DOI:
10.1039/D5RA02290H
(Paper)
RSC Adv., 2025,
15, 15724-15728
Mechanism of EnT-mediated amino-sulfonylation of alkenes with N-sulfonyl ketimine: a DFT investigation†
Received
2nd April 2025
, Accepted 6th May 2025
First published on 13th May 2025
Abstract
The sulfonylamination of alkenes using N-sulfonyl ketimines has previously been established as an efficient and straightforward strategy for synthesizing β-amino sulfone derivatives. In this study, density functional theory (DFT) calculations were performed to elucidate the free energy profiles of the plausible reaction pathways. Additionally, the local electrophilic index, percent buried volume around the spin-centered atom (% Vbur), and maximum spin density were analyzed to provide insights into the stability differences between the generated iminyl and tosyl radicals. This work aims to enhance the understanding of the reaction mechanism underlying this alkene amino-sulfonylation process and to guide the design of novel bifunctional reagents.
Introduction
Difunctionalization of alkenes offers an atom-economical approach to assemble two vicinal new bonds in a single step.1–3 Significant efforts have been devoted to exploring alkene difunctionalization strategies over the years.4–7 Among these, diphenylmethanone oxime esters have emerged as an important class of bifunctional reagents due to their low N–O bond dissociation energy, which facilitates homolysis under excited-state conditions.8–10 Recently, Du's,11 Zhang's,12 and our groups13 independently achieved energy-transfer (EnT)-mediated vicinal amino-sulfonylation of alkenes using bifunctional sulfonamides, enabling the synthesis of valuable β-amino sulfone derivatives. This methodology is featured with broad substrate scope, mild reaction conditions, and excellent regioselectivity (Fig. 1). Mechanistic studies indicate that the reaction proceeds through an energy-transfer-mediated process, involving homolysis of the weak N–S bond in the sulfonamide reagent to simultaneously generate tosyl and iminyl radicals. The transient tosyl radical selectively adds to the terminal position of the alkene substrate, followed by coupling with the persistent iminyl radical to yield the observed β-imino sulfone products. The success of this methodology hinges on the reactivity difference between the two generated radicals: the tosyl radical and the iminyl radical. Over the past few decades, density functional theory (DFT) calculations have become an indispensable tool in chemistry, aiding in the understanding of experimental observations and guiding the design of new reactions. To gain deeper insights into the reaction mechanism, particularly the high regioselectivity of this EnT-mediated amino-sulfonylation of alkenes, DFT investigations were conducted in this study.
 |
| | Fig. 1 EnT-mediated amino-sulfonylation of alkenes. | |
Computational methods
All calculations were conducted using Gaussian16 software package.14 Optimization of all stationary points were carried out at M062X/def2-SVP theoretical level.15–17 Frequency calculations were performed at the same level to verify the stationary points are minima (0 imaginary frequency) or saddle points (only 1 imaginary frequency). Single point energy calculations were carried out with Truhlar's M06-2X functional with def2-TZVPP basis set for all atoms. Solvation effects of dichloromethane for all calculations were considered using Truhlar's SMD solvent model.18 Computed structures were illustrated by CYLview software19 or Gaussview6.0.20 The minimum energy crossing points (MECPs) were located with the program developed by J. N. Harvey.21 The local electrophilicity index values (calculated at B3LYP/6-31G(d) theoretical level) and AIM analysis were performed with the assistance of Multiwfn software.22–24 In the buried volume calculations, the radical center was defined as atom with the maximum fractional spin, calculated by SambVca 2.1 platform.25
Results & discussion
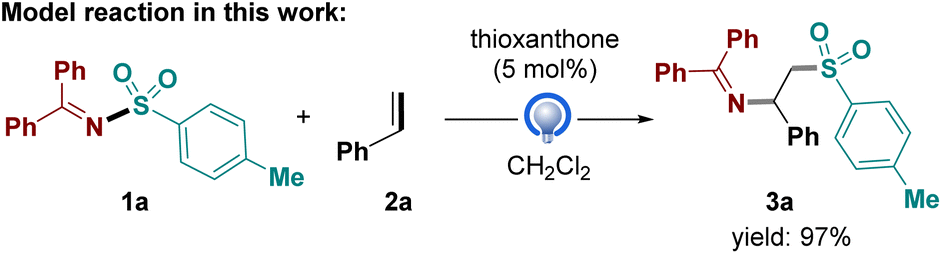
To elucidate the reaction mechanism and the origin of regioselectivity, the difunctionalization of styrene (2a) with Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–Ts (1a) was selected as the model reaction (Fig. 2). It should be noted CH2Cl2 (DCM) was chosen as the solvent in the optimal reaction conditions in our work,13 whereas EtOAc was used in Du's11 and Zhang's12 studies, yielding similar results. All calculations in this study were conducted in DCM, taking into account the comparable solvent parameters in the implicit solvent model applied. Initially, the free-energy profiles of the plausible reaction pathways were calculated and analyzed. Subsequently, the properties of the generated tosyl and iminyl radicals, particularly their stabilities, were compared to provide insights into the observed excellent regioselectivity.
