
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic synthesis of four stereoisomers of 3-substituted-4-hydroxypiperidines by carbonyl reductase-catalyzed reduction†
Jing Chenab,
Xi Chen *bc,
Jinsong Songbc,
Hongliu Zhangbc,
Hongjiang Yanga,
Jinhui Fengbc,
Qiaqing Wu*bc and
Dunming Zhubcd
*bc,
Jinsong Songbc,
Hongliu Zhangbc,
Hongjiang Yanga,
Jinhui Fengbc,
Qiaqing Wu*bc and
Dunming Zhubcd
aSchool of Biotechnology, Tianjin University of Science and Technology, Tianjin 300457, China
bTianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China. E-mail: chen_x@tib.cas.cn; wu_qq@tib.cas.cn
cNational Center of Technology Innovation for Synthetic Biology and Tianjin Engineering Research Center of Biocatalytic Technology, Tianjin 300308, China
dState Key Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin 300308, China
First published on 26th June 2025
Abstract
Chiral 3-substituted-4-hydroxypiperidines are highly valuable in pharmaceutical applications due to their diverse and potent biological activities. Biocatalytic ketone reduction by carbonyl reductases represents a promising approach for synthesizing these compounds. In this study, two structurally similar yet stereoselectively distinct carbonyl reductases, HeCR and DbCR, were identified. Both enzymes exhibited exceptional catalytic performance, with >99% enantiomeric excess (ee) and >99% conversion rate in the reduction of tert-butyl 4-oxo-3-phenylpiperidine-1-carboxylate (1a). We found that 1a exhibits a relatively low rate of racemization under the mild reaction conditions. Subsequently, analogs of 1a were synthesized and reduced with high enantioselectivity (ee > 99%) using HeCR and DbCR. Carbonyl reductases demonstrated excellent catalytic activity and stereoselectivity in the synthesis of 3-substituted-4-hydroxypiperidines with dual chiral centers, underscoring their potential for pharmaceutical applications.
Introduction
3-Substituted-4-hydroxypiperidines with two chiral centres play an important role in the synthesis of chiral drugs. As critical building blocks, 3-substituted-4-hydroxypiperidine have been used to synthesize GSK 484 and Compound WG, which both serve as enzyme inhibitors for the treatment of tumors – MIBT for the treatment of obesity-associated disorders, and ROMK-IN-2 for the treatment of hypertension and heart failure (Fig. 1A).1,2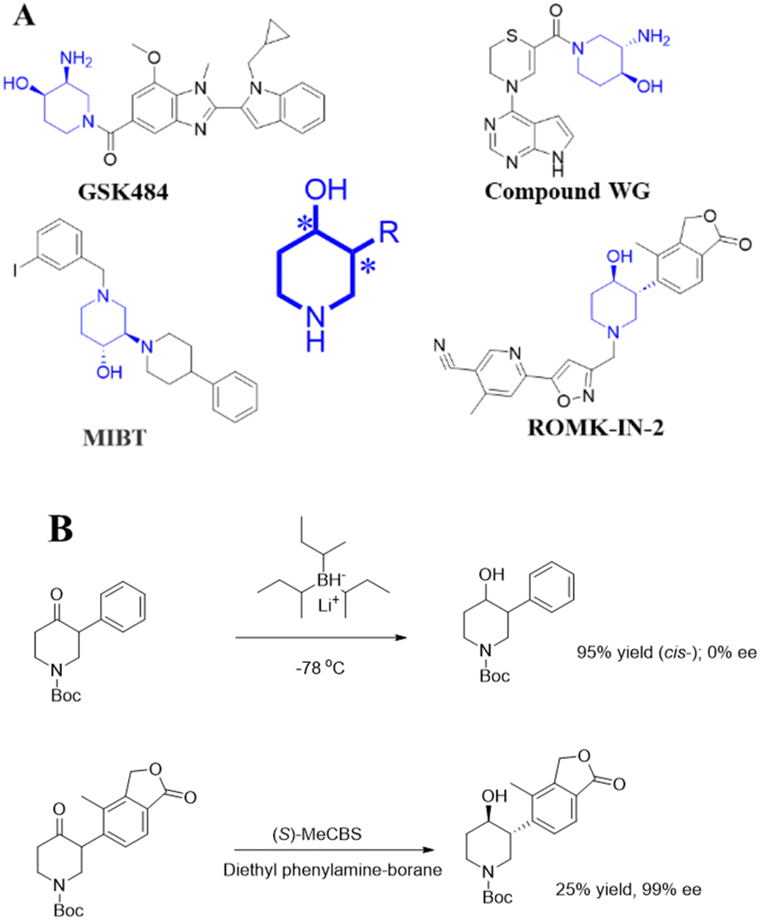 | ||
| Fig. 1 3-Substituted-4-hydroxypiperidine and its application (A); synthesis of chiral 3-substituted-4-hydroxypiperidine via chemical reduction (B). | ||
Despite the significance of 3-substituted-4-hydroxypiperidines, the precise construction of all four stereoisomers remains a significant challenge. While chemical asymmetric reduction is the predominant method, it often involves toxic and hazardous reagents, and/or harsh reaction conditions.3–6
For example, the reduction of piperidone at very low temperature by L-selectride gave the cis products with 98% yield and 0% ee.7 Using (S)-MeCBS as a catalyst, along with borane and N,N-diethylaniline as reducing agents, achieved an enantiomeric excess (ee) of >99% for the trans isomer, but with only 25% yield (Fig. 1B).3
Due to their mild reaction conditions and excellent stereoselectivity, carbonyl reductases have become the preferred catalysts for synthesizing chiral alcohols through ketone reduction.8–15 However, to date, no carbonyl reductase has been reported for the reduction of 3-substituted 4-oxo-piperidine. As part of our ongoing industrial enzymatic ketone reduction program,16–18 we tested our carbonyl reductase tool-box using tert-butyl 4-oxo-3-phenylpiperidine-1-carboxylate (1a) as the model substrate. Two carbonyl reductases with distinct stereoselectivity were selected for the reduction of a series of 1a analogs, affording all the four stereoisomers of 3-substituted-4-hydroxypiperidines by simple column chromatographic separation.
Results and discussion
To obtain a single isomer of 3-substituted-4-hydroxypiperidines through dynamic kinetic asymmetric reduction (DKAR) of 3-substituted-4-oxo-piperidines, three essential requirements must be met: first, the substrate must undergo in situ racemization (keto–enol tautomerism in 3-substituted-4-oxo-piperidines); second, the enzyme must exhibit substrate specificity (specificity in recognizing between (R)-1a with (S)-1a); third, the enzyme has excellent stereoselectivity for the reduction of the carbonyl group. We hypothesized that the keto–enol tautomerism of 1a would proceed rapidly due to stabilization of the enol tautomer by from the p-electron delocalization with 3-phenyl group. Initially, the carbonyl reductases expressed by available in our laboratory were screened using 1a as the substrate. In the initial screening using Escherichia coli wet cells expressing the target carbonyl reductase genes, a mixture of NAD+ and NADP+ and glucose dehydrogenase/glucose were added for cofactor recycling. Since the cis- and trans-products could be distinguished by thin-layer chromatography (TLC), both enzyme activity and diastereoselectivity were initially evaluated by TLC. Most of the tested enzymes showed low conversion at the substrate concentration of 10 mM (2.75 g L−1). The reactions showing obvious product formation were further analyzed by HPLC, and the results are summarized in ESI Table S1.† Subsequently, carbonyl reductases with high conversion rates were rescreened using NAD+ or NADP+ as cofactor, respectively, and the results of the reactions with >98% conversion rate are summarized in Table 1.
