DOI:
10.1039/D5RA02077H
(Paper)
RSC Adv., 2025,
15, 21142-21155
Internal electric field boosting visible photocatalytic degradation of antibiotics by flower-like CeO2/Bi2S3 S-scheme heterojunctions†
Received
24th March 2025
, Accepted 17th June 2025
First published on 23rd June 2025
Abstract
An internal electric field can be formed by constructing a heterojunction to achieve effective separation of photogenerated electrons and holes, which is able to solve the problem of easy compounding of photogenerated carriers in a single semiconductor photocatalyst. This research employs a hydrothermal synthesis technique to develop S-scheme heterojunction photocatalysts composed of cerium oxide and bismuth sulfide (CeO2/Bi2S3) and evaluates their efficacy in degrading TC under visible light. The formation of S-scheme heterojunctions was confirmed by X-ray photoelectron spectroscopy (XPS) and density functional theory (DFT) calculations showing that electrons migrate, and the internal electric field of the S-scheme heterojunctions achieves the separation of the electron–hole pairs, retaining the redox capacity of useful electrons and holes, which is responsible for the enhancement of the photocatalytic activity. The synthesized CeO2/Bi2S3-2 photocatalysts demonstrated a TC degradation rate of 82.43% after a duration of 120 minutes under visible light irradiation. The rate constant of this performance was two times greater than that of CeO2 alone and 2.75 times greater than that of Bi2S3. In addition, free radical trapping experiments and electron paramagnetic resonance results confirmed that ·O2− and h+ are active substances in the photocatalytic reaction process. Liquid chromatography-mass spectrometry (LC-MS) detected possible intermediates and suggested degradation pathways. This study has significant implications for the future development and enhancement of S-scheme heterojunction photocatalysts, contributing to advancements in photocatalytic materials.
1 Introduction
The discovery of antibiotics and their application in disease treatment, alongside the misuse of antibiotics such as tetracycline (TC), have resulted in significant environmental contamination.1,2 TC is present as a persistent organic pollutant in the environment, leading to the gradual accumulation and the potential development of microbial resistance, which poses risks to both ecological systems and human health.3,4 Consequently, the remediation of TC residues is critical, which might be achieved using various methods such as physical adsorption, biodegradation, and chemical techniques.5 Physical adsorption effectively removes contaminants but necessitates frequent replacement of the adsorbent material. Biodegradation employs microorganisms to degrade TC, offering economic advantages and effective performance; however, it often produces by-products that may be more toxic than TC, rendering it a less attractive choice.6,7 Chemical methods, such as photocatalytic degradation and precipitation, are also employed. Photocatalytic technology is an advanced oxidation process capable of completely degrading organic pollutants by generating reductive electrons and oxidative holes without producing secondary pollution, garnering significant attention in recent years.8
Cerium dioxide (CeO2), a widely-used n-type semiconductor, has significant applications in photocatalysis, fuel cells, and industrial catalysis, given its cost-effectiveness, high stability, and low environmental impact.9,10 However, its relatively broad energy bandgap of approximately 3.2 eV hinders its ability to absorb visible light effectively. As a result, the electrons generated upon photoexcitation are unable to move to the conduction band (CB) and recombine with holes, severely limiting its photocatalytic efficiency.11,12 Recent studies have addressed these limitations through various strategies, including morphological control,13,14 metal doping15–17 and the fabrication of heterojunctions.18–20 For instance, CeO2 nanoparticles with a precisely controlled size of 2.1 nm demonstrated a glyphosate decomposition rate 20 times greater than that of larger 4.8 nm nanoparticles, highlighting the significant effect of particle size on photocatalytic activity.21 The photocatalytic degradation of TC was markedly enhanced when using H2-reduced Mn-doped CeO2 compared to pure CeO2, with manganese doping facilitating a hierarchical structure and surface atomic arrangement that modifies the electronic properties of the material.16 Furthermore, Cu–CeO2/BiOBr Z-type heterojunction exhibited reduced electron–hole recombination, with copper doping lowering the energy bandgap of CeO2 and the heterojunction structure expanding the visible light absorption range, achieving a 92.3% degradation of sulfathiazole within 90 min.15 The construction of heterojunctions has emerged as an effective strategy to enhance photocatalytic performance, with common frameworks including type-II heterojunctions22 and Z-type heterojunctions.23,24 However, research indicates that traditional type-II heterojunctions face challenges for effective charge separation, while Z-type heterojunctions encounter issues such as redox pair interactions that reduce the available photogenerated electrons and holes, along with a charge transfer mechanism that undermines their purported benefits.25
To address these challenges, the step-structured heterojunction (S-scheme) has been proposed,26 which integrates an oxidized photocatalyst (OP) with a reduced photocatalyst (RP). This combination creates a catalytic system with enhanced performance, where the CB of the RP is elevated, along with its Fermi level (Ef), both of which are superior to those of the OP, establishing a significant driving force for electron transfer.27,28 Upon contact between the RP and OP, an internal electric field (IEF) is generated, facilitating electron migration and promoting the photocatalytic reaction. This IEF enables the recombination of excess electrons in the CB of the OP with the unwanted holes found in the valence band potential (VB) of the RP. This recombination serves to mitigate the losses associated with excess energy, reducing energy losses.29,30 Simultaneously, this mechanism ensures the preservation of beneficial electrons and holes, allowing them to maintain their active involvement in the essential redox reactions, thereby enhancing the efficiency of the photocatalytic system. Therefore, there is a need to find a suitable RP semiconductor to construct an S-scheme heterojunction with CeO2.
Bi2S3 functions as an n-type semiconductor characterized by a narrow bandgap, which exhibiting significant light absorption within the visible spectrum and allows for tunability in its bandgap energy.31,32 The Bi2S3 heterojunction, when combined with wide bandgap semiconductors, enhances effective electron transfer. In this heterojunction, electrons migrate from Bi2S3, which has a comparatively lower CB to a wide bandgap semiconductor with a higher CB. This mechanism not only improves electron mobility but also facilitates the separation of electron–hole pairs generated by light. Such a migration mechanism suggests enhanced visible light absorption when light strikes the heterojunction, ultimately leading to improved efficiency in the separation of photogenerated carriers.33 Hydrothermal synthesis of g-C3N4/Bi2S3 resulted in 95.6% degradation of Reactive Black 5 and 97.5% degradation of indigo cochineal after 120 min of light exposure.34 This research underscores the feasibility of the approach and hints that both CeO2 and Bi2S3 can be used to form highly efficient S-scheme heterojunctions.
In this work, CeO2/Bi2S3 S-scheme heterojunction photocatalysts were synthesized by combining the wide bandgap semiconductor CeO2 with the narrow bandgap semiconductor Bi2S3 using hydrothermal methods, and were used to degrade TC under light conditions. The assessment of the photodegradation efficiency of the CeO2/Bi2S3 S-scheme heterojunction photocatalysts facilitated an initial exploration of the mechanisms and pathways involved in TC degradation, informed by free radical trapping experiments, energy band structure analysis, and intermediate product characterization. The results showed that the CeO2/Bi2S3 S-scheme heterojunction photocatalyst not only enhanced the charge migration efficiency, but also effectively promoted the directional separation of the photogenerated electron–hole pairs through the establishment of an internal electric field, which significantly enhanced the photocatalytic degradation performance while maintaining the strong redox ability of the dominant carriers. The main text of the article should appear here with headings as appropriate.
2 Experimental section
2.1 Materials
Cerium nitrate (Ce(NO3)3·6H2O), bismuth nitrate (Bi(NO3)3·5H2O), L-ascorbic acid (LA), ethylene glycol (C2H6O2, EG), tetracycline (C22H24N2O8, TC) and thiourea (CH4N2S) were supplied by Shanghai Macklin Reagent Co. Isopropyl alcohol (IPA), sodium hydroxide (NaOH), and sodium ethylenediaminetetraacetic acid (EDTA-2Na) were purchased from Tianjin Kemel Chemical Reagent Co. The water used in the experiment was deionized water. All chemical reagents are directly used without further purification.
2.2 Preparation of CeO2
60.00 g NaOH in 87.5 mL of water was heated and stirred at 70 °C for 30 min to accelerate the dissolution, and 5.42 g Ce(NO3)3·6H2O in 12.5 mL of deionised water was stirred until it was completely dissolved, and then, the two solutions were mixed slowly and stirred magnetically for 30 min, and the mixture was transferred to 100 mL of a polytetrafluoroethylene-lined stainless steel autoclave, hydrothermal treatment at 100 °C for 24 h, and cooled to room temperature. The mixture was transferred to 100 mL PTFE-lined stainless steel autoclave, hydrothermally treated at 100 °C for 24 h, cooled to room temperature, and the resultant was centrifuged (9000 rpm, 5 min) and washed with water several times until the pH was 7. After drying at 100 °C for 4 h, the product was heated to 500 °C at an elevated rate of 5 °C min−1 and calcined in air for 5 h. The CeO2 nanorods were obtained by grinding into powder.