N–Ts (1a) was selected as the model reaction (Fig. 2). It should be noted CH2Cl2 (DCM) was chosen as the solvent in the optimal reaction conditions in our work,13 whereas EtOAc was used in Du's11 and Zhang's12 studies, yielding similar results. All calculations in this study were conducted in DCM, taking into account the comparable solvent parameters in the implicit solvent model applied. Initially, the free-energy profiles of the plausible reaction pathways were calculated and analyzed. Subsequently, the properties of the generated tosyl and iminyl radicals, particularly their stabilities, were compared to provide insights into the observed excellent regioselectivity.
 |
| | Fig. 2 Model reaction investigated in this work. | |
Free energy profiles of the model reaction
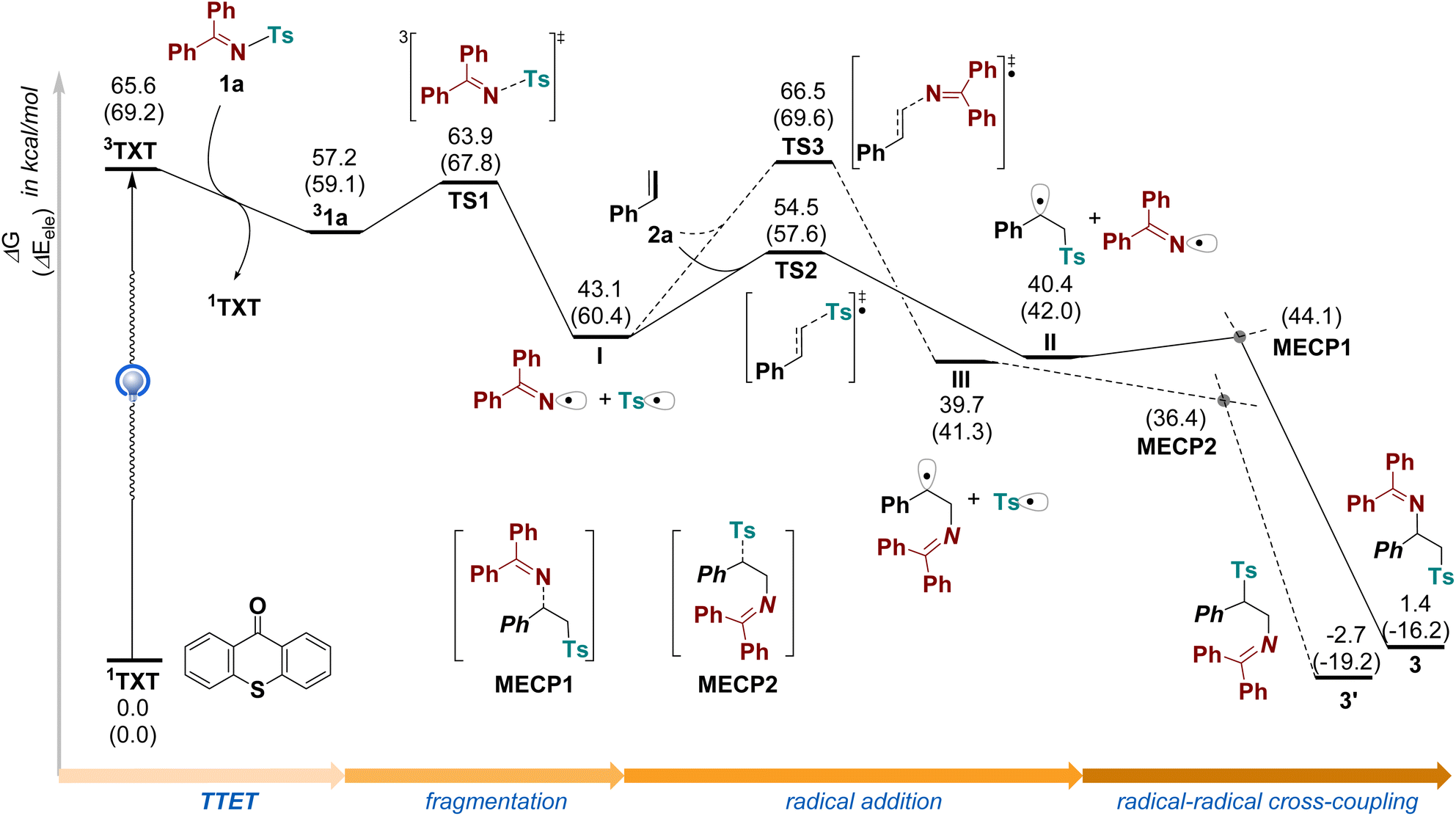
In light of the mechanistic studies in previous works,11–13 computational studies were carried out at M06-2X/def2TZVPP-SMD(DCM)//M06-2X/def2SVP-SMD (DCM) theoretical level to probe the mechanistic profile of the titled reaction between Ph2CN–Ts 1a and styrene 2a (Fig. 3). The reaction is initiated with the excitation of photocatalyst thioxanthone (TXT). After excitation of the initial bifunctional reagent 1a by the excited TXT via triplet–triplet energy transfer, the homolytic fission of the N–S bond in 1a takes place facilely, producing a transient sulfonyl radical and a persistent benzophenone iminyl radical. The homolysis of3 1a to radical pairs I is exergonic by −14.1 kcal mol−1, with a free energy barrier via TS1 only 6.7 kcal mol−1. A radical addition of the sulfonyl radical to the terminal carbon of styrene 2a occurs via TS2, delivering the addition intermediate II. The free energy barrier of this radical addition step is as high as 11.4 kcal mol−1, a