|
|||||
|---|---|---|---|---|---|
| Enzyme | Protein identifier | Cis-ee [%] | Trans-ee [%] | Cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Trans [%] :Trans [%] |
Conversion [%] |
| a Reaction conditions: potassium phosphate buffer (500 μL, 100 mM, pH 7.0) contained 10 mM substrate 1a, 20 mM glucose, 10 U mL−1 GDH (glucose dehydrogenase), 0.5 mg mL−1 NAD+ or NADP+, 5% DMSO (v/v) and 5 mg Escherichia coli whole cells with carbonyl reductases genes at 30 °C for 24 h; cis-:trans- and ee values were determined by HPLC.b NAD+ was added.c NADP+ was added. |
|||||
| HeCRb | WP_116436716.1 | >993R,4S | >993S,4S | 49:51 |
>99 |
| HeCRc | WP_116436716.1 | >993R,4S | >993S,4S | 47:53 |
>99 |
| DbCRc | TMB03420.1 | >993S,4R | >993R,4R | 45:55 |
>99 |
| NsCRc | WP_152591762.1 | >993S,4R | >993R,4R | 47:53 |
>99 |
| NsCRb | WP_152591762.1 | 973S,4R | >993R,4R | 47:53 |
>99 |
| SmADHc | ABB91667.1 | >993R,4S | 933S,4S | 48:52 |
98 |
| PcCR1c | KII83904.1 | >993S,4R | >993R,4R | 48:52 |
>99 |
| PcCR2b | KII83918.1 | 983S,4R | >993R,4R | 47:53 |
>99 |
| PcCR2c | KII83918.1 | 973S,4R | >993R,4R | 47:53 |
>99 |
| ScCRb | AGJ03541.1 | 893S,4R | >993R,4R | 38:62 |
98 |
| CgCRc | TWG88880.1 | 973R,4S | 823S,4S | 49:51 |
98 |

Product 2a was prepared using HeCR and DbCR as catalyst, respectively. The cis- and trans-products could be separated by thin-layer chromatography (TLC), enabling the isolation of all four isomers of 2a. The absolute configuration of (3R, 4S)-2a and (3R, 4R)-2a were determined by X-ray crystallography.
As shown in Table 1, the carbonyl reductases HeCR, DbCR, NsCR and PcCR1 exhibited high conversion rates and ee values with NADH or NADPH, respectively. Notably, only HeCR demonstrated excellent S-configuration selectivity toward the carbonyl group. In contrast, DbCR, NsCR and PcCR1 were quite similar, and the reduction gave (4R)-configurated products.
Although the enzymes exhibited excellent stereoselectivity for the reduction of the carbonyl group, they lacked the ability to differentiate between (R)-1a with (S)-1a. Then, rude enzyme activities and protein expression levels of HeCR, DbCR, NsCR and PcCR1 were examined, as shown in Fig. S1 and Table S2.† Since HeCR and DbCR showed higher enzyme activity and completely opposite stereoselectivity, HeCR and DbCR were selected for further investigation.
Using Discovery Studio, R-1a and S-1a were docked into the active sites of HeCR and DbCR to shed some insights in the observes stereoselectivity of these enzymes (Fig. S3–S6†).
Based on the catalytic mechanism of short-chain dehydrogenases, S137 (HeCR) stabilizes and polarizes the carbonyl group of the substrate, the hydride of C4 of the nicotinamide ring of NADPH attacks the carbonyl carbon of 1a, while the phenol group of Y150 donates the proton to the carbonyl oxygen, resulting in reduction of the carbonyl group (Fig. S3†).19 To verify the speculation, mutants S137A and Y150A were obtained, and the results showed these two mutants both lost the enzyme activities toward 1a, which was consistent with the catalytic mechanism. The catalytic efficiency of the enzyme was thus affected by the mean catalytic distance of hydride transfer (NADPH-1a) and the distances of S137-(R/S)-1a and Y150-(R/S)-1a.
Molecular docking analysis of (R)-1a and (S)-1a into the protein structures demonstrated that both enantiomers could be effectively accommodated by HeCR and DbCR. As illustrated in Fig. S1–S4,† the distances from (R)-1a to the key residues S137/Y150 (HeCR) and NADPH were comparable to those from or (S)-1a to S137/Y150 (HeCR) and NADPH, suggesting similar catalytic efficiency for both enantiomers. Similar computational results were obtained with DbCR. These results are aligned with the nearly 1:1 cis/trans ratio observed in the enzyme reactions. Therefore, we planned to enhance substrate specificity toward (R)-1a or (S)-1a through reshaping the enzyme substrate binding site. HeCR showed highest enzyme activity among the selected carbonyl reductase, considering that the mutation might affect the enzyme activity, HeCR was selected as the template for enzyme engineering.
Based on the docking results, specific amino acid residues (I91, A135, S136, I138, N140, Y147, L178, I187, and I188) interacting with (R/S)-1a were identified and selected for mutagenesis to enhance its specificity toward (R)-1a or (S)-1a (Fig. 2).
 | ||
| Fig. 2 Flexible docking (R)-1a into the active site of HeCR. | ||
Given the reversible nature of carbonyl reductase-catalyzed reactions, single stereoisomers of 2a were prepared with HeCR and DbCR, and employed as the substrate to screen enzyme activity and stereoselectivity by monitoring NADPH generation rate, respectively. Using this approach, all the saturation mutation libraries of the 9 amino acid residues were screened. Unexpectedly, among all the mutants, only HeCR Y147C exhibited significantly improved activity for the oxidation of the (3R,4S)-2a.
Subsequently, mutant Y147C was used as the catalyst for the reduction of 10 mM 1a, and the reaction progress was monitored using HPLC. Initially, the de and ee of (3R,4S)-2a were high (>99%), but the de value gradually decreased to 4% after 20 h (Table S4†). This indicated that mutant Y147C did not have good ability to differentiate between (R)-1a with (S)-1a. HPLC analysis revealed that the ee value of substrate 1a was not 0%, which attributed to the low racemization rate of 1a under the reaction conditions. To conform this observation, a preparative-scale reaction was conducted to obtain (S)-1a. After 10 minutes, the reaction mixture was extracted with ethyl acetate, and a mixture of (S)-1a (ee 81%) and (3R,4S)-2a (de 56%) was obtained. The racemization rate of (S)-1a (ee 81%) under carbonyl reduction conditions was monitored by HPLC, the ee of 1a decreased from 81% to 58% over 5 h (Table S5†). The results showed the in situ racemization of 1a was too slow, which rendered the efficient dynamic kinetic reduction impractical. Then the comparisons of the structures of HeCR with NsCR and PcCR1 were performed (Fig. S7–S9†), the corresponding site of HeCR for DbCR (F147), NsCR(Y151) selected for saturated mutation, however no mutant with significant improvement on the stereoselectivity between (R)/(S)-1a.
