Struct. Dyn., 2024, 42(13), 6925–6940, DOI:10.1080/07391102.2023.2236732.
- B. N. Socha, M. K. Patel, S. B. Pandya, U. H. Patel, R. H. Patel, B. Bhatt, S. Bhakhar, N. Vekariya and J. Valand, Qualitative and quantitative contributions of intermolecular interactions, quantum chemical calculation, biological activity and visible-light driven photo catalysts studies of nickel complex of 4-amino-N-(5-methyl-1,3,4-thiadiazol-2-yl)benzenesulfonamide, Inorg. Chem. Commun., 2023, 158, 111540, DOI:10.1016/j.inoche.2023.111540.
- G. Anand, M. Sivasubramanian, I. Manimehan, A. Ruby, R. Abinayashri and R. K. Asmitha, Synthesis, spectroscopic elucidation (FT-IR, FT-Raman, UV-vis), quantum chemical computation (PES, FMO, HOMO–LUMO, MEP, NLO, Hirshfeld) and molecular docking studies on 2-thiophenecarboxamide crystal, J. Mol. Struct., 2023, 1286, 135586, DOI:10.1016/j.molstruc.2023.135586.
- M. Aygün, D. Barut Celepci, Ö. Akgül and V. Pabuccuoglu, Single-crystal X-ray structure, theoretical (Hirshfeld, electronic properties, NBO, NLO, RDG), and molecular docking studies of three phthalimidoethanesulfonamide derivatives, J. Mol. Struct., 2024, 1317, 139094, DOI:10.1016/j.molstruc.2024.139094.
- S. B. Pandya, B. N. Socha, R. P. Dubey, U. H. Patel, R. H. Patel, B. S. Bhatt, P. Thakor, S. Bhakhar, N. Vekariya and J. Valand, Visible light-driven photocatalysts, quantum chemical calculations, ADMET-SAR parameters, and DNA binding studies of nickel complex of sulfadiazine, Sci. Rep., 2023, 13(1), 15275, DOI:10.1038/s41598-023-42668-z.
- N. Ayoub, A. Upadhyay, A. Tête, N. Pietrancosta, H. Munier-Lehmann and T. P. O'Sullivan, Synthesis, evaluation and mechanistic insights of novel IMPDH inhibitors targeting ESKAPEE bacteria, Eur. J. Med. Chem., 2024, 280, 116920, DOI:10.1016/j.ejmech.2024.116920.
- M. Bandyopadhyay, S. Bhadra, S. Pathak, A. M. Menon, D. Chopra, S. Patra, J. Escorihuela, S. De, D. Ganguly, S. Bhadra and M. K. Bera, An Atom-Economic Method for 1,2,3-Triazole Derivatives via Oxidative [3 + 2] Cycloaddition Harnessing the Power of Electrochemical Oxidation and Click Chemistry, J. Org. Chem., 2023, 88(22), 15772–15782, DOI:10.1021/acs.joc.3c01836.
- Y. Li, L. Geng, M. Zhang and Z. Zhang, Mechanisms and origins of stereoselectivity involved in NHC-catalyzed [3 + 3] Annulation of 2-bromoenals and β-ketothioamides: A DFT study, Mol. Catal., 2023, 542, 113135, DOI:10.1016/j.mcat.2023.113135.
- R. Li, T. Gao, P. Zhang and A. Li, Non-IRC Mechanism of Bimolecular Reactions with Submerged Barriers: A Case Study of Si+ + H2O Reaction, J. Phys. Chem. A, 2024, 128(12), 2376–2382, DOI:10.1021/acs.jpca.4c00787.
- S. Kuzhanthaivelan, S. Sunitha and B. Rajakumar, Computational investigation of thermochemistry and kinetics of the reaction between n-butyl peroxy and hydroperoxyl radicals, Chem. Phys. Lett., 2023, 810, 140191, DOI:10.1016/j.cplett.2022.140191.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2337812. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra02166a |
| This journal is © The Royal Society of Chemistry 2025 |