little higher than the first step, suggesting it is rate-determining during the formation of the desired product 3. Furthermore, the relative Gibbs free energy of II is close to that of radical pairs I, suggesting this step is reversible and the addition carbon-centered radical is as stable as the tosyl radical. Finally, a radical–radical cross coupling between addition intermediate II and iminyl radical affords the final product 3 via the minimum energy crossing point (MECP1). This step via MECP1 is highly exergonic by −39.0 kcal mol−1, driving the reaction forward. In contrast, the benzophenone iminyl radical added to styrene would generate another addition intermediate III via TS3, followed by radical–radical cross coupling with sulfonyl radical, to produce the side product 3′. The free energy barrier via TS3 is as high as 23.4 kcal mol−1, much higher than that via TS2, suggesting the side product via this pathway is unlikely to be observed. In fact, the free energy difference between TS2 and TS3 is as large as 12.0 kcal mol−1, suggesting the remarkable activity difference between sulfonyl radical and iminyl radical. As the radical addition step is the selectivity-determining step for this protocol, the formation of isomer 3′ could be kinetically suppressed, which agrees well with the excellent regioselectivity observed in experiments. Remarkably, the free energy of product 3 is a little higher than the starting material by 1.4 kcal mol−1, indicating a thermodynamically unfavorable process. Thus, the success of this photochemical transformation also demonstrates the unparalleled advantage of photo-induced reaction mode over regular thermal mode, offering an alternative pathway to access thermodynamically unfavorable transformations under mild conditions.