Because the cis- and trans-2a could be separated by column chromatography, HeCR and DbCR were used as catalysts to obtain the four isomers of 3-substituted-4-hydroxypiperidines. According to the literature,20–23 substrates (1b–1e) bearing para-substituted phenyl group were synthesized. However, attempts to synthesize the substrates with ortho- and meta-substituent on the phenyl group was unsuccessful. Using HeCR and DbCR as biocatalyst, reductions 1b–1e at 5 mM substrate concentration were conducted at 30 °C for 24 h, and the conversion and product ratio were determined by HPLC analysis. As show in Table 2, these two enzymes showed excellent stereoselectivity toward the ketone reduction with >99% ee, but the 3-chirality could not be differentiated. The preparative-scale reactions were performed at 10 mM substrate concentration (100 mL) using HeCR and DbCR as biocatalyst, respectively. All the four enantiomers of 2a–2e were isolated by column chromatography with >99% de and >99 ee with 24–48% yield. We tried to crystalize all of the products to determine their absolute configurations, but only the absolute configurations of the isomers of 2d were determined by X-ray crystallography.
|
||||||
|---|---|---|---|---|---|---|
| Substrates | Cis-ee [%] | Trans-ee [%] | Cis-:Trans- [%] |
Conversion [%] | Yield of (3R,4S)b [%] | Yield of (3S,4S)b [%] |
| a Reaction condition: potassium phosphate buffer (500 μL, 100 mM, pH 7.0) contained 5 mM substrate 1a–1e, 10 mM glucose, 0.1 mg mL−1 GDH, 0.5 mg mL−1 NADP+, 5% DMSO (v/v) and 10 mg E. coli whole cells of HeCR and DbCR at 30 °C for 24 h, respectively. De and ee values were determined by HPLC.b The isolated yields were from the preparative reactions: potassium phosphate buffer (100 mL, 100 mM, pH 7.0) contained 10 mM substrate 1a–1e, 25 mM glucose, 0.5 mg mL−1 GDH, 0.5 mg mL−1 NADP+, 5% DMSO (v/v) and 40 mg mL−1 whole cells of HeCR and DbCR at 30 °C for 48 h. | ||||||
| HeCR | ||||||
| 1a | >99 | >99 | 47:53 |
>99 | 48 | 47 |
| 1b | >99 | >99 | 59:40 |
87 | 41 | 37 |
| 1c | >99 | >99 | 50:50 |
>99 | 46 | 44 |
| 1d | >99 | >99 | 52:48 |
97 | 29 | 35 |
| 1e | >99 | >99 | 49:51 |
99 | 43 | 32 |

Conclusions
In summary, given the significance of chiral 3-substituted-4-hydroxypiperidines, we developed a method for the preparation of four stereoisomers of 3-substituted-4-hydroxypiperidines via reduction of tert-butyl 4-oxo-3-arylpiperidine-1-carboxylate (1a–1e) using HeCR or DbCR, respectively. These enzymes lack the ability to differentiate the chirality at 3-position, an attempt to improve this by protein engineering has failed. In addition, a relatively low racemization rate of 1a was observed under the reaction conditions. These two factors prevented the achievement of DKAR to obtain a single stereoisomer. As such, further studies are needed to achieve DKAR by improving the enzyme capability of recognizing the chirality at 3-position and realizing deracemization of the substrate under relatively mild reaction conditions.Experiment section
Materials
The materials used for culture media, including peptone, yeast extract and agar, were purchased from Becton, Dickinson and Company. The LB medium contained 10 g L−1 tryptone, 5 g L−1 yeast extract, and 10 g L−1 NaCl. Ampicillin were purchased from Beijing Probe Bioscience Co. Ltd. Isopropyl-β-D-thiogalactopyranoside (IPTG) was purchased from AMRESCO Inc. The NMR spectra were measured on a Bruker Avance 400 using CDCl3 as the solvent. Commercial chemicals were purchased from Aladdin Co. HPLC analysis was performed on an Agilent 1200 series HPLC system. The racemic alcohol standard samples were prepared by the reduction of the corresponding ketones with sodium borohydride.Screening of carbonyl reductases using 1a as the substrate
The carbonyl reductases available in our laboratory were screened with 1a as the substrate. The strains were inoculated into 96-well plate which contained 400 μL LB medium per well (50 μg mL−1 Ampicillin). After being cultivated for 12 h in a 37 °C shaker, 100 μL seed culture was transferred to a new 96 deep-well plate, containing 900 μL LB medium per well (50 μg mL−1 Ampicillin and 0.1 mM IPTG). The new 96 deep-well plate was placed into a 25 °C shaker for further cultivation for 15 h, and then centrifuged for 15 min at 4 °C and 4629 rcf. to collect the cell pellets. They were then frozen at −80 °C for more than 4 h. Then the frozen wet cells were used to construct 1 mL reaction system. Into potassium phosphate buffer (1 mL, 100 mM, pH 7.0) containing 5% DMSO (v/v), 10 mM substrate, 20 mM glucose, and 0.5 mg mL−1 NAD(P)+, 10 mg mL−1 of wet E. coli whole cells expressing the target enzymes and 10 U mL−1 GDH were added. The reaction was performed at 30 °C for 24 h and then stopped by adding an equal amount of ethyl acetate to extract. To extract completely, the plates were continued shaken at 25 °C for about 30 min. TLC detection is used to preliminarily determine the reaction conversion rate and de value. The enzymes showing high conversion rates were rescreened with NAD+ and NADP+ respectively while keeping other components unchanged. The reaction mixture was extracted with an equal volume of ethyl acetate. After the solvent was evaporated, the residue was dissolved in a mixture of isopropanol and hexane, dried over anhydrous Na2SO4. The conversion and stereoisomeric ratio of the product were determined by HPLC analysis using a Chiralcel AS-H column, 0.9 mL min−1, n-hexane:iso-propanol = 98:2, 220 nm.
Screening of crude enzyme activities of HeCR, DbCR, NsCR and PcCR1
Reaction system: potassium phosphate buffer (150 μL, 100 mM, pH 7.0) contained 2 mM substrate 1a, 0.5 mg mL−1 NADPH, 10% DMSO (v/v) and 50 μL of crude enzyme extract (supernatant after centrifugation of ultrasonically lysed 10 mg E. coli cells expressing the carbonyl reductase gene). Relative enzymatic activities were measured at 340 nm by monitoring NADPH consumption.Preparation of racemate 2a
Compound 1a (200 mg) was dissolved in 5 mL methanol, and sodium borohydride (28 mg, 1 eq.) was added. The reaction was monitored by TLC until no substrate remained. The solvent was evaporated under reduced pressure. The residue was treated with 5 mL of saturated ammonium chloride, followed by extraction with ethyl acetate (5 mL × 3). The combined extract was concentrated under reduced pressure. The crude product was dissolved in dichloromethane, and purified by column chromatography (petroleum ether/ethyl acetate = 9:1).
cis-2a: 1H NMR (400 MHz, CDCl3) δ 7.35 (t, J = 7.5 Hz, 2H), 7.25 (dd, J = 12.8, 6.2 Hz, 3H), 4.10 (d, J = 3.4 Hz, 1H), 3.95 (s, 1H), 3.49 (s, 1H), 3.21 (d, J = 12.3 Hz, 1H), 2.89 (d, J = 11.9 Hz, 1H), 1.86 (d, J = 4.0 Hz, 2H), 1.46 (s, 9H).
anti-2a: 1H NMR (400 MHz, CDCl3) δ 7.36 (t, J = 7.4 Hz, 2H), 7.31–7.24 (m, 3H), 4.41–4.02 (m, 2H), 3.86 (td, J = 10.4, 4.4 Hz, 1H), 2.93–2.69 (m, 2H), 2.58 (td, J = 10.9, 4.1 Hz, 1H), 2.10–2.00 (m, 1H), 1.69 (s, 1H), 1.57 (qd, J = 12.9, 12.4, 3.8 Hz, 1H), 1.46 (s, 9H).