2.3 Preparation of CeO2/Bi2S3
A total of 0.97 g of Bi(NO3)3·5H2O was combined with 25 mL of EG and stirred for 30 min, resulting in what is referred to as solution A. Solution B was also prepared by incorporating 0.34 g of CeO2 and 0.23 g of CH4N2S was also stirred in 25 mL of EG. Once solution B was successfully prepared, it was gradually combined with solution A and stirred for 30 min to guarantee proper mixing of the two solutions. The ultimate product is subsequently moved to a stainless steel autoclave that has a lining made of polytetrafluoroethylene, and subjected to hydrothermal treatment at 160 °C for 14 h. The resultant products were washed multiple times with water through centrifugation (9000 rpm for 5 min) and then dried at 80 °C for 12 h in a vacuum oven. The CeO2/Bi2S3 composites were ground into powders, and the specific procedure of the experiment is shown in Scheme 1. Based on the different applied molar ratios of CeO2 to Bi2S3 in the composite photocatalysts were of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1, 1:1 and 1:2, the obtained photocatalysts were named CeO2/Bi2S3-1, CeO2/Bi2S3-2, CeO2/Bi2S3-3, and CeO2/Bi2S3-4, respectively. The Bi2S3 photocatalysts were also prepared without doping CeO2, via same procedure as described above.
:1, 2:1, 1:1 and 1:2, the obtained photocatalysts were named CeO2/Bi2S3-1, CeO2/Bi2S3-2, CeO2/Bi2S3-3, and CeO2/Bi2S3-4, respectively. The Bi2S3 photocatalysts were also prepared without doping CeO2, via same procedure as described above.
 |
| | Scheme 1 Schematic illustration of the synthesis of CeO2/Bi2S3 photocatalysts. | |
2.4 Characterization
A D/max-2500PC (Rigaku) instrument was used to perform powder X-ray diffraction to evaluate the physical phases and crystal structure of the photocatalysts. Concurrently, the morphology of the photocatalysts was assessed using a cold field emission scanning electron microscope (SEM, Hitachi S-4800), enabling the observation of the samples' morphological features at the microscopic scale. Additionally, a high-resolution transmission electron microscope (TEM, JEM-2100F), complemented by an energy dispersive spectrometer (EDS), was utilized to provide further insights into the internal structures and compositions of the photocatalysts. Moreover, to thoroughly explore the chemical composition of the samples and their respective oxidation states, an X-ray photoelectron spectrometer (XPS, Shimadzu AXIS Supra+) was employed to characterize the oxidation states. The photocatalysts' light absorption characteristics were evaluated using a UV-Vis Diffuse Reflectance Spectrometer (UV-Vis DRS, V-750 (JASCO)), which provide a quantitative information of their light absorption capabilities. Finally, the investigation of the photogenerated carrier complexation in the photocatalysts was carried out using photoluminescence spectroscopy (PL), with a fluorescence photometer (FLS-1000) excited at 365 nm. Determination of degraded TC intermediates by liquid chromatography-mass spectrometry (Bruker ESI-Q-TOF).
2.5 Photoelectrochemical analysis
The transient photocurrent response and electrochemical impedance were assessed with a conventional three-electrode setup utilizing the CHI660E electrochemical workstation. The counter electrode was represented by a Pt electrode, whereas the reference electrode was Ag|AgCl (3 M KCl). A mixture of 10 mg of photocatalysts, 800 μL of ethanol, and 200 μL of Nafion was sonicated for 10 min and applied to a 1 × 1 cm2 FTO glass, which was dried and then the FTO glass electrode was clamped to a copper-violet electrode holder as the working electrode. It was submerged in 0.2 M Na2SO4 solution and placed under the radiation of a 300 W xenon lamp.
2.6 Evaluation of photocatalytic performance
The photocatalytic degradation of TC was examined using a 300 W xenon light source (CEL-PF300-T6) equipped with a 420 nm filter to simulate solar light irradiation. A 100 mL solution of TC, with an initial concentration C0 = 10 mg L−1 was transferred into a 200 mL jacketed beaker, into which 20 mg of photocatalyst was introduced. The temperature of the beaker was maintained at 25 °C throughout the experiment using a circulating cooling water system, and the solution was homogenized via magnetic stirring. Adsorption equilibrium was established by stirring the solution in the dark for 40 min. Following this, the light source was activated for the photoreaction. After every 20-min intervals, approximately 4 mL of the solution was abstracted with a syringe and filtered through a 0.45 μm water filter to remove the photocatalysts. The reactive radicals produced during photocatalysis were investigated using electron paramagnetic resonance (EPR, Bruker A300, USA). The TC concentration was measured with a UV-visible spectrophotometer (YOKE T3202S) at a designated wavelength of 357 nm, and the degradation rate of TC was assessed in accordance with Beer–Lambert law by utilizing the absorbance measured at that wavelength. The degradation rate of TC (D) is computed using eqn (1).| |
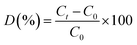 | (1) |
where Ct denotes the concentration at time t, while C0 signifies the initial concentration.
2.7 Theoretical calculations
The Vienna Ab initio Simulation Package (VASP) was employed to perform all the density functional theory (DFT) calculations employing the projector-augmented wave method (PAW).35,36 To enhance the precision and dependability of our calculations, we utilized the Perdew–Burke–Ernzerhof (PBE) exchange–correlation function.37 This combination effectively represents electron behavior and associated forces, providing a solid theoretical foundation for our research. Additionally, we employed a plane-wave basis set with a kinetic energy cut-off of 400 eV to ensure the adequacy of our computational approach. We adopted the lattice parameters a = 11.771 Å, b = 11.324 Å and c = 32.670 Å for the CeO2/Bi2S3 heterostructure, which consists of a single layer of Bi2S3 on the CeO2 (111) substrate. In this research, a vacuum layer thickness of 20 Å is established. To accurately characterize the energetic behavior of the system, a cut-off energy of 520 eV was employed for the plane-wave basis set. Brillouin zone integration was conducted using Monkhorst–Pack k-point sampling.38 Throughout the self-consistent calculations, a convergence energy threshold of 10−5 eV was maintained, minimizing the potential influence of numerical instability on the results. Additionally, the maximum allowable stress on each atom was constrained to 0.05 eV Å−1.
3 Results and discussion
3.1 Structural and morphological analysis
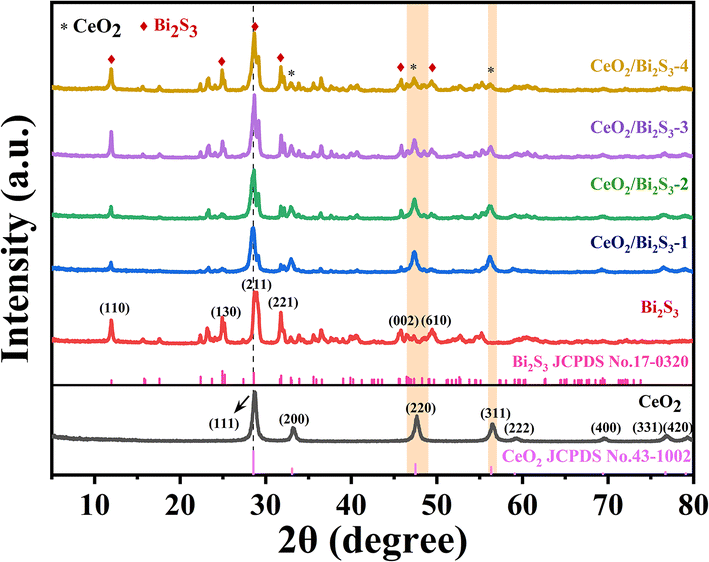
Fig. 1 presents the powder XRD diffraction patterns for the pure CeO2, Bi2S3 and CeO2/Bi2S3 photocatalysts at varying molar ratios. The distinctive diffraction peaks of the synthesized CeO2 (No. 43-1002) appear at 2θ = 28.72°, 33.16°, 47.62°, 56.40°, 59.34°, 69.64°, 76.94° and 79.26°, which correlate with the (111), (200), (220), (311), (222), (400), (331) and (420) crystal planes. The absence of impurity peaks indicates successful synthesis of pure CeO2. The characteristic diffraction peaks of the as-prepared Bi2S3 located at 2θ = 11.94°, 24.92°, 28.96°, 31.76°, 45.82°, and 49.36° correspond to the peaks of Bi2S3 (No. 17-0320) at the (110), (130), (211), (221), (002), and (610) crystal faces. The powder XRD patterns of the CeO2/Bi2S3 photocatalysts displayed distinctive peaks corresponding to both CeO2 and Bi2S3. As the ratio of CeO2/Bi2S3 in the material decreases, the diffraction peaks matching the (200), (220), and (311) crystal planes gradually decrease, while the diffraction peaks at the (110), (130), (221), and (610) crystal planes gradually enhance. The shift of the diffraction peaks to a lower angle is due to the doping of Bi into the crystal structure of CeO2, which leads to lattice expansion and the shift of the diffraction peaks to a lower angle.17,39,40 These observations confirm the successful fabrication of the CeO2/Bi2S3 photocatalysts.