 |
| | Fig. 3 Free energy profile for the reaction of N-sulfonyl ketimine 1a and styrene 2a calculated at the M06-2X/def2TZVPP-SMD(DCM)//M06-2X/def2SVP-SMD(DCM) level of theory. All energies are calculated relative to separate starting materials, shown in kcal mol−1. | |
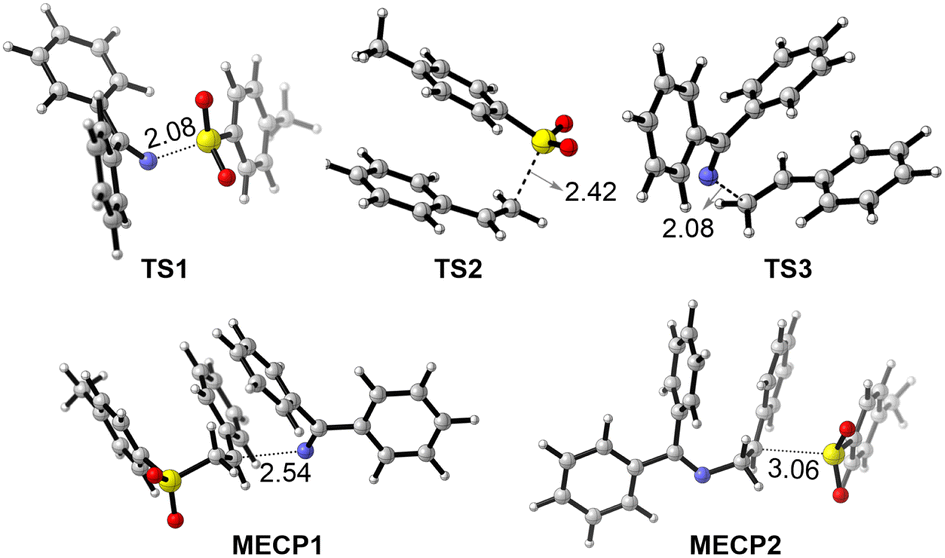
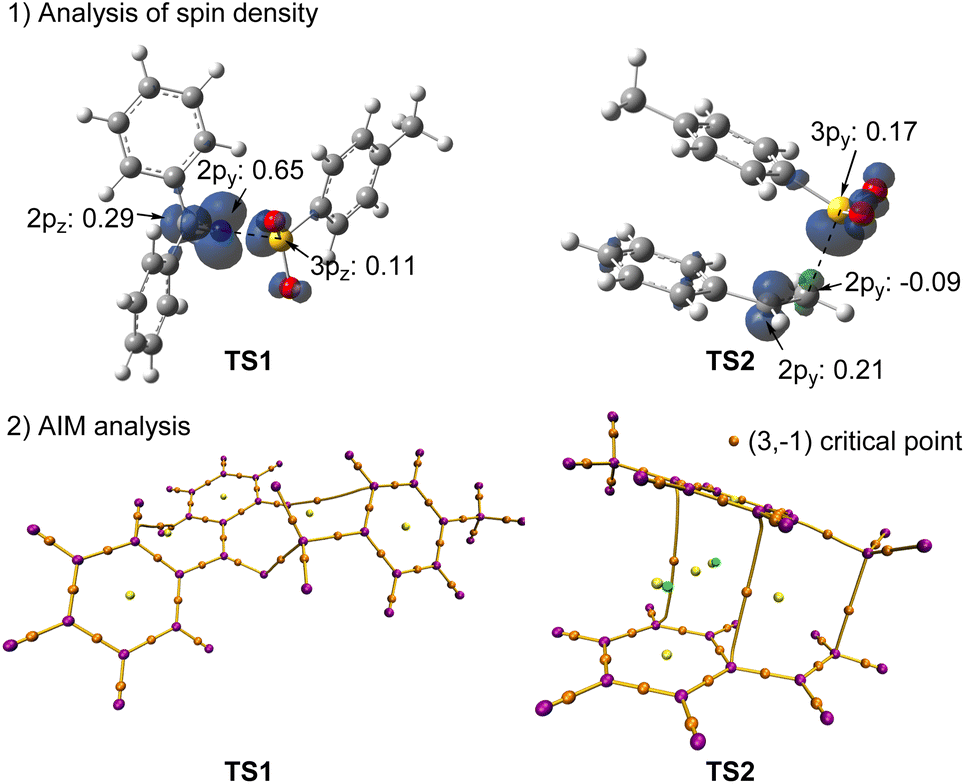
To better understand the reaction mechanism, the 3D structures of selected species in the potential energy surfaces were then scrutinized (Fig. 4). In TS1, the length of the breaking N–S bond is 2.08 Å, a little longer than the corresponding bond length in,3 1a 1.73 Å, suggesting an early transition state. This observation is also consistent with the exergonic nature of the process from3 1a to I. In TS2, the two phenyl rings in parallel displaced configuration forms favorable π–π stacking interaction, with a forming C–S bond at 2.42 Å distance. In TS3, the forming C–N bond is 2.08 Å in length, that is 0.65 Å longer than the C–N bond in product 3a. In the radical–radical crossing points MECP1 and MECP2, the distances of the forming bonds are 2.54 and 3.06 Å, respectively, suggesting that there is only weak interaction between the approaching fragments. These structural details provide valuable insights into the geometric changes occurring during the reaction. Additionally, spin density analyses were performed to examine the key transition states, TS1 and TS2 (Fig. 5). In TS1, the spin density is predominantly located on the 2py natural atomic orbital of nitrogen and the 2pz orbital of the imine carbon atom. In contrast, in TS2, the spins on both alkene atoms are oriented in opposite directions, suggesting that the tosyl spin radical interacts with the LUMO orbital of the alkene to form the C–S bond. Furthermore, AIM analysis was conducted to gain deeper insights into both transition states. In TS1, a weak interaction is observed between the C–H bond of the iminyl group and the phenyl ring of the tosyl radical, whereas in TS2, π–π stacking interactions are observed between the two phenyl rings.
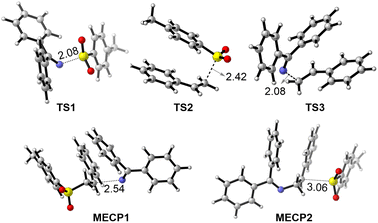 |
| | Fig. 4 3D-structures of transition states and minimum-energy crossing points. Distances were shown in Angstrom. | |