Preparation of chiral 2a
E. coli BL21 (DE3) cells expressing HeCR or DbCR gene were cultivated in 5 mL of LB medium containing 50 μg mL−1 ampicillin overnight at 37 °C. The overnight culture was inoculated into 800 mL of LB medium containing 50 μg mL−1 ampicillin and grown at 37 °C in 200 rpm shaker until the culture's optical density OD600 reached 0.6–0.8. The expression of the enzyme was induced by the addition of 0.1 mM IPTG and then continue to culture for 16 h at 25 °C in 200 rpm shaker. The cells were harvested by centrifugation (4629 rcf, 10 min, 4 °C), and added into 100 mL potassium phosphate buffer (100 mM, pH 7.0) containing 10 mM 1a, 25 mM glucose, 0.5 mg mL−1 GDH, 0.5 mg mL−1 NADP+, and 5% DMSO (v/v). The reaction was performed at 30 °C for 24 hours, followed by extraction with ethyl acetate (3 × 100 mL). The solvent was removed under reduced pressure, the product was dissolved in dichloromethane and purified by column chromatography (petroleum ether/ethyl acetate = 9:1). The pure product was dissolved in ethyl acetate, then place the solution in a cool, windless place, and the solvent was slowly evaporated to afford crystals. The deposition number in CCDC were 2426453 ((3R,4S)-2a) and 2214401 ((3R,4R)-2a).
Homology modeling and molecular docking of HeCR, DbCR and NsCR
The structures of HeCR and DbCR were modeled using ColabFold according to the literature,24 and the coenzyme NADPH was retrieved form carbonyl reductase SSCR (PDB: 1Y1P). Flexible dockings of (R)- and (S)-1a into HeCR and DbCR using a flexible docking protocol of Discovery studio. Other parameters were kept on default settings in flexible docking. The suitable conformations with the lower energy were chosen for the analysis of substrate–enzyme interactions and the distance between the carbonyl group of the substrate and C4–H of NADPH. Amino acid residues within the active sites of the two proteins were compared. Based on these comparisons, specific residues I91, A135, S136, I138, N140, Y147, L178, I187, and I188 were selected as mutation sites to investigate their roles in substrate binding and catalysis.Construction of HeCR mutant libraries
The mutant libraries of single site were generated by site-saturation mutagenesis (168 clones of each library). First, the template plasmids were extracted with Plasmid Extraction Kit and the concentrations were measured by BioSpectrometer and the appropriate primers were designed (Table S2†).Mutations were introduced by PCR using designed primers and Super Pfx DNA Polymerase. The PCR conditions for the short fragment were as follows: 98 °C, 2 min; 32 cycles (98 °C, 10 s; 58 °C, 30 s; 72 °C, 30 s); final extension at 72 °C for 5 min, while for mega-PCR: 98 °C, 2 min (98 °C, 10 s; 58 °C, 30 s; 72 °C, 4 min 30 s) × 29 cycles, and 72 °C for 10 min. The template was digested with restriction endonuclease Dpn I for 2 h at 37 °C and the PCR product was used to chemically transform competent cells of E. coli BL21(DE3). The recombinant cells were spread on LB agar plates containing 50 mg L−1 Ampicillin and cultured overnight at 37 °C.
Screening of HeCR mutant libraries
After successfully obtaining the mutant libraries, 168 colonies from each mutant library were picked into two 96-well plates which contained 400 μL LB medium per well (50 μg mL−1 ampicillin). After being cultivated for 12 h in a 37 °C shaker, 100 μL seed culture was transferred to a new 96 deep-well plate, containing 900 μL LB medium per well (50 μg mL−1 ampicillin and 0.1 mM IPTG). The new 96 deep-well plates were placed into a 25 °C shaker for further cultivation for 12 h, and then centrifuged for 15 min, at 4 °C and 4629 rcf. to collect the cell pellets. The cell pellets were resuspended in 500 μL glycine-sodium hydroxide buffer (100 mM, pH 11) with 1 mg mL−1 lysozyme and incubated at 37 °C for 2 h with shaking. They were then frozen at −80 °C for 4 h to obtain the cell-free extract. Four reaction mixtures were prepared, each containing 10% DMSO (v/v) of 2 mM 2a in distinct configurations, 0.5 mg mL−1 NADP+, and formulated in a sodium glycinate-sodium hydroxide buffer (100 mM, pH 11). The hydrolysis reactions were carried out in a final volume of 200 μL containing 100 μL of cell-free extract and 100 μL reaction mixture. The activity was measured by the amount of generated NADPH, which was monitored by the absorbance at 340 nm. The high-selective enzymes were screened by determining NADPH production rates, which reflected the reverse reaction rates of mutant enzymes toward distinct configurational product.Measuring the racemization rate of 1a
Into potassium phosphate buffer (30 mL, 100 mM, pH 7.0) containing 5% DMSO (v/v), 20 mM substrate, 50 mM glucose, and 0.5 mg mL−1 NADP+, 20 mg mL−1 of wet E. coli whole cells expressing HeCR and 10 U mL−1 GDH were added. The reaction was performed at 30 °C for 10 minutes, and then the mixture was extracted with ethyl acetate (30 mL ×3), and the combined organic phase was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the obtained solid was dissolved in 3 mL DMSO. 50 μL of DMSO solution was mixed with 450 μL of potassium phosphate buffer (100 mM, pH 7.0). The mixture was shaken at 30 °C, and samples were taken at 0, 30 minutes, 1, 1.5, 2, 3, and 5 h, and extracted by ethyl acetate. The extract was subjected to HPLC analysis.General method for the preparation of 3-substituted phenyl-4-piperidinones
Ethyl p-methylphenylacetate (5 g, 28 mmol) and 1,1-diethoxy-N,N-dimethylmethanamine (3.63 g, 30.5 mmol) were added in DMF (40 mL), and the reaction mixture was heated at 140 °C for 6 h. After the complete consumption of the substrate, ethyl β-alaninate hydrochloride (4.7 g, 30.5 mmol) was added gradually to the reaction mixture over 15 min, and the reaction was allowed to proceed at 80 °C for 2 h. The reaction mixture was cooled on ice, and 40 mL of acetic acid was added. Sodium triacetoxyborohydride (21.4 g, 101 mmol) was then added over 15 minutes at room temperature. The reaction was stirred at 50 °C for 2 h. After 80 mL ethyl acetate was added, the mixture was cooled on ice, and 5 M NaOH (100 mL) was added. The organic phase was separated, and the aqueous phase was further extracted with ethyl acetate (80 mL × 2). The combined organic phases were washed with saturated sodium chloride (NaCl, 200 mL) and dried over anhydrous Na2SO4. The organic phase was concentrated and redissolved in dichloromethane and purified by column chromatography (petroleum ether/ethyl acetate = 6:1) to obtain ethyl 3-((3-ethoxy-3-oxopropyl) amino)-2-(p-tolyl) propanoate.