 |
| | Fig. 1 Powder XRD diffraction patterns of CeO2, Bi2S3, and CeO2/Bi2S3 samples. | |
The morphology of the photocatalysts was examined using SEM and TEM. The pure form of CeO2 manifests as nanorods averaging 200 and 500 nm in length, as illustrated in Fig. 2(a) and (d). In contrast, Fig. 2(b) and (e) reveal that Bi2S3 displays a flower-like microarchitecture, roughly 4 μm in size. Fig. 2(c) and (f) demonstrate the doping of nanorods on the surface of the flower-like Bi2S3, with high-resolution TEM images confirming the strong binding of CeO2 and Bi2S3, indicating successful construction of the heterogeneous junction. Elemental distribution mapping via EDS for the CeO2/Bi2S3-2 photocatalyst is illustrated in Fig. 2(g)–(j). The result reveals a uniform distribution of Bi, Ce, S, and O on the surface of CeO2/Bi2S3-2, supporting the effective formation of the heterojunction as well as the successful complexation of CeO2/Bi2S3.
 |
| | Fig. 2 SEM images of (a) CeO2; (b) Bi2S3; (c) CeO2/Bi2S3-2, TEM images of (d) CeO2; (e) Bi2S3; (f) CeO2/Bi2S3-2; EDS elemental distribution mapping of (g) Bi; (h) Ce; (i) S; (j) O in CeO2/Bi2S3-2. | |
3.2 XPS analysis
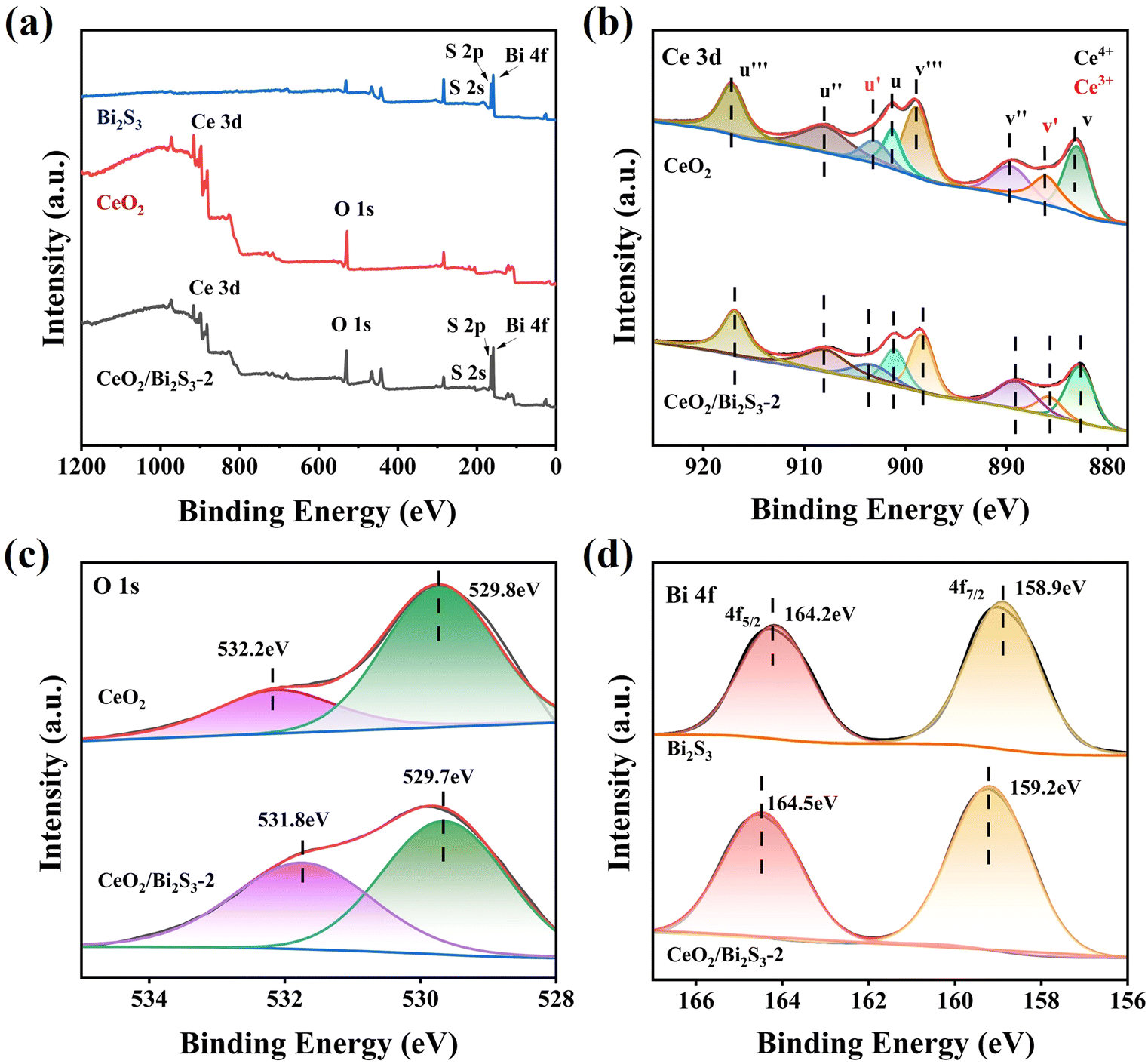
The elemental composition and oxidation states of the photocatalysts were analyzed using XPS. As shown in Fig. 3(a), the elemental peaks for both CeO2 and Bi2S3 are present in CeO2/Bi2S3-2, confirming the successful synthesis of the heterojunction. The Ce 3d spectrum, depicted in Fig. 3(b), exhibits both Ce4+ and Ce3+ components, with eight characteristic peaks, detailed in Table S1.† In this context, the variables u corresponds to the spin orbitals of Ce 3d3/2 and v corresponds to the spin orbitals of Ce 3d5/2, while u′ and v′ pertain to Ce3+. Conversely, the variables u′′′, u′′, u, v′′′, v′′, and v are associated with Ce4+. The binding energy of the Ce 3d electron in CeO2 is higher than that observed in CeO2/Bi2S3-2 photocatalysts. The O 1s of CeO2 in Fig. 3(c) has two strong peaks at 529.8 eV and 532.2 eV, corresponding to the lattice oxygen and surface hydroxyl oxygen of CeO2, respectively.41,42 In contrast, a decrease in the binding energies of Ce 3d and O 1s occurs in CeO2/Bi2S3-2. The Bi 4f spectrum of Bi2S3 (Fig. 3(d)) has two strong peaks at 158.9 eV and 164.2 eV belonging to Bi 4f7/2 and Bi 4f5/2, which suggests that the Bi exists in the form of Bi3+.43 The binding energy of CeO2/Bi2S3-2 is enhanced relative to the Bi 4f spectrum of Bi2S3. When electrons are lost or gained, the binding energy of the constituent elements increases or decreases accordingly.29 This indicates that electron transfer occurs between the contact surfaces of CeO2 and Bi2S3, where the binding energy of Ce and O decreases and electrons are gained, the binding energy of Bi increases and electrons are lost, and electrons are transferred from Bi2S3 to CeO2, leading to the creation of heterojunctions.