 |
| | Fig. 5 Spin density and atoms in molecules (AIM) analysis of transition states TS1 and TS2. | |
Comparing the properties of the tosyl and iminyl radicals
Interestingly, the iminyl radical is often regarded as a persistent radical, allowing it to remain stable for a longer duration and exhibit lower reactivity compared to transient radical species. To better understand the stability differences between the iminyl and tosyl radicals, key properties such as the local electrophilicity index, percent buried volume around the spin-centered atom (% Vbur), and spin density at the radical center were calculated. Recently, Paton and co-workers demonstrated that the percent buried volume around the radical center and the maximum spin density can serve as effective descriptors to quantify radical stability, using the following metric (eqn (1)).26| | |
Radical stability score = Vbur + 50 × (1 − max. spin)
| (1) |
For Ph2CN˙ radical, the radical stability score is 43, while for Ts˙ radical, this value is 81.6. This suggests the Ts˙ radical is more stable than Ph2CN˙ radical, which appears contradictory to the experimental observation. Therefore, the radical stability score alone cannot be used to explain the stability differences between these radicals in this study. To be noted, the buried volume around the spin-centered atom of Ph2CN˙ radical (% Vbur = 44.5%) is smaller than that of the Ts˙ radical (% Vbur = 49.6%) and the maximum spin density of the former (1.03) is higher than that of the later (0.36). These findings suggest that the Ph2CN˙ radical is more reactive than the Ts˙ radical, which also seems to be inconsistent with established knowledge and experimental results (Fig. 6). Given that radical addition to styrene can be considered an electrophilic process, the local electrophilicity index (ω(r)) of the radical centers was calculated. It was found that the Ts˙ radical (ω(r) = 0.46 eV) exhibits a higher local electrophilicity index compared to the Ph2CN˙ radical (ω(r) = 0.37 eV), indicating that the Ts˙ radical is more reactive. This result aligns with the experimentally observed regioselectivity, providing a more consistent explanation for the reactivity differences.