Ethyl 3-((3-ethoxy-3-oxopropyl) amino)-2-(p-tolyl) propanoate (1 g, 3.3 mmol) was added slowly into THF (100 mL) which containing sodium tert-butoxide (17.3 g 18 mmol). After stirring under ice cooling for 30 minutes, the mixture was stirred at room temperature for 3 hours. Water (12 mL) was then added, and the mixture was stirred at 80 °C for 5 hours, followed by cooling. The reaction was quenched with saturated sodium bicarbonate, and the resulting mixture was extracted with ethyl acetate. The solvent was evaporated under reduced pressure to afford a mixture containing 3-(p-tolyl) piperidin-4-one.
A round-bottom flask equipped with a dropping funnel containing di-tert-butyl dicarbonate (Boc2O) (3.49 g 17.3 mmol) in THF (20 mL) was connected to a nitrogen balloon. 3-(p-tolyl)piperidin-4-one (3.27 g, 17.3 mmol) was dissolved in THF (100 mL) and triethylamine (3.49 g, 34.6 mmol). The flask was placed in an ice-water bath, and the Boc2O solution was added dropwise over 30 min. After an additional 10 min of stirring, the ice-water bath was removed, and the reaction mixture was stirred at room temperature for 4 h, followed by heating at 50 °C for 3 h. The reaction was quenched by adding saturated sodium bicarbonate solution (100 mL), and the product was extracted with ethyl acetate (100 mL × 3). The solvent was removed under reduced pressure, and the crude product was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 9:1) to obtain tert-butyl 4-oxo-3-(p-tolyl) piperidine-1-carboxylate.
tert-Butyl 3-(p-tolyl)-4-oxopiperidine-1-carboxylate (1b) (1.7 g white solid, 21% yield). 1H NMR (400 MHz, CDCl3) δ 7.15 (s, 2H), 7.07 (d, J = 7.8 Hz, 2H), 4.26 (s, 1H), 4.21–4.12 (m, 1H), 3.66 (t, J = 7.7 Hz, 1H), 3.51 (td, J = 15.5, 14.0, 8.5 Hz, 2H), 2.55 (d, J = 6.2 Hz, 2H), 2.34 (s, 3H), 1.50 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 207.25, 137.27, 132.50, 129.41, 128.46, 80.65, 56.03, 49.15, 43.62, 40.65, 28.44, 21.15. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H23NO3Na, 312.1576; found, 312.1567.
tert-Butyl 3-(4-methoxyphenyl)-4-oxopiperidine-1-carboxylate (1c) (1.1 g, white solid, 14% yield). 1H NMR (400 MHz, CDCl3) δ 7.10 (d, J = 8.2 Hz, 2H), 6.89 (d, J = 8.1 Hz, 2H), 4.25 (s, 1H), 4.17 (dd, J = 12.6, 6.2 Hz, 1H), 3.79 (s, 3H), 3.65 (dd, J = 10.0, 5.8 Hz, 1H), 3.50 (dq, J = 13.9, 8.1, 7.0 Hz, 2H), 2.55 (t, J = 5.9 Hz, 2H), 1.50 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 207.37, 158.96, 154.46, 129.64, 127.55, 114.15, 80.65, 55.58, 55.29, 49.28, 43.68, 40.64, 28.44. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H23NO4Na, 328.1525; found, 328.1550.
tert-Butyl 3-(4-fluorophenyl)-4-oxopiperidine-1-carboxylate (1d) (1.38 g white solid, 17% yield). 1H NMR (400 MHz, CDCl3) δ 7.15 (dd, J = 8.4, 5.6 Hz, 2H), 7.04 (t, J = 8.6 Hz, 2H), 4.28 (s, 1H), 4.20 (dt, J = 11.8, 5.1 Hz, 1H), 3.69 (dd, J = 10.2, 5.9 Hz, 1H), 3.47 (ddd, J = 14.2, 9.0, 4.9 Hz, 2H), 2.56 (q, J = 6.7, 4.7 Hz, 2H), 1.50 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 206.79, 154.39, 131.21, 130.28, 130.20, 115.70, 115.49, 80.80, 55.63, 49.33, 43.74, 40.70, 28.42. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H20FNO3Na, 316.1325; found, 316.1358.
tert-Butyl 3-(4-chlorophenyl)-4-oxopiperidine-1-carboxylate (1e) (2.05 g white solid, 13% yield). 1H NMR (400 MHz, CDCl3) δ 7.36–7.29 (m, 2H), 7.12 (dd, J = 8.4, 2.3 Hz, 2H), 4.28 (s, 1H), 4.20 (d, J = 13.4 Hz, 1H), 3.68 (t, J = 7.8 Hz, 1H), 3.53–3.41 (m, 2H), 2.56 (d, J = 6.2 Hz, 2H), 1.50 (d, J = 2.5 Hz, 9H). 13C NMR (100 MHz, CDCl3) δ 206.52, 154.37, 133.92, 133.52, 130.01, 128.86, 80.86, 55.77, 49.12, 43.74, 40.70, 28.42. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H20ClNO3Na, 332.1029; found, 332.1038.
Substrate scope study of HeCR and DbCR
Into potassium phosphate buffer (500 μL, 100 mM, pH 7.0) containing 5% DMSO (v/v), 10 mM substrate, 20 mM glucose, and 0.5 mg mL−1 NADP+, 20 mg mL−1 of wet E. coli whole cells expressing HeCR or DbCR and 10 U mL−1 GDH were added. The reaction was performed at 30 °C for 24 h, and the reaction mixture was extracted with an equal amount of ethyl acetate. After ethyl acetate was completely evaporated, the residue was redissolved using isopropanol and hexane. The resulting solution was dried with anhydrous Na2SO4. The conversion and stereoisomeric ratio of the product were determined by HPLC analysis using a Chiralcel AS-H column, 0.9 mL min−1, n-hexane:iso-propanol = 98:2, 220 nm.
Preparation of chiral 2b–2e
Into potassium phosphate buffer (100 mL, 100 mM, pH 7.0) containing 5% DMSO (v/v), 10 mM 2b/2c/2d/2e, 25 mM glucose, and 0.5 mg mL−1 NADP+, 40 mg mL−1 of wet E. coli whole cells expressing HeCR or DbCR and 10 U mL−1 GDH were added. The reaction was performed at 30 °C for 48 h, followed by extraction of the mixture with ethyl acetate (3 × 100 mL). The solvent was removed under reduced pressure, and the crude product was dissolved in dichloromethane and purified by column chromatography (petroleum ether/ethyl acetate = 9:1).
tert-Butyl (3R,4S)-4-hydroxy-3-(p-tolyl)piperidine-1-carboxylate ((3R,4S)-2b) (121 mg, white solid, 41% yield). 1H NMR (400 MHz, CDCl3) δ 7.16 (s, 2H), 7.14 (s, 2H), 4.07 (s, 1H), 4.06–4.00 (m, 1H), 3.94 (d, J = 13.4 Hz, 1H), 3.46 (t, J = 12.4 Hz, 1H), 3.19 (ddd, J = 15.2, 12.0, 3.6 Hz, 1H), 2.91–2.81 (m, 1H), 2.34 (s, 3H), 1.46 (d, J = 1.8 Hz, 9H). 13C NMR (100 MHz, CDCl3) δ 154.98, 137.38, 136.72, 129.48, 127.74, 79.56, 68.49, 46.08, 42.54, 41.67, 38.01, 32.10, 28.53, 21.06. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H25NO3Na, 314.1732; found, 314.1751. [α]20D 104.9 (c 1.0, CH3OH).