 |
| | Fig. 3 XPS spectra of CeO2, Bi2S3, and CeO2/Bi2S3-2: (a) survey scan spectra; (b) Ce 3d; (c) O 1s; (d) Bi 4f. | |
3.3 Photocatalytic performance test
The prepared photocatalysts effectively degraded TC when exposed to light from a 300 W xenon lamp, and it should be noted that the adsorption of TC molecules by the photocatalysts is not negligible, with adsorption equilibrium reached for all samples after 40 min in the dark. In Fig. 4(a), it is evident that the concentration of TC remained almost the same with no addition of photocatalysts. Pure CeO2 and Bi2S3 achieved photocatalytic degradation rates of 54.85% and 50.78%, respectively, after 120 min. Compared with the pristine materials, the CeO2/Bi2S3 photocatalysts were significantly improved, and the degradation rates of TC by CeO2/Bi2S3-1, CeO2/Bi2S3-2, CeO2/Bi2S3-3 and CeO2/Bi2S3-4 were 72.31%, 82.43%, 73.23% and 68.51% within 120 min, respectively. It is evident that the degradation efficiencies of the CeO2/Bi2S3 photocatalysts initially increased and then decreased with higher Bi2S3 content, with CeO2/Bi2S3-2 exhibiting the highest performance. Quasi-primary kinetic fitting was performed using eqn (2)where k represents the apparent reaction rate constant. The kinetic curve indicates that CeO2/Bi2S3-2 exhibits a k value of 0.01417 min−1, which is higher than all other samples. (Fig. 4(b)). The value observed is significantly greater, demonstrating an increase of two times compared to pure CeO2, which has a rate of 0.00709 min−1. Additionally, it is 2.75 times higher than the rate for pure Bi2S3, which is recorded at 0.00516 min−1.
 |
| | Fig. 4 (a) Photocatalytic degradation of TC by as-prepared photocatalysts under simulated solar illumination and (b) its kinetic fitting curves; 20 mg CeO2/Bi2S3-2 degradation of (c) different initial concentrations of TC solution and (d) its kinetic fitting curves; photocatalytic degradation of tetracycline solution by CeO2/Bi2S3-2 at (e) different catalyst dosage (f) different initial pHs. | |
To examine how the initial concentration influences the photodegradation of TC, experiments were conducted using varying initial concentrations of TC solutions, with 20 mg of CeO2/Bi2S3-2 employed for each trial. The results are illustrated in Fig. 4(c) and (d). Clearly, an increase in the starting concentration of TC leads to an overall decrease in degradation efficiency, which declines from 82.43% to 73.19%. Additionally, the primary kinetic constant decreased from 0.01417 min−1 to 0.01145 min−1. At higher initial concentrations, a greater number of TC molecules compete with one another, resulting in fewer molecules being captured and adsorbed by the photocatalysts.44,45 In contrast, lower initial concentrations reduce this competitive effect, allowing a greater number of TC molecules to adhere to the photocatalyst's surface.
The quantity of photocatalysts is a significant element that influences the effectiveness of photocatalytic degradation. An investigation was conducted to assess the degradation impact of varying amounts of CeO2/Bi2S3-2 on TC at a concentration of 10 mg L−1. Fig. 4(e) reveals that the efficiency of TC degradation increases and subsequently declines as the catalyst amount rises from 10 mg to 50 mg. During the dark reaction phase, an increase in catalyst dosage provides more adsorption sites, thereby enhancing the adsorption capacity. However, the degradation rate of TC exhibits a pattern of initial increase followed by a decrease during the light reaction phase. The peak degradation of TC was observed at an optimized catalyst dosage of 20 mg. This decline in degradation rate at higher catalyst concentrations may be attributed to increased turbidity in the solution, which impairs the catalyst's ability to absorb light, thus reducing the degradation rate of the photoreactive component.46,47 Therefore, a dosage of 20 mg is selected as the optimum for subsequent studies.
The pH of the solution is a critical factor influencing the photocatalytic degradation of TC. Variations in pH can significantly alter the interactions between the photocatalyst and the pollutant throughout the reaction process. Consequently, it is essential to optimize the solution's pH by selecting appropriate concentrations of acids and bases, such as 0.1 M HCl and NaOH. In Fig. 4(f), the degradation rates of TC at pH of 3, 5, 7, 9 and 11, the final degradation rate of TC was recorded at 20.31%, 31.86%, 82.43%, 64.96% and 57.60%, respectively. The degradation rate increases as the pH rises from 3 to 7 but declines when the pH exceeds 7. At lower pH levels, ·O2− reacts with H+ to form H2O2 and ·O2− is consumed in large quantities, resulting in a much lower degradation rate.48 At pH 7, photogenerated electrons react more readily with dissolved oxygen to form ·O2−, resulting in easier separation of carriers, higher electron transfer efficiency and enhanced photocatalytic degradation rates.49 In alkaline conditions, the adsorption of TC by the catalyst increases, and the excess TC adsorption blocks the visible light from reaching the catalyst surface, which reduces the light absorption rate of the catalyst, which in turn leads to a lower degradation rate.50 Furthermore, in alkaline conditions, h+ reacts with OH− to form ·OH, and the h+ content decreases, resulting in a lower rate of TC degradation.51 In the subsequent free radical trapping experiments, it was demonstrated that the contribution of h+ to TC degradation was greater than that of ·OH. Therefore, the degradation rate of TC was reduced under alkaline conditions. The performance of photocatalytic degradation of TC by CeO2/Bi2S3-2 photocatalytic material was compared with previous studies, and as shown in Table 1, the degradation performance of the prepared CeO2/Bi2S3-2 photocatalytic material was superior to that of most of the other reported photocatalytic materials.
Table 1 Comparison of TC degradation rate of CeO2/Bi2S3-2 with other photocatalysts
| Sample |
TC (mg L−1) |
Irradiation time (minutes) |
Degradation (%) |
Ref. |
| CeO2/Bi2S3-2 |
10 |
120 |
82.43 |
This work |
| CeO2/Bi2O2CO3 |
20 |
90 |
79.50 |
12 |
| β-Bi2O3@CeO2 |
10 |
180 |
100 |
19 |
| CeO2/CNNS |
10 |
120 |
79.6 |
42 |
| Bi2O2CO3/Ti3C2 |
20 |
120 |
81.00 |
54 |
| g-C3N4/CeO2 |
10 |
160 |
77.95 |
55 |
| Bi2WO6 |
30 |
180 |
79.68 |
56 |
| 2% Au/CeO2 |
20 |
90 |
86.40 |
57 |
| CeO2/Co3O4 |
20 |
30 |
85.35 |
58 |
| CeO2/Co3O4 |
20 |
60 |
90 |
59 |
| PDIs/C, N, S-CeO2 |
20 |
30 |
80.10 |
60 |
3.4 Photoelectrochemical analysis
This research examined the effectiveness of separating photogenerated carriers through photoluminescence spectroscopy. The PL spectra for CeO2, Bi2S3, and CeO2/Bi2S3-2 were recorded with an excitation wavelength of 368 nm. An inverse relationship was identified between PL intensity and the efficiency of photogenerated electron–hole pair separation; specifically, lower PL intensity correlates with a reduced likelihood of electron–hole complex formation, thereby enhancing separation.52 The PL intensity of the CeO2/Bi2S3-2 photocatalyst, as shown in Fig. 5(a), is significantly lower when compared to that of both pure CeO2 and pure Bi2S3, indicating effective suppression of electron–hole complexation. This suppression may be attributed to the development of an IEF within the heterojunction that promotes electron–hole pair separation. The electrochemical impedance spectrum (EIS) presented in Fig. 5(b) shows that a smaller curve radius indicates a reduced level of electrochemical impedance.53 The impedance curve of CeO2/Bi2S3-2 exhibits a smaller radius when compared to the curves of the pure CeO2 and the pure Bi2S3, suggesting that the heterojunction formed by the composite materials facilitates charge transfer by lowering resistance. Furthermore, findings from the transient photocurrent response of all samples exposed to visible light (Fig. 5(c)) corroborate the EIS results. This evidence underscores the significant advantages of heterojunctions over individual CeO2 and Bi2S3 in terms of the effectiveness of photogenerated carrier separation, thereby enhancing photocatalytic performance.
 |
| | Fig. 5 (a) PL, (b) EIS Nyquist plot, and (c) transient photocurrent response of CeO2, Bi2S3 and CeO2/Bi2S3-2. | |
3.5 Stability and cyclic testing
The stability of the CeO2/Bi2S3-2 photocatalysts was assessed through cycling experiments. In these experiments, the CeO2/Bi2S3-2 photocatalysts were collected by filtration after each degradation test, after repeated washing with anhydrous ethanol, and subsequently evaporated excess water in an oven for 12 h at 60 °C before being subjected to cyclic operation again under identical conditions. As depicted in Fig. 6(a), following five cycles of photocatalytic degradation, the rate of photocatalytic degradation dropped from 82.43% to 76.26%, representing a reduction of 6.17%. This decrease is attributed to material loss during the recycling process. Additionally, powder XRD patterns before and after the use of the photocatalyst (Fig. 6(b)) indicated that the structure of CeO2/Bi2S3-2 remained largely unchanged throughout the reaction process, demonstrating the excellent stability and recyclability of the CeO2/Bi2S3-2 photocatalyst.