 |
| | Fig. 6 Stability analysis of iminyl and tosyl radicals: local electrophilicity index of the radical center (ω(r)), percent buried volume around spin-centered atom (% Vbur), and spin density at radical center were shown. | |
Further, the spin orbitals of the iminyl and tosyl radicals were analyzed to better understand their reactivities. As shown in Fig. 7, the spin orbital on the nitrogen atom is nearly perpendicular to the π electron orbital of the iminyl group. Additionally, the presence of adjacent phenyl groups partially buries the spin orbital on the nitrogen atom, limiting its accessibility. In contrast, the spin orbital on the sulfur atom in the Ts˙ radical is nearly perpendicular to the phenyl ring, making it more exposed and readily accessible to nucleophilic partners. These structural and electronic features provide further insight into the higher persistency of the iminyl radical compared to the transient tosyl radical, consistent with their observed reactivity differences.
 |
| | Fig. 7 Top and side view of the spin orbitals of iminyl and tosyl radicals. Mulliken spin density values larger than 0.01 were given near the corresponding atoms. | |
Conclusions
EnT-mediated 1,2-sulfonylamination of alkenes with bifunctional reagent sulfonylamide has previously been demonstrated to be a straightforward and atom-economic way to access β-amino sulfone derivatives. To elucidate the reaction mechanism and the stability differences between the key iminyl and tosyl radicals, density functional theory (DFT) calculations were performed. The reaction initiates with the photoexcitation of the photocatalyst thioxanthone, followed by triplet–triplet energy transfer to generate the excited state of the bifunctional reagent (1a*). Homolysis of the weak N–S bond in 1a* produces a pair of radicals: the iminyl radical and the tosyl radical. The stability and reactivity differences between these radicals govern their subsequent interactions with the alkene substrate, leading to the formation of the product with excellent regioselectivity. The local electrophilicity index (ω(r)) was identified as a more effective descriptor for explaining the reactivity differences between the two radicals, compared to the percent buried volume around the spin-centered atom (% Vbur) and the maximum spin density. Additionally, the shapes of the spin orbitals in both radicals provide further insights into their reactivity differences. This study not only enhances the understanding of experimental observations in EnT-mediated sulfonylamination of alkenes but also offers valuable guidance for the design of new bifunctional reagents.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Conceptualization, P. C. and K. C.; methodology, P. C.; software, P. C.; validation, P. C., X. J. and K. C.; formal analysis, P. C.; investigation, P. C.; resources, K. C.; data curation, P. C.; writing—original draft preparation, P. C. and X. J.; writing—review and editing, K. C.; visualization, K. C.; supervision, X. J. and K. C.; project administration, P. C.; funding acquisition, X. J. and K. C. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The High Performance Computing Center of Central South University was appreciated for computational source support.
Notes and references
- H.-M. Huang, P. Bellotti, J. Ma, T. Dalton and F. Glorius, Bifunctional reagents in organic synthesis, Nat. Rev. Chem., 2021, 5, 301–321 CrossRef CAS.
- Q. Cheng, Z. Bai, S. Tewari and T. Ritter, Bifunctional sulfilimines enable synthesis of multiple N-heterocycles from alkenes, Nat. Chem., 2022, 14, 898–904 CrossRef CAS.
- G. Tan, F. Paulus, Á. Rentería-Gómez, R. F. Lalisse, C. G. Daniliuc, O. Gutierrez and F. Glorius, Highly selective radical relay 1,4-Oxyimination of two electronically differentiated olefins, J. Am. Chem. Soc., 2022, 144, 21664–21673 CrossRef CAS PubMed.
- G. R. Mathi, Y. Jeong, Y. Moon and S. Hong, Photochemical carbopyridylation of alkenes using N-alkenoxypyridinium salts as bifunctional reagents, Angew. Chem., Int. Ed., 2019, 59, 2049–2054 CrossRef.