tert-Butyl (3S,4R)-4-hydroxy-3-(p-tolyl)piperidine-1-carboxylate ((3S,4R)-2b) (123 mg, white solid, 41% yield). 1H NMR (400 MHz, CDCl3) δ 7.14 (s, 2H), 4.07 (d, J = 3.2 Hz, 1H), 4.01 (s, 1H), 3.92 (s, 1H), 3.46 (t, J = 12.5 Hz, 1H), 3.19 (ddd, J = 15.2, 12.1, 3.6 Hz, 1H), 2.91–2.82 (m, 1H), 2.34 (s, 3H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 154.98, 137.39, 136.72, 129.48, 127.74, 79.56, 68.49, 46.09, 42.56, 41.64, 37.93, 32.11, 28.53, 21.06. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H25NO3Na, 314.1732; found, 314.1754. [α]20D – 117.1 (c 1.0, CH3OH).
tert-Butyl (3S,4S)-4-hydroxy-3-(p-tolyl)piperidine-1-carboxylate ((3S,4S)-2b) (108 mg, white solid, 37% yield). 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 2.3 Hz, 4H), 4.29–4.19 (m, 1H), 4.15 (d, J = 13.3 Hz, 1H), 3.83 (td, J = 10.4, 4.2 Hz, 1H), 2.82 (dt, J = 25.3, 13.1 Hz, 2H), 2.54 (td, J = 11.5, 11.0, 4.0 Hz, 1H), 2.34 (s, 3H), 2.04 (ddd, J = 10.3, 5.1, 2.5 Hz, 1H), 1.64–1.50 (m, 1H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 154.69, 137.22, 136.15, 129.71, 128.03, 79.84, 73.03, 51.06, 48.69, 42.79, 33.25, 29.75, 28.48, 21.10. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H25NO3Na, 314.1732; found, 314.1754. [α]20D – 27.8 (c 1.0, CH3OH).
tert-Butyl (3R,4R)-4-hydroxy-3-(p-tolyl)piperidine-1-carboxylate ((3R,4R)-2b) (128 mg, white solid, 43% yield). 1H NMR (400 MHz, CDCl3) δ 7.19–7.11 (m, 4H), 4.31–4.19 (m, 1H), 4.14 (s, 1H), 3.86–3.76 (m, 1H), 2.83 (d, J = 12.4 Hz, 2H), 2.59–2.48 (m, 1H), 2.34 (s, 3H), 2.10–1.98 (m, 1H), 1.55 (dd, J = 12.3, 3.4 Hz, 1H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 154.70, 137.21, 136.17, 129.71, 128.04, 79.85, 73.02, 51.04, 48.71, 42.77, 33.25, 29.74, 28.48, 21.11. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H25NO3Na, 314.1732; found, 314.1757. [α]20D 29.3 (c 1.0, CH3OH).
tert-Butyl (3R,4S)-4-hydroxy-3-(4-methoxyphenyl)piperidine-1-carboxylate ((3R,4S)-2c) (127 mg, yellow solid, 46% yield). 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 8.2 Hz, 2H), 6.89 (d, J = 8.2 Hz, 2H), 4.06 (d, J = 3.1 Hz, 1H), 4.00 (dd, J = 8.6, 5.3 Hz, 1H), 3.92 (d, J = 13.5 Hz, 1H), 3.80 (s, 3H), 3.43 (t, J = 12.4 Hz, 1H), 3.19 (ddd, J = 15.1, 12.1, 3.7 Hz, 1H), 2.85 (dt, J = 12.3, 3.2 Hz, 1H), 1.91–1.76 (m, 2H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 158.66, 154.97, 132.37, 128.83, 114.20, 79.57, 68.53, 55.30, 45.61, 42.62, 32.05, 28.52, 1.07. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H25NO4Na, 330.1681; found, 330.1697. [α]20D 97.4 (c 1.0, CH3OH).
tert-Butyl (3S,4R)-4-hydroxy-3-(4-methoxyphenyl)piperidine-1-carboxylate ((3S,4R)-2c) (156 mg, yellow solid, 46% yield). 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 8.3 Hz, 2H), 6.93–6.85 (m, 2H), 4.06 (d, J = 3.0 Hz, 1H), 4.01 (dd, J = 12.6, 3.3 Hz, 1H), 3.92 (d, J = 13.5 Hz, 1H), 3.80 (s, 3H), 3.43 (t, J = 12.4 Hz, 1H), 3.19 (td, J = 12.6, 3.7 Hz, 1H), 2.85 (dt, J = 12.1, 3.3 Hz, 1H), 1.83 (dt, J = 14.2, 3.0 Hz, 2H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 158.66, 154.97, 132.36, 128.83, 114.21, 79.57, 68.54, 55.30, 45.61, 42.69, 32.05, 28.52, 1.07. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H25NO4Na, 330.1681; found, 330.1699. [α]20D – 91.1 (c 1.0, CH3OH).
tert-Butyl (3S,4S)-4-hydroxy-3-(4-methoxyphenyl)piperidine-1-carboxylate ((3S,4S)-2c) (128 mg, yellow solid, 44% yield). 1H NMR (400 MHz, CDCl3) δ 7.19 (d, J = 8.2 Hz, 2H), 6.90 (d, J = 8.1 Hz, 2H), 4.24 (d, J = 13.7 Hz, 1H), 4.14 (d, J = 13.4 Hz, 1H), 3.80 (s, 4H), 2.85 (t, J = 13.3 Hz, 1H), 2.76 (t, J = 12.6 Hz, 1H), 2.59–2.47 (m, 1H), 2.13–1.98 (m, 1H), 1.63–1.51 (m, 1H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 158.97, 154.69, 131.13, 129.10, 114.42, 79.85, 73.06, 55.32, 50.57, 48.75, 42.77, 33.25, 28.48. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H25NO4Na, 330.1681; found, 330.1697. [α]20D – 31.6 (c 1.0, CH3OH).
tert-Butyl (3R,4R)-4-hydroxy-3-(4-methoxyphenyl)piperidine-1-carboxylate ((3R,4R)-2c) (125 mg, yellow solid, 37% yield). 1H NMR (400 MHz, CDCl3) δ 7.24–7.14 (m, 2H), 6.94–6.85 (m, 2H), 4.24 (d, J = 13.7 Hz, 1H), 4.14 (d, J = 13.5 Hz, 1H), 3.81 (s, 4H), 2.87 (d, J = 13.2 Hz, 1H), 2.78 (dd, J = 22.9, 10.5 Hz, 1H), 2.53 (ddd, J = 13.9, 10.5, 4.0 Hz, 1H), 2.09–1.99 (m, 1H), 1.64–1.49 (m, 1H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 158.99, 154.72, 131.17, 129.14, 114.45, 79.87, 73.09, 55.34, 50.59, 48.78, 42.76, 33.29, 28.51. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H25NO4Na, 330.1681; found, 330.1698. [α]20D 27.7 (c 1.0, CH3OH).