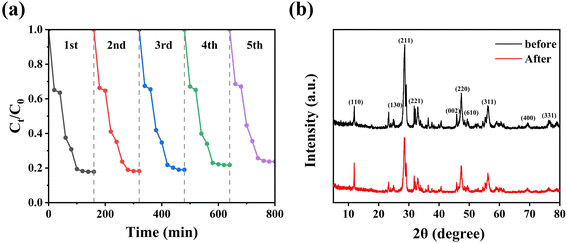 |
| | Fig. 6 Powder XRD spectra of CeO2/Bi2S3-2 catalyst for (a) 5 cycles of TC degradation experiment and (b) before and after the reaction. | |
3.6 Photocatalytic mechanism analysis
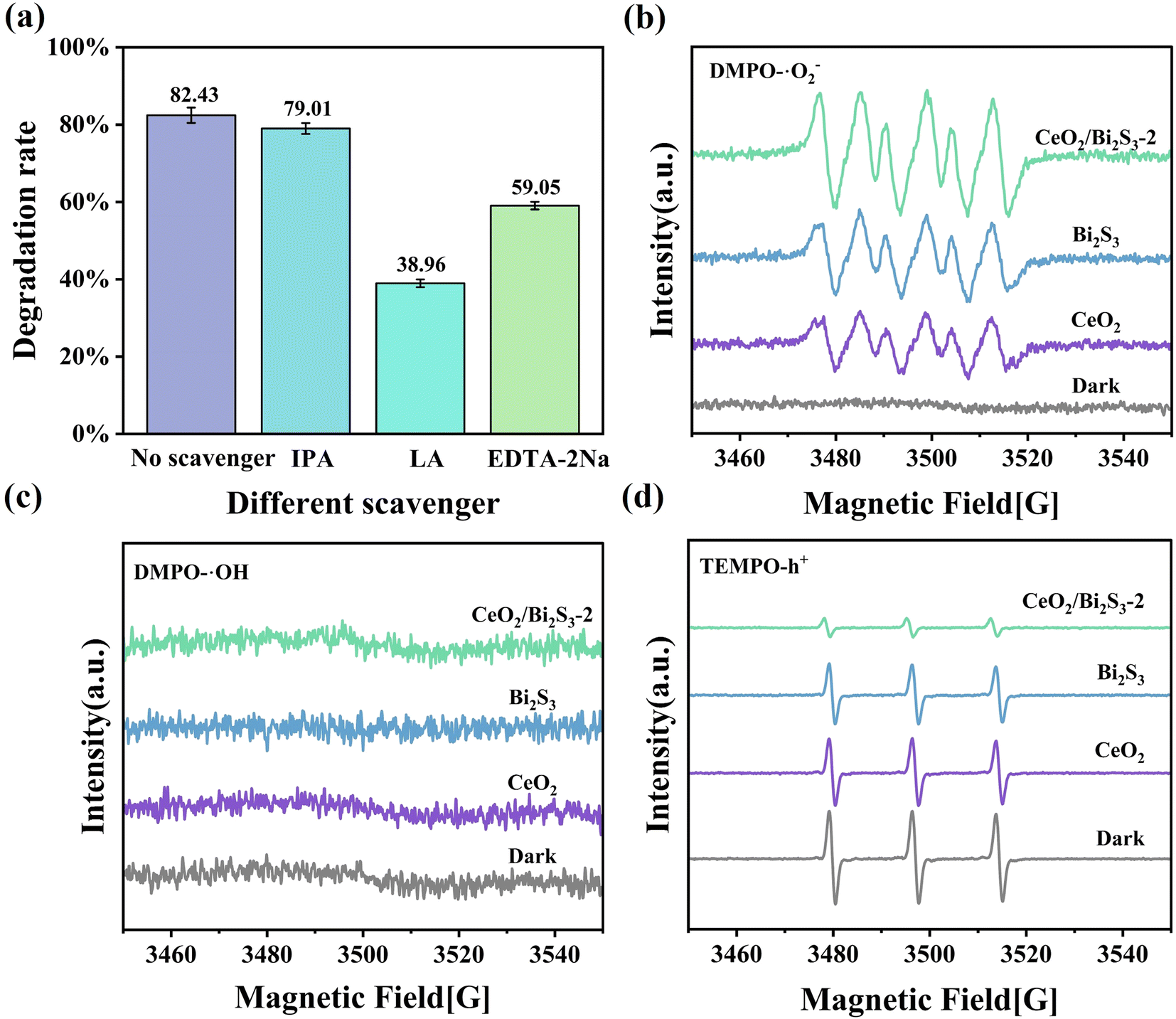
Experiments were conducted to capture and identify free radicals generated during the process as a way to investigate the fundamental mechanisms responsible for the photocatalytic degradation of TC when utilizing CeO2/Bi2S3-2. EDTA-2Na, LA,61 and IPA were utilized as scavengers for various reactive species, specifically h+, ·O2−, and ·OH. The effectiveness of these scavengers in modulating the degradation of TC was evaluated. As depicted in Fig. 7(a), the introduction of IPA resulted into a negligible change in the degradation rate of TC, suggesting that ·OH has a somewhat limited impact on the photodegradation process of TC. Our observations indicated that the inclusion of LA and EDTA-2Na significantly hindered the photocatalytic degradation process of TC. Specifically, we noted a substantial decline in the degradation rate of TC, which plummeted from 82.43% to 38.96% following the addition of LA. Furthermore, the degradation rate was also impacted by the addition of EDTA-2Na, leading to a decrease to 59.05%. These results suggest that the active species ·O2− and h+ play a primary role in the degradation process, while ·OH has a minimal impact. To further confirm whether ·O2−, ·OH, and h+ are indeed present in the photocatalytic process, we performed a qualitative detection of free radicals using EPR. The ·O2− and ·OH radicals were detected using the reagent 5,5-dimethyl-1-pyrroline N-oxide (DMPO), and the presence of h+ was detected by 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO). As shown in Fig. 7(b), no signal of DMPO–·O2− was detected under dark conditions, while a significant DMPO–·O2− signal was detected after light exposure, which proved that ·O2− was produced during the photocatalytic process. The order of DMPO–·O2− signal intensity is CeO2 < Bi2S3 < CeO2/Bi2S3-2, which indicates that CeO2/Bi2S3-2 generates more ·O2−.Whereas ·OH had no obvious signal under both dark and light conditions (Fig. 7(c)), indicating that no ·OH was produced during the whole reaction. From Fig. 7(d), it can be seen that the signal peak of TEMPO decreases in intensity after light exposure, which is due to the reaction between h+ and TEMPO, where h+ increases and TEMPO consumed by h+ increases, resulting in a weakening of the TEMPO signal.46 This proves that h+ does exist. The intensity of the signal peak for TEMPO–h+ was weakest for CeO2/Bi2S3-2, which generated more h+. The above results demonstrate that CeO2/Bi2S3 S-scheme heterojunction generates more ·O2− and h+ radicals involved in the degradation of TC.
 |
| | Fig. 7 (a) Effect of different scavengers on the photocatalytic degradation of TC by CeO2/Bi2S3-2; EPR signals of (b) DMPO–·O2−; (c) TEMPO–h+; (d) DMPO–·OH. | |
To provide additional insight into the mechanism by which CeO2/Bi2S3-2 degrades TC, we analysed the light absorption characteristics of the samples that we had prepared using UV-Vis DRS. The findings presented in Fig. 8(a) indicate that the pure CeO2 demonstrates significant light absorption capabilities in the ultraviolet spectrum, whereas it shows minimal absorption in the visible region. Additionally, pure Bi2S3 shows a robust ability to absorb light within the visible region. The formation of a heterojunction that combines CeO2 with Bi2S3 has led to significant improvements in light absorption properties for CeO2/Bi2S3-2. When comparing its performance to that of pure CeO2 and pure Bi2S3, CeO2/Bi2S3-2 exhibits superior capability to absorb light within the visible spectrum, attributable to the light-absorbing of Bi2S3 within the heterojunction. The flat band potentials (Efb) of Bi2S3 and CeO2 were assessed using the Mott–Schottky test. The slopes of the Mott–Schottky curves for both are positive, which proves that both CeO2 and Bi2S3 are n-type semiconductors.39,44 The results in Fig. 8(b) and (c) reveal that the Efb values for CeO2 and Bi2S3 were −0.21 eV and −0.59 eV, respectively, when measured against the Ag/AgCl reference electrode at a pH 7. The energy levels of Efb for both CeO2 and Bi2S3 were evaluated from eqn (3) to be −0.01 eV for CeO2 and −0.39 eV (vs. NHE) for Bi2S3.62
| | |
ENHE = EAg/AgCl + 0.197
| (3) |
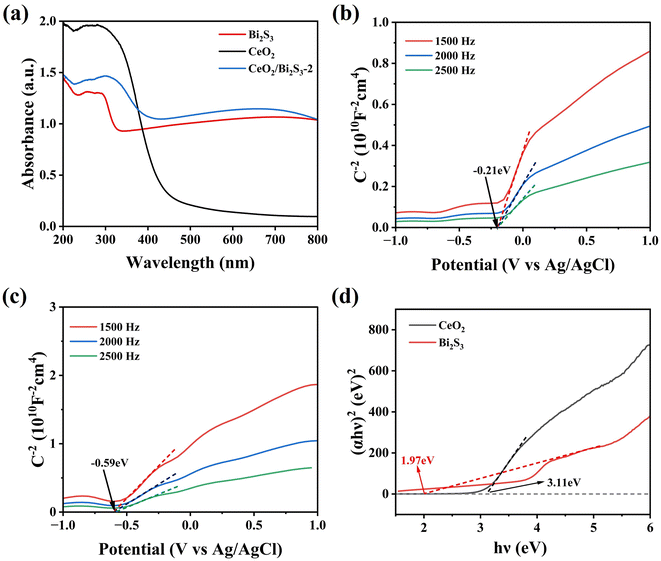 |
| | Fig. 8 (a) DRS spectra. Mott–Schottky spectra of (b) CeO2 and (c) Bi2S3. (d) Tauc plot of CeO2 and Bi2S3. | |
The Efb of n-type semiconductors is near the Ef,63 and is situated about 0.1 V more positive than the CB.64,65 Consequently, the CB levels of CeO2 and Bi2S3 are measured to be −0.11 eV and −0.49 eV (vs. NHE), respectively. The bandwidth of the sample is calculated from eqn (4).