- J.-H. Ye, P. Bellotti, T. O. Paulisch, C. G. Daniliuc and F. Glorius, Visible-light-induced cycloaddition of α-ketoacylsilanes with imines: facile access to β-lactams, Angew. Chem., Int. Ed., 2021, 60, 13671–13676 CrossRef CAS.
- P. Bellotti, M. Koy, C. Gutheil, S. Heuvel and F. Glorius, Three-component three-bond forming cascade via palladium photoredox catalysis, Chem. Sci., 2021, 12, 1810–1817 RSC.
- J. E. Erchinger, R. Hoogesteger, R. Laskar, S. Dutta, C. Hümpel, D. Rana, C. G. Daniliuc and F. Glorius, EnT-Mediated N-S Bond Homolysis of a Bifunctional Reagent Leading to Aliphatic Sulfonyl Fluorides, J. Am. Chem. Soc., 2023, 145, 2364–2374 CrossRef CAS.
- X.-L. Luo, D.-J. Luo, L.-L. Qin, Z.-P. Ye and P.-J. Xia, Recent Advances in Diphenylmethanone Oxime Reagent Chemistry for Unsaturated Bond Bifunctionalization via Energy Transfer Catalysis, Eur. J. Org Chem., 2025, 28, e202401186 CrossRef CAS.
- T. Patra, M. Das, C. G. Daniliuc and F. Glorius, Metal-free photosensitized oxyimination of unactivated alkenes with bifunctional oxime carbonates, Nat. Catal., 2021, 4, 54–61 CrossRef CAS.
- Y. Zheng, Z.-J. Wang, Z.-P. Ye, K. Tang, Z.-Z. Xie, J.-A. Xiao, H.-Y. Xiang, K. Chen, X.-Q. Chen and H. Yang, Regioselective Access to Vicinal Diamines by Metal-Free Photosensitized Amidylimination of Alkenes with Oxime Esters, Angew. Chem., Int. Ed., 2022, 61, e202212292 CrossRef CAS.
- L. Wang, Y. Yu, L. Deng and K. Du, Photochemical and Atom-Economical Sulfonylimination of Alkenes with Bifunctional N-Sulfonyl Ketimine, Org. Lett., 2023, 25, 2349–2354 CrossRef CAS.
- J.-W. Sang, P. Du, D. Xia, Y. Zhang, J. Wang and W.-D. Zhang, EnT-Mediated Amino-Sulfonylation of Alkenes with Bifunctional Sulfonamides: Access to β-Amino Sulfone Derivatives, Chem.–Eur. J., 2023, 29, e202301392 CrossRef CAS PubMed.
- Z.-J. Chen, C. Huang, Z.-Z. Xie, Z.-J. Wang, Y. Zheng, H.-Y. Xiang, K. Chen and H. Yang, Photosensitized 1, 2-Sulfonylamination of Alkenes With Sulfonylamide, ChemistrySelect, 2024, 9, e202404815 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16 Rev. A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- C. Y. Legault, CYL View, 1.0b, Université de Sherbrooke, 2009 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6.1, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- J. N. Harvey, M. Aschi, H. Schwarz and W. Koch, The singlet and triplet states of phenyl cation. A hybrid approach for locating minimum energy crossing points between non-interacting potential energy surfaces, Theor. Chem. Acc., 1998, 99, 95–99 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu, A comprehensive electron wavefunction analysis toolbox for chemists, Multiwfn, J. Chem. Phys., 2024, 161, 082503 CrossRef CAS.
- T. Lu and Q. Chen, Realization of Conceptual Density Functional Theory and Information-Theoretic Approach in Multiwfn Program, in Conceptual Density Functional Theory, WILEY-VCH GmbH, Weinheim, 2022, pp. 631–647 Search PubMed.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Towards the online computer-aided design of catalytic pockets, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- S. V. S. Sowndarya, P. C. S. John and R. S. Paton, A quantitative metric for organic radical stability and persistence using thermodynamic and kinetic features, Chem. Sci., 2021, 12, 13158–13166 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b