tert-Butyl (3R,4S)-3-(4-fluorophenyl)-4-hydroxypiperidine-1-carboxylate ((3R,4S)-2d) (126 mg, white solid, 29% yield). 1H NMR (400 MHz, CDCl3) δ 7.26 (s, 1H), 7.22 (t, J = 6.8 Hz, 2H), 7.04 (t, J = 8.3 Hz, 2H), 4.08 (s, 1H), 4.00 (d, J = 12.9 Hz, 1H), 3.93 (d, J = 13.5 Hz, 1H), 3.44 (t, J = 12.3 Hz, 1H), 3.21 (t, J = 12.4 Hz, 1H), 2.87 (d, J = 11.8 Hz, 1H), 1.83 (d, J = 10.3 Hz, 2H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 154.94, 136.21, 129.45, 129.38, 115.66, 115.45, 79.71, 68.41, 45.79, 42.26, 38.23, 32.27, 28.50. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H22FNO3Na, 318.1481; found, 318.1492. [α]20D 91.8 (c 1.0, CH3OH).
tert-Butyl (3S,4R)-3-(4-fluorophenyl)-4-hydroxypiperidine-1-carboxylate ((3S,4R)-2d) (106 mg, white solid, 24% yield). 1H NMR (400 MHz, CDCl3) δ 7.26 (d, J = 1.9 Hz, 1H), 7.25–7.18 (m, 2H), 7.05 (d, J = 8.5 Hz, 2H), 4.08 (d, J = 3.3 Hz, 1H), 4.04–3.97 (m, 1H), 3.93 (d, J = 13.5 Hz, 1H), 3.44 (t, J = 12.4 Hz, 1H), 3.21 (tt, J = 13.0, 10.6, 4.6 Hz, 1H), 2.87 (d, J = 11.9 Hz, 1H), 1.84 (ddd, J = 8.9, 5.3, 2.6 Hz, 2H), 1.47 (d, J = 2.1 Hz, 9H) 13C NMR (100 MHz, CDCl3) δ 154.94, 136.19, 129.45, 129.38, 115.65, 115.44, 79.71, 68.40, 45.79, 42.27, 38.22, 32.28, 28.50. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H22FNO3Na, 318.1481; found, 318.1494. [α]20D 88.2 (c 1.0, CH3OH).
tert-Butyl (3S,4S)-3-(4-fluorophenyl)-4-hydroxypiperidine-1-carboxylate ((3S,4S)-2d) (156 mg, white solid, 35% yield). 1H NMR (400 MHz, CDCl3) δ 7.29–7.19 (m, 2H), 7.06 (d, J = 8.6 Hz, 2H), 4.35–4.00 (m, 2H), 3.82 (td, J = 10.4, 4.1 Hz, 1H), 2.98–2.67 (m, 2H), 2.57 (td, J = 11.4, 10.9, 3.8 Hz, 1H), 2.20–1.86 (m, 1H), 1.64 (s, 1H), 1.60–1.53 (m, 1H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 154.63, 135.09, 129.61, 129.54, 115.94, 115.73, 79.98, 72.97, 50.60, 48.65, 42.76, 33.42, 28.46. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H22FNO3Na, 318.1481; found, 318.1489. [α]20D – 21.3 (c 1.0, CH3OH).
tert-Butyl (3R,4R)-3-(4-fluorophenyl)-4-hydroxypiperidine-1-carboxylate ((3R,4R)-2d) (106 mg, white solid, 24% yield). 1H NMR (400 MHz, CDCl3) δ 7.28–7.17 (m, 2H), 7.05 (s, 2H), 4.24 (d, J = 13.8 Hz, 1H), 4.14 (d, J = 13.0 Hz, 1H), 3.81 (dt, J = 10.4, 5.3 Hz, 1H), 2.86 (t, J = 13.4 Hz, 1H), 2.76 (t, J = 12.7 Hz, 1H), 2.58 (td, J = 11.0, 3.9 Hz, 1H), 2.10–2.00 (m, 1H), 1.70 (d, J = 16.1 Hz, 1H), 1.64–1.56 (m, 1H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 154.63, 135.08, 129.61, 129.53, 115.94, 115.73, 79.98, 72.97, 50.60, 48.66, 42.75, 33.41, 28.45. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H22FNO3Na, 318.1481; found, 318.1489. [α]20D 19.3 (c 1.0, CH3OH).
tert-Butyl (3R,4S)-3-(4-chlorophenyl)-4-hydroxypiperidine-1-carboxylate ((3R,4S)-2e) (134 mg, white solid, 43% yield). 1H NMR (400 MHz, CDCl3) δ 7.35–7.28 (m, 2H), 7.19 (d, J = 8.1 Hz, 2H), 4.09 (s, 1H), 4.00 (d, J = 12.9 Hz, 1H), 3.93 (d, J = 13.5 Hz, 1H), 3.44 (t, J = 12.4 Hz, 1H), 3.20 (ddd, J = 14.3, 10.4, 4.6 Hz, 1H), 2.86 (d, J = 11.8 Hz, 1H), 1.46 (d, J = 2.9 Hz, 9H). 13C NMR (100 MHz, CDCl3) δ 154.92, 139.02, 132.94, 129.32, 128.82, 79.75, 68.30, 45.96, 42.10, 38.21, 32.33, 28.50. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H22ClNO3Na, 334.1186; found, 334.1197. [α]20D 104.2 (c 1.0, CH3OH).
tert-Butyl (3S,4R) 3-(4-chlorophenyl)-4-hydroxypiperidine-1-carboxylate ((3S,4R)-2e) (137 mg, white solid, 44% yield). 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 8.1 Hz, 2H), 7.19 (d, J = 8.2 Hz, 2H), 4.08 (s, 1H), 4.00 (d, J = 13.2 Hz, 1H), 3.93 (d, J = 13.6 Hz, 1H), 3.44 (t, J = 12.4 Hz, 1H), 3.21 (s, 1H), 2.86 (d, J = 12.0 Hz, 1H), 1.83 (d, J = 9.8 Hz, 2H), 1.46 (d, J = 2.7 Hz, 9H). 13C NMR (100 MHz, CDCl3) δ 154.92, 139.02, 132.94, 129.33, 128.82, 79.76, 68.30, 45.96, 42.11, 38.22, 32.33, 28.50. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H22ClNO3Na, 334.1186; found, 334.1197. [α]20D – 105.5 (c 1.0, CH3OH).
tert-Butyl (3S,4S)-3-(4-chlorophenyl)-4-hydroxypiperidine-1-carboxylate ((3S,4S)-2e) (100 mg, white solid, 32% yield). 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.2 Hz, 2H), 4.24 (d, J = 13.2 Hz, 1H), 4.20–4.10 (m, 1H), 3.88–3.69 (m, 2H), 2.81 (dt, J = 37.2, 13.0 Hz, 2H), 2.57 (ddd, J = 13.6, 10.6, 3.7 Hz, 1H), 2.09–2.00 (m, 1H), 1.67 (s, 1H), 1.57 (qd, J = 12.8, 12.2, 7.4 Hz, 1H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 154.68, 138.05, 133.30, 129.52, 129.16, 80.10, 72.89, 50.81, 48.54, 42.82, 33.53, 28.52. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H22ClNO3Na, 334.1186; found, 334.1198. [α]20D – 35.7 (c 1.0, CH3OH).
tert-Butyl (3R,4R)-3-(4-chlorophenyl)-4-hydroxypiperidine-1-carboxylate ((3R,4R)-2e) (140 mg, white solid, 45% yield). 1H NMR (400 MHz, CDCl3) δ 7.36–7.30 (m, 2H), 7.23–7.17 (m, 2H), 4.28–4.19 (m, 1H), 4.14 (d, J = 11.8 Hz, 1H), 3.88–3.72 (m, 1H), 2.81 (dt, J = 37.1, 13.0 Hz, 2H), 2.57 (ddd, J = 13.9, 10.5, 3.7 Hz, 1H), 2.09–1.99 (m, 1H), 1.69 (s, 1H), 1.60 (s, 1H), 1.46 (d, J = 2.3 Hz, 9H). 13C NMR (100 MHz, CDCl3) δ 138.04, 129.54, 129.19, 80.12, 77.49, 72.94, 50.84, 48.55, 42.83, 33.54, 28.54. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H22ClNO3Na, 334.1186; found, 334.1194. [α]20D – 35.9 (c 1.0, CH3OH).