where the absorption coefficient, Planck's constant, optical frequency and absorbance are represented by
α,
h,
ν, and
A, respectively. As direct bandgap semiconductors, CeO
2 and Bi
2S
3 have an
n value of 2. From
Fig. 8(d), the forbidden bandwidths for CeO
2 and Bi
2S
3 are 3.11 eV and 1.97 eV, respectively. Additionally, the VB for CeO
2 and Bi
2S
3 are identified as 2.99 eV and 1.48 eV, respectively, as indicated by
eqn (5) (
vs. NHE).
The difference in charge density of photocatalysts was calculated using DFT. Fig. 9(a) and (c) show the electron cloud densities of CeO2 and Bi2S3 before contact, respectively. Fig. 9(c) reveals the charge density difference of CeO2/Bi2S3. The yellow areas indicate regions of electron accumulation, while the green areas signify electron depletion. It is clear that electrons accumulated on the CeO2 surface, while the Bi2S3 surface exhibited charge loss. This charge loss is due to the migration of electrons from Bi2S3 to CeO2 following contact, indicating the formation of an IEF between the two surfaces, with the electric field directed from Bi2S3 towards CeO2.
 |
| | Fig. 9 Charge distribution of (a) CeO2 and (b) Bi2S3 before contacting (c) difference in charge density of CeO2/Bi2S3. | |
Based on the experimental results presented in this study, we propose the energy band configurations and charge transfer mechanisms for CeO2 and Bi2S3, both before and after their interaction, as well as the processes related to the photocatalytic breakdown of TC (Fig. 10). Prior to contact, the energy bands of CeO2 and Bi2S3 are interleaved, and the reduced semiconductor Bi2S3 has a higher Ef than the oxidized semiconductor CeO2. Thus, when the two materials come into contact, the variation in Fermi levels causes the natural movement of electrons from Bi2S3 to CeO2. This process continues until the Fermi levels equalize, resulting in the creation of an IEF at the interface directed from Bi2S3 towards CeO2. Simultaneously, the energy band at the Bi2S3 interface loses electrons, causing it to curve upwards, while the CeO2 interface acquires electrons, which leads to a downward bend. This modification hinders the transfer of electrons between the CB of Bi2S3 and that of CeO2. Additionally, this alteration also restricts the flow of holes, preventing them from moving from the VB of CeO2 to the VB of Bi2S3.29,66 Upon solar illumination, the holes and electrons generated by the excitation of CeO2 and Bi2S3, in the presence of IEF, the electrons in the CB of CeO2 are moved to the interface and recombine with the holes from the VB of Bi2S3, which are also transferred to the interface. By eliminating unwanted electrons and holes through recombination, holes on CeO2 VB and electrons on Bi2S3 CB are retained and accumulated, which are strongly oxidizing and reducing, respectively.25 And a more direct degradation of TC by holes on CeO2 VB. The CB of Bi2S3, measured at −0.49 eV, which is higher compared to the −0.33 eV of O2/·O2− (vs. NHE). In this process, the electrons in the CB facilitate the reduction of dissolved O2 in water to produce ·O2−, which subsequently reacts with TC to contribute to its degradation. Specific reactions are represented by eqn (6)–(10).
| | |
CeO2 + hv → e−(CeO2) + h+(CeO2)
| (6) |
| | |
Bi2S3 + hv → e−(Bi2S3) + h+(Bi2S3)
| (7) |
| | |
h+(CeO2) + TC → products
| (8) |
| | |
e−(Bi2S3) + O2 → ·O2−
| (9) |
| | |
·O2− + TC → products
| (10) |
 |
| | Fig. 10 Schematic energy band structure and photocatalytic mechanism of CeO2/Bi2S3-2 photocatalysts. | |
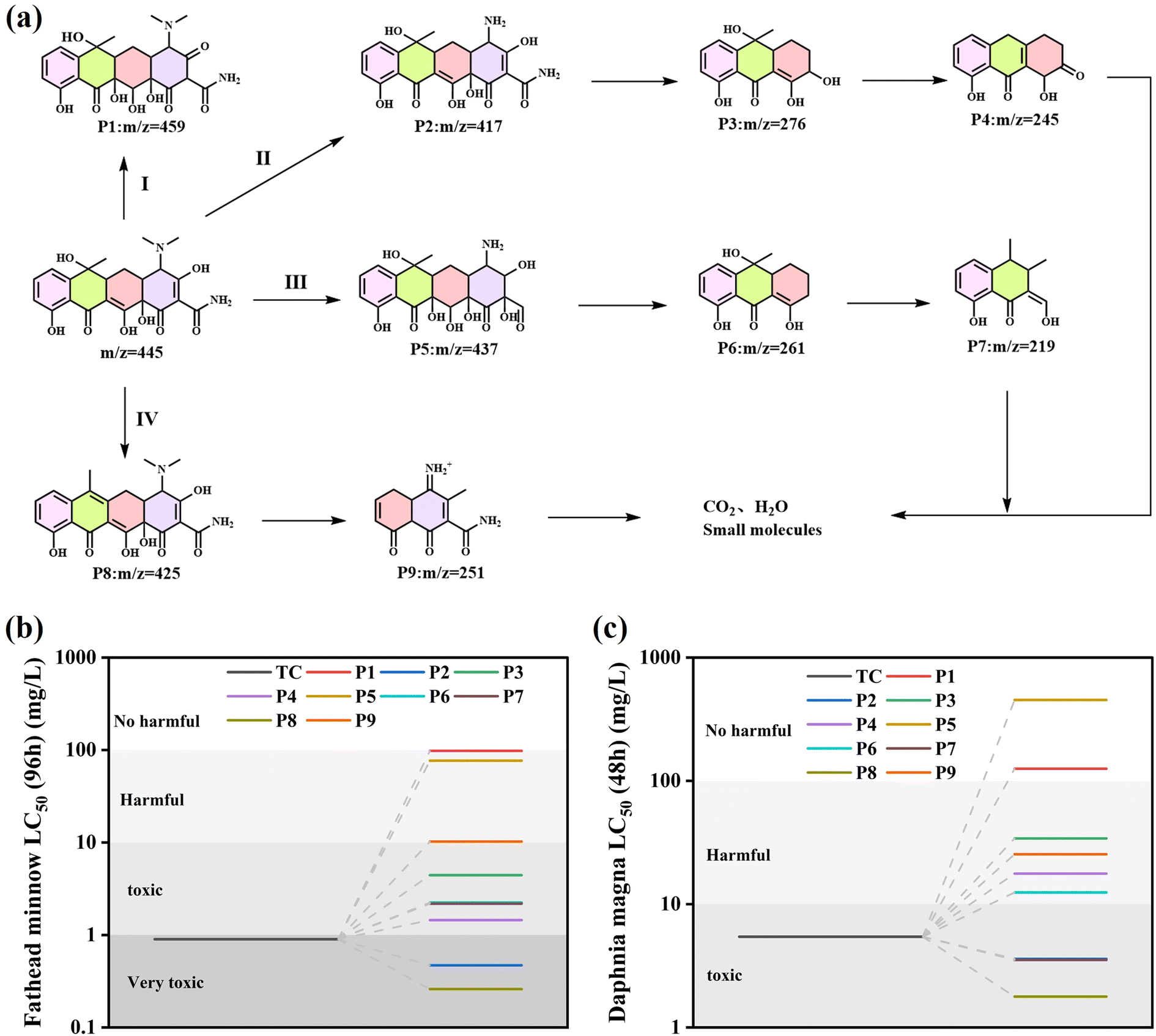
The intermediates of TC degradation by CeO2/Bi2S3-2 at different light durations were explored by mass spectrometry (MS). The peak that was recorded at an m/z value of 445 was linked to the original molecule TC. As the duration of light exposure increased, the intensity at m/z = 445 significantly diminished. In addition, peaks at m/z values of 459, 437, 425, 417, 276, 261, 251, 245 and 219 were identified, with the related MS spectra displayed in Fig. S1(a)–(f).† Based on the detected intermediates and available literature, a pathway for TC degradation was raised (Fig. 11(a)). In pathway I, the TC molecule undergoes a process known as hydroxylation, which results in the formation of a new compound designated as P1 (m/z = 459). In pathway II, the N–C bond's low binding energy makes it susceptible to destruction, leading to demethylation that produces P2 (m/z = 417). Subsequently, P2 experiences a ring-opening reaction, yielding P3 (m/z = 276), which further experiences dehydroxylation and dehydration to form P4 (m/z = 245). In pathway III, the TC molecule experiences several hydroxylation and demethylation processes to produce P5 (m/z = 437). This process subsequently entails a ring-opening reaction followed by dehydroxylation, resulting in the generation of a product designated as P6 (m/z = 261). Subsequently, P6 also undergoes ring-opening and dehydroxylation, resulting in the formation of P7 (m/z = 219). For pathway IV, the TC molecule dehydroxylates one hydroxyl group to produce P8 (m/z = 425), which in turn generates P9 (m/z = 251) by ring opening and demethylation and dehydroxylation. TC is broken down into intermediates with smaller molecular weights, which are further oxidized to small molecular compounds or even to CO2 and H2O.
 |
| | Fig. 11 (a) Possible intermediates and pathways for TC degradation by CeO2/Bi2S3-2 photocatalysts; (b) fathead minnow LC50 (96 h); (c) Daphnia magna LC50 (48 h). | |
In addition, the toxicity of TC and intermediates was evaluated using the Toxicity Estimation Software (T.E.S.T), and the specific values are shown in Table S2.† TC was evaluated to be highly toxic to fathead minnow (LC50 = 0.9 mg L−1) and toxic to Daphnia magna (LC50 = 5.44 mg L−1). The LC50 of all products except P8 was higher than that of TC (Fig. 11(b)), and the LC50 of most products was higher than that of TC for Daphnia magna (Fig. 11(c)), which indicated that CeO2/Bi2S3-2 degraded TC into a low toxicity compound.
4 Conclusions
In this study, flower-shaped CeO2/Bi2S3 composite photocatalysts were synthesized using hydrothermal methods. Characterization using SEM, TEM and XRD confirmed that the CeO2/Bi2S3 is a heterojunction. Through XPS analysis, DRS analysis, DFT calculation and Mott–Schottky tests, the CeO2/Bi2S3 heterojunction was found to function according to a charge transfer mechanism of the S-scheme type, which enhances charge mobility and generates an internal electric field that effectively separates electron–hole pairs. This process maintains the redox capacity of the resultant beneficial electrons and holes, thereby improving photocatalytic degradation efficiency. Under optimal reaction conditions, a significant degradation of TC was observed following 120 min of exposure to light. The results indicated that the degradation rate reached an impressive 82.43%. Free radical trapping experiments and LC-MS analysis of degradation products revealed that TC degradation primarily involves ·O2− and h+ radicals, leading to the formation of CO2, H2O, and other small molecules through a series of ring-opening, demethylation, and dehydroxylation reactions. This research provides robust evidence for the photocatalytic breakdown of antibiotics through S-scheme heterojunctions and presents a viable approach for developing heterojunctions that integrate wide bandgap and narrow bandgap semiconductors.
Data availability
The data that has been used is confidencial.
Author contributions
Shanlin He: writing – original draft, investigation, writing – review & editing, formal analysis, data curation. Yawei Du: formal analysis, visualization. Chen Li: resources, supervision. Claudia Li: writing – review & editing, supervision. Jingde Li: writing – review & editing, supervision. Jaka Sunarso: writing – review & editing, supervision. Sibudjing Kawi: writing – review & editing, supervision. Yinhui Li: writing – review & editing, supervision, project administration, funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the China Scholarship Council (Grant No. 202106705005), A*STAR under its Low-Carbon Energy Research (LCER) Funding Initiative (FI) Project (WBS: A-8000278-00-00) and the National Research Foundation, Singapore, (U2102d2011).
References
- X. Jiang, S. Lai, W. Xu, J. Fang, X. Chen, J. Beiyuan, X. Zhou, K. Lin, J. Liu and G. Guan, J. Alloys Compd., 2019, 809, 151804 CrossRef CAS
 .
. - C. Deng, S. Li, K. N. Khattak, F. Li, H. Yang, F. Tang and X. Yang, J. Environ. Chem. Eng., 2023, 11, 110433 CrossRef CAS .
- H. Wang, B. Liao, T. Lu, Y. Ai and G. Liu, J. Hazard. Mater., 2020, 385, 121552 CrossRef CAS .
- A. Mohammad, M. E. Khan, M. H. Cho and T. Yoon, Appl. Surf. Sci., 2021, 565, 150337 CrossRef CAS .
- X. Liu, R. Ma, L. Zhuang, B. Hu, J. Chen, X. Liu and X. Wang, Crit. Rev. Environ. Sci. Technol., 2020, 51, 751–790 CrossRef .
- Q. Liao, H. Rong, M. Zhao, H. Luo, Z. Chu and R. Wang, Sci. Total Environ., 2021, 757, 143981 CrossRef CAS PubMed .
- Y. Tao, T. Xiao, Q. Fu, B. Miao, S. Hou, G. Peng, Y. Xiong and M. Tang, RSC Adv., 2025, 15, 7307–7317 RSC .
- N. El Messaoudi, Z. Ciğeroğlu, Z. M. Şenol, M. Elhajam and L. Noureen, J. Water Process Eng., 2023, 55, 104150 CrossRef .
- H. Li, Q. Guo, Y. Li, M. Fu, D. Tian and T. Qi, J. Environ. Chem. Eng., 2021, 9, 106252 CrossRef CAS .
- C. Chen, M. Li, Y. Jia, R. Chong, L. Xu and X. Liu, J. Colloid Interface Sci., 2020, 564, 442–453 CrossRef CAS .
- S. Sekar, C. Bathula, I. Rabani, J. W. Lee, S. H. Lee, Y. S. Seo and S. Lee, Ultrason. Sonochem., 2022, 90, 106177 CrossRef CAS .
- C. Lai, F. Xu, M. Zhang, B. Li, S. Liu, H. Yi, L. Li, L. Qin, X. Liu, Y. Fu, N. An, H. Yang, X. Huo, X. Yang and H. Yan, J. Colloid Interface Sci., 2021, 588, 283–294 CrossRef CAS PubMed .
- D. P.-H. Tran, S.-J. You and Y.-F. Wang, J. Environ. Chem. Eng., 2024, 12, 112667 CrossRef CAS .
- H. Pei, Q. Jia, R. Guo, T. Zhang, N. Liu and Z. Mo, Colloids Surf., A, 2022, 648, 129256 CrossRef CAS .
- X. Chen, Y. Wu, Y. Tang, P. Li, S. Gao, Q. Wang, W. Liu and S. Zhan, Chin. Chem. Lett., 2024, 35, 109245 CrossRef CAS .
- A. Wang, Z. Zheng, H. Wang, Y. Chen, C. Luo, D. Liang, B. Hu, R. Qiu and K. Yan, Appl. Catal., B, 2020, 277, 119171 CrossRef CAS .
- S. Zhang, D. Han, Z. Wang and F. Gu, Small, 2024, 20, e2309656 CrossRef .
- L. Wolski, K. Grzelak, M. Muńko, M. Frankowski, T. Grzyb and G. Nowaczyk, Appl. Surf. Sci., 2021, 563, 150338 CrossRef CAS .
- X. Yang, Y. Zhang, Y. Wang, C. Xin, P. Zhang, D. Liu, B. B. Mamba, K. K. Kefeni, A. T. Kuvarega and J. Gui, Chem. Eng. J., 2020, 387, 124100 CrossRef CAS .