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for (3R, 4S)-2a has been deposited at the CCDC under deposition number 2423453; (3R, 4R)-2a has been deposited at the CCDC under deposition number 2214401; (3R,4S)-2d has been deposited at the CCDC under deposition number 2433005; (3S,4S)-2d has been deposited at the CCDC under deposition number 2433010; (3R,4R)-2d has been deposited at the CCDC under deposition number 2433256.Author contributions
J. C, X. C., Q. W. and D. Z. conceived and designed the project, J. C. performed the main content of the experiments, J. S. , H. Z. provided guidance on biological experimental methods, and X. C. provided guidance on chemical synthesis, J. C. and X. C. wrote the manuscript, D. Z. and H. Z. obtained the financial support for the project leading to this publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R & D Program of China (No. 2021YFC2102000), National Natural Science Foundation of China (No. 22207120), and Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (No. TSBICIP-KJGG-031 and TSBICIP-CXRC-078). The authors thank Jie Zhang (Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences) for technical help with X-ray singlecrystal diffraction analysis.Notes and references
- P. R. Neithnadka, M. Kaspady Abdulla, C. Panja, A. Arumugam, M. Kannan, S. Krishnamoorthy, C. M. Gopala, P. Gunaga, J. M. Richter, M. Vetrichelvan, A. Mathur and A. Gupta, Org. Process Res. Dev., 2025, 29, 389–400 CrossRef CAS.
- A. Lobkovich, P. Kale-Pradhan and M. Lipari, Ann. Pharmacother., 2023, 58, 398–406 CrossRef PubMed.
- I. Yoshinori, H. Tadatoshi, T. Naoki, S. Junya and Y. Masayuki, EP pat. 03733151A, 2005 Search PubMed.
- F. M. Carmen, G.-G. M. Rosario, L. Bin, P. L. Allen, WO pat. 2013112323A1, 2013 Search PubMed.
- S. Sun, Q. Jia, A. Y. Zenova, S. Lin, A. Hussainkhel, J. Mezeyova, E. Chang, S. J. Goodchild, Z. Xie, A. Lindgren, G. de Boer, R. Kwan, K. Khakh, L. Sojo, P. Bichler, J. P. Johnson, J. R. Empfield, C. J. Cohen, C. M. Dehnhardt and R. Dean, Bioorg. Med. Chem. Lett., 2021, 45, 128133 CrossRef CAS PubMed.
- J. M. Richter, P. Gunaga, N. Yadav, R. O. Bora, R. Bhide, N. Rajugowda, K. Govindrajulu, S. Godesi, N. Akuthota, P. Rao, A. Sivaraman, M. Panda, M. Kaspady, A. Gupta, A. Mathur, P. C. Levesque, J. Gulia, M. Dokania, M. Ramarao, P. Kole, S. Chacko, K. A. Lentz, S. Sivaprasad Lvj, R. P. Thatipamula, S. Sridhar, S. Kamble, A. Govindrajan, S. I. Soleman, D. A. Gordon, R. R. Wexler and E. S. Priestley, J. Med. Chem., 2024, 67, 9731–9744 Search PubMed.
- Y. H. T. Ikeura, H. Nishida, J. Shirai, N. Sakauchi, Piperidine derivative, process for producing the same, and use, EP pat. 1553084A1, 2005 Search PubMed.
- S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Chem. Rev., 2022, 122, 3637–3710 CrossRef CAS PubMed.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 Search PubMed.
- F. Hollmann, D. J. Opperman and C. E. Paul, Angew. Chem., Int. Ed., 2021, 60, 5644–5665 CrossRef CAS PubMed.
- R. N. Patel, Biomolecules, 2013, 3, 741–777 CrossRef PubMed.
- J. J. Sangster, J. R. Marshall, N. J. Turner and J. Mangas-Sanchez, ChemBioChem, 2022, 23, e202100464 CrossRef CAS PubMed.
- U. Hanefeld, F. Hollmann and C. E. Paul, Chem. Soc. Rev., 2022, 51, 594–627 Search PubMed.
- B.-S. Chen and F. Z. Ribeiro de Souza, RSC Adv., 2019, 9, 2102–2115 RSC.
- G. Ni, J. Tang, J. Zou, S. Chen, D. Ju and F. Zhang, Chin. J. Org. Chem., 2019, 39, 339–349 CrossRef CAS.
- H. Zhang, L. Zhu, J. Feng, X. Liu, X. Chen, Q. Wu and D. Zhu, Catal. Sci. Technol., 2022, 12, 5841–5849 RSC.
- X. Chen, H. Zhang, M. A. Maria-Solano, W. Liu, J. Li, J. Feng, X. Liu, S. Osuna, R.-T. Guo, Q. Wu, D. Zhu and Y. Ma, Nat. Catal., 2019, 2, 931–941 CrossRef CAS.
- L. Zhu, X. Chen, J. Feng, Y. Cui, Q. Wu and D. Zhu, J. Org. Chem., 2023, 88, 11905–11912 Search PubMed.
- T. Nobutada, N. Takamasa, T. N. Kazuo and H. Akira, SDR Structure, Mechanism of Action, and Substrate Recognition, Curr. Org. Chem., 2001, 5, 89–111 CrossRef.
- B. Zhao and K. Burgess, ACS Med. Chem. Lett., 2018, 9, 155–158 CrossRef CAS PubMed.
- K. Prabhat, M. Anookh, A. Navnath, H. Chakradhar, M. Appaji, G. Ramesh, D. Shailesh, J. Prashant and J. Poonam, WO pat. 2009004650A1, 2009 Search PubMed.
- J. T. Gupton, E. Crawford, M. Mahoney, E. Clark, W. Curry, A. Lane, A. Shimozono, V. Moore-Stoll, K. Elofson, W. Juekun, M. Newton, S. Yeudall, E. Jaekle, R. Kanters and J. A. Sikorski, Tetrahedron, 2018, 74, 7408–7420 Search PubMed.
- I. Yoshinori, H. Tadatoshi, S. Junya, T. Yoshikawa, N. Hiroshi, Y. Mitsuhisa, M. Masahiro and I. Hiroyuki, US pat., US2006142337A1, 2006 Search PubMed.
- G. Kim, S. Lee, E. Levy Karin, H. Kim, Y. Moriwaki, S. Ovchinnikov, M. Steinegger and M. Mirdita, Nat. Protoc., 2025, 20, 620–642 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2423453, 2214401, 2433005, 2433010 and 2433256. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra02485d |
| This journal is © The Royal Society of Chemistry 2025 |