- Y. Zhu, H. Qian, A. M. Alnuqaydan, S. M. Siddeeg, B. S. Abdullaeva and A. Shawabkeh, J. Water Process Eng., 2024, 57, 104703 CrossRef .
- H. Wu, Q. Sun, J. Chen, G.-Y. Wang, D. Wang, X.-F. Zeng and J.-X. Wang, Chem. Eng. J., 2021, 425, 130640 CrossRef CAS .
- C.-H. Shen, X.-J. Wen, Z.-H. Fei, Z.-T. Liu and Q.-M. Mu, Chem. Eng. J., 2020, 391, 123612 CrossRef CAS .
- C. Ayappan and A. Mani, J. Water Process Eng., 2023, 51, 103373 CrossRef .
- H. Yu, D. Wang, B. Zhao, Y. Lu, X. Wang, S. Zhu, W. Qin and M. Huo, Sep. Purif. Technol., 2020, 237, 116365 CrossRef CAS .
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS .
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS .
- Y. Xu, X. Tang, Y. Xiao, H. Tang, H. Lin, Y. Lv and H. Zhang, Chemosphere, 2023, 331, 138765 CrossRef CAS PubMed .
- Q. Xu, S. Wageh, A. A. Al-Ghamdi and X. Li, J. Mater. Sci. Nanotechnol., 2022, 124, 171–173 CrossRef .
- C. Feng, J. Rong, Y. Zhang, X. Zheng, X. Li, S. Xu and Z. Li, Appl. Catal., B, 2023, 337, 123005 CrossRef CAS .
- R. Ning, H. Pang, Z. Yan, Z. Lu, Q. Wang, Z. Wu, W. Dai, L. Liu, Z. Li, G. Fan and X. Fu, J. Hazard. Mater., 2022, 435, 129061 CrossRef CAS .
- W. Wang, H. Cheng, B. Huang, X. Lin, X. Qin, X. Zhang and Y. Dai, J. Colloid Interface Sci., 2013, 402, 34–39 CrossRef CAS .
- J. Huang, B. Zhu, D. Song, B. Wang, L. Chen, L. Lu, Q. Chen, L. Gai, C. Zhai, L. Chen and H. Tao, Chem. Eng. J., 2023, 464, 142784 CrossRef CAS .
- P. Latifian, S. F. Hosseini, M. S. Seyed Dorraji and M. H. Rasoulifard, J. Mol. Liq., 2023, 376, 121445 CrossRef CAS .
- R. Rajendran, O. Rojviroon, P. Arumugam, K. Natchimuthu, V. Vasudevan, J. Kannupaiyan, R. Muangmora, P. Phouheuanghong and T. Rojviroon, J. Alloys Compd., 2024, 976, 173116 CrossRef CAS .
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS .
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS .
- J. P. Perdew, B. Kieron and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed .
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef .
- Y. Wang, Z. Xing, H. Zhao, S. Song, M. Liu, Z. Li and W. Zhou, Chem. Eng. J., 2022, 431, 133355 CrossRef CAS .
- M. Guo, Z. Xing, T. Zhao, Z. Li, S. Yang and W. Zhou, Appl. Catal., B, 2019, 257, 117913 CrossRef CAS .
- K. Saravanakumar, R. Karthik, S. M. Chen, J. Vinoth Kumar, K. Prakash and V. Muthuraj, J. Colloid Interface Sci., 2017, 504, 514–526 CrossRef CAS .
- D. Yang, Q. Ye, C. Qu, F. Meng, L. Wang and Y. Li, J. Environ. Chem. Eng., 2024, 12, 112563 CrossRef CAS .
- W. Liu, D. Zhong, Z. Dai, Y. Liu, J. Wang, Z. Wang and J. Pan, J. Alloys Compd., 2019, 780, 907–916 CrossRef CAS .
- P. Peng, Z. Chen, X. Li, Y. Wu, Y. Xia, A. Duan, D. Wang and Q. Yang, Sep. Purif. Technol., 2022, 291, 120901 CrossRef CAS .
- L. Liang, S. Gao, J. Zhu, L. Wang, Y. Xiong, X. Xia and L. Yang, Chem. Eng. J., 2020, 391, 123599 CrossRef CAS .
- F. Zhao, X. Li, T. Xiong, M. Zuo, L. Luo, P. Qin, M. Lei, Y. Liang, X. Gong, D. Zou and Z. Wu, Sep. Purif. Technol., 2023, 314, 123533 CrossRef CAS .
- V. V, J. K, M. Alsawalha, Z. Zhang, M. L. Fu and B. Yuan, J. Environ. Manage., 2023, 348, 119246 CrossRef CAS PubMed .
- K. Divakaran, A. Baishnisha, V. Balakumar, K. N. Perumal, C. Meenakshi and R. S. Kannan, J. Environ. Chem. Eng., 2021, 9, 105560 CrossRef CAS .
- X. Chen, J. Yao, B. Xia, J. Gan, N. Gao and Z. Zhang, J. Hazard. Mater., 2020, 383, 121220 CrossRef CAS .
- F. Chen, Q. Yang, X. Li, G. Zeng, D. Wang, C. Niu, J. Zhao, H. An, T. Xie and Y. Deng, Appl. Catal., B, 2017, 200, 330–342 CrossRef CAS .
- H. Zhang, J. Xu, Y. Yuan, Y. Guo, X. Tan, H. Wang, X. Hu and C. Tang, Sep. Purif. Technol., 2024, 330, 125520 CrossRef CAS .
- S. Li, S. Hu, W. Jiang, Y. Liu, Y. Zhou, J. Liu and Z. Wang, J. Colloid Interface Sci., 2018, 530, 171–178 CrossRef CAS .
- J. Chen, Y. Xiao, N. Wang, X. Kang, D. Wang, C. Wang, J. Liu, Y. Jiang and H. Fu, Sci. China Mater., 2023, 66, 3165–3175 CrossRef CAS .
- B. Tan, Y. Fang, Q. Chen, X. Ao and Y. Cao, J. Colloid Interface Sci., 2021, 601, 581–593 CrossRef CAS .
- M. Kaur, S. Singh, S. K. Mehta, S. K. Kansal, A. Umar, A. A. Ibrahim and S. Baskoutas, J. Alloys Compd., 2023, 960, 170637 CrossRef CAS .
- L. Chen, B. Xu, M. Jin, L. Chen, G. Yi, B. Xing, Y. Zhang, Y. Wu and Z. Li, J. Mol. Struct., 2023, 1278, 134911 CrossRef CAS .
- J. Huang, M. Lu, M. Liu, Y. Xie, P. Wei, H. Xie, L. Li, W. Han, Z. Zhang and Y. Qi, J. Environ. Manage., 2025, 379, 124893 CrossRef CAS .
- B. Wu, C. Shan, X. Zhang, H. Zhao, S. Ma, Y. Shi, J. Yang, H. Bai and Q. Liu, Appl. Surf. Sci., 2021, 543, 148677 CrossRef CAS .
- Y. Liu, J. Yang, B. Wu, W. Zhang, X. Zhang, C. Shan and Q. Liu, Colloids Surf., A, 2020, 586, 124193 CrossRef CAS .
- L. Jing, Y. Xu, M. Xie, Y. Liu, X. Du and J. Hu, J. Alloys Compd., 2024, 979, 173568 CrossRef CAS .
- B. Caglar, E. K. Guner, S. Ersoy, S. Caglar, A. O. Özdemir, K. V. Özdokur, B. Doğan, F. İçer and Ç. Çırak, J. Alloys Compd., 2021, 885, 160964 CrossRef CAS .
- M. Chen, X. Cai, Q. Yang, W. Lu, Z. Huang, T. Gan, H. Hu and Y. Zhang, Sep. Purif. Technol., 2023, 314, 123546 CrossRef CAS .
- Z. Lu, G. Zhou, M. Song, D. Wang, P. Huo, W. Fan, H. Dong, H. Tang, F. Yan and G. Xing, J. Mater. Chem. A, 2019, 7, 13986–14000 RSC .
- Z. Shang, T. Wang, A. Ren, Y. Yu, Y. Zheng, Y. Tao, P. Feng, Y. Xiao and X. Wang, Appl. Surf. Sci., 2023, 619, 156718 CrossRef CAS .
- Y. Liu, X. Zhang, X. Li and Z. Zhou, J. Water Process Eng., 2024, 57, 104588 CrossRef .
- H. Zhang, Y. H. Sun, S. L. An, R. H. Guo, R. F. Wang and Y. W. Ma, RSC Adv., 2025, 15, 8541–8552 RSC .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Yawei Du‡a,
Chen Li*b,
Claudia Li
a,
Yawei Du‡a,
Chen Li*b,
Claudia Li